
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Wittig bioconjugation of maleimide derived, water soluble phosphonium ylides to aldehyde-tagged proteins†
Rafael W.
Hartmann
 ab,
Matthijs
Pijnappel
a,
Johan
Nilvebrant
c,
Hildur Run
Helgudottir
d,
Arni
Asbjarnarson
d,
Gunnhildur Asta
Traustadottir
d,
Thorarinn
Gudjonsson
de,
Per-Åke
Nygren
c,
Fredrik
Lehmann
a and
Luke R.
Odell
*b
ab,
Matthijs
Pijnappel
a,
Johan
Nilvebrant
c,
Hildur Run
Helgudottir
d,
Arni
Asbjarnarson
d,
Gunnhildur Asta
Traustadottir
d,
Thorarinn
Gudjonsson
de,
Per-Åke
Nygren
c,
Fredrik
Lehmann
a and
Luke R.
Odell
*b
aRecipharm OT Chemistry, Virdings allé 16, 75450 Uppsala, Sweden
bDepartment of Medicinal Chemistry, Uppsala University, Uppsala Biomediciniska Centrum, Husargatan 3, 75123 Uppsala, Sweden. E-mail: luke.odell@ilk.uu.se
cDepartment of Protein Science, Division of Protein Engineering, KTH School of Engineering Sciences in Chemistry, Biology and Health, AlbaNova Universitetscentrum, Roslagsvägen 30B, 10961 Stockholm, Sweden
dStem Cell Research Unit, Biomedical Center, University of Iceland, 101 Reykjavik, Iceland
eDepartment of Laboratory Hematology, Landspítali-University Hospital, Reykjavik, Iceland
First published on 17th November 2021
Abstract
Herein we disclose the transformation of maleimides into water-soluble tris(2-carboxyethyl)phosphonium ylides and their subsequent application in the bioconjugation of protein- and peptide-linked aldehydes. The new entry into Wittig bioconjugate chemistry proceeds under mild conditions and relies on highly water soluble reagents, which are likely already part of most biochemists’ inventory.
Introduction
Bioconjugation has been a central topic at the interface between chemistry and biology ever since the emergence of commercial biotechnology and widespread availability of proteins and peptides. Antibody–drug conjugates (ADCs), in which a small molecule payload is covalently linked to a monoclonal antibody, serve to showcase the therapeutic utility of bioconjugation. To date, nine ADCs have received FDA-approval for cancer treatment and dozens more are undergoing clinical development.1 From a synthetic chemist's point-of-view, bioconjugation is challenging due to the high density of various functional groups on the solvent-accessible surface of proteins, which brings about regio- and chemoselectivity issues. The relatively low natural abundance of cysteine and the exceptional nucleophilicity of its side chain thiol residue have consequently made it a widely exploited amino acid.2 Conversely, maleimides are the most ubiquitous electrophile in bioconjugate chemistry and react with cysteine to form thioethers by means of a Michael addition.3 Aside from their omnipresence as building blocks for the synthesis of ADCs,1N-derivatized maleimides have found utility in all areas of protein chemistry, including their fluorescent labelling,4,5 immobilization,6 and crosslinking.7 However, cysteine-based bioconjugation is associated with a number of drawbacks: firstly, access to the nucleophilic thiol moiety typically necessitates the reductive cleavage of disulfide bonds, which often make up the only covalent bond between protein subunits and prevent their dissociation.8 Albeit far better than in analogous lysine-based methods, the regioselectivity of the conjugation to antibody-bound cysteines is confounded by the presence of several interchain disulfides.9 Moreover, the exceptional electrophilicity of maleimides necessitates their incorporation toward the end of the linker-payload synthesis.10 Lastly, although long considered stable, succinimido thioethers have since been shown to decompose in and ex vivo via hydrolysis, thiol exchange and retro-Michael processes.11,12 Several synthetic strategies have been developed to address thiosuccinimide instability, but none are generally applicable.3Due to their electrophilicity, absence in the natural proteome and straightforward synthetic accessibility, numerous methods for the conjugation of small molecules to protein-bound aldehydes have been developed (Scheme 1A). They rely on a variety of chemistries including aldol,13,14 Wittig,15 Pictet–Spengler,16 and Henry reactions,17 among others. An ADC based on a regioselectivity aldehyde-labelled antibody (TRPH-222) has recently entered clinical development, validating the general approach. The 2-(hydrazinomethyl)indole head group required for payload ligation however necessitates a complex multistep synthesis.18 The recent report of a maleimide based Wittig reaction applicable to the bioconjugation setting (Scheme 1B) stands out as an exception in this regard and is ideally suited for in vitro applications.19 Even more recently, the utility of such Wittig ylides with electrophiles other than aldehydes was demonstrated in vitro.20 However, because the synthesis of bioconjugates generally proceeds at higher concentrations and thus requires greater aqueous solubility of the components, the formation of highly lipophilic triphenylphosphorane intermediates limits the utility of the approach in this context as many clinically relevant payloads are hydrophobic small molecules.21
 | ||
| Scheme 1 Methods for the derivatization of aldehyde-labelled proteins. | ||
Herein we examine the synthetic utility of phosphonium ylides derived from commercially available tris(2-carboxyethyl)-phosphine (1, Scheme 2) and N-functionalized maleimides. Like conventional triphenylphosphonium ylides,22 they were postulated to react with peptide- and protein-derived aldehydes to form alkenes in high yields. Importantly, as demonstrated in another publication,19 this alkene linkage does not suffer from the traditional stability problems associated with thioethers, providing a superior bioconjugation method. Building on the original work by Kalia et al., the method outlined herein proceeds via a hydrophilic intermediate and relies on reactants that are already commonplace in the biochemist's toolbox, making the reaction highly practical for use in the context of preparative bioconjugate chemistry.
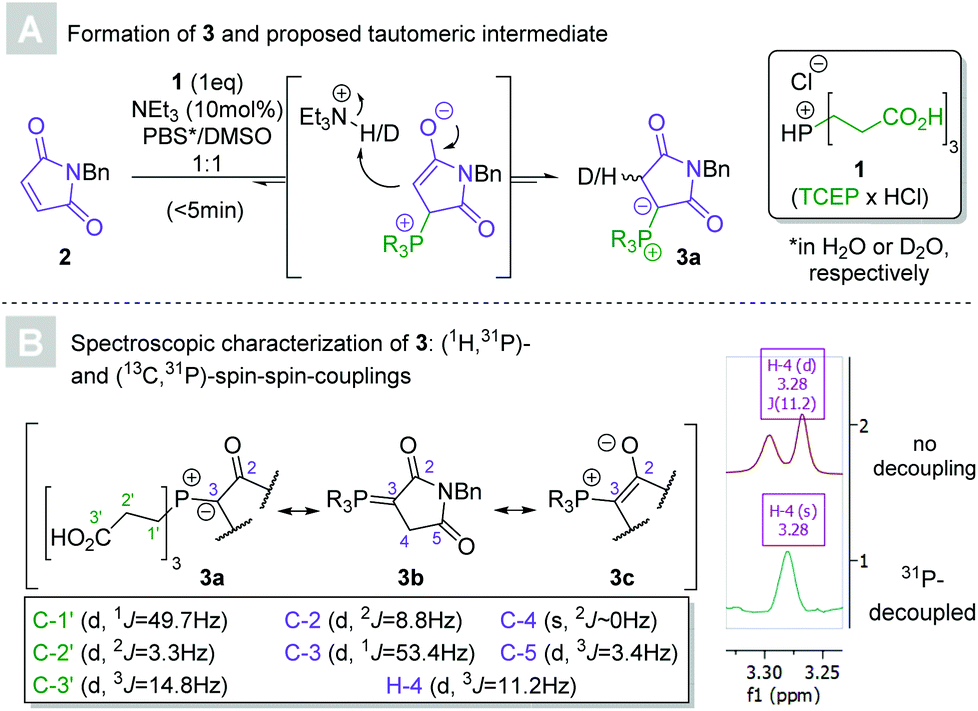 | ||
| Scheme 2 (A) Proposed mechanisms for the equilibration between maleimide 2 and TCEP (1) to tris(2-carboxyethyl)phosphonium ylide 3. (B) NMR spectroscopic evidence in support of the tautomeric identity of 3 and most accurate resonance structure 3c. | ||
Results and discussion
In 2016, Kalia and co-workers demonstrated that the reaction between maleimides and triphenylphosphine in the presence of catalytic para-nitrophenol yielded ylides, which subsequently underwent Wittig reactions with aldehydes. The so-furnished exocyclic Michael acceptors have potential as handles for bioconjugation in their own right.23 We explored the applicability of the Wittig reaction for the conjugation step itself, a feat also since accomplished by the authors of the original paper.19 An attractive alternative to the hydrophobic triphenylphosphine presented itself in tris(2-carboxyethyl)phosphine hydrochloride (TCEP × HCl, 1, see Scheme 2). The biocompatible, commercially available, crystalline solid is a common reducing agent in aqueous media. Thorough research of the literature revealed that the reaction of TCEP with maleimides had indeed been previously reported, but only identified as a potential confounder in bioconjugate chemistry.24,25 Importantly, the synthetic utility of TCEP-derived phosphonium ylides remains unexplored.Preliminary experiments revealed that an equimolar mixture of N-benzylmaleimide 2 and 1 in aqueous DMSO afforded the putative 1,2-ylide 3a (according to HPLC-MS analysis) after several hours. Comparative experiments confirmed the anticipated superior aqueous solubility of the reactants when TCEP was used instead of conventional triphenylphosphine (see ESI†), indicating the method's potentially greater suitability for synthetic applications. The addition of a catalytic amount (10 mol%) of triethylamine sped up the reaction and yielded full conversion within minutes, eliminating the necessity for another proto-base such as para-nitrophenol. Kinetic nuclear magnetic resonance experiments (using maleimide 7, see Scheme 3, as the starting material for solubility reasons) confirmed the triethylamine's acceleratory effect on the formation of the phosphonium ylide (see ESI†).
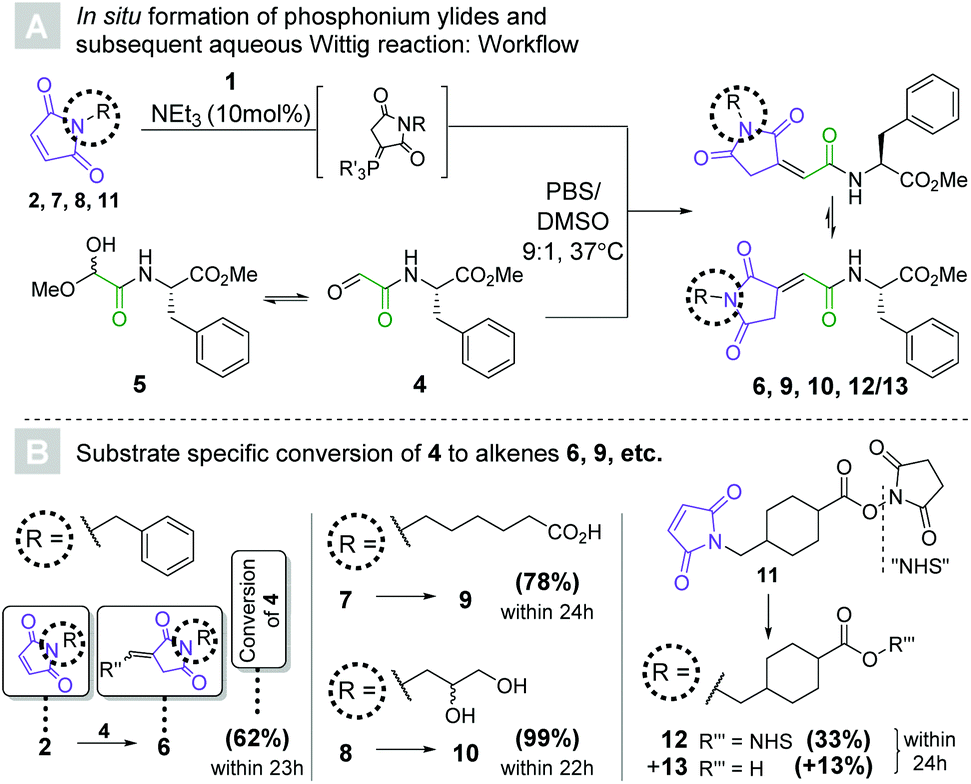 | ||
| Scheme 3 (A) workflow for the in situ activation of maleimides (2, 7, 8, 11) to tris(2-carboxyethyl)phosphonium ylides, followed by their reaction with peptide-derived glyoxylate 4 to yield alkenes. c(peptide) = 2 mM, c(ylide) = 6 mM (3 mol. eq.). (B) Conversions towards alkenes (6, 9, 10, 12/13) as determined by HPLC-UV (λ = 254 nm) after the specified reaction times (see ESI†). | ||
Several attempts at the isolation of 3 failed, presumably due to the reversibility of the reaction. The characterization of the ylide was nevertheless accomplished without the need for purification and confirmed its anticipated structure. Immediate incorporation of a single deuterium atom was observed by HRMS when the ylide was generated in heavy water, supporting the involvement of a 1,3-betaine intermediate. The methylene group bound to the ylide carbon appeared as a doublet (3J = 11.2 Hz) in 1H NMR as a result of (1H,31P)-spin–spin coupling. C-3 coupled to the phosphorous atom with an unusually low coupling constant (1J = 53.4 Hz),26 indicating that phosphonium enolate 3c may mirror the ylide's electronic structure more closely than those of the (1,2)-betaine (3a) or phosphorane (3b).
Having established a convenient route towards maleimide derived tris(2-carboxyethyl)phosphonium ylides, we sought to test their Wittig reactivity (see Scheme 3). Aldehyde 4 was synthesized from Ser-Phe methyl ester by means of Malaprade oxidation (see ESI†),27 and isolated in the form of masked aldehyde 5. The latter was reacted with in situ generated phosphonium ylides for 22 to 24 hours. A 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of aqueous phosphate buffered saline (PBS, pH = 7.4) and DMSO was chosen as the reaction medium to mimic bioconjugation conditions.
:1 mixture of aqueous phosphate buffered saline (PBS, pH = 7.4) and DMSO was chosen as the reaction medium to mimic bioconjugation conditions.
The first of these experiments once again involved maleimide 2, whose relative lipophilicity was meant to mimic that of highly hydrophobic payloads often used in antibody–drug conjugate generation.21 To our delight, 62% conversion was achieved within 23 hours as three molar equivalents of the corresponding ylide were reacted with aldehyde 4 to yield alkene 6. HPLC-MS revealed that two isomeric products had formed, which were presumed to be the two double bond isomers. To our surprise, only one isomer remained after extraction of the product into chloroform, indicating an exceptionally low energetic barrier for isomerization and a solvent effect on the equilibrium. The same observation was made for all other alkenes synthesized in this fashion, although the ratio of isomers in the aqueous medium differed. Seeing as the double bond geometry was solvent-dependent and had no bearing on the utility of the method, no further investigation was undertaken.
The more polar substrates ω-(N-maleimido)caproic acid (7) and 3-(N-maleimido)-1,2-propylene glycol (8) gave 78% and 99% of the respective alkenes (9 and 10) in 24 hours and 22 hours, respectively. Maleimide 7 represents the protein-facing handle in several marketed antibody–drug conjugates,28 indicating that the methodology may be applicable to the generation of structural analogues of these therapeutic modalities. The choice of glycol 8 was motivated by its hydrophilicity and the host of options it presents for bioorthogonal derivatization.29
Despite the presence of the activated ester, commercially available succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC, 11) reacted with TCEP selectively to form the desired phosphonium ylide. The Wittig reaction with 4 afforded 33% of 12 along with its hydrolyzed congener 13. This particularly noteworthy finding suggests that our Wittig bioconjugation method may be applicable to the challenging heterodimerization of peptides. Finally, to interrogate potential disulfide cleavage and thiol alkylation, a control experiment was performed in which three molar equivalents of the ylide derived from 7 were incubated with (Cbz-Cys-OH)2 for 24 h. Pleasingly, only traces (<5%) of S–S-cleavage were observed and importantly, no subsequent thiol alkylation was detected by LCMS analysis (see ESI†).
Encouraged by the moderate to excellent conversions of 4 to the desired peptide-derived alkenes, we next endeavored to test the applicability of the novel Wittig methodology to the modification of proteins. To this end, we directed our attention to albumin binding domain-derived affinity protein 6 (ADAPT6), a probe with nanomolar affinity for human epidermal growth factor receptor 2 (HER2) and a molecular weight of 7 kDa.30 We expressed a variant of the protein containing a C-terminal His6-tag and an N-terminal Ser-residue. Oxidation of the latter's α-hydroxylamino motif by sodium periodate yielded the corresponding glyoxylate (ADAPT-CHO, Scheme 4A, top).27 Three of the initial maleimide substrates (N-benzyl 2, N-(ω-caproyl) 7, and N-(2,3-dihydroxy-1-propyl) 8) as well as one (14) obtained by derivatization of the chemotherapeutic doxorubicin with SMCC were treated with TCEP, yielding the corresponding phosphonium ylides in situ. 100 Molar equivalents were added to ADAPT-CHO at 37 °C and the reactions were allowed to progress for a total of 24 hours. With no intermittent purification, the reaction mixtures were analyzed by HPLC-MS and conversions were calculated based on the AUCs of the deconvoluted mass spectra.31 All reactions yielded a single conjugate, whose deconvoluted masses were in agreement with the respective desired alkenes (ADAPT = 2, = 7, = 8, = 14, respectively, see ESI†). Moreover, only in two cases (ADAPT = 2 and = 7, respectively) were trace amounts of ADAPT-CHO detectable, indicating excellent conversions at or above 97% in all cases.
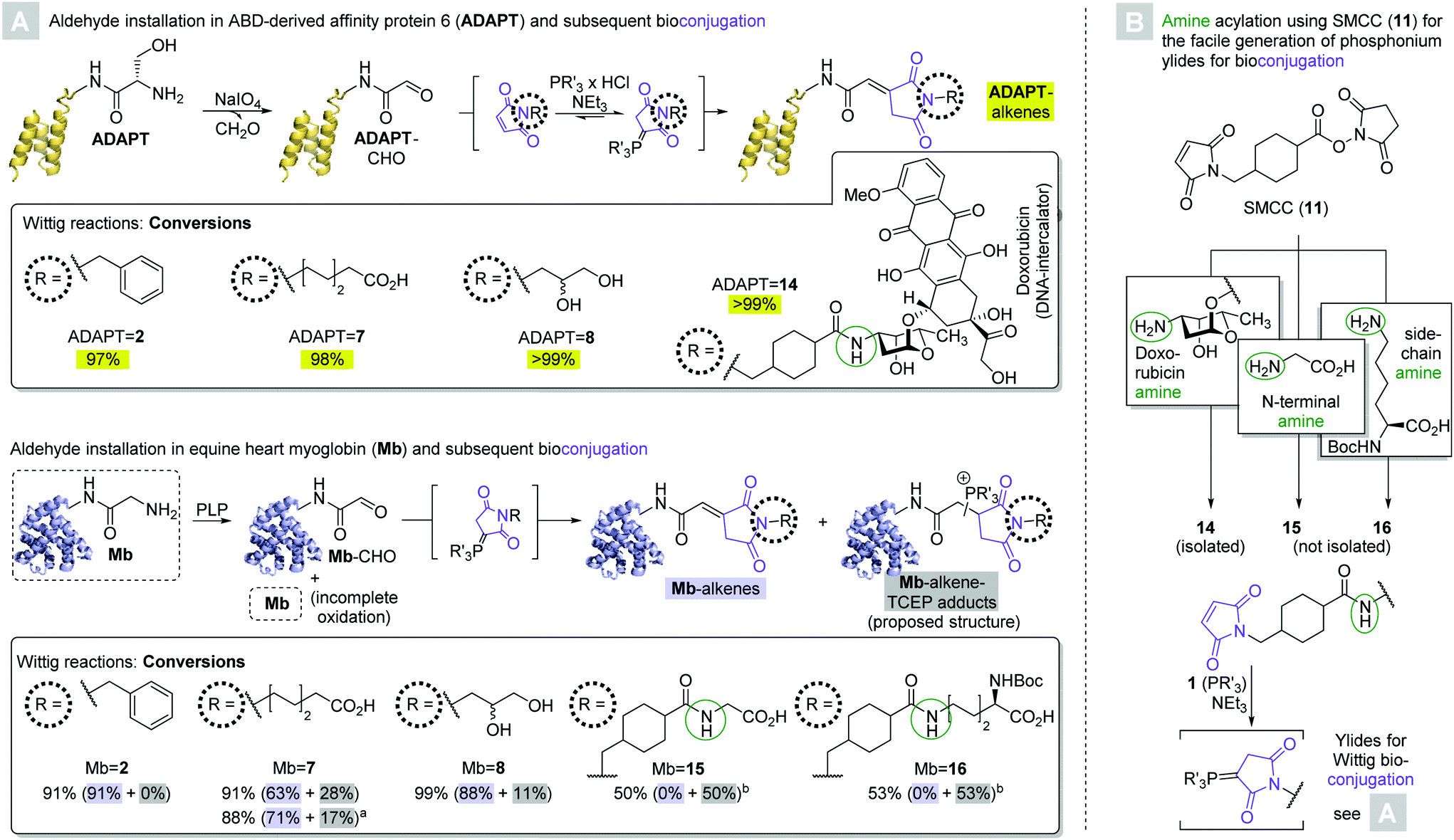 | ||
| Scheme 4 (A) Generation of and Wittig reactions between ADAPT6 aldehyde (ADAPT-CHO, top), myoglobin aldehyde (Mb-CHO, bottom) and maleimide derived phosphonium ylides in PBS/DMSO 9:1 (cProtein = 30 μM). Percentages refer to conversions of protein, which were calculated based on AUCs in the deconvoluted mass spectra after 24 hours of reaction time at 37 °C. Unless otherwise noted, 100 mol. eq. of phosphonium ylide were used. Where applicable, percentages in parentheses refer to the quantities of Mb-alkene (shaded blue) and the respective TCEP addition product (shaded gray). a20 mol. eq. of phosphonium ylide were added to the protein aldehyde at the outset of the reaction, and another 20 mol. eq. were added after 6 hours. The total reaction time remained 24 hours. bA telescoped approach was used for the synthesis, ylide formation and bioconjugation of the maleimides (see B). (B) Workflow for the maleimide derivatization of amines with commercially obtained SMCC (11). SMCC-derivatized doxorubicin (14) was purified prior to ylide formation and Wittig bioconjugation, whereas glycine and Boc-lysine derived maleimides (15 and 16, respectively) were used in crude form. | ||
Next, we chose a larger substrate in equine heart myoglobin (Mb, Mw = 17 kDa) to verify the method's applicability. The protein's N-terminal Gly-residue was oxidized with pyridoxal-6-phosphate (PLP, Scheme 4A, bottom) to yield the corresponding aldehyde (Mb-CHO).13 A recently published protocol for the derivatization of aldehyde-labelled peptides by means of a Henry reaction with nitromethane was employed to assess the outcome of the reaction (see ESI†),17 and revealed that the protein contained substantial amounts of unoxidized starting material in accordance with previous studies. The reaction of Mb-CHO with 100 molar equivalents of in situ generated N-benzyl-maleimide (2) derived ylide yielded 91% conversion to olefin Mb = 2 within 24 hours.
The analogous reaction with N-(ω-caproyl)maleimide (7) derived phosphonium ylide gave 63% conversion to the desired alkene Mb = 7. MS also revealed the presence of 28% of a side product, whose mass was in agreement with that of a TCEP-adduct. The presence of unbound phosphine, on one hand, is likely a consequence of the reversibility of ylide formation. Alkylidene succinimides, on the other hand, readily undergo Michael additions with thiols even in the absence of another strongly electron withdrawing carbonyl group on the other end of the C–C double bond.23 It therefore appears likely that the site of phosphine addition is the newly formed α,β-unsaturated δ-dicarbonyl, albeit the regiochemistry beyond that remains elusive. Although unintended, the formation of the phosphine addition product may be preferrable to that of the alkene in some cases, owing to its presumably enhanced aqueous solubility over the alkene. The degree of TCEP-addition could partly be controlled via changes in the stoichiometry: the addition of only 40 molar equivalents of ylide (20 mol. eq. added at outset, another 20 mol. eq. after six hours) yielded 71% conversion to Mb = 7 and only 17% of the side product within a total of 24 hours.
The reaction between Mb-CHO and 100 equivalents of N-(2,3-dihydroxy-1-propyl) maleimide (8) derived ylide gave conversions of 88% to alkene Mb = 8 and 11% to the respective TCEP addition product within 24 hours, respectively.
Maleimides 15 and 16 were obtained by SMCC derivatization of glycine and Nα-Boc-L-lysine, which were treated with one molar equivalent of TCEP and 100 molar equivalents of the resultant ylides were reacted with Mb-CHO in a telescoped fashion. Moderate conversions of 50% and 53% toward the TCEP addition products of bioconjugates Mb = 15 and Mb = 16, respectively, were observed within 24 hours, and no trace of the respective alkenes were detectable. These observations appear to be the result of the telescoped generation and bioconjugation of 15 and 16: since neither reaction with SMCC yielded quantitative conversion, the addition of one molar equivalent of TCEP relative to the amino acid starting material amounted to an excess of phosphine. It may therefore be inferred that if the formation of the TCEP addition product is to be avoided, it is crucial to isolate the maleimide prior to ylide formation. If, on the other hand, the formation of a phosphine addition product is desirable, intermittent purification can be omitted. Moreover, the formation of Mb = 15 and Mb = 16 indicates the applicability of the method at hand to achieve the heterodimerization of proteins, exploiting amine residues on either the N-terminus or a lysine side chain on one of the coupling partners.
All nine bioconjugates were subsequently subjected to SDS-PAGE analysis to confirm their integrity. The conjugates’ molecular weight was not significantly different than those of the protein aldehydes from which they originate (ADAPT-CHO and Mb-CHO, respectively, see ESI†), indicating that no meaningful degree of aggregation or degradation occurred.
Lastly, the cytotoxicity of ADAPT = 14 and its two components was assessed (see ESI†). The relative viability of D492 breast epithelial progenitor cells32,33 was not significantly different 24 hours after treatment with 12 μM solutions of either ADAPT (57 ± 16%) or the conjugate (54 ± 3.5%), respectively, while only 26 ± 7.4% of cells survived the application of doxorubicin at the same concentration. At a concentration of 1.5 μM, most cells remained viable despite treatment with ADAPT (88 ± 27%) or conjugate (65 ± 13%), while 51 ± 9% of cells succumbed to doxorubicin treatment. Similar trends were observed for an engineered D492 cell line characterized by increased HER2 expression,34 which appeared less sensitive to treatment with either of the three analytes under analogous experimental parameters.
The weaker antineoplastic effect of the conjugate compared to the unconjugated cytotoxin may indicate suboptimal cellular uptake of the former, that amine acylation decreases the latter's ability to engage its intracellular target (DNA),35,36 or both. Using the experimental parameters at hand, doxorubicin's IC50-value was approximately 1.5 μM, which is too high to qualify it as an effective payload in targeted chemotherapy against the cell lines at hand37 even in the absence of these apparent shortfalls.38,39 Another drawback of the anthracycline is its pronounced hydrophobicity (especially after amine acylation), which limited the bioconjugate's solubility and prohibited us from performing cytotoxicity experiments at concentrations high enough to obtain a complete dose–response curve.
The development of therapeutically useful bioconjugates requires the iterative refinement of all of its components (the protein carrier, linker and payload),40–42 and it is not surprising that ADAPT = 14 does not qualify as such. Future endeavors aimed at its optimization should involve a payload several orders of magnitude more potent in the cell line to be targeted, and the choice of a cleavable linker (as opposed to SMCC)43 may increase the chance of success. Nevertheless, ADAPT = 14's successful synthesis using the methodology outlined herein showcases the applicability of Wittig chemistry to the generation of potentially cytotoxic protein conjugates and hence, the novel reaction represents a useful addition to bioconjugate chemistry's toolkit.
Conclusions
Maleimides have been found to spontaneously react with tris(2-carboxyethyl)phosphine (TCEP, 1) in the presence of a catalytic amount of triethylamine under aqueous conditions to form the corresponding water soluble phosphonium ylides. These intermediates can be exploited in the bioconjugation of peptide and protein derived aldehydes. The commercially available heterobifunctional linker SMCC (11) allowed for the facile introduction of maleimide handles onto amines under aqueous conditions, extending the method's substrate scope substantially. The reaction between TCEP-ylides derived from purified maleimides and protein aldehydes gave excellent (88% to >99%) conversions within 24 hours in all cases. Moreover, we report the first example of the use of Wittig chemistry for the conjugation of a cytotoxic payload to a protein, showcasing the clinical potential of the methodology.Despite the emergence of superior methods in recent years, clinically applied bioconjugation still frequently relies on conventional maleimide chemistry. Due to their excellent aqueous solubility and accessibility from a ubiquitous handle for bioconjugation, tris(2-carboxyethyl)phosphonium ylides provide a superior alternative for the derivatization of protein-bound aldehydes. We envisage their widespread application in bioconjugate chemistry.
Experimental
General information
Reactions containing only small molecule components were monitored using an Agilent (US) 1100 series LC/MS (single quadrupole) system equipped with an electrospray interface, a UV diode array detector and an ACE3 C8 (3.0 × 50 mm) column (ACE, UK) with a gradient of acetonitrile in 0.1% aqueous trifluoroacetic acid over 3 min and a flow of 1 mL min−1.Upon derivatization, proteins and conjugates were analyzed using an Agilent (US) 1290 Infinity II series LC system equipped with a UV diode array detector and an Agilent (US) 6550 iFunnel q-ToF mass spectrometer. An Acquity UPLC Protein BEH C4 (2.1 × 50 mm) column (Waters, US) and unless otherwise noted, a gradient of 0.1% formic acid in acetonitrile in 0.1% aqueous formic acid over 10 min and a flow of 1 mL min−1 were used.
Preparative HPLC was performed using a Gilson HPLC System (US) equipped with a UV diode array detector and an ACE3 C18-HL (250 × 21.2 mm) column (ACE, UK) with a gradient of acetonitrile in 0.1% aqueous trifluoroacetic acid over 10 min and a flow of 25 mL min−1.
Please refer to the ESI† for more in-depth general information as well as additional experimental procedures, analytical and biological data.
Procedure for the preparation of TCE-phosphonium ylides in situ
The formation of N-benzyl maleimide (2) derived ylide serves as a representative example:A stock solution of maleimide (2, 3.7 mg, 20 μmol, 1.0 eq.) in dimethylsulfoxide (268 μmol, c = 73.8 mM) was diluted with 2.41 mL of a solution of TCEP hydrochloride (1, 23.4 mg, 81.6 μmol) and triethylamine (1.2 μL, 8.2 μmol) in aqueous phosphate buffered saline (10 mL, 10 mM, pH = 7.4), thus producing a solvent ratio of 9:1 (PBS/DMSO), equimolar ratio between maleimide and TCEP hydrochloride and 10 mol% triethylamine. The mixture was subsequently shaken for one hour, whereupon it was frozen at −20 °C until immediately prior to its use in the subsequent Wittig reaction.
Procedure for the Wittig bioconjugation of TCE-phosphonium ylides
The bioconjugation of N-benzyl maleimide (2) derived ylide to ADAPT-CHO serves as a representative example:An aliquot of ADAPT-CHO (102 μL, c = 0.98 mg mL−1, 13.7 nmol, 1.0 eq.) in aqueous phosphate buffered saline (10 mM, pH = 7.4) was pipetted into a tainted glass vial, diluted with PBS (10 mM, pH = 7.4, 136 μL), DMSO (26 μL) and a solution of N-benzylmaleimide (2) derived ylide in aqueous dimethylsulfoxide (186 μL, 1.37 μmol, 100 eq.) as prepared above was added. The reaction mixture was then gently agitated and incubated at 37 °C without stirring for 24 hours. The reaction mixture was then frozen at −80 °C and only thawed immediately before analysis.
Purification and biological evaluation of ADAPT = 14
A single batch of conjugate was synthesized from 2.05 mg (281 nmol) of ADAPT-CHO in the fashion described above. After 24 hours of reaction time, the homogenous reaction mixture was directly purified by preparative HPLC (25 to 50% gradient of acetonitrile in 0.1% aqueous TFA over 10 minutes). These conditions presumably denatured the protein, but other ADAPT-conjugates have been shown to refold upon chemical or thermal denaturation.30 Product containing fractions were pooled and freeze-dried to yield ADAPT = 14 (1.91 mg, 238 nmol) as a faintly red solid (85% isolated yield) in 90% purity.The latter was taken up in 0.3 M aqueous acetic acid (2 mL) and the mixture was gently homogenized with the help of a micropipette. It was then diluted with aqueous phosphate buffered saline (10 mL) and the pH was adjusted to 7.4 using 4 M sodium hydroxide solution. The solution was then diluted with more phosphate buffered saline to yield a total volume of 20 mL (c = 12 μM).
The conjugate's cytotoxicity in D492 and D492HER2 cells was then determined as follows: the cells were cultured in H14 media supplemented with penicillin (100 U mL−1) and streptomycin (100 μg mL−1) in culture flasks coated with collagen I as described previously.44 Cells were seeded in 96 well plates at a density of 10′000 cells per well and cultivated overnight prior to addition of the analyte. ADAPT = 14, ADAPT or doxorubicin were added at concentrations of 12 μM or 1.5 μM and the treated cells were incubated for 24 hours. PrestoBlue™ viability indicator was added to each well, and after two hours, the absorptivity at λ = 570 nm and 595 nm was determined. Cell viability was then compared to untreated cells. Results are based on four technical replicates. Please refer to the ESI† for more details.
Author contributions
RWH contributed to the conceptualization, investigation and development of the methodology outlined herein, visualized the results, wrote and revised the original manuscript. MP contributed to formal analysis and investigation. JN contributed to conceptualization, investigation and reviewed the manuscript. HRH contributed to formal analysis and investigation and reviewed the manuscript. AA contributed to formal analysis and investigation and reviewed the manuscript. GAT contributed to formal analysis and investigation and reviewed the manuscript. TG contributed to the acquisition of resources, formal analysis and investigation and reviewed the manuscript. PÅN contributed to the conceptualization, acquisition of resources and review of the manuscript. FL contributed to the conceptualization, acquisition of resources and review of the manuscript as well as providing supervision. LRO contributed to the conceptualization and development of the methodology, reviewed the manuscript, and provided supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by Uppsala University and the Swedish Research Council (Vetenskapsrådet 2018-05133).Notes and references
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- A. Miseta and P. Csutora, Mol. Biol. Evol., 2000, 17, 1232–1239 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- Y. Ling-Ling, C. Zhen-Zhen, T. Li-Li, X. Ke-Hua and T. Bo, Chin. J. Anal. Chem., 2009, 37, 1073–1081 Search PubMed.
- K. Renault, J. W. Fredy, P. Y. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS PubMed.
- J. Schartner, J. Güldenhaupt, S. K. Gaßmeyer, K. Rosga, R. Kourist, K. Gerwert and C. Kötting, Analyst, 2018, 143, 2276–2284 RSC.
- A. Sinz, Mass Spectrom. Rev., 2006, 25, 663–682 CrossRef CAS PubMed.
- M. Trivedi, J. Laurence and T. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614–625 CrossRef CAS PubMed.
- M. Källsten, R. Hartmann, K. Artemenko, S. B. Lind, F. Lehmann and J. Bergquist, Analyst, 2018, 143, 5487–5496 RSC.
- D. Mondal, J. Ford and K. G. Pinney, Tetrahedron Lett., 2018, 59, 3589–3642 CrossRef.
- N. Fishkin, E. K. Maloney, R. V. J. Chari and R. Singh, Chem. Commun., 2011, 47, 10752–10754 RSC.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- J. Alam, T. H. Keller and T. P. Loh, J. Am. Chem. Soc., 2010, 132, 9546–9548 CrossRef CAS PubMed.
- P. Wang, S. Zhang, Q. Meng, Y. Liu, L. Shang and Z. Yin, Org. Lett., 2015, 17, 1361–1364 CrossRef CAS PubMed.
- M. J. Han, D. C. Xiong and X. S. Ye, Chem. Commun., 2012, 48, 11079–11081 RSC.
- P. Agarwal, J. Van Der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 Search PubMed.
- S. Mahesh, V. Adebomi, Z. P. Muneeswaran and M. Raj, Angew. Chem., Int. Ed., 2020, 59, 2793–2801 CrossRef CAS PubMed.
- S. I. Lim, Drug Discovery Today, 2020, 25, 168–176 CrossRef CAS PubMed.
- S. Parmar, S. P. Pawar, R. Iyer and D. Kalia, Chem. Commun., 2019, 55, 14926–14929 RSC.
- Y. Shi, L. Fu, J. Yang and K. S. Carroll, Nat. Chem., 2021, 13, 1140–1150 CrossRef PubMed.
- I. Pysz, P. J. M. Jackson, D. J. Barlow, K. M. Rahman and D. E. Thurston, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1146, 122075 CrossRef CAS PubMed.
- G. Wittig and W. Haag, Chem. Ber., 1955, 88, 1654–1666 CrossRef CAS.
- D. Kalia, P. V. Malekar and M. Parthasarathy, Angew. Chem., Int. Ed., 2016, 55, 1432–1435 CrossRef CAS PubMed.
- D. E. Shafer, J. K. Inman and A. Lees, Anal. Biochem., 2000, 282, 161–164 CrossRef CAS PubMed.
- T. Kantner and A. G. Watts, Bioconjugate Chem., 2016, 27, 2400–2406 CrossRef CAS PubMed.
- G. A. Gray, J. Am. Chem. Soc., 1973, 95, 5092–5094 CrossRef CAS.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 Search PubMed.
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS PubMed.
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- J. Garousi, S. Lindbo, J. Nilvebrant, M. Astrand, J. Buijs, M. Sandström, H. Honarvar, A. Orlova, V. Tolmachev and S. Hober, Cancer Res., 2015, 75, 4364–4371 CrossRef CAS PubMed.
- K. S. Palla, L. S. Witus, K. J. Mackenzie, C. Netirojjanakul and M. B. Francis, J. Am. Chem. Soc., 2015, 137, 1123–1129 CrossRef CAS PubMed.
- E. Briem, S. Ingthorsson, G. A. Traustadottir, B. Hilmarsdottir and T. Gudjonsson, J. Mammary Gland Biol. Neoplasia, 2019, 24, 139–147 CrossRef PubMed.
- T. Gudjonsson, R. Villadsen, H. L. Nielsen, L. Rønnov-Jessen, M. J. Bissell and O. W. Petersen, Genes Dev., 2002, 16, 693–706 CrossRef CAS PubMed.
- S. Ingthorsson, K. Andersen, B. Hilmarsdottir, G. M. Maelandsmo, M. K. Magnusson and T. Gudjonsson, Oncogene, 2016, 35, 4244–4255 CrossRef CAS PubMed.
- C. Tan, H. Tasaka, K.-P. Yu, L. Murphy and D. Karnofsky, Cancer, 1967, 20, 333–353 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- K. Uhr, W. J. C. Prager-van der Smissen, A. A. J. Heine, B. Ozturk, M. Smid, H. W. H. Göhlmann, A. Jager, J. A. Foekens and J. W. M. Martens, SpringerPlus, 2015, 4, 611 CrossRef PubMed.
- S. C. Jeffrey, M. T. Nguyen, J. B. Andreyka, D. L. Meyer, S. O. Doronina and P. D. Senter, Bioorg. Med. Chem. Lett., 2006, 16, 358–362 CrossRef CAS PubMed.
- N. Stefan, R. Gébleux, L. Waldmeier, T. Hell, M. Escher, F. I. Wolter, U. Grawunder and R. R. Beerli, Mol. Cancer Ther., 2017, 16, 879–892 CrossRef CAS PubMed.
- P. R. Hamann, L. M. Hinman, I. Hollander, C. F. Beyer, D. Lindh, R. Holcomb, W. Hallett, H. R. Tsou, J. Upeslacis, D. Shochat, A. Mountain, D. A. Flowers and I. Bernstein, Bioconjugate Chem., 2002, 13, 47–58 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. Deblanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer, P. D. Senter, L. Zhang, M. F. Miles and K. D. Aldape, Nat. Biotechnol., 2003, 21, 2003 Search PubMed.
- R. Y. Zhao, S. D. Wilhelm, C. Audette, G. Jones, B. A. Leece, A. C. Lazar, V. S. Goldmacher, R. Singh, Y. Kovtun, W. C. Widdison, J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2011, 54, 3606–3623 CrossRef CAS PubMed.
- R. J. Blaschke, A. R. Howlett, P. Y. Desprez, O. W. Petersen and M. J. Bissell, Methods Enzymol., 1994, 245, 535–556 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01155c |
| This journal is © The Royal Society of Chemistry 2021 |