Design and synthesis of a new orthogonally protected glutamic acid analog and its use in the preparation of high affinity polo-like kinase 1 polo-box domain – binding peptide macrocycles†
Received
9th June 2021
, Accepted 16th July 2021
First published on 2nd August 2021
Abstract
Targeting protein – protein interactions (PPIs) has emerged as an important area of discovery for anticancer therapeutic development. In the case of phospho-dependent PPIs, such as the polo-like kinase 1 (Plk1) polo-box domain (PBD), a phosphorylated protein residue can provide high-affinity recognition and binding to target protein hot spots. Developing antagonists of the Plk1 PBD can be particularly challenging if one relies solely on interactions within and proximal to the phospho-binding pocket. Fortunately, the affinity of phospho-dependent PPI antagonists can be significantly enhanced by taking advantage of interactions in both the phospho-binding site and hidden “cryptic” pockets that may be revealed on ligand binding. In our current paper, we describe the design and synthesis of macrocyclic peptide mimetics directed against the Plk1 PBD, which are characterized by a new glutamic acid analog that simultaneously serves as a ring-closing junction that provides accesses to a cryptic binding pocket, while at the same time achieving proper orientation of a phosphothreonine (pT) residue for optimal interaction in the signature phospho-binding pocket. Macrocycles prepared with this new amino acid analog introduce additional hydrogen-bonding interactions not found in the open-chain linear parent peptide. It is noteworthy that this new glutamic acid-based amino acid analog represents the first example of extremely high affinity ligands where access to the cryptic pocket from the pT-2 position is made possible with a residue that is not based on histidine. The concepts employed in the design and synthesis of these new macrocyclic peptide mimetics should be useful for further studies directed against the Plk1 PBD and potentially for ligands directed against other PPI targets.
Introduction
It has been estimated that there are anywhere from 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 600000 protein–protein interactions (PPIs) in human cells comprising what is referred to as the “interactome”.1–3 Many of these PPIs are the objects of intense interest, due to the central roles they play in regulating cellular function.4 Proteins can associate with each other in highly specific ways that are often transient and related to the spatiotemporal control of cell cycle regulation and cell division.5–7 Accordingly, aberrations in PPI – dependent processes can contribute to a diversity of pathologies related to cancer.8,9 In this light, it is not surprising that targeting PPIs has emerged as an important area of discovery for anticancer therapeutic development.2,10,11
000 to 600000 protein–protein interactions (PPIs) in human cells comprising what is referred to as the “interactome”.1–3 Many of these PPIs are the objects of intense interest, due to the central roles they play in regulating cellular function.4 Proteins can associate with each other in highly specific ways that are often transient and related to the spatiotemporal control of cell cycle regulation and cell division.5–7 Accordingly, aberrations in PPI – dependent processes can contribute to a diversity of pathologies related to cancer.8,9 In this light, it is not surprising that targeting PPIs has emerged as an important area of discovery for anticancer therapeutic development.2,10,11
In designing agents that target PPIs, a key consideration is that they must bind strongly enough to flat protein interfaces to overcome protein–protein interaction energies.12 Although PPIs occur via multiple contacts over extended shallow binding surfaces, smaller binding regions, termed “hot spots” can be taken advantage of, which contribute disproportionately to the overall binding energy of the complex.13–15 In phospho-dependent PPIs, the presence of a phosphorylated protein residue (typically tyrosine, serine or threonine) enables high-affinity recognition and binding to target protein hot spots. This class of PPIs is often mediated by modular protein units that are of fundamental importance in cellular signal transduction.16–20 Developing antagonists of the phospho-dependent class of PPIs can be particularly challenging if one relies solely on interactions within and proximal to the phospho-binding pocket. Yet proteins are dynamic and hidden “cryptic” pockets may be revealed while interacting with binding partners.21,22 Designing phospho-dependent PPI antagonists can be significantly advanced by taking into consideration interactions in both the phospho-binding and proximal cryptic pockets. In our current paper, we describe the design and synthesis of phospho-dependent PPI-directed macrocyclic peptide mimetics. These employ a hereto unreported non-natural amino acid that provides accesses to a cryptic binding pocket, while serving as a ring-closing junction. This residue induces conformational constraint through peptide macrocyclization that provides correct orientation for binding of a key phosphoamino acid residue. The work reported herein provides new ways of accessing the phospho-binding pocket and a proximal cryptic pocket of an important signal transduction module.
Results and discussion
Design of a new genre of PBD – binding peptide macrocycles
Starting point for developing PBD – binding peptides.
The serine/threonine specific cell cycle regulator polo-like kinase 1 (Plk1) is recognized as bonafide molecular target for anti-cancer therapy development.23–25 Plk1 requires the coordinated actions of both an N-terminal catalytic kinase domain (KD) and a C-terminal polo-box domain (PBD), which engages in phospho-dependent PPIs with sequences having a phosphoserine (pS) or phosphothreonine (pT) residue. The Plk1 PBD recognizes sequences of the general form, [P/F]-[Φ/P]-[Φ]-[T/Q/H/M]-S-[pT/pS]-[P/Φ] (where Φ designates a hydrophobic residue), with the dipeptide segment “S-pT” representing a critical element that is necessary for high binding affinity.26,27 Peptides built around the S-pT motif that maximize binding interactions in the most efficient manner represent attractive platforms for developing PBD-binding antagonists. A starting point for these efforts is provided by the pentapeptide PBD-binding ligand PLHSpT (1), which was determined to be a minimal recognition sequence derived from the region of the polo-box interacting protein 1 (PBIP1) proximal to the phosphorylated pT78 residue. The PBD-binding affinity of 1 has variously been found to provide IC50 values from tens to hundreds of micromolar in ELISA assays using full-length Plk1.28,29
Accessing a cryptic binding pocket enhances PBD – binding affinity.
An importance advance in the design of PBD – binding antagonists was provided by Abell, who made the observation based on crystal packing, that a hydrophobic binding channel on the surface of the Plk1 PBD formed by Y417, Y421, Y481, F482, Y485 and L478 could be revealed by a more than 100° rotation of the Y481 side chain in the presence of ligands capable of accessing the pocket.30 It turns out that this “cryptic pocket” may have significance in the physiological functions of the PBD. For example, Polo kinase can be localized to microtubules by binding of its PBD to the microtubule associated protein Map205.31 The nature of this interaction has been shown by X-ray crystallography to involve insertion of the side chain of the F304 residue of Map205 into the pocket.32 It has also been shown that dimerization can be induced by the insertion of a hydrophobic residue of one PBD into the cryptic pocket of another.33
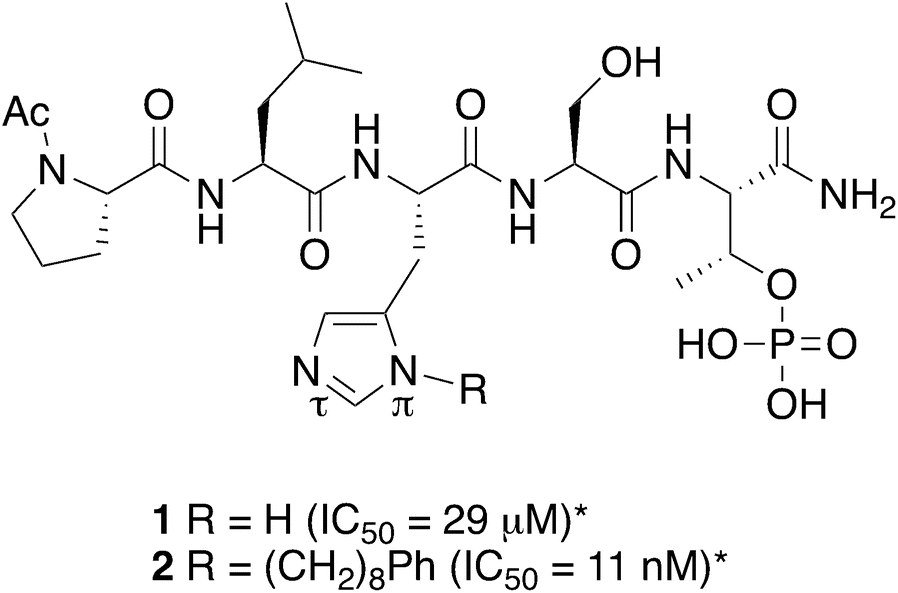
Taking advantage of interactions within the cryptic pocket can have profound effects on the affinities of PBD-binding peptides. Following Abell's disclosure that the N-terminal Phe residue of the PBIP1 pT78-derived nonamer sequence, FDPPLHSpTA makes significant contributions to binding affinity by accessing this pocket,30 we independently found that the cryptic binding site could be accessed and achieve up to three-orders-of-magnitude enhancement in PBD-binding affinity by tethering alkylphenyl groups from different positions on the much shorter pentamer sequence PLHSpT (1).34–36 Of particular note, we were able to reach the pocket from peptides of the form PLH*SpT (2), where H* indicates the presence of a –(CH2)8Ph group on the His N3(π) nitrogen [i.e., His-[N(π)-(CH2)8Ph].
The pT-2 position is “privileged”.
The pT-2 position can be considered to be “privileged”, since “SpT” forms an essential core Plk1 PBD recognition element.27 Accordingly “XSpT” may be viewed as a minimal motif that can efficiently accesses both the phospho-binding pocket and the cryptic pocket. To date, the highest affinity PBD – binding constructs have relied on tethering long chain alkyl–aryl groups from the pT-2 His N3(π) nitrogen.34–42 We have also found by tethering long chain alkylphenyl groups from a variety of amino acids at the pT-2 position, that although these showed increased PBD-binding affinities relative to the unsubstituted parent residues, the His N3(π) nitrogen remained the optimal site of attachment (Fig. 1).37
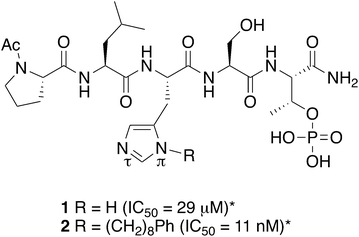 |
| | Fig. 1 Structures of peptides discussed in the text (*ref. 34). | |
Accessing the cryptic pocket while reducing conformational flexibility.
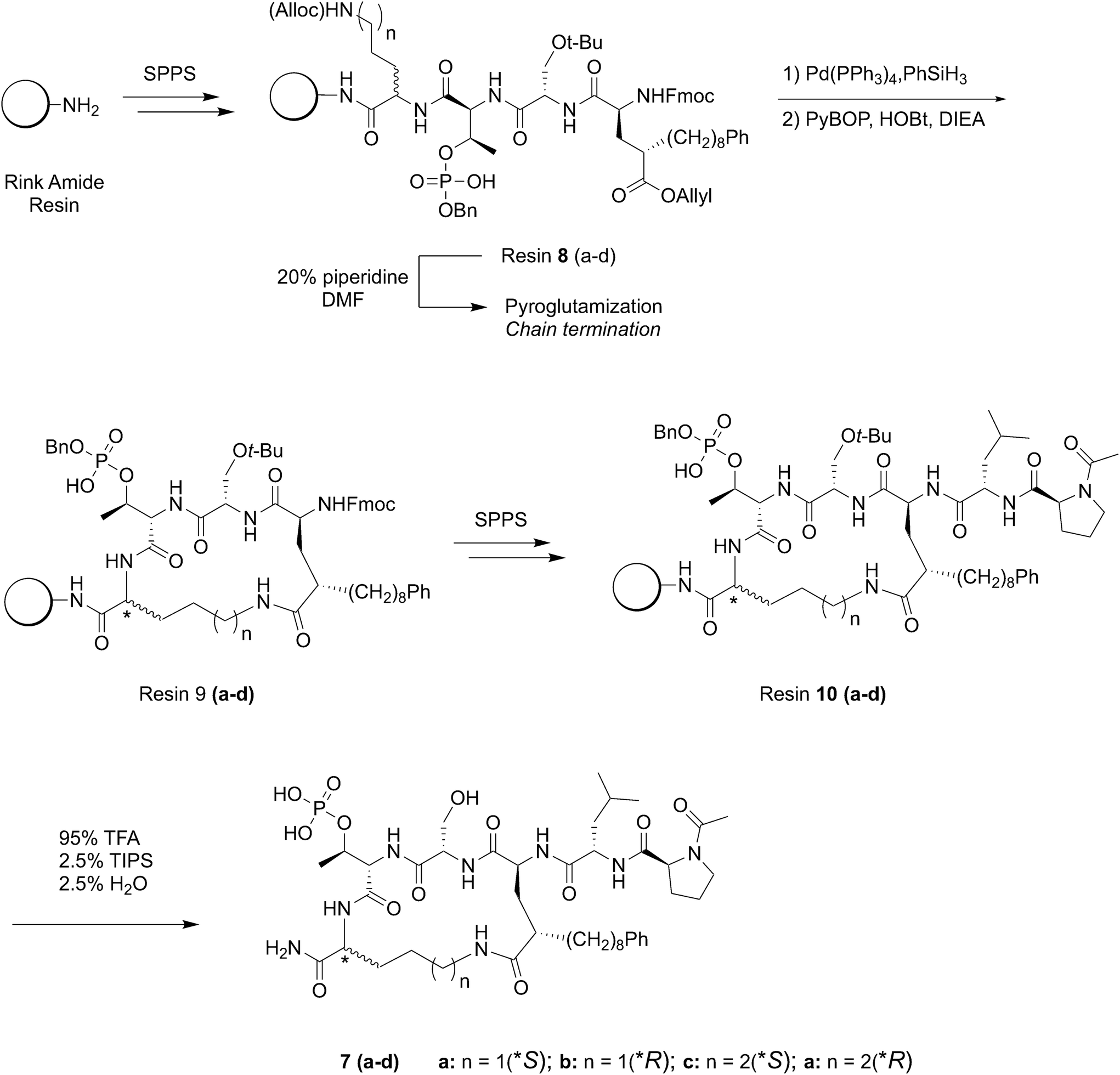
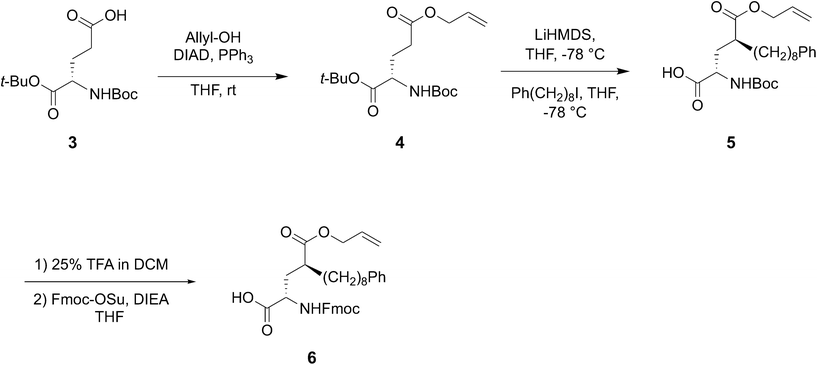
Reduction of conformational flexibility through macrocylization can be a powerful means of increasing the binding affinity of peptide ligands by reducing entropy penalties incurred by transitioning from high conformational flexibility in solution to the fixed geometries required by binding. Data obtained from bis-alkyl histidine cyclic ligands suggested that cyclization between the C-terminus and the pT-2 position is compatible with maintenance of PBD-binding geometries.39,43 With this as background, we chose to investigate a new amino acid analog, which would introduce the long-chain alkylphenyl functionality at the pT-2 critical for high-affinity ligands, while at the same time presenting orthogonal protection suitable for on-resin macrocyclization with the C-terminus. In this approach, macrocyclic ligands could be synthesized directly on-resin using solid-phase peptide synthesis (SPPS) resin without the need for subsequent solution phase cyclization – deprotection steps. For this purpose we designed the SPPS-compatible intermediate Fmoc-4(S)-octylphenyl-Glu(OAllyl)-OH (6, Scheme 1) as a ring-forming residue that contained a critical tethered alkylphenyl chain of defined stereochemistry at the γ-position of the glutamic acid side chain.
 |
| | Scheme 1 Synthesis of key macrocycle junction – forming residue 6. | |
Synthesis
Synthesis of the key ring-forming residue 6.
The globally protected glutamic acid derivative 4 was synthesized from the commercially available acid 3 by Mitsunobu esterification using allyl alcohol (Scheme 1). The dianion formed by treatment of 4 with LiHMDS at −78 °C was alkylated with 8-phenyloctyl iodide37 under stereo control induced by the chair conformation of the intermediate lithium enolate.44 This provided diastereomerically pure 1-allyl 5-(tert-butyl) (2S,4S)-4-((tert-butoxycarbonyl)amino)-2-(8-phenyloctyl) pentanedioate (5) as a clear viscous oil in 71% yield (Scheme 1). Deprotection of tert-butyl groups (25% TFA) followed by conversion of the resulting free amine to its N-Fmoc – protected form, yielded the desired 6 in 69% yield as a clear semi-solid (Scheme 1).
Use of new amino acid residue 6 to construct C-terminal macrocycles on-resin.
Macrocyclic ligands of type 7 were constructed using Fmoc-based SPPS protocols on Rink amide resin (Scheme 2). The approach employs C-terminal Alloc-protected Orn or Lys residues at the pT + 1 position, which could be cyclized to the side chain of the synthetic Glu analog on-resin following orthogonal deprotection of all allyl groups using Pd(PPh3)4 and PhSiH3 (Scheme 2).45 The use of C-terminal Orn and Lys residues varies the resulting macrocycle ring size from n = 18 units for Orn (peptides 7a and 7b) to n = 19 units for Lys (peptides 7c and 7d) following on-resin C-terminal macrocyclization. In addition, we prepared peptides using both S- and R-forms of Orn and Lys, since it was not clear what the preferred stereochemistry would be at this ring junction. The N-terminal Ac-Pro-Leu segment was retained as projecting out from the macrocycle ring in order to access a previously identified “pyrrolidine-binding” region.25 Our original approach was to prepare the fully protected hexapeptides and then selectively remove Alloc and allyl ester protection and macrocyclize side chains on-resin. However, we found that chain termination occurred during Fmoc-deprotection of the amino – terminal 8-phenyloctyl-containing Glu residues in resins 8(a–d), due to facile pyroglutamyl formation.46 In order to circumvent this, we subjected the resins 8(a–d) to Alloc and allyl ester removal [Pd(PPh3)4, PhSiH3] and then macrocyclized to yield resins 9(a–d) (PyBOP, HOBt, DIEA) prior to N-Fmoc deprotection of the Glu residue (Scheme 2). The remaining two amino-terminal residues were then added without complications. The peptides were cleaved from the resin (95% TFA, 2.5% H2O and 2.5% (i-Pr)3SiH and purified by reverse phase HPLC to yield the final macrocycles 7(a–d) (Scheme 2). The resulting ligands represent hexapeptides having similar molecular weights as the linear and cyclic bis-alkyl His pentapeptides.39,43
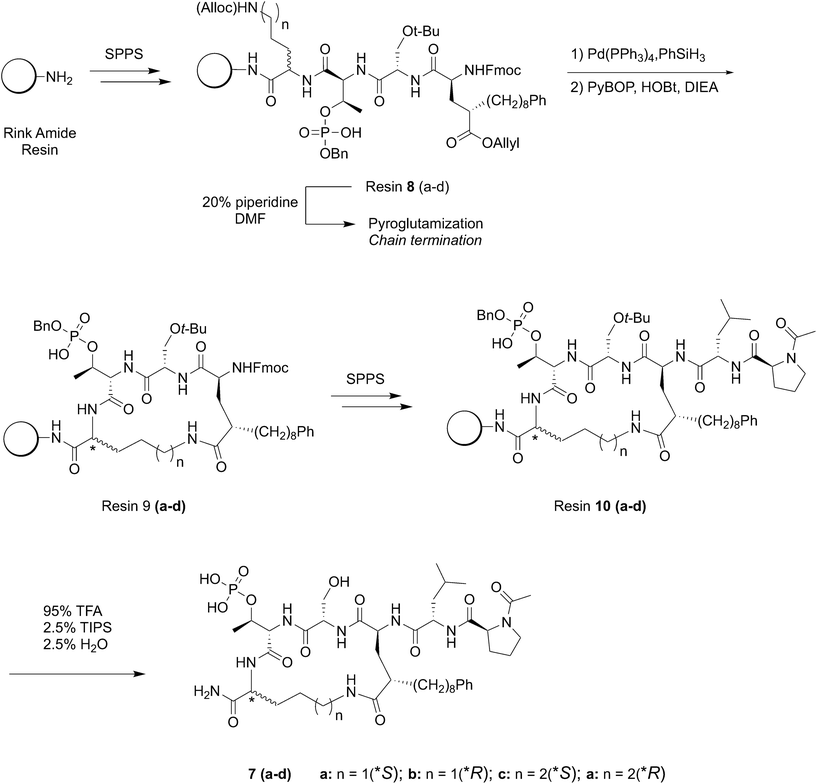 |
| | Scheme 2 Synthesis of chain-to-chain cyclic PBD hexapeptides of type 7. | |
Biological evaluation
Determination of Plk1 PBD – binding affinities using ELISA assays.
In order to determine PBD – binding affinities, we employed our previously described ELISA competition assays, which measure the ability of our synthetic constructs to compete with full-length Plk1 for binding to plate-bound PMQS(pT)PLN-NH2.29,40 In this assay, the parent linear peptide 2 showed an IC50 value of 110 nM (Table 1). This value is approximately 10-fold higher than our previously reported ELISA IC50 values for the same peptide.34–36,47 We attribute this to differences in binding affinities of the immobilized competitor peptides used for these assays. While our current assays employ native Plk1, the previous assays employed green-fluorescent protein (GFP)-labeled Plk1, potentially perturbing inhibitory interactions between the PBD and KD and in so doing, affecting equilibrium binding to phosphopeptides.27 We found that macrocycles having an S-configuration at the pT + 1 ring-forming residue exhibited potencies similar to parent 2, with the S-Orn and S-Lys containing peptides (7a and 7c, respectively) displaying IC50 values of 103 and 136 nM, respectively (Table 1). Surprisingly, the corresponding R-amino acid analogs 7b (R-Orn) and 7d (R-Lys) displayed dramatically reduced potencies (IC50 ≥ 10 μM). The potential conformational bases for these differences are revealed by X-ray co-crystal structures of PBD-bound 7a (see below).
Table 1 Determination of PBD-binding affinities
| Compd. |
ELISA IC50a (nM) |
Fluorescence polarization IC50b (nM) |
| Full-length Plk1 |
Plk1 PBD |
Plk2 PBD |
Plk3 PBD |
|
Determined as described.29,40
Determined as described.29,48,49
Fold selectivity relative to Plk1 PBD.
Not determined.
|
|
2
|
110 ± 16 |
4 ± 0.4 |
570 ± 20 (142×)c |
1200 ± 20 (300×) |
|
7a
|
103 ± 29 |
5 ± 0.4 |
700 ± 30 (140×) |
980 ± 60 (190×) |
|
7b
|
≈10000 |
n.d.d |
n.d. |
n.d. |
|
7c
|
136 ± 45 |
5 ± 0.4 |
600 ± 20 (120×) |
1100 ± 60 (220×) |
|
7d
|
≈10000 |
n.d. |
n.d. |
n.d. |
Determination of affinities against the isolated PBDs of Plks 1, 2 and 3.
An important aspect of developing peptidomimetic inhibitors targeting the Plk1 PBD is to achieve binding selectivity for Plk1 versus the highly homologous PBDs of Plk2 and Plk3. This is due to the possible tumor-suppressor functions of these Plks.50,51 We utilized fluorescence polarization (FP) assays to measure the abilities of our most potent inhibitors to compete with optimized pThr-containing fluorescence probes for binding to the isolated PBDs of Plk1, 2 and 3.29,48,49 Isolated PBDs were utilized, since no PBD-binding assays are currently available that employ full-length Plk2 or Plk3. Linear peptide 2 and macrocycles 7a and 7c showed similar single-digit IC50 values against the Plk1 PBD. These values were approximately 20-fold lower than values observed against full-length Plk1 (Table 1). The differences may potentially be attributed to inhibitory interactions between the PBD and KD in full-length Plk1 that are missing in assays using isolated PBDs.27,29 The parent linear peptide 2 showed selectivity values for Plk1 versus Plks 2 and 3 of 140- and 300-fold, respectively (Table 1). These are slightly better than our previously reported selectivity values of 26-fold and 56-fold against these two PBDs.41 Macrocyclic peptides 7a and 7c displayed similar selectivity as 2 against the PBDs of Plk2 and Plk3, with values of between 120- and 220-fold (Table 1).
Determination of Plk1 PBD-binding interactions of macrocycle 7a by X-ray crystallography
To observe the binding effects of macrocyclization using (2S,4S)-2-amino-4-(8-phenyloctyl)pentanedioic acid at the pT-2 position onto S-Orn at the pT + 1 position, we performed X-ray co-crystal analysis of 7a bound to the isolated Plk1 PBD. As shown in (Fig. 2, panel A), we superimposed the 7a structure onto the crystal structure of 2 bound to the PBD (PDB code: 3RQ7)34 and in so doing we observed that macrocycle 7a overlays very well with the linear parent peptide, maintaining critical access to the phosphate-binding pocket, the cryptic pocket and the pyrrolidine-binding region. In macrocycle 7a, the 4(S)-configuration of the alkylated Glu residue can effectively direct the key 8-octylphenyl aryl ring to the aromatic-rich cryptic binding pocket responsible for achieving high affinity. A close up of this region shows the manner in which the 5-carboxamide and the 4(S)-stereochemistry work together to replicate key aspects of the His N3(π) alkyl portions of parent peptide 2 (Fig. 2, panel B). Even more fortunately, 7a appears to engage in two additional hydrogen bonding interactions with the PBD. The first occurs between the Glu side chain 5-carboxamide and the phenolic hydroxyl of Tyr485 (Fig. 2, panel B) and the second occurs between the C-terminal carboxamide and the backbone carbonyl of His489 (2.54 Å distance, Fig. 2, panel C). A close up of the C-terminal region shows that the pT carboxamide carbonyl of 7a hydrogen bonds with the Leu491 residue in a fashion similar to the parent 2. This is also apparently why the S-configuration of the C-terminal Orn residue would be favored over the R-enantiomer (Fig. 2, panel C). The N-terminal Leu-Pro residues of 7a replicate the hydrogen bonds with the guanidinium group of Arg518 and the backbone amide of Asp416 as is found with the linear peptide 2. Overall, macrocycle 7a binds in a fashion that is highly similar to the linear parent peptide in spite of the fact that it lacks the critical His N3(π) alkyl functionality at the pT-2 position. This represents the first example where access of the cryptic binding pocket from the pT-2 position can be achieved with such fidelity to His N3(π)-based constructs. The electron density map for 7a is shown in Fig. 3 and the molecular replacement electron density is shown in Fig. S9.†
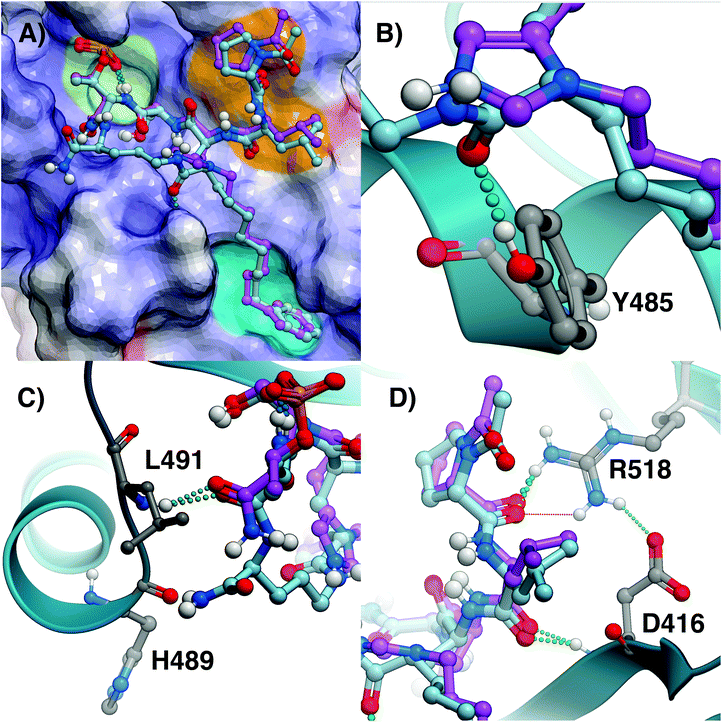 |
| | Fig. 2 Comparison of crystal structures of 2 and 7a bound the Plk1 PBD. (A) Crystal structure of PBD-bound 2 (PDB: 3RQ7; carbons purple) superimposed onto the crystal structure of PBD-bound 7a (carbons cyan). The protein surface is rendered as electrostatic potential (blue = positive; red = negative; white = neutral), with key binding regions color-highlighted (phosphate-binding pocket = lime; cryptic pocket = cyan; pyrrolidine-binding region = orange); (B) closeup of the critical region where the alkylphenyl groups join the peptides; (C) closeup of the C-terminal residues; (D) closeup of N-terminal interactions. | |
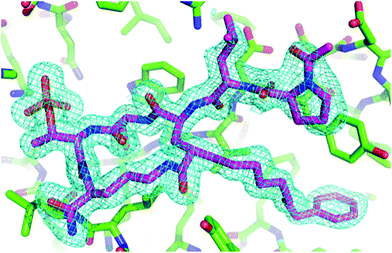 |
| | Fig. 3 Electron density for 7a macrocycle. The final 2Fo − Fc electron density map surrounding the macrocycle is shown at a contour level of 1.25σ. The hydrogens in the final refined model have been omitted for clarity. | |
Replacement of the pT residue in peptide 7a with a phosphatase-stable mimetic
(2S,3R)-2-Amino-3-methyl-4-phosphono-butanoic acid (Pmab, 11) is a pT mimetic, in which the phosphoryl moiety has been replaced by a phosphonic acid group (Fig. 4).52 This has been used in a variety of biological contexts, including peptidomimetic ligands that bind to the Plk1 PBD with affinities approaching that of the corresponding pT-containing peptides.28,34,35 In our current work, we prepared Pmab in its orthogonally protected form, [(2S,3R)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-(di-tert-butoxyphosphoryl)-3-methylbutanoic acid (N-Fmoc-Pmab(Ot-Bu)2-OH, 12)53] using our previously reported synthetic protocol.29 We then employed this reagent to prepare the Pmab-containing macrocycle 13 using solid-phase chemistry similar to what we used to prepare the parent peptide 7a. In full-length Plk1 ELISA assays, we observed that the affinity of the resulting Pmab-containing macrocycle 13 was very similar to the parent linear pT-containing peptide 2 (IC50 = 360 nM for 13versus 220 nM for 2, see ESI Fig. S2†). The retention of high affinity for the Pmab-containing macrocycle is consistent with the crystal structure of PBD-bound 7a, which shows that the phosphoryl ester oxygen does not engage in hydrogen bonding interactions that would be lost on replacement with a methylene group (Fig. 5).
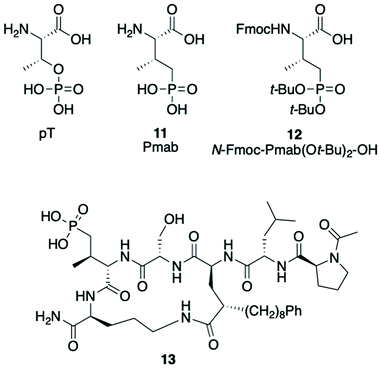 |
| | Fig. 4 Structures of pT, Pmab analogs and Pmab-containing macrocycle 13. | |
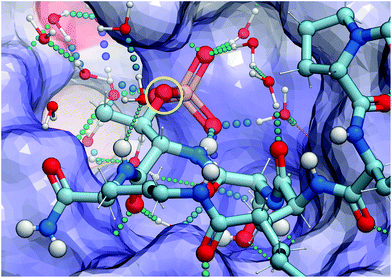 |
| | Fig. 5 X-ray crystal structure of PDB – 7a highlighting hydrogen bonding interactions in the phosphate – binding pocket and the lack of interactions with the phosphoryl ether oxygen (shown in yellow circle). | |
Conclusion
Targeting PPIs has emerged as important area of discovery for anticancer therapeutic development, with a key consideration being that PPI inhibitors must bind strongly enough to relatively flat protein interfaces to overcome PPI association energies. Multivalent interactions that simultaneously access both binding hot spots and proximal cryptic pockets on the protein surface can be an attractive way to achieve desired overall high binding affinities. In phospho-dependent PPIs, a phosphorylated protein residue provides recognition and binding to a key protein hot spot. A challenge is to design antagonists that can take into consideration interactions in both the phospho-binding and proximal transiently revealed cryptic pockets. In our current paper, we describe the design and synthesis of macrocyclic peptide mimetics directed against the Plk1 PBD that are characterized by a new non-natural amino acid, which simultaneously serves as a ring-closing junction while providing accesses to a cryptic binding pocket and at the same time achieving proper orientation of a pT residue for optimal interaction in the signature phospho-binding pocket. Macrocycles prepared with this new amino acid analog maintain the protein – ligand hydrogen bonds of the parent linear peptide, while introducing additional interactions. It is noteworthy that this new Glu-based amino acid analog represents the first example where access to the cryptic pocket from the pT-2 position is made possible with a non-His residue that achieves retention of affinity equal to the parent His-[N(π)-(CH2)8Ph] construct. We also demonstrate that the pT residue can be replaced by a hydrolytically-stable mimetic with retention of binding affinity. Our work discloses a new genre of high affinity Plk1-binding peptide mimetic. The concepts employed in the design and synthesis of these constructs should be useful for further studies directed against the Plk1 PBD and potentially for ligands directed against other PPI targets.
Experimental
Synthetic
General methods.
All experiments involving moisture-sensitive compounds were conducted under anhydrous conditions (positive argon pressure) using standard syringe, cannula, and septa apparatus. Commercial reagents were purchased from Sigma, TCI America, Acros, Aapptec, or Chem-Impex. Fmoc-Lys(Alloc)-OH, Fmoc-Orn(Alloc)-OH, Fmoc-D-Lys(Alloc)-OH, Fmoc-D-Orn(Alloc)-OH, Fmoc-Ser(Trt)-OH, Fmoc-Ser(t-Bu), Fmoc-His(Trt)-OH, Fmoc-Leu-OH, Fmoc-Pro-OH and Fmoc-Thr[PO(OH)(OBn)]-OH were purchased from Chem-Impex. The intermediates tert-butyl (4-iodobutyl)carbamate,54tert-butyl (5-iodopentyl)carbamate,55tert-butyl (6-iodohexyl)carbamate,56 and Fmoc-His[N(π)-(CH2)8-Ph]-OH57 were synthesized as previously described. The final peptide PLH*SpT (2) was synthesized by SPPS as previously described to >95% purity.34 All solvents were purchased in anhydrous form (Aldrich) and used directly. HPLC-grade hexanes, EtOAc, DCM, and MeOH were used in chromatography. Analytical TLCs were performed using Analtech precoated plates (Uniplate, silica gel GHLF, 250 nm) containing a fluorescence indicator. Silica column chromatography employed a Teledyne CombiFlash Rf 200i instrument with either hexane/EtOAc or DCM/MeOH gradients. Microwave reactions were conducted in a Biotage Initiator microwave synthesis apparatus. NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. Coupling constants are reported in Hertz, and peak shifts are reported in δ (ppm) relative to CDCl3 (1H 7.26 ppm, 13C 77.16 ppm). Low-resolution mass spectra (ESI) were measured with either an Agilent 260 1200 LC/MSD-SL system or a Shimadzu 2020 LC – MS system. High-resolution mass spectra (HRMS) were obtained by positive ion, ESI analysis on a Thermo Scientific LTQ-XL Orbitrap mass spectrometer with HPLC sample introduction using a short narrow-bore C 1s reversed-phase column with MeCN/H2O gradients. Preparative HPLC of final peptides was performed using a Waters 2545 binary pump equipped with a reverse-phase Gemini C18 column (Phenomenex Inc., pore size: 110 Å, particle size: 10 μm, 250 μ × 21.2 mm) with a gradient of 5–99% MeCN/H2O containing 0.1% (TFA) over 30 minutes at a flow rate of 20 mL min−1 and monitored with a UV detector at 210 and 254 nm. Semi-preparative HPLC purification was performed using an Agilent 1200 series quaternary pump (MeCN/H2O gradient containing 0.1% TFA) with a with a reverse phase Phenomenex Kinetix-C18 column (pore size: 110 Å, particle size: 5 μm, 250 × 10.0 mm) at a flow rate 3 mL min−1 and monitored with a UV detection at 210 nm. Fractions containing pure peptide were combined and lyophilized to obtain a white powder. Analytical HPLC of final peptides was performed using an Agilent 1200 series quaternary pump (MeCN/H2O gradient containing 0.1% TFA) with a Phenomenex Gemini-C18 column (pore size: 110 Å, particle size:5 μm, 250 × 4 mm), at a flow rate 1 mL min−1, and UV detection at 210 nm.
General solid-phase peptide synthesis (SPPS) procedures.
SPPS resin (Rink amide MBHA LL) was pre-swollen in DMF (4 mL) for 1 h with shaking. HMPB MBHA resin was also utilized for certain peptides and the loading procedure is described where applicable. Fmoc-protected amino acids (2–4 equivalents based on resin) were dissolved in DMF (3–4 mL) containing 4% N,N-diisopropylethylamine (DIEA) and pre-activated by the addition of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluoro-phosphate (HATU, 0.95 mol equivalents relative to the amino acid) for 5 min with gentle agitation and added to the resin. Coupling reactions were shaken in an automated shaker at room temperature and allowed to proceed from 3–16 hours depending on the equivalents used and steric bulk of each amino acid. The Fmoc groups were removed by treating the resin twice with 20% piperidine in DMF for 30 min. After each coupling and deprotection step, the resin was sequentially washed 3–4 times with DMF, MeOH and DCM. Coupling reactions were routinely checked for completion using the Kaiser test. Once completed, the resin was filtered and washed 4 times with DMF (6–8 mL), followed by Fmoc-deprotection using 20% piperidine in DMF (4 mL, 2× 10 minutes each). Cleavage from Rink amide LL resin and global deprotection was done using a cocktail of TFA/triisopropylsilane (TIPS)/H2O (95/2.5/2.5) (4 mL × 2, 2 h each). Crude peptides were purified using preparative reverse-phase HPLC with gradient elution (89.9/10/0.1 H2O/MeCN/TFA to 99.9/0.1 MeCN/TFA over 30 minutes).
General protocol for cleavage of aliquots of resin-bound peptide for analysis by LC – MS.
An aliquot of resin-bound peptide (3–5 mg) was treated with a freshly made solution of TFA/H2O/triethylsilane (TES) (95:2.5:2.5, v/v/v, 0.5 mL) for 30–45 min at room temperature. The resin was removed by filtration. The filtrate was collected in a 1.5 mL Eppendorf tube. The filtrate volume was concentrated, and the crude peptide was precipitated with cold ether (1.5 mL). After agitation on a vortex shaker, the mixture was spun in a centrifuge. The supernatant was decanted leaving a pellet, which was dissolved in MeOH (or H2O, 1 mg mL−1) and subjected to LC – MS analysis.
5-Allyl 1-(tert-butyl) (tert-butoxycarbonyl)-L-glutamate (4).
N-Boc-L-glutamic acid α-tert-butyl ester (3) (670 mg, 2.21 mmol), triphenylphosphine (770 mg, 2.94 mmol), and allyl alcohol (0.20 mL, 2.94 mmol) were dissolved in anhydrous THF (11 mL) and cooled in an ice bath. Diisopropylazodicarboxylate (DIAD, 0.57 mL, 2.94 mmol) was added dropwise to the mixture with stirring. The reaction was allowed to warm to room temperature over 3 h, then directly adsorbed to Celite for flash chromatography. Purification by CombiFlash silica gel column chromatography (gradient elution of 0–30% EtOAc in hexane) provided 4 (695 mg, 92% yield) as a clear viscous oil. 1H-NMR (400 MHz, CDCl3) δ 5.98–5.83 (m, 1H), 5.35–5.19 (m, 2H), 5.07 (d, J = 7.2 Hz, 1H), 4.58 (d, J = 5.7 Hz, 2H), 4.26–4.16 (m, 1H), 2.52–2.31 (m, 2H), 2.22–2.08 (m, 1H), 1.98–1.86 (m, 1H), 1.48–1.41 (m, 18H); 13C-NMR δ (101 MHz, CDCl3) d 172.64, 171.45, 155.50, 132.23, 118.45, 82.34, 79.92, 77.48, 77.16, 76.84, 65.44, 53.54, 30.43, 28.46, 28.23, 28.14; HR-MS (ESI+) calculated for C17H29NO6: 344.2068 [M + H]+; found: 344.2075.
1-Allyl 5-(tert-butyl) (2S,4S)-4-((tert-butoxycarbonyl)amino)-2-(8-phenyloctyl) pentanedioate (5).
Compound 4 (1.05 g, 3.06 mmol) was dissolved in anhydrous THF (6 mL) and cooled to −78 °C. A 0.9 M solution of lithium bis(trimethylsilyl)amide (LiHMDS, 7.5 mL, 6.73 mmol, 2.2 equiv.) was added dropwise with stirring at −78 °C. The reaction was allowed to stir for an additional 2 h at −78 °C. After 2 h, 8-phenyloctyl iodide37 (1.45 g, 4.59 mmol, 1.5 equiv.) was added dropwise as a solution in anhydrous THF (3 mL). The alkylation step was allowed to proceed overnight at −78 °C. Following completion, the reaction was quenched with 5% aqueous citric acid (5 mL) and allowed to warm to room temperature. The THF was removed by rotovap and the resulting solution was diluted with H2O (25 mL) and extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine, dried over sodium sulfate, and concentrated. The resulting residue was purified by CombiFlash silica gel column chromatography (gradient elution of 0–80% EtOAc in hexane) to provide 5 (1.16 g, 71% yield) as a clear viscous oil. 1H-NMR (400 MHz, CDCl3) δ 7.33–7.12 (m, 5H), 5.99–5.85 (m, 1H), 5.37–5.19 (m, 2H), 4.87 (d, J = 8.6 Hz, 1H), 4.67–4.49 (m, 2H), 4.18 (q, J = 8.1 Hz, 1H), 2.64–2.55 (m, 2H), 2.48 (m, 1H), 1.99–1.84 (m, 2H), 1.66–1.52 (m, 4H), 1.52–1.38 (m, 18H), 1.37–1.18 (m, 10H); 13C-NMR (101 MHz, CDCl3) δ 175.69, 171.85, 155.55, 143.02, 132.38, 128.51, 128.34, 125.68, 118.49, 82.10, 79.84, 77.16, 65.34, 53.09, 42.51, 36.11, 34.79, 32.50, 31.63, 29.53, 29.50, 29.48, 29.41, 28.46, 28.11, 27.07; HR-MS (ESI+) calculated for C31H49NO6: 532.3633 [M + H]+; found: 532.3626.
(2S,4S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-((allyloxy)carbonyl)-12-phenyldodecanoic acid (6).
Compound 5 (1.0 g, 1.88 mmol) was dissolved in 25% trifluoroacetic acid (TFA) in DCM (v/v, 10 mL). TIPS (0.96 mL, 4.70 mmol) was added to the mixtue and the reaction was allowed to stir for 3 h at room temperature. Following removal of the tert-butyl protection, the reaction was concentrated to dryness. The resulting residue was dissolved in anhydrous THF (12 mL), and DIEA (0.98 mL, 5.64 mmol) was added with stirring. Fmoc-NHS ester (0.80 g, 2.50 mmol) was added and the reaction was allowed to stir at room temperature overnight. The reaction was concentrated under vacuum, diluted with 0.1 M HCl (30 mL), and extracted into EtOAc (3 × 50 mL). The combined organic phases were washed with brine, dried over sodium sulfate, and concentrated. The resulting residue was purified by CombiFlash silica gel column chromatography (gradient elution of 0–10% MeOH in DCM) to provide 6 (0.78 g, 69% yield) as a clear semi-solid. 1H-NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.59 (t, J = 7.2 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.35–7.23 (m, 5H), 7.22–7.12 (m, 3H), 5.94–5.80 (m, 1H), 5.38–5.15 (m, 2H), 4.63–4.37 (m, 4H), 4.36–4.27 (m, 1H), 4.22 (t, J = 7.0 Hz, 1H), 2.64–2.49 (m, 3H), 2.19–1.99 (m, 2H), 1.72–1.49 (m, 4H), 1.37–1.17 (m, 10H); 13C-NMR (101 MHz, CDCl3) δ 176.47, 175.73, 156.31, 143.72, 143.00, 141.43, 132.03, 128.52, 128.34, 127.90, 127.22, 125.68, 125.30, 125.17, 120.13, 118.72, 77.16, 67.50, 65.56, 53.05, 47.20, 42.71, 36.10, 33.98, 32.67, 31.61, 29.52, 29.46, 29.40, 26.97, 17.82; HR-MS (ESI+) calculated for C37H43NO6: 598.3163 [M + H]+; found: 598.3150.
General procedure C (on-resin peptide cyclization).
Using Rink amide MBHA LL resin, the peptide chain containing Alloc/allyl ester amino acids are installed using the general SPPS procedure described above (Scheme 2). Once the synthetic glutamic acid analog had been installed to generate resins 8(a–d), the Alloc/allyl ester groups were deprotected using Pd(PPh3)4 (0.5 equiv.) and phenylsilane (20 equiv.) in DCM (3 × 3 mL, 30 minutes each) under argon gas. Excess palladium was scavenged by treating the resin with 0.5% sodium diethyl dithiocarbamate in DMF (2 × 3 mL, 20 minutes each). Cyclization was then performed using PyBOP (5 equiv.), HOBt (5 equiv.), and DIEA (10 equiv.) in DMF (4 mL) for 4 h. The cyclization was checked for completion using the Kaiser test. Any unreacted amines were capped using 3 mL of acetic anhydride:pyridine:DMF (1:1:4) for 30 minutes. The resulting resins 9(a–d), containing cyclized peptides were further elaborated using the general SPPS procedure to provide resins 10(a–d). [Note: attempts to cyclize the peptide side chains following completion of the peptide backbone all resulted in chain termination due to pyroglutamate formation of the synthetic glutamic acid analog (Scheme 2, data not shown). Therefore, cyclization was performed prior to removal of the Fmoc protecting group.]
Acetyl-prolyl-leucyl-cyclo[glutamyl-[4S-octyl-8-phenyl]-seryl-(O-phospho)-threonyl-L-ornithinylamide] (7a).
Compound 7a was synthesized according to the general SPPS procedure (0.060 mmol resin scale) and cyclized on-resin using General Procedure C. Cleavage and purification by preparative RP-HPLC provided 7a (24 mg, 42% overall yield) as a lyophilized white powder. LR-MS (ESI+) calculated for C44H71N8O13P: 951.5 [M + H]+; found: 951.6.
Acetyl-prolyl-leucyl-cyclo[glutamyl-[4S-octyl-8-phenyl]-seryl-(O-phospho)-threonyl-D-ornithinylamide] (7b).
Compound 7b was synthesized according to the general SPPS procedure (0.060 mmol resin scale) and cyclized on-resin using General Procedure C. Cleavage and purification by preparative RP-HPLC provided 7b (15 mg, 26% overall yield) as a lyophilized white powder. LR-MS (ESI+) calculated for C44H71N8O13P: 951.5 [M + H]+; found: 951.5.
Acetyl-prolyl-leucyl-cyclo[glutamyl-[4S-octyl-8-phenyl]-seryl-(O-phospho)-threonyl-L-lysylamide] (7c).
Compound 7c was synthesized according to the general SPPS procedure (0.060 mmol resin scale) and cyclized on-resin using General Procedure C. Cleavage and purification by preparative RP-HPLC provided 7c (25 mg, 43% overall yield) as a lyophilized white powder. LR-MS (ESI+) calculated for C45H73N8O13P: 965.5 [M + H]+; found: 965.6.
Acetyl-prolyl-leucyl-cyclo[glutamyl-[4S-octyl-8-phenyl]-seryl-(O-phospho)-threonyl-D-lysylamide] (7d).
Compound 7d was synthesized according to the general SPPS procedure (0.060 mmol resin scale) and cyclized on-resin using General Procedure C. Cleavage and purification by preparative RP-HPLC provided 7d (17 mg, 29% overall yield) as a lyophilized white powder. LR-MS (ESI+) calculated for C45H73N8O13P: 965.5 [M + H]+; found: 965.5.
Acetyl-prolyl-leucyl-cyclo[glutamyl-[4S-octyl-8-phenyl]-seryl-[(2S,3R)-2-amino-3-methyl-4-phosphonobutanoic acid]-L-Lysylamide] (13).
Compound 12 [(N-Fmoc-Pmab(Ot-Bu)2-OH] was prepared as per the synthetic protocol reported in literature.29 Compound 13 was synthesized according to the general SPPS procedure (0.036 mmol resin scale) and cyclized on-resin using General Procedure C. Cleavage and purification by preparative RP – HPLC provided 13 (1.6 mg, 5% overall yield) as a lyophilized white powder. LR-MS (ESI+) calculated for C45H73N8O12P: 949.5 [M + H]+; found: 949.3.
Biological methods
ELISA inhibition versus full-length Plk1.
ELISA assays to test for inhibitory potency versus full-length Plk1 were run as previously described.29 Briefly, a biotinylated phosphopeptide (sequence: Biotin-Ahx-PMQS(pT)PLN-NH2) was diluted into PBS (pH 7.4) to 1 μM (from a 2 mM DMSO stock solution) and loaded onto the wells of a 96-well Neutravidin-coated plate (Pierce Biotechnology) at 100 μL per well for 1 h. The wells were washed once with 150 mL PBST (PBS + 0.05% Tween-20), and then 100 μL of 1% BSA in PBS (blocking buffer) were added for 1 h. The cytosolic lysate containing myc-tagged Plk1 protein was diluted to 300 μg mL−1 in PBS containing protease/phosphatase inhibitors (Pierce Biotechnology), mixed with competitive inhibitor (from a 10× stock in 5% DMSO/PBS), and allowed to pre-incubate for 1 h (100 μL per well in a 96-well plate, 30 μg total protein). The blocked ELISA plate was washed 2× with PBST (150 μL) and the pre-incubated lysates were added to the plate to incubate for 1 h. The wells were washed 4× with PBST (150 μL), then probed with anti-myc primary antibody (1:1500 dilution, mouse monoclonal, Pierce Biotechnology) for 1 h. The wells were then washed 4× with PBST (150 μL) and incubated with rabbit anti-mouse secondary antibody HRP conjugate (1:3000 dilution, Pierce Biotechnology) for 1 h. The wells were then washed 5× with PBST (150 μL) and incubated with Turbo TMB-ELISA solution (Pierce Biotechnology) until the desired absorbance was reached (5–10 minutes). The reaction was quenched by the addition of 2 N H2SO4 aq. and the absorbance was measured at 450 nm using a BioTek Synergy 2 plate reader. Absorbance was plotted versus concentration (log M) and fitted to a non-linear regression using GraphPad Prism 7 software (model: log(inhibitor) vs. response – Variable slope (four parameters)) to provide IC50 values. IC50 values from multiple independent experiments were normalized and averaged to provide values ± standard error of the mean (SEM).
Selectivity screening versus isolated PBDs.
Selectivity screening was performed as previously described.29 Briefly, isolated PBD proteins from Plks 1–3 were diluted to a 2× working dilution in assay buffer (HEPES-buffered saline with 0.05% Tween-20, 1 mM DTT, and 1 mM EDTA). The following final protein concentrations were used: 100 nM for Plk1 PBD, 200 nM for Plk2 PBD, and 500 nM for Plk3 PBD. These concentrations represent the approximate Kd values determined for the respective fluorescence polarization (FP) probe sequences. Inhibitors were serially diluted to generate 4× working dilutions in assay buffer containing 4% DMSO. 20 μL of 2× PBD solution was added to each well of a 384-well plate (0% binding controls received 20 μL of assay buffer). 10 μL of the 4× inhibitor solution (or DMSO blank) was added to corresponding wells and allowed to pre-incubated at room temperature for 30 minutes with shaking. The following sequences were utilized as fluorescent probes: 5CF-GPMQSpTPLNG-NH2 for Plk1 PBD, 5CF-GPMQTSpTPKNG-NH2 for Plk2 PBD, and 5CF-PLATSpTPKNG-NH2 for Plk3 PBD.48,49 Fluorescent probes were diluted to 40 nM (4×) in assay buffer, then 10 μL was added to each well. The plate was allowed to equilibrate at room temperature for 30 minutes with shaking. Fluorescence polarization was read using a BioTek Synergy 2 plate reader with 485/20 excitation and 528/20 emission. The FP values were obtained in triplicate and normalized to 100% (no inhibitor) and 0% binding (no protein) controls. Normalized values were plotted versus concentration and analyzed using non-linear regression in GraphPad Prism 7 [log(inhibitor) vs. response – variable slope (four parameter) model]. IC50 values represent average ± SEM.
X-ray crystallography
Crystallization of the PBD – macrocycle complex.
Plk1 PBD protein (residues 371–603), purified as previously described,28 was provided by Dr Dan Lim. The complex was formed and crystals were grown using the same methods and under similar conditions to the complex with a related macrocycle.58 Briefly, frozen PBD stock at 37 mg mL−1 was thawed and diluted to 10 mg mL−1. A stock solution of the macrocycle at 100 mM in DMSO was added directly to the diluted protein to a final concentration of 1 mM, then 4 M ammonium acetate was added to a final 0.4 M concentration. Crystals were grown by hanging drop vapor diffusion against a well solution 0.2 M. CaCl2, 15% PEG-3350. Crystals were cryo-protected by dipping briefly in well solution supplemented with additional PEG-3350 added to a final concentration of 37.5%.
Structure solution and refinement.
X-ray diffraction data were collected at the Advance Proton Source (APS) using the Q315 detector at the NE-CAT 24-ID-E beam line. HKL200059 was used to index, integrate and scale the diffraction data to 1.65 Å resolution. The space group of the crystal was P21 with two PBD-macrocycle complexes in the asymmetric unit. The structure was solved by molecular replacement with PHENIX60 implementation of PHASER61 using the A chain of PDB entry 4DFW as the search model. The model was refined in PHENIX with COOT62 as the model building tool. A refinement constraints file for the entire macrocycle ligand was generated by the GRADE web server (http://grade.globalphasing.org) and used through most of the refinement. At the end of the refinement ELBOW63 was used to create a constraints library for the peptide modifications and the peptide scaffold of the macrocycle was replaced by the sequence (ACE)-PRO-LEU-ALA-SER-(TPO). Data collection and refinement statistics are shown in Table 2. The electron density map for 7a is shown in Fig. 3 and the molecular replacement electron density is shown in Fig. S9.†
Table 2 Data collection and refinement statistics
| Wavelength (Å) |
0.97917 |
| Space group |
P1211 |
| Resolution range (Å) |
49.13–1.64 (1.70–1.64) |
| Unit cell |
57.71 Å; 64.96 Å; 71.67 Å |
| 90°;101.62°; 90° |
| Unique reflections |
62415 (5560) |
| Multiplicity |
7.4 (7.2) |
| Mean I/Σ(I) |
17.86 (1.62) |
| Completeness (%) |
98.78 (88.52) |
| Wilson B-factor |
25.12 |
|
R
merge
|
0.071 (1.158) |
|
R
meas
|
0.076 (1.249) |
|
R
pim
|
0.028 (0.462) |
| CC1/2 |
1.000 (0.687) |
| Reflections used in refinement |
62415 (5559) |
| Reflections used for Rfree |
2000 (178) |
|
R
work
|
0.1754 (0.2651) |
|
R
free
|
0.2041 (0.2851) |
| Number of non-hydrogen atoms |
4208 |
| Macromolecules |
3680 |
| Ligands |
58 |
| Solvent |
470 |
| Protein residues |
442 |
| RMS (bonds) |
0.004 |
| RMS (angles) |
0.80 |
| Ramachandran favored (%) |
98.64 |
| Ramachandran allowed (%) |
1.36 |
| Ramachandran outliers (%) |
0 |
| Rotamer outliers (%) |
0.49 |
| Clashscore |
1.49 |
| Average B-factor |
41.40 |
| Macromolecules |
40.76 |
| Ligands |
41.72 |
| Solvent |
46.37 |
| Number of TLS groups |
21 |
Data availability statement
Atomic coordinates and structure factors for Plk1 PBD-bound 7a have been deposited in the RCSB Protein Data Bank (PDB code 7MX1). Authors will release the atomic coordinates and experimental data upon article publication.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Our studies are supported by the NIH Intramural Program, Center for Cancer Research (ZIA BC 006198), National Cancer Institute (DH, KT, RC, DK and TB) and NIH grants ES015339 and GM104047 (RG and MY). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the members of the Drennan lab (MIT) for collecting the data. This work was supported in part by a JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (KT).
References
- T. Rolland, M. Taşan, B. Charloteaux, S. J. Pevzner, Q. Zhong, N. Sahni, S. Yi, I. Lemmens, C. Fontanillo, R. Mosca, A. Kamburov, S. D. Ghiassian, X. Yang, L. Ghamsari, D. Balcha, B. E. Begg, P. Braun, M. Brehme, M. P. Broly, A.-R. Carvunis, D. Convery-Zupan, R. Corominas, J. Coulombe-Huntington, E. Dann, M. Dreze, A. Dricot, C. Fan, E. Franzosa, F. Gebreab, B. J. Gutierrez, M. F. Hardy, M. Jin, S. Kang, R. Kiros, G. N. Lin, K. Luck, A. MacWilliams, J. Menche, R. R. Murray, A. Palagi, M. M. Poulin, X. Rambout, J. Rasla, P. Reichert, V. Romero, E. Ruyssinck, J. M. Sahalie, A. Scholz, A. A. Shah, A. Sharma, Y. Shen, K. Spirohn, S. Tam, A. O. Tejeda, S. A. Trigg, J.-C. Twizere, K. Vega, J. Walsh, M. E. Cusick, Y. Xia, A.-L. Barabási, L. M. Iakoucheva, P. Aloy, J. De Las Rivas, J. Tavernier, M. A. Calderwood, D. E. Hill, T. Hao, F. P. Roth and M. Vidal, Cell, 2014, 159, 1212–1226 CrossRef CAS PubMed.
- Y. Feng, Q. Wang and T. Wang, BioMed. Res. Int., 2017, 1289259, DOI:10.1155/2017/1289259.
- O. Slater, B. Miller and M. Kontoyianni, Curr. Top. Med. Chem., 2020, 20, 855–882 CrossRef CAS PubMed.
- M. A. Ghadie, J. Coulombe-Huntington and Y. Xia, Curr. Opin. Struct. Biol., 2018, 50, 42–48 CrossRef CAS PubMed.
- X. Wang, X. Wei, B. Thijssen, J. Das, S. M. Lipkin and H. Yu, Nat. Biotechnol., 2012, 30, 159–164 CrossRef CAS PubMed.
- J. Westermarck, J. Ivaska and G. L. Corthals, Mol. Cell. Proteomics, 2013, 12, 1752–1763 CrossRef CAS.
- C. S. H. Tan, K. D. Go, X. Bisteau, L. Dai, C. H. Yong, N. Prabhu, M. B. Ozturk, Y. T. Lim, L. Sreekumar, J. Lengqvist, V. Tergaonkar, P. Kaldis, R. M. Sobota and P. Nordlund, Science, 2018, 359, 1170–1177 CrossRef CAS PubMed.
- J. De Las Rivas, D. Alonso-Lopez and M. M. Arroyo, Adv. Protein Chem. Struct. Biol., 2018, 111, 263–282 CAS.
- T.-Y. Yoon and H.-W. Lee, Curr. Opin. Chem. Biol., 2019, 53, 75–81 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS.
- E. Stefan, J. Troppmair and K. Bister, Adv. Protein Chem. Struct. Biol., 2018, 111, 101–132 CAS.
- L. Laraia, G. McKenzie, D. R. Spring, A. R. Venkitaraman and D. J. Huggins, Chem. Biol., 2015, 22, 689–703 CrossRef CAS PubMed.
- T. Clackson and J. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- A. A. Bogan and K. S. Thorn, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS PubMed.
- I. S. Moreira, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Bioinf., 2007, 68, 803–812 CrossRef CAS.
- M. B. Yaffe and A. E. H. Elia, Curr. Opin. Cell Biol., 2001, 13, 131–138 CrossRef CAS PubMed.
- M. B. Yaffe, Nat. Rev. Mol. Cell Biol., 2002, 3, 177–186 CrossRef CAS.
-
T. Pawson and P. Nash, in Handbook of Cell Signaling, ed. R. A. Bradshaw and E. A. Dennis, Academic Press, San Diego, 2nd edn, 2010, pp. 399–411, DOI:10.1016/B978-0-12-374145-5.00057-7.
- B. T. Seet, I. Dikic, M.-M. Zhou and T. Pawson, Nat. Rev. Mol. Cell Biol., 2006, 7, 473–483 CrossRef CAS PubMed.
- M. J. Lee and M. B. Yaffe, Cold Spring Harbor Perspect. Biol., 2016, 8, 8:a005918 Search PubMed.
- D. Beglov, D. R. Hall, A. E. Wakefield, L. Luo, K. N. Allen, D. Kozakov, A. Whitty and S. Vajda, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, e3416–e3425 CrossRef CAS PubMed.
- L. Raich, F. Noe, S. Olsson, K. Meier, J. Gunther, C. D. Christ and F. Noe, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017427118 CrossRef CAS PubMed.
- K. S. Lee, T. R. Burke Jr., J.-E. Park, J. K. Bang and E. Lee, Trends Pharmacol. Sci., 2015, 36, 858–877 CrossRef CAS.
- V. Archambault, G. Lepine and D. Kachaner, Oncogene, 2015, 34, 4799–4807 CrossRef CAS PubMed.
- J. E. Park, D. Hymel, T. R. Burke Jr. and K. S. Lee, F1000Research, 2017, 6, 1024, DOI:10.12688/f11000research.11398.12681.
- A. E. H. Elia, L. C. Cantley and M. B. Yaffe, Science, 2003, 299, 1228–1231 CrossRef CAS PubMed.
- A. E. Elia, P. Rellos, L. F. Haire, J. W. Chao, F. J. Ivins, K. Hoepker, D. Mohammad, L. C. Cantley, S. J. Smerdon and M. B. Yaffe, Cell, 2003, 115, 83–95 CrossRef CAS PubMed.
- S.-M. Yun, T. Moulaei, D. Lim, J. K. Bang, J.-E. Park, S. R. Shenoy, F. Liu, Y. H. Kang, C. Liao, N.-K. Soung, S. Lee, D.-Y. Yoon, Y. Lim, D.-H. Lee, A. Otaka, E. Appella, J. B. McMahon, M. C. Nicklaus, T. R. Burke Jr., M. B. Yaffe, A. Wlodawer and K. S. Lee, Nat. Struct. Mol. Biol., 2009, 16, 876–882 CrossRef CAS PubMed.
- D. Hymel and T. R. Burke Jr., ChemMedChem, 2017, 12, 202–206 CrossRef CAS.
- P. Sledz, C. J. Stubbs, S. Lang, Y.-Q. Yang, G. J. McKenzie, A. R. Venkitaraman, M. Hyvoenen and C. Abell, Angew. Chem., Int. Ed., 2011, 50, 4003–4006 CrossRef CAS.
- V. Archambault, P. P. D'Avino, M. J. Deery, K. S. Lilley and D. M. Glover, Genes Dev., 2008, 22, 2707–2720 CrossRef CAS PubMed.
- J. Xu, C. Shen, T. Wang and J. Quan, Nat. Struct. Mol. Biol., 2013, 20, 1047–1053 CrossRef CAS PubMed.
- K. Zhu, Z. Shan, L. Zhang and W. Wen, Structure, 2016, 24, 1110–1119 CrossRef CAS PubMed.
- F. Liu, J.-E. Park, W.-J. Qian, D. Lim, M. Graber, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., Nat. Chem. Biol., 2011, 7, 595–601 CrossRef CAS PubMed.
- F. Liu, J.-E. Park, W.-J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., ACS Chem. Biol., 2012, 7, 805–810 CrossRef CAS PubMed.
- F. Liu, J.-E. Park, W.-J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., ChemBioChem, 2012, 13, 1291–1296 CrossRef CAS PubMed.
- W.-J. Qian, J.-E. Park, K. S. Lee and T. R. Burke Jr., Bioorg. Med. Chem. Lett., 2012, 22, 7306–7308 CrossRef CAS PubMed.
- W. Qian, J.-E. Park, F. Liu, K. S. Lee and T. R. Burke, Bioorg. Med. Chem., 2013, 21, 3996–4003 CrossRef CAS PubMed.
- W.-J. Qian, J.-E. Park, D. Lim, C. C. Lai, J. A. Kelley, S.-Y. Park, K. W. Lee, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., Biopolymers, 2014, 102, 444–455 CrossRef CAS.
- X. Z. Zhao, D. Hymel and T. R. Burke Jr., Bioorg. Med. Chem. Lett., 2016, 26, 5009–5012 CrossRef CAS.
- X. Z. Zhao, D. Hymel and T. R. Burke Jr., Bioorg. Med. Chem., 2017, 25, 5041–5049 CrossRef CAS PubMed.
- X. Z. Zhao, K. Tsuji, D. Hymel and T. R. Burke Jr., Molecules, 2019, 24, 1488 CrossRef CAS.
- W.-J. Qian, J.-E. Park, R. Grant, C. C. Lai, J. A. Kelley, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., Biopolymers, 2015, 104, 663–673 CrossRef CAS.
- S. Hanessian and R. Margarita, Tetrahedron Lett., 1998, 39, 5887–5890 CrossRef CAS.
- V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2000, 39, 4348–4352 CrossRef CAS.
- T. R. Burke Jr., M. Knight and B. Chandrasekhar, Tetrahedron Lett., 1989, 30, 519–522 CrossRef.
- W.-J. Qian, J.-E. Park, D. Lim, S.-Y. Park, K.-W. Lee, M. B. Yaffe, K. S. Lee and T. R. Burke Jr., Chem. Biol., 2013, 20, 1255–1264 CrossRef CAS.
- W. Reindl, K. Strebhardt and T. Berg, Anal. Biochem., 2008, 383, 205–209 CrossRef CAS PubMed.
- W. Reindl, M. Graeber, K. Strebhardt and T. Berg, Anal. Biochem., 2009, 395, 189–194 CrossRef PubMed.
- N. Syed, P. Smith, A. Sullivan, L. C. Spender, M. Dyer, L. Karran, J. O'Nions, M. Allday, I. Hoffmann, D. Crawford, B. Griffin, P. J. Farrell and T. Crook, Blood, 2006, 107, 250–256 CrossRef CAS PubMed.
- Y. Yang, J. Bai, R. Shen, S. A. N. Brown, E. Komissarova, Y. Huang, N. Jiang, G. F. Alberts, M. Costa, L. Lu, J. A. Winkles and W. Dai, Cancer Res., 2008, 68, 4077–4085 CrossRef CAS.
- V. Ojea, M. Ruiz, G. Shapiro and E. Pombo-Villar, Tetrahedron Lett., 1994, 35, 3273–3276 CrossRef CAS.
- F. Liu, J.-E. Park, K. S. Lee and T. R. Burke Jr., Tetrahedron, 2009, 65, 9673–9679 CrossRef CAS.
- A. Niida, H. Tanigaki, E. Inokuchi, Y. Sasaki, S. Oishi, H. Ohno, H. Tamamura, Z. Wang, S. C. Peiper, K. Kitaura, A. Otaka and N. Fujii, J. Org. Chem., 2006, 71, 3942–3951 CrossRef CAS.
- G. M. Maharvi, A. E. Bharucha and A. H. Fauq, Bioorg. Med. Chem. Lett., 2013, 23, 2808–2811 CrossRef CAS PubMed.
- M. Culcasi, G. Casano, C. Lucchesi, A. Mercier, J.-L. Clément, V. Pique, L. Michelet, A. Krieger-Liszkay, M. Robin and S. Pietri, J. Med. Chem., 2013, 56, 2487–2499 CrossRef CAS.
- W. Qian, F. Liu and T. R. Burke, J. Org. Chem., 2011, 76, 8885–8890 CrossRef CAS.
- D. Hymel, R. A. Grant, K. Tsuji, M. B. Yaffe and T. R. Burke Jr., Bioorg. Med. Chem. Lett., 2018, 28, 3202–3205 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- N. W. Moriarty, R. W. Grosse-Kunstleve and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1074–1080 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob01120k |
| ‡ Current address: Discovery Chemistry, Novo Nordisk Research Center Seattle, Seattle, WA 98109, USA |
| § Current address: Department of Medicinal Chemistry, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, Tokyo 101-0062, Japan |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Robert A.
Grant
a,
Robert A.
Grant