
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled density glycodendron microarrays for studying carbohydrate–lectin interactions†
Antonio
Di Maio
 a,
Anna
Cioce
b,
Silvia
Achilli
c,
Michel
Thépaut
c,
Corinne
Vivès
c,
Franck
Fieschi
c,
Javier
Rojo
*a and
Niels-C.
Reichardt
*bd
a,
Anna
Cioce
b,
Silvia
Achilli
c,
Michel
Thépaut
c,
Corinne
Vivès
c,
Franck
Fieschi
c,
Javier
Rojo
*a and
Niels-C.
Reichardt
*bd
aGlycosystems Laboratory, Instituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, Av. Américo Vespucio 49, 41092 Seville, Spain. E-mail: a.di-maio@imperial.ac.uk; javier.rojo@iiq.csic.es
bCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramon 182, 20014, Donostia San Sebastián, Spain. E-mail: anna.cioce@crick.ac.uk; nreichardt@cicbiomagune.es
cUniv. Grenoble Alpes, CNRS, CEA, Institut de Biologie Structurale, 38100 Grenoble, France. E-mail: franck.fieschi@ibs.fr
dCIBER-BBN, Paseo Miramón 182, 20009 San Sebastian, Spain
First published on 13th August 2021
Abstract
Glycodendron microarrays with defined valency have been constructed by on-chip synthesis on hydrophobic indium tin oxide (ITO) coated glass slides and employed in lectin–carbohydrate binding studies with several plant and human lectins. Glycodendrons presenting sugar epitopes at different valencies were prepared by spotwise strain-promoted azide–alkyne cycloaddition (SPAAC) between immobilised cyclooctyne dendrons and azide functionalised glycans. The non-covalent immobilisation of dendrons on the ITO surface by hydrophobic interaction allowed us to study dendron surface density and SPAAC conversion rate by in situ MALDI-TOF MS analysis. By diluting the dendron surface density we could study how the carbohydrate–lectin interactions became exclusively dependant on the valency of the immobilised glycodendron.
Glycan microarrays are now well established high-throughput research tools for studying the specificity of novel lectins and carbohydrate processing enzymes.1–4
Monovalent carbohydrate–glycan interactions are usually weaker than most specific protein–protein interactions but the lectin binding affinity of glycans can be enhanced by over 3 orders of magnitude by the dense and multivalent presentation of glycans immobilised on a microarray surface.5 The surface display of glycans can favour the rapid rebinding of ligands to receptor domains and the chelation of multiple ligands with multivalent receptors leading to an overall increase in avidity. In nature, glycolipid clustering, N-glycan branching and repetition of recognition motifs on extended biopolymers like mucins or polysaccharides are mechanisms to achieve a similar increase in glycan-protein avidity.
For the preparation of microarrays, glycan ligands are either isolated from natural sources or prepared by chemical and enzymatic synthesis, and conveniently derivatized for printing onto microarray surfaces by a variety of chemical or physical methods.4
Glycans and their conjugates are usually synthesised in solution before immobilisation or isolated from natural sources but on-chip chemo-enzymatic assembly of glycan ligands as pioneered by our group and others can rapidly provide high density glycan microarrays with potentially very high savings in time and resources.9–11 Monitoring the conversion of surface-based reactions is important for assessing ligand homogeneity but at the same time challenging due to the limited number of compatible available analytical methods.
Fluorescently labelled lectins that bind specifically to starting materials and products can be used to qualitatively monitor the progress of enzymatic glycosylations using recombinant glycosyltransferases.11
With the development of surfaces that are compatible with analysis by mass spectrometry and microarray technology, the analysis of surface based reactions including the construction of glycan arrays, was finally possible.12–17As a variable surface density can affect the strength of interaction between glycans and lectins, several groups have developed strategies to homogenize and control glycan surface density.18,19
A number of approaches employ macromolecules like polymers, cyclic peptides, peptide nucleic acids, proteins or dendrons that have been functionalized with glycans at a discrete inter-glycan spacing.20–22 Immobilisation of these well-defined glyco-macromolecules however, results in variable inter-glycan distances due to the non-homogenous functionalisation of the surface with the glycoconjugates.23
Glycan surface densities have been assessed using a fluorescent linker for attachment24 for direct quantification of immobilised species, by wash off experiments with fluorescent ligands5or by autoradiography after enzymatic extension of ligands with a tritium labelled probe.6Via tritium imaging we have previously visualized how printing of different glycans at the same concentration lead to a variable surface density presumably due to differences in size, hydrophobicity and structure dependant kinetics for the immobilisation reaction.6–8
With the exception of the tritium imaging method, the mentioned methods only provide a rough estimation of ligand density as a function of the ligand printing concentration but no data on individual ligand densities. Being able to control and assess glycan ligand density however is key for the development of well-defined and reproducible glycan microarrays for quantitative applications in clinical diagnostics and glycomics research. In addition, it is helpful for evaluating multivalent display systems for solution phase applications by ensuring that the multivalent presentation on the surface matches those used in solution phase, eliminating the congestion surface effect.19,25 Here we present a strategy for preparing glycan arrays with a homogenous and constant ligand density by a process comprising (1) the non-covalent coating of a slide with a layer of a NHS-functionalized lipid, (2) attachment of a dendron spacer carrying a bioorthogonal cyclooctyne group and (3) complete functionalisation of the spacer with different glycans by SPAAC.
Recently Mende et al. published a similar strategy based on the on-chip synthesis of multivalent alkyne-functionalized peptides which were coupled with azido glycans by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC).26
The authors prepared a homogenous surface layer of alkyne-functionalized tetrapeptides by an on-chip laser assisted printing process termed cLIFT. While the described process is likely to produce more homogenous neoglycopeptide arrays than by printing of pre-synthesized multivalent ligands, the surface homogeneity or density was not directly assessed.
Our approach towards glycan arrays with constant glycan density made use of a previously developed microarray platform where glycan ligands are attached via hydrophobic interactions to a lipophilic indium tin oxide (ITO) glass slide.12 Microarrays prepared in this manner are fully compatible with readout both by fluorescence imaging and MALDI-TOF MS which allowed us to monitor all reactions carried out on the slide. For the glycan microarray employed in this study, we prepared a novel class of cyclooctyne activated mono-bi- and tetravalent dendrons (1–3) based on a bis-MPA (2,2-bismethylpropionic acid) synthetic strategy.12 These dendrons are functionalised with an amine group at the focal point allowing the immobilisation on the surface, a bis-MPA skeleton and cyclooctyne groups at the terminal points, ready to be involved for the SPAAC conjugation with a selection of simple azido ethylglycosides (4–7)27,28 (Fig. 1).
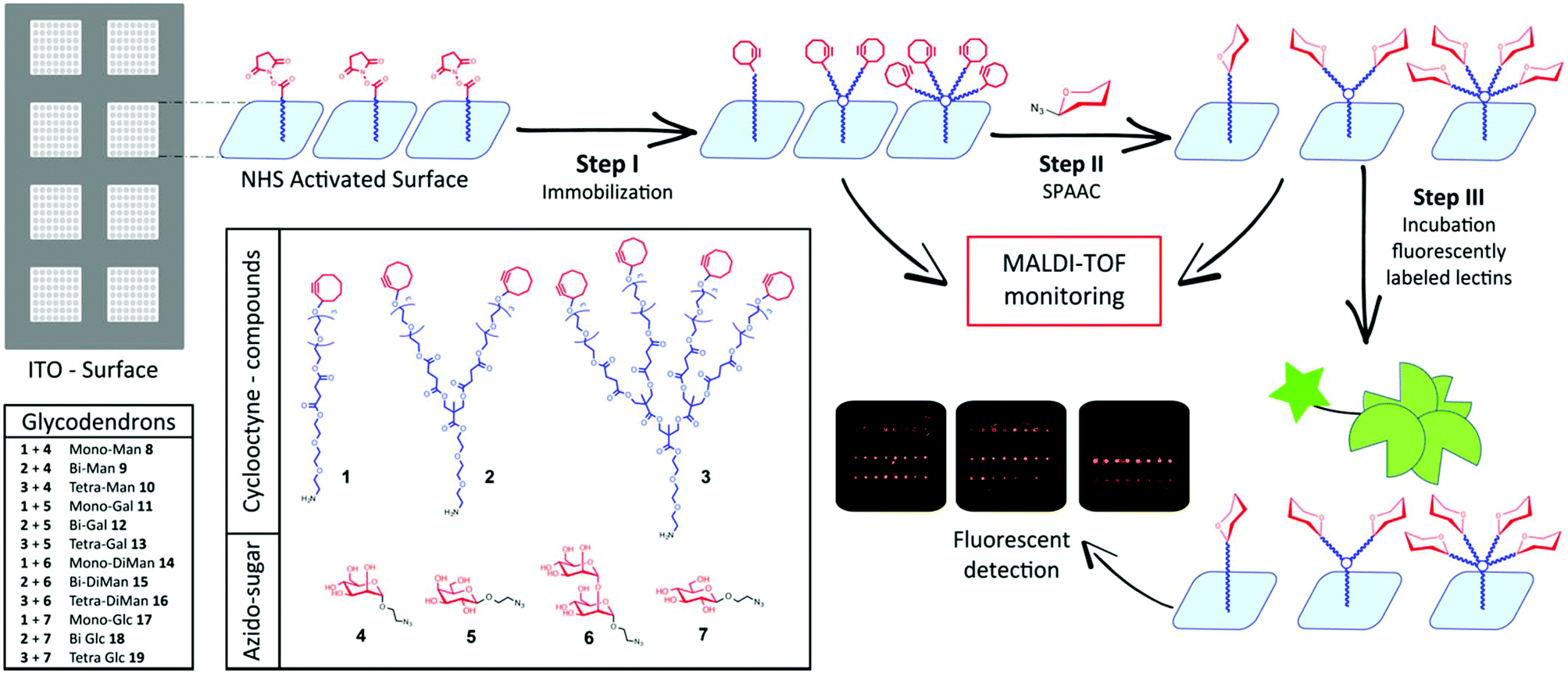 | ||
| Fig. 1 General outline of the glycodendron array strategy. | ||
For the assembly of the glycodendron arrays we functionalised commercial indium tin oxide slides first with a layer of octadecylphosphonate onto a NHS-activated carbamate linker (Fig. 2a) by spray-coating as reported previously.13
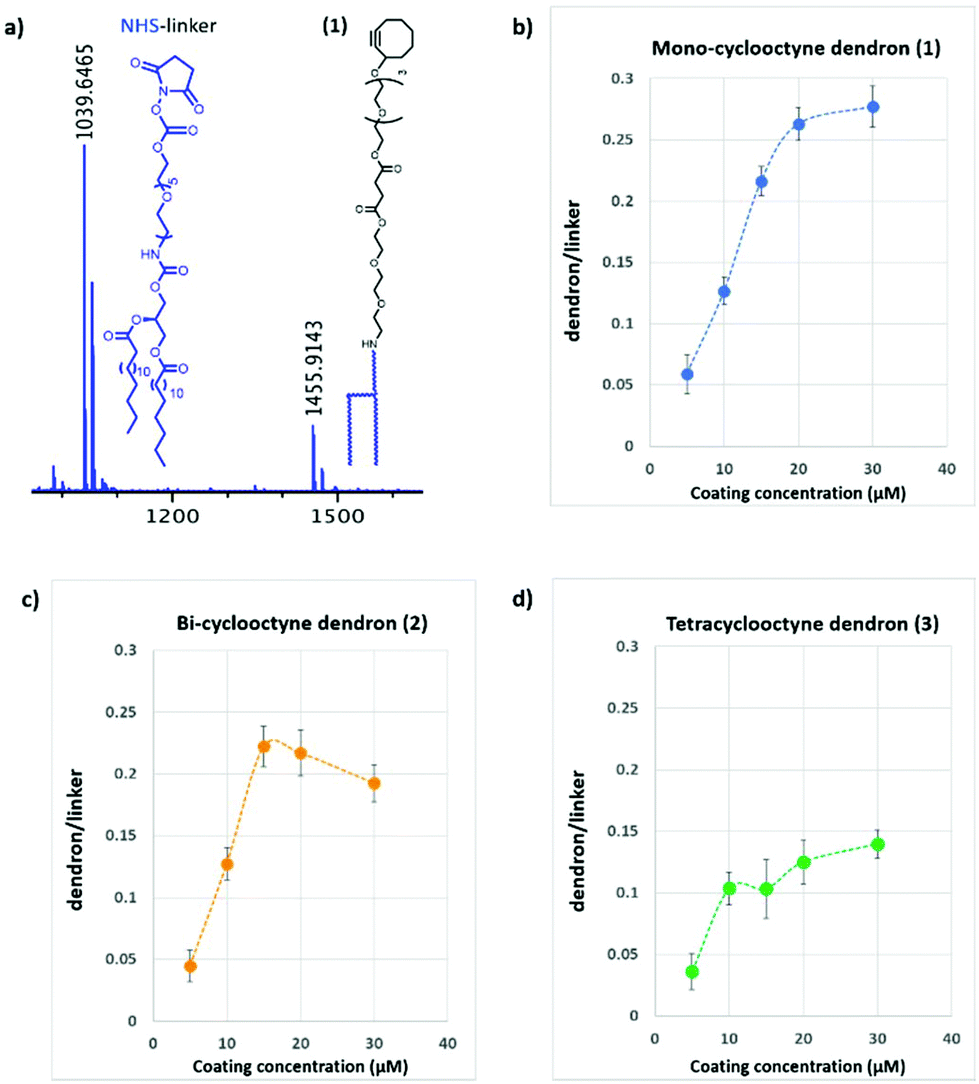 | ||
| Fig. 2 (a) MALDI-TOF spectrum showing immobilized Mono-cyclooctyne (1) mass peak (m/z 1456) vs unreacted NHS-linker mass peak (m/z 1040). (b–d) Graphs of the dendron/linker ratio against coating concentration. | ||
This hydrophobic bilayer was then functionalised with cyclooctyne-dendrons (1–3), at different concentrations and in separate wells (Step I, Fig. 1). To eliminate any congestion surface effects we explored surface functionalisation at low dendron concentrations between 5–30 μM (Fig. 2). At higher dendron dilution we reasoned that the glycan surface density would be easily adjustable by employing dendron spacers with one, two or four terminal cyclooctyne groups (Fig. 1).
The coating density with cyclooctyne dendrons 1–3 was determined by mass spectrometry as the ratio of dendron-conjugate and unreacted free NHS activated linker (m/z 1040) which was used as internal standard. The effect of linker concentration used for coating on the surface density measured as dendron/linker ratio is depicted in Fig. 2 for mono-, bi- and tetravalent cyclooctyne dendrons 1–3. As a general trend, we observed a linear increase of the relative surface density with increasing dendron concentration up to a maximum where saturation in surface density occurred. We found a compound-dependant surface saturation at 20 μM for the monovalent ligand 1 while the bulkier bi- and tetra-valent dendrons 2 and 3 saturated at lower coating concentrations around 15 and 10 μM respectively (Fig. 2b–d).
The variation observed in dendron/linker ratios, for the different dendrons, is likely to reflect both the structure-dependent ionization and the spatial requirements during immobilisation.
It is of note that at lower concentration (5–10 μM) the three dendrons showed a similar density while at higher coating concentrations the dendron structures strongly affect the immobilization ratio.
These results suggest that a comparable ligand density for the three multivalent scaffolds (1–3) can be achieved for a coating concentration between 5–10 μM. Moreover, surfaces functionalized with dendrons at high dilution lead to low surface densities where the binding event is likely determined by dendron valency and architecture alone and with minimal “bridging” of lectins with 2 more immobilised ligand molecules.
To study the effect of the ligand density on multivalent binding, dendron surfaces (1–3) were prepared at three coating concentrations (5, 10, and 15 μM), coupled to azido linked mannose (4) and α1,2-dimannose (6) via spot-wise printing and screened against fluorescently labelled Concanavalin A (Fig. 3).
 | ||
| Fig. 3 Fluorescence quantification of Concanavalin A binding to Man (8–10) and αMan1,2Man-based (14–16) glycodendrons at different dendrons coating concentration (5, 10 and 15 μM). | ||
At lower density (5–10 μM) tetravalent glycodendrons functionalized either with α1,2-dimannose (6) or mannose (4) bound Con A stronger than the mono- and bivalent scaffolds. The dimannoside ligand bound stronger to ConA than the mannose monosaccharide at all dendron valencies studied (Fig. 3). At higher glycodendron densities (15 μM), tetravalent mannose (10) and α1,2-dimannose (16) showed similar binding strength to ConA. Moreover, no differences in binding between the mono- (8) and the bivalent (9) mannose scaffolds were observed.
Based on these initial results, we chose to functionalize the microarray surface with dendrons at a low printing concentration of 5 μM. Arrays of mono, bi- and tetravalent glycodendrons were then prepared via SPAAC by spotting azido-ethyl glycosides (4–7) solutions onto cyclooctyne surfaces (1–3) (Step II, Fig. 1). On-chip SPAAC reactions were evaluated by MALDI TOF MS to ensure complete conversion for all printed glycosides. The surface coating at low dendron densities, together with the complete conversion in SPAAC conjugation (ESI†), provided homogeneous glycodendron presentations for the study of valency dependant carbohydrate lectin binding, excluding the multivalent effect from the array. For our binding study (Step III, Fig. 1) we chose the two commercially available plant lectins Pisum sativum (PSA) and Wisteria floribunda (WFA) and the extracellular domains (ECD) of the human lectins receptors DC-SIGN, DC-SIGNR, Langerin and Dectin-2 (Fig. 4).
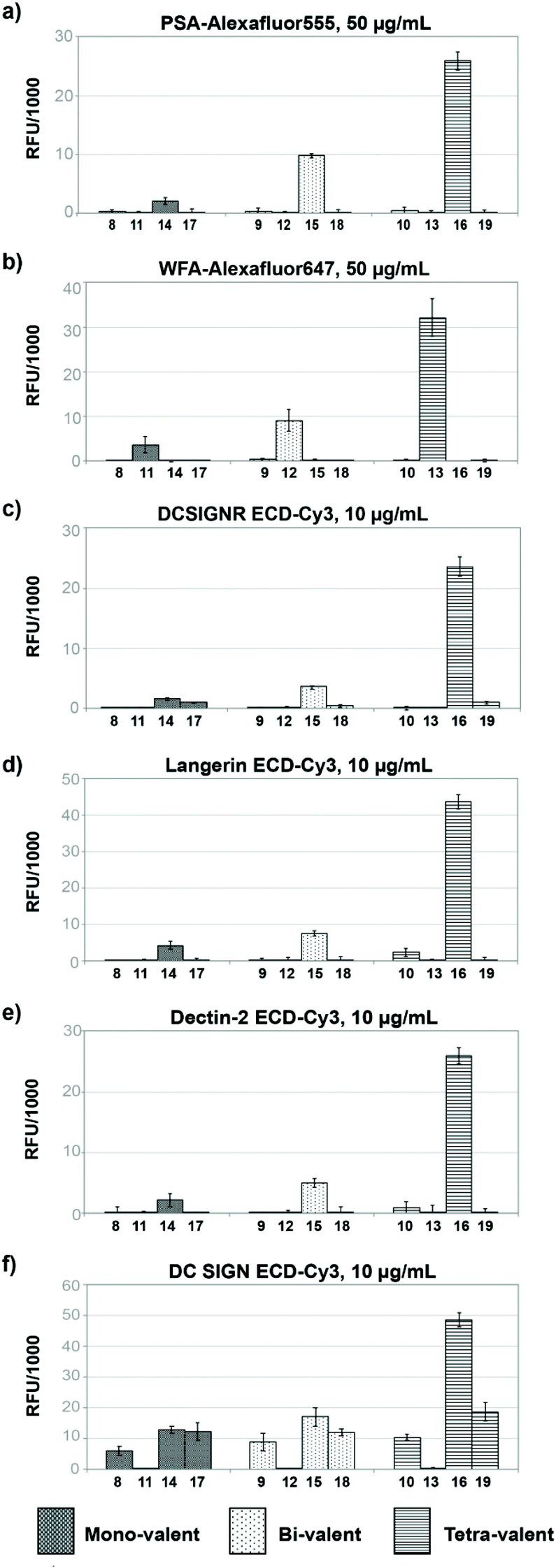 | ||
| Fig. 4 Glycodendron binding profiles to (a) PSA, (b) WFA, (c) DC-SIGNR ECD, (d) Langerin ECD, (e) Dectin-2 ECD, (f) DC-SIGN ECD. | ||
Most glycan arrays, currently employed in glycomics research, are printed at ligand concentrations between 50–100 μM for which saturated ligand immobilisation was observed for the majority of glycans. Here, we chose to functionalize surfaces with the dendrons at a highly dilute concentration of 5 μM to deliberately achieve low surface densities where the binding event would be largely determined by the dendron valency and architecture thus avoiding crosslinking of lectins to two or more immobilised ligand molecules.
Plant lectins
In our assay dimeric PSA29 with preference for binding Fucα1,6GlcNAc and α-Man residues showed no binding to the mannose (8–10), galactose (11–13) or glucose derivatives (17–19) independent of their valency. Only structures presenting disaccharide Manα1,2Man (14–16) were bound by the lectin with a strong increase in binding when moving from the mono to the tetravalent species (Fig. 4a). Dendrons functionalised with mannose monosaccharide were not bound at this low surface concentration.In the case of monomeric WFA,30 a lectin with preference for terminal GalNAc structures, the lectin bound the galactoside derivatives (11–13) with a strong effect of ligand valency in the binding (Fig. 4b).
Human lectins
The human tetravalent C-type lectin DC-SIGNR ECD,31,32 trivalent Langerin ECD33,34 and the monomeric Dectin-2 ECD35 showed highly selective binding only to constructs presenting the Manα1,2Man epitope, (14–16) with increasing binding intensity for higher valency. As with PSA lectin, no binding was observed towards dendrons functionalised with mannose monosaccharide (8, 9, 10).For DC-SIGN ECD,13 we observed similar binding strength towards mono- and divalent dendrons displaying mannose monosaccharide (8,9), Manα1,2Man (14,15) and glucose (17,18) but strongest binding to the Manα1,2Man disaccharide (16) in the tetravalent form (Fig. 4f). In line with the known binding specificity of DC-SIGN, no binding was observed for the dendrons presenting galactose residues (11–13).
This nearly 5-fold increase in affinity for the Manα1,2Man in a tetravalent presentation shows the gain in selectivity through a particular multivalent presentation in an otherwise more promiscuous lectin. Brewer et al. have explained this behaviour with a threshold epitope density required to produce a productive interaction between lectins and glycans that is capable to trigger a biological signalling event e.g. in cellular immunity.23
As the lectins show a different degree of labelling (DOL) the fluorescence values (RFU) obtained should not be compared directly between each other without prior normalisation (see ESI†).
In conclusion, we have developed a rapid screening platform to study multivalent carbohydrate lectin interactions as a function of dendron valency and carbohydrate epitope. The glycodendron on-chip synthesis was assisted by MALDI-TOF MS analysis to monitor the conversion of surface based SPAAC reactions.
The dilute ligand presentation on the array surface implied that ligand valency was mainly determined by the dendron architecture, and in consequence allowed us to identify matched glycan epitope/dendron combinations with high avidity and much-improved selectivity over other structurally similar compounds. We anticipate this type of glycan array, which includes defined ligand presentation as an additional parameter, to be a valuable tool for the discovery of multivalent glycoconjugates as antagonists to interfere in clinically relevant carbohydrate–lectin interactions such as pathogen infection or glycoimmune therapy.
Conflicts of interest
No conflicts of interest are declared.Acknowledgements
A.D., A.C., S.A., J.R., N.C.R and F.F. were supported by the EU Horizon 2020 Research and Innovation Program (Marie Sklodowska-Curie Grant 642870, ETN-Immunoshape). N.C.R additionally acknowledges funding from the Ministry of Science and Education (MINECO) Grant No. CTQ2017-90039-R and RTC-2017-6126-1 and the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency-Grant No. MDM-2017-0720. Also, J.R. acknowledges funding the MINECO, Grant No. CTQ2017-86265-P and ISCIII RETICS ARADyAL (RD16/0006/0011). These grants were co-funded by the European Regional Development Fund (ERDF). This work used the Multistep Protein Purification Platform of the Grenoble Instruct-ERIC center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology, supported by FRISBI (ANR-10-INBS-05-02) and GRAL, within the Univ. Grenoble Alpes graduate school CBH-EUR-GS (ANR-17-EURE-0003). F.F. also acknowledges the French Agence Nationale de la Recherche (ANR) PIA for Glyco@Alps (ANR-15-IDEX-02).Notes and references
- C. Ruprecht, A. Geissner, P. H. Seeberger and F. Pfrengle, Carbohydr. Res., 2019, 481, 31–35 CrossRef CAS PubMed.
- C. Gao, M. Wei, T. R. McKitrick, A. M. McQuillan, J. Heimburg-Molinaro and R. D. Cummings, Front. Chem., 2019, 7, 1–18 CrossRef PubMed.
- A. Geissner and P. H. Seeberger, Annu. Rev. Anal. Chem., 2016, 9, 223–247 CrossRef CAS PubMed.
- C. D. Rillahan and J. C. Paulson, Annu. Rev. Biochem., 2011, 80, 797–823 CrossRef CAS PubMed.
- P.-H. Liang, S.-K. Wang and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 11177–11184 CrossRef CAS PubMed.
- S. Serna, C. H. Hokke, M. Weissenborn, S. Flitsch, M. Martin-Lomas and N. C. Reichardt, ChemBioChem, 2013, 14, 862–869 CrossRef CAS PubMed.
- J. Y. Hyun, J. Pai and I. Shin, Acc. Chem. Res., 2017, 50, 1069–1078 CrossRef CAS PubMed.
- M. Mende, V. Bordoni, A. Tsouka, F. F. Loeffler, M. Delbianco and P. H. Seeberger, Faraday Discuss., 2019, 219, 9–32 RSC.
- L. Ban and M. Mrksich, Angew. Chem., Int. Ed., 2008, 47, 3396–3399 CrossRef CAS PubMed.
- J. Voglmeir, R. Sardzík, M. J. Weissenborn and S. L. Flitsch, OMICS, 2010, 14, 437–444 CrossRef CAS PubMed.
- S. Serna, J. Etxebarria, N. Ruiz, M. Martin-Lomas and N.-C. Reichardt, Chemistry, 2010, 16, 13163–13175 CrossRef CAS PubMed.
- A. Beloqui, J. Calvo, S. Serna, S. Yan, I. B. H. Wilson, M. Martin-Lomas and N. C. Reichardt, Angew. Chem., Int. Ed., 2013, 52, 7477–7481 CrossRef CAS PubMed.
- A. Cioce, M. Thépaut, F. Fieschi and N. C. Reichardt, Chem. – Eur. J., 2020, 26, 12809–12817 CrossRef CAS PubMed.
- A. Sanchez-Ruiz, S. Serna, N. Ruiz, M. Martin-Lomas and N. C. Reichardt, Angew. Chem., Int. Ed., 2011, 50, 1801–1804 CrossRef CAS PubMed.
- S. Y. Tseng, C.-C. Wang, C.-W. Lin, C.-L. Chen, W.-Y. Yu, C.-H. Chen, C.-Y. Wu and C.-H. Wong, Chem. – Asian J., 2008, 3, 1395–1405 CrossRef CAS PubMed.
- L. Ban, N. Pettit, L. Li, A. D. Stuparu, L. Cai, W. Chen, W. Guan, W. Han, P. G. Wang and M. Mrksich, Nat. Chem. Biol., 2012, 8, 769–773 CrossRef CAS PubMed.
- C. J. Gray, L. G. Migas, P. E. Barran, K. Pagel, P. H. Seeberger, C. E. Eyers, G.-J. Boons, N. L. B. Pohl, I. Compagnon, G. Widmalm and S. L. Flitsch, J. Am. Chem. Soc., 2019, 141, 14463–14479 CrossRef CAS PubMed.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 15732–15742 CrossRef CAS PubMed.
- O. Oyelaran, Q. Li, D. Farnsworth and J. C. Gildersleeve, J. Proteome Res., 2009, 8, 3529–3538 CrossRef CAS PubMed.
- K. T. Huang, K. Gorska, S. Alvarez, S. Barluenga and N. Winssinger, ChemBioChem, 2011, 12, 56–60 CrossRef CAS PubMed.
- N. Parera Pera, H. M. Branderhorst, R. Kooij, C. Maierhofer, M. Van Der Kaaden, R. M. J. Liskamp, V. Wittmann, R. Ruijtenbeek and R. J. Pieters, ChemBioChem, 2010, 11, 1896–1904 CrossRef CAS PubMed.
- C. Tiertant, D. Goyard and O. Renaudet, ACS Omega, 2018, 3, 1403–14020 CrossRef PubMed.
- T. K. Dam and C. F. Brewer, Glycobiology, 2010, 20, 270–279 CrossRef CAS PubMed.
- X. Song, B. Xia, Y. Lasanajak, D. F. Smith and R. D. Cummings, Glycoconjugate J., 2008, 25, 15–25 CrossRef CAS PubMed.
- N. V. Shilova, O. E. Galanina, T. V. Pochechueva, A. a. Chinarev, V. a. Kadykov, A. B. Tuzikov and N. V. Bovin, Glycoconjugate J., 2005, 22, 43–51 CrossRef CAS PubMed.
- M. Mende, A. Tsouka, J. Heidepriem, G. Paris, D. S. Mattes, S. Eickelmann, V. Bordoni, R. Wawrzinek, F. F. Fuchsberger, P. H. Seeberger, C. Rademacher, M. Delbianco, A. Mallagaray and F. F. Loeffler, Chem. – Eur. J., 2020, 26, 9954–9963 CrossRef CAS PubMed.
- E. Arce, P. M. Nieto, V. Díaz, R. García Castro, A. Bernad and J. Rojo, Bioconjugate Chem., 2003, 14, 817–823 CrossRef CAS PubMed.
- J. J. Reina, A. Di Maio, J. Ramos-Soriano, R. C. Figueiredo and J. Rojo, Org. Biomol. Chem., 2016, 14, 2873–2882 RSC.
- I. J. Goldstein and C. E. Hayes, Adv. Carbohydr. Chem. Biochem., 1978, 35, 127–340 CrossRef CAS PubMed.
- T. Kurokawa, M. Tsuda and Y. Sugino, J. Biol. Chem., 1976, 251, 5686–5693 CrossRef CAS.
- E. J. Soilleux, R. Barten and J. Trowsdale, J. Immunol., 2000, 165, 2937–2942 CrossRef CAS PubMed.
- D. A. Mitchell, A. J. Fadden and K. Drickamer, J. Biol. Chem., 2001, 276, 28939–28945 CrossRef CAS PubMed.
- L. De Witte, A. Nabatov, M. Pion, D. Fluitsma, M. A. W. P. De Jong, T. De Gruijl, V. Piguet, Y. Van Kooyk and T. B. H. Geijtenbeek, Nat. Med., 2007, 13, 367–371 CrossRef CAS PubMed.
- M. A. W. P. de Jong, L. E. M. Vriend, B. Theelen, M. E. Taylor, D. Fluitsma, T. Boekhout and T. B. H. Geijtenbeek, Mol. Immunol., 2010, 47, 1216–1225 CrossRef CAS PubMed.
- S. Saijo, S. Ikeda, K. Yamabe, S. Kakuta, H. Ishigame, A. Akitsu, N. Fujikado, T. Kusaka, S. Kubo, S.-H. Chung, R. Komatsu, N. Miura, Y. Adachi, N. Ohno, K. Shibuya, N. Yamamoto, K. Kawakami, S. Yamasaki, T. Saito, S. Akira and Y. Iwakura, Immunity, 2010, 32, 681–691 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, microarray analysis, conditions for the production of C-type lectin receptors and spectroscopic data for all novel compounds. See DOI: 10.1039/d1ob00872b |
| This journal is © The Royal Society of Chemistry 2021 |