Fluorescence resonance energy transfer-based screening for protein kinase C ligands using 6-methoxynaphthalene-labeled 1,2-diacylglycerol-lactones†
Received
26th April 2021
, Accepted 21st July 2021
First published on 21st July 2021
Abstract
Protein kinase C (PKC) is associated with a central cellular signal transduction pathway and disorders such as cancer and Alzheimer-type dementia and is therefore a target for the treatment of these diseases. The development of simple methods suitable for high-throughput screening to find potent PKC ligands is desirable. We have developed an assay based on fluorescence-quenching screening with a solvatochromic fluorophore attached to a competitive probe and its alternative method based on Förster/fluorescence resonance energy transfer (FRET) phenomena. Here, an improved FRET-based PKC binding assay using a diacylglycerol (DAG) lactone labeled with a donor fluorescent dye, 6-methoxynaphthalene (6MN), was developed. The 6MN-labeled DAG-lactone has a higher binding affinity for the PKCδ C1b domain and the fluorescent PKCδ C1b domain labeled by fluorescein as an acceptor fluorescent dye (Fl-δC1b) than the diethylaminocoumarin (DEAC)-labeled DAG-lactone. The combination of the 6MN-labeled DAG-lactone and Fl-δC1b showed a change in fluorescence response larger than that of the DEAC-labeled DAG-lactone and Fl-δC1b. The IC50 values of known PKC ligands calculated by the present FRET-based method using 6MN-labeled DAG-lactone agree well with the Ki values obtained by the conventional radioisotope-based assays. Some false positive compounds, identified by the previous solvatochromic fluorophore-based method, were found to be negative by this method. The present FRET-based PKC binding assay is more sensitive and could be more useful.
Introduction
Protein kinase C (PKC) is involved in cellular signal transduction and can lead to proliferation, differentiation, migration and apoptosis.1–9 PKC is a significant target of therapeutic agents because it is linked to several disorders such as diabetes, cancer, heart diseases, autoimmune diseases and Alzheimer's disease.10–13 The PKC isozymes are classified into conventional PKCs (cPKC) (α, βI/II, and γ), novel PKCs (nPKC) (δ, ε, η, and θ) and atypical PKCs (aPKC) (ζ, ι/λ).8 The cPKCs and nPKCs are regulated by ligand binding through their tandem C1 (C1a, C1b) domains, but the aPKCs contain an atypical C1 domain. The endogenous ligand for PKC is sn-1,2-diacylglycerol (DAG), which is a second messenger,14 generated downstream from receptor tyrosine kinases and G-protein coupled receptors. DAG is produced at the inner face of the plasma membranes and its binding to the C1 domain of PKC causes a conformational change of PKC into the active form and translocation,15,16 which is followed by signaling through multiple downstream pathways.17–19 Except in the case of aPKC, which fails to bind to DAG,20 the binding of DAG to the C1 domain of PKC is an important step in the activation process. Many PKC ligands targeting the C1 domains have been found by radioisotope (RI) assays21–24 to be pharmacological probes or drug leads. Among these synthetic PKC ligands, we have focused on DAG-lactones, which are conformationally constrained cyclic DAG derivatives containing a γ-lactone ring. Many DAG-lactones have been found to provide potent PKC binding affinity as well as selectivity between classes of targets containing a C1 domain.25,26 RI assays are highly sensitive and have been widely used for the evaluation of ligand binding, but fluorescence-based assays are more suitable for high-throughput screening because they are free of problems associated with radioactive chemicals. For this reason, we have developed fluorescence-based ligand screening assays27,28 as alternatives to RI assays. A fluorescence-based assay for PKC-ligand binding affinity using a synthetic PKCδ C1b domain labeled with a solvatochromic fluorophore on the edge of its ligand-binding pocket can detect ligand binding through changes in the surrounding environment.29 However, this assay is not suitable for high-throughput screening for several reasons. The change of fluorescence intensity is affected by the different properties of test compounds. A fluorescent dye on the edge of PKCδ C1b might interrupt the correct folding and limit the accuracy of assay systems. The development of Förster/fluorescence resonance energy transfer (FRET)-based PKC assays which monitor kinase activity or ligand interactions by recombinant proteins with a combination of genetically encoded fluorescent proteins has been reported. Such assays involve cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP), or green fluorescent protein 2 (GFP2) and enhanced yellow fluorescent protein (EYFP).30,31 These assays detect the interactions between fluorescent proteins and cannot detect PKC-binding compounds that do not cause conformational changes. The interaction of small fluorescent compounds with each other is critical for the detection of compounds that bind to PKC. Originally, FRET is useful to monitor this interaction because it involves the transfer of excitation energy from a fluorescence donor to an acceptor and its efficiency and sensitivity depend on the separation of the donor and the acceptor. The FRET phenomenon is a purely spectroscopic measurement32–34 but a FRET-mediated competitive assay can be used for studies of the binding between two molecules.35,36 In our previous work, FRET phenomena between the DAG-lactone labeled by a FRET donor diethylaminocoumarin (DEAC)37–40 at the α-alkylidene position of the sn-2 DEAC-type DAG-lactone (1) and a PKCδ C1b domain containing a FRET acceptor fluorescein at the position 235 (Fl-δC1b) were examined (Fig. 2),41 and the competitive binding of PKC ligands such as PDBu to the δC1b domain was measured. This FRET-based competitive assay does not require the introduction of a fluorescent dye on the edge of PKCδ C1b, and thus it has some advantages. The correct folding of the fluorescein-labeled δC1b domain is not interrupted, and the FRET intensity is not correlated with the fluorescence properties of the test compounds. In this study, a FRET donor fluorophore, which was introduced in a DAG-lactone, was refined and the development of a new applicable FRET-based competitive assay was undertaken.
Results and discussion
Adoption of FRET-donor molecules
As FRET-donor molecules (Fig. 1), the previously used sn-2 DEAC-type DAG-lactone (1)41 and the DAG-lactone labeled with 6-methoxynaphthalene (6MN)42 at the sn-2 position (sn-2 6MN-type DAG-lactone) (2), which has been used as a fluorescent probe in PKC ligand screening assays,28 were used, and their fluorescence spectra in MeOH, 50% MeOH/H2O, or H2O were measured (Fig. 2). MeOH/H2O systems were used because probes in MeOH might be considered to be in a hydrophobic environment, whereas those in H2O might be in a hydrophilic environment. sn-2 DEAC-type DAG-lactone (1) showed the highest fluorescence intensity in MeOH, while sn-2 6MN-type DAG-lactone (2) showed the highest fluorescence intensity in 50% MeOH/H2O. Under these conditions, sn-2 6MN-type DAG-lactone 2 showed less solvatochromic response than sn-2 DEAC-type DAG-lactone 1. To suppress the changes of the fluorescence properties of probes by non-specific hydrophobic interactions with competitors, less sensitivity of solvatochromic properties is considered to be more suitable for FRET assay probes.
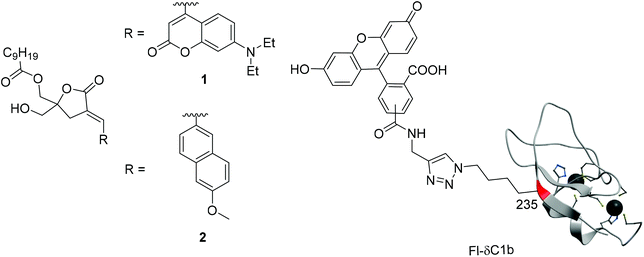 |
| | Fig. 1 Structures of a FRET donor (left) and a FRET acceptor (right). Left: sn-2 DEAC-type DAG-lactone (1) and sn-2 6MN-type DAG-lactone (2); right: Fl-δC1b(231–281); Val235 (red) is replaced by a norleucine (Nle) derivative labeled with fluorescein at the ε-position. | |
 |
| | Fig. 2 Fluorescence spectra of 50 nM sn-2 DEAC-type DAG-lactone (1) (a) and 50 nM sn-2 6MN-type DAG-lactone (2) (b) in MeOH (green), 50% MeOH/H2O (red), and H2O (blue), measured by excitation of 1 at 380 nm or 2 at 340 nm. | |
In the previous FRET-based competitive assay, sn-2 DEAC-type DAG-lactone (1) and Fl-δC1b were used as a FRET donor and acceptor, respectively.41 In this study, the δC1b(231–281) binding affinity of sn-2 6MN-type DAG-lactone (2) (Ki = 35.9 nM, Table 1) was higher than that of sn-2 DEAC-type DAG-lactone (1) (Ki = 182 nM).28,41 Thus, the binding affinity to Fl-δC1b(231–281) of these labeled DAG-lactones was investigated by the conventional RI assay using [3H]PDBu because the Kd value of Fl-δC1b for [3H]PDBu binding (0.75 nM) is comparable to that of δC1b (0.41 nM).41 The sn-2 6MN-type DAG-lactone (2) showed a higher binding affinity for the Fl-δC1b domain (Ki = 46.3 nM, Table 1) than the sn-2 DEAC-type DAG-lactone (1) (Ki = 1050 nM). The sn-2 6MN-type DAG-lactone (2) showed almost the same level of binding affinity with the Fl-δC1b domain or the δC1b domain, while the sn-2 DEAC-type DAG-lactone (1) showed a remarkably lower binding affinity with the Fl-δC1b domain than with the δC1b domain. The use of the sn-2 6MN-type DAG-lactone (2) as a FRET donor would therefore lead to a larger FRET phenomenon than that of the sn-2 DEAC-type DAG-lactone (1) (Fig. 3).
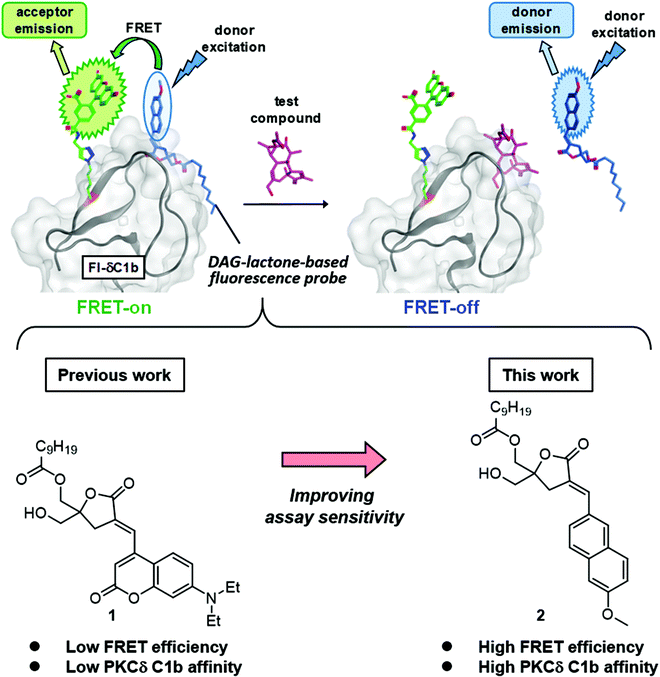 |
| | Fig. 3 FRET-based assays of PKC ligands. The excitation of the donor fluorescent dyes (DEAC (previous work) and 6MN (this work)), introduced in a DAG-lactone (1 and 2, respectively), causes the fluorescence emission of the acceptor fluorescent dye, fluorescein, which was introduced in Fl-δC1b (FRET-on). The 6MN-type DAG-lactone 2 can show greater FRET efficiency than DEAC-type DAG-lactone 1. Replacement of the probes 1 or 2 by a test compound causes a decrease in the fluorescence intensity of fluorescein (FRET-off). | |
Table 1
K
i values of fluorescent DAG-lactones in specific [3H]PDBu binding to δC1b and Fl-δC1b
| Ligand |
δC1b |
K
i (nM)a Fl-δC1b |
|
Inhibition constant for the synthetic ligand binding to synthetic δC1b or Fl-δC1b. Mean ± SEM.
Ref. 41.
Ref. 28.
|
|
sn-2 DEAC-type DAG-lactone (1) |
182 ± 18b |
1050 ± 193 |
|
sn-2 6MN-type DAG-lactone (2) |
35.9 ± 1.6c |
46.3 ± 5.7 |
FRET experiments
The C1b domain, which contains residues 231–281 of PKCδ, was used as the targeted moiety. Val235 was replaced by a fluorescein-labeled norleucine (Nle) derivative because Val235 is located on the surface of the domain. Its side chain is oriented toward the outside of the domain and it was presumed that the introduced fluorescein moiety would not affect the folding of the domain (PDB ID: 1PTR).41 Fl-δC1b (231–281) was then synthesized by Fmoc solid-phase peptide synthesis. Cycloaddition of the alkyne unit attaches fluorescein to the azide unit at the ε-position of the Nle235 residue, and subsequent native chemical ligation was achieved as described previously (Fig. 1).41 In our previous research, the distance between Fl-δC1b and sn-2 DEAC-type DAG-lactone (1) was found to be sufficient to support the FRET phenomena.41 In this study, the addition of Fl-δC1b (acceptor) to an assay solution containing sn-2 6MN-type DAG-lactone (2) (donor) caused a remarkable increase of the fluorescence intensity of the fluorescein acceptor. The addition of 2 equiv. of Fl-δC1b achieved saturation of the fluorescence intensity. In comparison with the combination of Fl-δC1b and sn-2 DEAC-type DAG-lactone (1), the combination of Fl-δC1b and sn-2 6MN-type DAG-lactone (2) showed a higher increase of the fluorescence intensity of the acceptor molecule in all the Fl-δC1b concentrations and thus a remarkably larger FRET phenomenon (Fig. 4a). Observation of the final FRET phenomenon was followed by the addition of PDBu as a competitive inhibitor. This led to a concentration-dependent decrease of the fluorescence intensity (Fig. 4b). The combination of Fl-δC1b and sn-2 6MN-type DAG-lactone (2) showed a larger change of the fluorescence intensity the acceptor molecule according to the PDBu concentrations than the combination of Fl-δC1b and sn-2 DEAC-type DAG-lactone (1). The increase of this ratio reveals the cancellation of the FRET phenomenon (FRET-off) by PDBu. Practically, the fluorescence spectra of sn-2 6MN-type DAG-lactone (2) with addition of the acceptors showed a large change (Fig. 4c), whereas the spectra of sn-2 DEAC-type DAG-lactone (1) with addition of the acceptors showed a modest change (Fig. 4d). The cancellation of the FRET phenomenon (FRET-off) by the addition of PDBu was remarkably observed in the case of sn-2 6MN-type DAG-lactone (2) (Fig. 4e), and barely in those of sn-2 DEAC-type DAG-lactone (1) (Fig. 4f). Consequently, the sn-2 6MN-type DAG-lactone (2) appears to be superior as a FRET donor to sn-2 DEAC-type DAG-lactone (1).
 |
| | Fig. 4 (a) Fluorescence intensity changes following the addition of the acceptor Fl-δC1b (0, 1, 2, 3, 4, and 5 equivalents (eq.)) to the donor sn-2 DEAC-type DAG-lactone (1) (blue square, λex = 380 nm) or sn-2 6MN-type DAG-lactone (2) (orange rhombus, λex = 340 nm) (1 eq.) (FRET-on), and (b) fluorescence intensity changes on addition of PDBu (0, 10, 20, 30 and 40 eq.) to the mixture of the acceptor Fl-δC1b (5 eq.) and the donor sn-2 DEAC-type DAG-lactone (1) (blue square) or sn-2 6MN-type DAG-lactone (2) (orange rhombus) (1 eq.) (FRET-off) are plotted. (c and d) Fluorescence spectra of sn-2 6MN-type DAG-lactone (2) or sn-2 DEAC-type DAG-lactone (1) with addition of the acceptors, respectively. (e and f) Fluorescence spectra of sn-2 6MN-type DAG-lactone (2) or sn-2 DEAC-type DAG-lactone (1) with addition of PDBu, respectively. | |
FRET-based competitive assays
A binding analysis of known PKC ligands was performed to assess the reliability of the FRET-based competitive assay. The IC50 values of PDBu, phorbol 12,13-diacetate (PDA) and DAG-lactone 3 were determined by this assay as 21.7, 156.3, and 71.4 nM, respectively (Fig. 5). The Ki values of PDBu, PDA, and 3, which were previously determined by the RI-based competitive assay using [3H]PDBu,28,29 were 0.72, 68.9, and 2.7 nM, respectively. The order of the IC50 values of these PKC ligands is identical to that of the Ki values.
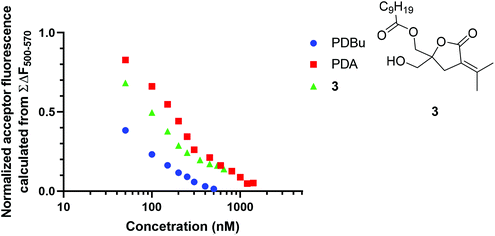 |
| | Fig. 5 Results of the FRET-based competitive assay of known PKC ligands: PDBu (blue round), phorbol 12, 13-diacetate (PDA) (red square) and DAG-lactones 3 (green triangle). IC50 (nM): 21.7, 156.3, and 71.4 (the order of PDBu, PDA, and 3). | |
Our previous fluorescence-quenching screening based on the competition with sn-2 6MN-type DAG-lactone (2) revealed 6 compounds (Fig. 6) as false positive hits from a chemical library, from the Tokyo Medical and Dental University Screening Center.28 The binding affinity of these 6 compounds was evaluated by the present FRET-based competitive assay (Fig. 6). Compounds 4, 6, 7, and 8 were successfully identified as negative compounds, whereas compounds 5 and 9 remained false positive hits. Compounds 5 and 9 caused the termination of the FRET phenomena (FRET-off) as observed with PKC ligands, possibly because of some effects of compounds 5 and 9 on Fl-δC1b or sn-2 6MN-type DAG-lactone (2). It is suggested that the present FRET-based competitive assay system could offer an evaluation that is more reliable than that in our previous fluorescence-quenching screening system, although the possibility of false positive hits must still be considered.
 |
| | Fig. 6 FRET-based competitive assay of PDBu (blue round), compounds 3 (red square), 4 (green triangle), 5 (purple triangle), 6 (orange rhombus), 7 (black round), 8 (brown square), and 9 (indigo triangle). The IC50 values (nM) of PDBu, 3, 5, and 9 were determined as 260, 130, 55, and 140, respectively. | |
Conclusions
In this study, a novel FRET-based competitive assay using sn-2 6MN-type DAG-lactone (2) as a donor molecule and Fl-δC1b as an acceptor molecule has been developed. This is an improvement over the previous assay which used sn-2 DEAC-type DAG-lactone (1) as a donor molecule. The sn-2 6MN-type DAG-lactone (2) probe possesses a binding affinity for the Fl-δC1b domain higher than that of sn-2 DEAC-type DAG-lactone (1). In comparison with the combination of Fl-δC1b and sn-2 DEAC-type DAG-lactone (1), the combination of Fl-δC1b and sn-2 6MN-type DAG-lactone (2) showed larger FRET phenomena (FRET-on) and also a larger change in the cancellation of FRET phenomena (FRET-off) by addition of PDBu. This suggests that sn-2 6MN-type DAG-lactone (2) is a FRET donor that is superior to sn-2 DEAC-type DAG-lactone (1). The present FRET-based competitive assay is compatible with the conventional RI-based competitive assay using [3H]PDBu when screening known PKC ligands. Some compounds, which were previously detected as false positive hits from a chemical library, were successfully identified as negative compounds by the present assay. The FRET-based competitive assay is applicable to PKC ligand screening, and this FRET donor/acceptor combination might be applicable to the screening of small compounds targeting other proteins.
Experimental
Preparation of peptides
The δC1b(231–281) domain and the Fl-δC1b(231–281) domain [δC1b(231–281)Val235Nle(ε-Fl)], in which Val235 is replaced by a norleucine (Nle) derivative labeled with fluorescein at the ε-position, were synthesized according to published procedures.41,43 The optimized native chemical ligation (NCL) conditions [Ac-δC1b(231–246)V235Nle(ε-Fl)-S(CH2)2CO2Et (1.85 mg, 0.592 μmol as the 4TFA salt) and H-δC1b(247–281)-NH2 (2.92 mg, 0.616 μmol as the 8TFA salt) in 50 mM sodium phosphate buffer (pH 7.4) containing 6 M guanidine hydrochloride, 20 mM 4-mercaptophenylacetic acid, and 10 mM tris(2-carboxyethyl)phosphine hydrochloride (final peptide concentration: 1 mM), and incubation for 8 h at 37 °C] were used to improve the yield of the NCL reaction (from 13%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 to 69% (3.10 mg)) in the synthesis of the Fl-δC1b(231–281) domain.
41 to 69% (3.10 mg)) in the synthesis of the Fl-δC1b(231–281) domain.
Preparation of DAG-lactone derivatives
sn-2 DEAC-type DAG-lactone (1), sn-2 6MN-type DAG-lactone (2), and DAG-lactone 3 were synthesized previously.25,26,28,41,44,45
[3H]PDBu binding assay
The dissociation constant (Kd) of synthetic PKCδ C1b in [3H]PDBu binding and the inhibitory constant (Ki) of fluorescent-labeled DAG-lactone for binding of the δC1b domain were assessed by a poly(ethylene glycol) precipitation assay as described previously.29,46,47
Fluorescence analysis
Solutions of 50 nM sn-2 DEAC-type DAG-lactone (1) and 50 nM sn-2 6MN-type DAG-lactone (2) in MeOH, 50% MeOH/H2O and H2O were prepared. Fluorescence spectra were recorded by excitation at 380 nm for sn-2 DEAC-type DAG-lactone (1) or at 340 nm for sn-2 6MN-type DAG-lactone (2) on a JASCO FP-6600 spectrofluorometer using a quartz cell with a 1.0 cm path length.
FRET experiments41
A phosphatidylserine (PS) solution (500 μg mL−1) was prepared: 50 μL of PS (10 mg mL−1) in CHCl3 was transferred to a 2 mL tube and evaporated under N2. To the residue, 1 mL of 50 mM Tris·HCl (pH 7.4) was added, and the mixture was sonicated with a probe sonicator (5 s × 3). A FRET assay solution (1 mL) containing 50 nM FRET donor molecule, 100 μg mL−1 PS and 50 mM Tris·HCl (pH 7.4) was prepared. After the addition of the FRET acceptor solution to the assay solution, the assay solution was mixed by pipetting up and down followed by incubation for 5 min in the dark. The fluorescence intensity was measured on a JASCO FP-6600 spectrofluorometer (λex = 380 nm, [448 nm/518 nm] for sn-2 DEAC-type DAG-lactone (1) or λex = 340 nm, [397 nm/522 nm] for sn-2 6MN-type DAG-lactone (2)). The concentration of the FRET acceptor started at 0 nM and increased in steps to 250 nM (0, 50, 100, 150, 200, and 250 nM). The competitive assay solution contains a 50 nM FRET donor, a 250 nM FRET acceptor, 100 μg mL−1 PS, and 50 mM Tris·HCl (pH 7.4) (Fig. 5b). The concentration of PDBu started at 0 μM and increased stepwise to 2 μM (0, 0.5, 1, 1.5, and 2 μM). The corrected spectral data with the assay solution containing each concentration of the acceptor were used to calculate the spectrum areas of the acceptor (fluorescein)-related fluorescence from 500 to 570 nm (F = ΣF500–570). The change of the fluorescence intensity was calculated using ΔF = (F − F0)/F0.
FRET-based competitive assay
The competitive assay solution (1 mL) contains a 50 nM FRET donor (sn-2 6MN-type DAG-lactone (2)), a 100 nM FRET acceptor (Fl-δC1b(231–281)), 100 μg mL−1 PS, and 50 mM Tris·HCl (pH 7.4). A candidate ligand (100 μM in DMSO) was serially added to the compound 2-Fl-δC1b domain complex solution (final ligand concentration of 0–800 nM), and fluorescence spectra (350–650 nm) were recorded on a JASCO FP-6600 spectrofluorometer (λex = 340 nm). The spectrum areas of the acceptor (fluorescein)-related fluorescence (500–570 nm) were calculated using ΣΔF500–570 (ΔF = Fblank − Fcompetitor) and normalized using the following formula: Normalized acceptor fluorescence = {ΣΔF500–570 (100% inhibition) − ΣΔF500–570 (each point of competitor concentrations)}/ΣΔF500–570 (100% inhibition). The resulting numbers were plotted and fitted using non-linear regression in GraphPad Prism 8.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank Dr Kenji Maeda, National Center for Global Health and Medicine Research Institute, for valuable discussion on PKC ligands, and Chemical Biology Screening Center, Tokyo Medical and Dental University for its supply of chemical library compounds. This work was supported in part by JSPS KAKENHI Grant Numbers 20H03362 (H. T.), 19K22488 (H. T.) and 20K15951 (K. T.); the Research Program on HIV/AIDS, Japan Agency for Medical Research and Development (AMED) under Grant Number JP20fk0410015 (H. T.); AMED JP20am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) (H. T.); TMDU President's Young Researchers Award 2014 from Tokyo Medical and Dental University (TMDU) (N. O.). This research is based on the Cooperative Research Project of the Research Center for Biomedical Engineering.
References
- Y. Nishizuka, Science, 1992, 258, 607 CrossRef CAS PubMed.
- T. Watanabe, Y. Ono, Y. Taniyama, K. Hazama, K. Igarashi, K. Ogita, U. Kikkawa and Y. Nishizuka, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10159 CrossRef CAS PubMed.
- H. Mischak, J. H. Pierce, J. Goodnight, M. G. Kazanietz, P. M. Blumberg and J. F. Mushinski, J. Biol. Chem., 1993, 268, 20110 CrossRef CAS.
- C. Li, E. Wernig, M. Leitges, Y. Hu and Q. Xu, FASEB J., 2003, 17, 2106 CAS.
- C. Brodie and P. M. Blumberg, Apoptosis, 2003, 8, 19 CrossRef CAS PubMed.
- T. Ghayur, M. Hugunin, R. V. Talanian, S. Ratnofsky, C. Quinlan, Y. Emoto, P. Pandey, R. Datta, Y. Huang, S. Kharbanda, H. Allen, R. Kamen, W. Wong and D. Kufe, J. Exp. Med., 1996, 184, 2399 CrossRef CAS PubMed.
- M. J. Humphries, K. H. Limesand, J. C. Schneider, K. I. Nakayama, S. M. Anderson and M. E. Reyland, J. Biol. Chem., 2006, 281, 9728 CrossRef CAS PubMed.
- A. C. Newton, J. Biol. Chem., 1995, 270, 28495 CrossRef CAS PubMed.
- A. Toker, Front. Biosci., 1998, 3, D1134 CrossRef CAS PubMed.
- C. E. Antal, A. M. Hudson, E. Kang, C. Zanca, C. Wirth, N. L. Stephenson, E. W. Trotter, L. L. Gallegos, C. J. Miller, F. B. Furnari, T. Hunter, J. Brognard and A. C. Newton, Cell, 2015, 160, 489 CrossRef CAS PubMed.
- N. Warwar, S. Efendic, C. G. Ostenson, E. P. Haber, E. Cerasi and R. Nesher, Diabetes, 2006, 55, 590 CrossRef CAS PubMed.
- N. Bowling, R. A. Walsh, G. Song, T. Estridge, G. E. Sandusky, R. L. Fouts, K. Mintze, T. Pickard, R. Roden, M. R. Bristow, H. N. Sabbah, J. L. Mizrahi, G. Gromo, G. L. King and C. J. Vlahos, Circulation, 1999, 99, 384 CrossRef CAS PubMed.
- R. Etcheberrigaray, M. Tan, I. Dewachter, C. Kuipéri, I. Van der Auwera, S. Wera, L. Qiao, B. Bank, T. J. Nelson, A. P. Kozikowski, F. Van Leuven and D. L. Alkon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11141 CrossRef CAS PubMed.
- A. Kishimoto, Y. Takai, T. Mori, U. Kikkawa and Y. Nishizuka, J. Biol. Chem., 1980, 255, 2273 CrossRef CAS.
- Y. Ogawa, Y. Takai, Y. Kawahara, S. Kimura and Y. Nishizuka, J. Immunol., 1981, 127, 1369 CAS.
- N. Sakai, K. Sasaki, N. Ikegaki, Y. Shirai, Y. Ono and N. Saito, J. Cell Biol., 1997, 139, 1465 CrossRef CAS PubMed.
- P. K. Majumder, P. Pandey, X. Sun, K. Cheng, R. Datta, S. Saxena, S. Kharbanda and D. Kufe, J. Biol. Chem., 2000, 275, 21793 CrossRef CAS PubMed.
- T. A. DeVries, M. C. Neville and M. E. Reyland, EMBO J., 2002, 21, 6050 CrossRef CAS PubMed.
- Q. J. Wang, Trends Pharmacol. Sci., 2006, 27, 317 CrossRef CAS PubMed.
- A. C. Newton, Am. J. Physiol. Endocrinol. Metab., 2010, 298, E395 CrossRef CAS PubMed.
- V. E. Marquez and P. M. Blumberg, Acc. Chem. Res., 2003, 36, 434 CrossRef CAS PubMed.
- K. Irie, R. C. Yanagita and Y. Nakagawa, Med. Res. Rev., 2012, 32, 518 CrossRef CAS PubMed.
- R. C. Yanagita, Y. Nakagawa, N. Yamanaka, K. Kashiwagi, N. Saito and K. Irie, J. Med. Chem., 2008, 51, 46 CrossRef CAS PubMed.
- Y. Baba, Y. Ogoshi, G. Hirai, T. Yanagisawa, K. Nagamatsu, S. Mayumi, Y. Hashimoto and M. Sodeoka, Bioorg. Med. Chem. Lett., 2004, 14, 2963 CrossRef CAS PubMed.
- K. Nacro, B. Bienfait, J. Lee, K. C. Han, J. H. Kang, S. Benzaria, N. E. Lewin, D. K. Bhattacharyya, P. M. Blumberg and V. E. Marquez, J. Med. Chem., 2000, 43, 921 CrossRef CAS PubMed.
- H. Tamamura, B. Bienfait, K. Nacro, N. E. Lewin, P. M. Blumberg and V. E. Marquez, J. Med. Chem., 2000, 43, 3209 CrossRef CAS PubMed.
- W. Nomura, Y. Tanabe, H. Tsutsumi, T. Tanaka, K. Ohba, N. Yamamoto and H. Tamamura, Bioconjugate Chem., 2008, 19, 1917 CrossRef CAS PubMed.
- W. Nomura, N. Ohashi, Y. Okuda, T. Narumi, T. Ikura, N. Ito and H. Tamamura, Bioconjugate Chem., 2011, 22, 923 CrossRef CAS PubMed.
- N. Ohashi, W. Nomura, T. Narumi, N. E. Lewin, K. Itotani, P. M. Blumberg and H. Tamamura, Bioconjugate Chem., 2011, 22, 82 CrossRef CAS PubMed.
- D. C. Braun, S. H. Garfield and P. M. Blumberg, J. Biol. Chem., 2005, 280, 8164 CrossRef CAS PubMed.
- J. Brumbaugh, A. Schleifenbaum, A. Gasch, M. Sattler and C. Schultz, J. Am. Chem. Soc., 2006, 128, 24 CrossRef CAS PubMed.
- L. Stryer and R. P. Hauglnad, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719 CrossRef CAS PubMed.
- L. Stryer, J. Biol. Chem., 2012, 287, 15164 CrossRef CAS PubMed.
- C. E. Antal, J. D. Violin, M. T. Kunkel, S. Skovsø and A. C. Newton, Chem. Biol., 2014, 21, 459 CrossRef CAS PubMed.
- S. Kumar, P. Kellish, W. E. Robinson Jr., D. Wang, D. H. Appella and D. P. Arya, Biochemistry, 2012, 51, 2331 CrossRef CAS PubMed.
- J. M. Zwier, T. Roux, M. Cottet, T. Durroux, S. Douzon, S. Bdioui, N. Gregor, E. Bourrier, N. Oueslati, L. Nicolas, N. Tinel, C. Boisseau, P. Yverneau, F. Charrier-Savournin, M. Fink and E. Trinquet, J. Biomol. Screening, 2010, 15, 1248 CrossRef CAS PubMed.
- D. L. Matlock and T. Heyduk, Anal. Biochem., 1999, 270, 140 CrossRef CAS PubMed.
- A. Kakio, S. Nishimoto, K. Yanagisawa, Y. Kozutsumi and K. Matsuzaki, J. Biol. Chem., 2001, 276, 24985 CrossRef CAS PubMed.
- S. L. Timofeevski, J. J. Prusakiewicz, C. A. Rouzer and L. J. Marnett, Biochemistry, 2002, 41, 9654 CrossRef CAS PubMed.
- S. Sakamoto and K. Kudo, J. Am. Chem. Soc., 2008, 130, 9574 CrossRef CAS PubMed.
- N. Ohashi, W. Nomura, N. Minato and H. Tamamura, Chem. Pharm. Bull., 2014, 62, 1019 CrossRef CAS PubMed.
- B. List, C. F. Barbas III and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15351 CrossRef CAS PubMed.
- N. Ohashi, W. Nomura, M. Kato, T. Narumi, N. E. Lewin, P. M. Blumberg and H. Tamamura, J. Pept. Sci., 2009, 15, 642 CrossRef CAS PubMed.
- H. Tamamura, D. M. Sigano, N. E. Lewin, P. M. Blumberg and V. E. Marquez, J. Med. Chem., 2004, 47, 644 CrossRef CAS PubMed.
- H. Tamamura, D. M. Sigano, N. E. Lewin, M. L. Peach, M. C. Nicklaus, P. M. Blumberg and V. E. Marquez, J. Med. Chem., 2004, 47, 4858 CrossRef CAS PubMed.
- M. G. Kazanietz, L. B. Areces, A. Bahador, H. Mischak, J. Goodnight, J. F. Mushinski and P. M. Blumberg, Mol. Pharmacol., 1993, 44, 298 CAS.
- N. A. Sharkey and P. M. Blumberg, Cancer Res., 1985, 45, 19 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00814e |
| ‡ These authors contributed equally. |
| § Present address: Showa Pharmaceutical University, Machida, Tokyo 194–8543, Japan. |
| ¶ Present address: School of Pharmaceutical Sciences and Graduate School of Biomedical & Health Sciences, Hiroshima University, Minami-ku, Hiroshima 734–8553, Japan. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Takahiro
Ishii‡
,
Takuya
Kobayakawa
,
Nami
Ohashi§
,
Wataru
Nomura¶
,
Takahiro
Ishii‡
,
Takuya
Kobayakawa
,
Nami
Ohashi§
,
Wataru
Nomura¶





