
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions promoted by hypervalent iodine reagents and boron Lewis acids
Ayan
Dasgupta
,
Christian
Thiehoff
,
Paul D.
Newman
 ,
Thomas
Wirth
* and
Rebecca L.
Melen
*
,
Thomas
Wirth
* and
Rebecca L.
Melen
*
Cardiff University, School of Chemistry, Park Place, Main Building, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: wirth@cf.ac.uk; MelenR@Cardiff.ac.uk
First published on 13th May 2021
Abstract
Understanding the role of boranes in hypervalent iodine chemistry will open up new reactivities which can be utilised in organic synthesis. Due to similar reactivities, λ3-iodanes have presented themselves as viable alternatives for many transformations dominated by transition metals whilst mitigating some of the associated drawbacks of metal systems. As showcased by recent reports, boranes can adopt a dual role in hypervalent iodine chemistry that surpasses mere activation of the hypervalent iodine reagent. Increased efforts to harness this potential with diverse boranes will uncover exciting reactivity with high applicability across various disciplines including adoption in the pharmaceutical sciences. This review will be relevant to the wider synthetic community including organic, inorganic, materials, and medicinal chemists due to the versatility of hypervalent iodine chemistry especially in combination with borane activation or participation. We aim to highlight the development of hypervalent iodine compounds including their structure, bonding, synthesis and utility in metal-free organic synthesis in combination with Lewis acidic boranes.
 Ayan Dasgupta | Dr Ayan Dasgupta received his PhD degree from IIT Madras (India) in 2016 where he worked under the supervision of Prof. S. Sankararaman. He subsequently worked with Prof. H. Hopf as an Indigo exchange student at TU Braunschweig (Germany) in 2013. He later joined TU Munich (Germany) as a postdoctoral research associate in 2017 and worked on hypervalent iodine catalysis under the supervision of Prof. T. Gulder. Ayan joined the Melen research group as a postdoctoral research associate in 2018. His research focus is on the use of main group elements as catalysts in organic synthesis. |
 Christian Thiehoff | Dr Christian Thiehoff studied chemistry at the Westfälische Wilhelms–Universität Münster (Germany) and received his M.Sc. in 2014. During this time, he spent several months in the group of Prof. R. A. Batey at the University of Toronto (Canada) in 2013. He completed his doctoral studies under the supervision of Prof. R. Gilmour at the Westfälische Wilhelms–Universität Münster (Germany) where his research focused on the use of stereoelectronic effects in the design of functional small molecules. After a postdoctoral stay in the research group of Dr R. L. Melen at Cardiff University, he is currently working at Evotec (UK) Ltd. |
 Paul D. Newman | Dr Paul D. Newman obtained his PhD in 1991 under the guidance of Prof. P. Williams at Cardiff University (UK). He then worked as a PDRA with Dr R. Cross and Dr R. Peacock at the University of Glasgow (UK) followed by a return to Cardiff to work with Prof. P. G. Edwards. From 2003–2009 he was a research fellow in the Cardiff Catalysis Institute to which he remains affiliated in his current senior Lecturer role. He has a longstanding interest in asymmetric coordination compounds and their application notably with phosphine and/or NHC-based ligands and has authored or co-authored 66 publications in the field. |
 Thomas Wirth | Dr Thomas Wirth is professor of organic chemistry at Cardiff University (UK). He received his PhD after studying chemistry at Bonn (Germany) and the Technical University of Berlin (Germany). After a postdoctoral stay at Kyoto University (Japan) as a JSPS fellow, he worked independently at the University of Basel (Switzerland) before taking up his current position at Cardiff University in 2000. He was invited as a visiting professor to a number of places: Toronto (1999), Tokyo (2000), Osaka (2004, 2008), Kyoto (2012). He was awarded the Werner-Prize from the New Swiss Chemical Society (2000), the Furusato Award from JSPS London (2013), the Wolfson Research Merit Award from the Royal Society (2016) and the Bader-Award from the Royal Society of Chemistry (2016). In 2016, he was elected as a fellow of The Learned Society of Wales. His main interests of research concern stereoselective electrophilic reactions, oxidative transformations with hypervalent iodine reagents and flow chemistry performed in microreactors. |
 Rebecca Melen | Dr Rebecca Melen studied for her PhD degree at the University of Cambridge (UK). Following Postdoctoral studies in Toronto (Canada) with Prof. D. W. Stephan and Heidelberg (Germany) with Prof. L. H. Gade, she took up a position at Cardiff University (UK) in 2014 where she is now a Reader in Inorganic Chemistry. In 2018, she was awarded an EPSRC early career fellowship and she was the 2019 recipient of the RSC Harrison Meldola Memorial Prize. Her research interests include diverse aspects of main group reactivity and catalysis, including the applications of main group chemistry in organic synthesis. |
Introduction
Since the discovery of dichloro(phenyl)-λ3-iodane (1) by Willgerodt,1 chemical curiosity regarding hypervalent iodine compounds has resulted in numerous publications concerning their preparation, structure and application. Aside from Willgerodt's reagent, the initial spark that fuelled interest was the discovery of reagents 2 and 6, known as Koser's reagent2 and Dess–Martin periodinane (DMP)3 respectively, that are useful in various oxidative processes of organic molecules.4–6 Further prominent representatives include (diacetoxyiodo)benzene (PIDA, 3), [bis(trifluoroacetoxy)iodo]benzene (PIFA, 4), Togni reagent II (5), and 2-iodoxybenzoic acid (IBX, 7) (Fig. 1) are also commercially available and remarkably useful for metal-free synthesis due to their high nucleofugality, controllable reactivity, high stability and easy handling. Hypervalent iodine compounds are valuable reagents, having broad applicability in organic synthesis,6 and present themselves as non-toxic and environmentally benign alternatives to heavy metals.7 Moreover, hypervalent iodine compounds are commonly used in oxidation reactions,8–10 they have also found widespread utilisation in functionalisations, and are ideally suited for applications in total synthesis as well as the pharmaceutical industry.11,12 When combined with boranes, typically Lewis acid, activation of the hypervalent iodine compound occurs as shown in Scheme 1.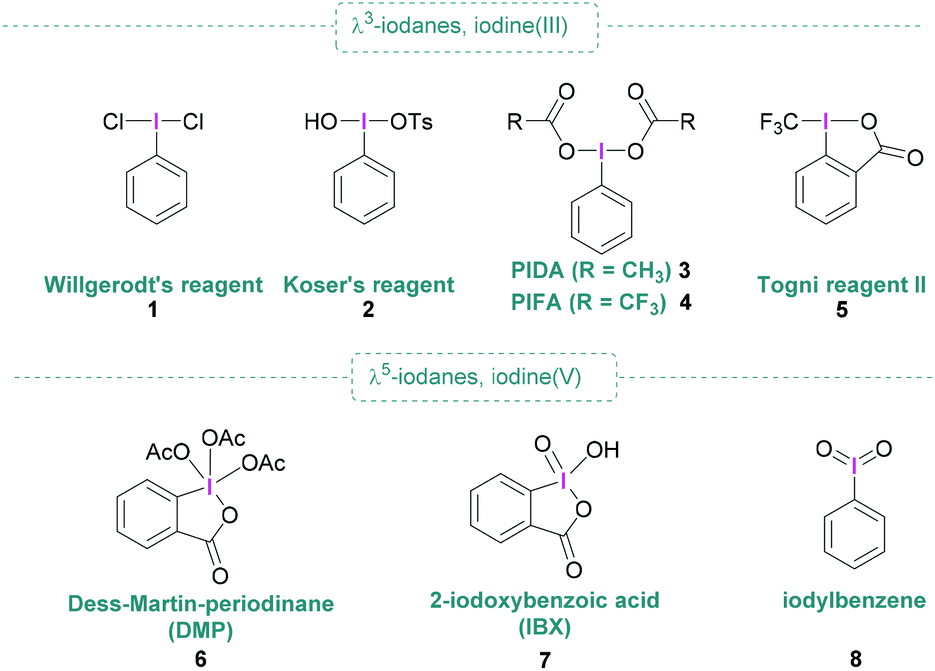 | ||
| Fig. 1 Representative examples of common λ3 and λ5-iodanes. | ||
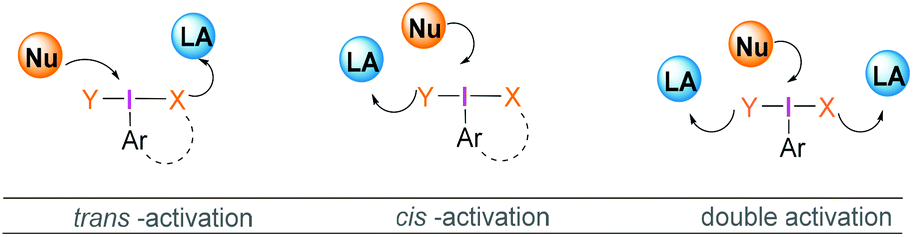 | ||
| Scheme 1 Different mode of acid activation of λ3-iodane. | ||
However, in some cases, the borane can also play a dual role that goes beyond mere activation, for example by transferring one of its substituents to the hypervalent iodine compound.
Reactivities of hypervalent iodine compounds
The different types of hypervalent iodine compounds reported to date can be classified according to the oxidation state of the iodine atom.13 Two oxidation states, +3 and +5, are commonly found in these compounds which are generally represented as λ3- and λ5-iodanes.Here the superscript states the number of bonds to an atom in comparison with its real or hypothetical parent hydride. λ7-Iodanes are also known in which the iodine atom has a +7 oxidation state. The unique structural features of hypervalent iodine compounds is the formation of linear, polarisable, formal 3-centre 4-electron bonds (3c–4e− bonds)14 between the iodine atom and its two trans ligands. These bonds are composed of two electrons in the unhybridised 5p-orbital of the iodine atom and an electron of each ligand. This bonding type renders the iodine atom partially positively charged (polarised hypervalent bond) and highly electrophilic making it susceptible towards nucleophilic attack.
The typical reactivity of λ5- and λ7-iodanes involves oxidative processes, whereas λ3-iodanes display two types of reactivity which are determined by the number of heteroatom and carbon-ligands.15 The first group of compounds within λ3-iodanes are of the type RIL2 and can act as potent oxidising agents. The presence of two heteroatom ligands is an essential requirement for this reactivity. The oxidation reaction consists of a two step process of initial ligand exchange followed by reductive elimination. These two processes constitute the fundamental reactivity behaviour of hypervalent iodine compounds. The second group, of the type R2IL, comprises two carbon-based ligands and one heteroatom ligand on the iodine atom. These compounds act as group transfer reagents of one carbon ligand (R) to a range of nucleophiles via reductive elimination of RI and are poor oxidising reagents.15
The exact mechanism of the ligand exchange reaction of λ3-iodanes is still unclear however, a dissociative or an associative pathway (Scheme 2) can be operative, depending on the nature of the λ3-iodanes.15,16 The former would proceed via the dissociation of a ligand L from λ3-iodane 9 generating iodonium ion 10 as an intermediate followed by nucleophilic attack to give compound 11. For the latter, the order of events would be reversed. The coordination of a nucleophile (Nu−) to the electrophilic iodine centre in 9 gives the square planar [12-I-4] species trans-12 with a trans-arrangement of the ligands (L) which is in equilibrium with its cis-form (cis-12). Subsequent loss of a ligand L yields the iodine(III) compound 11.
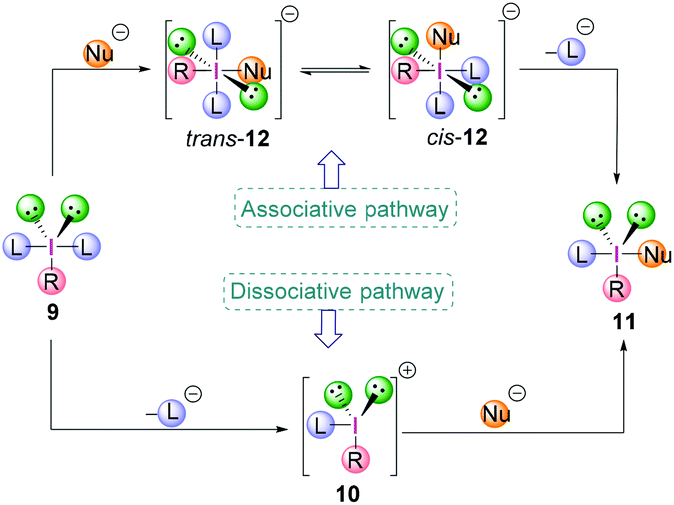 | ||
| Scheme 2 Ligand exchange reaction of λ3-iodanes via the associative or dissociative pathway. | ||
Although the dicoordinated intermediate 10 has been observed in gas phase experiments,17 solution phase based experimental data indicate the presence of trivalent species due to coordination by anions or solvent molecules. On the other hand, isolation and X-ray crystallographic analysis of stable tetracoordinated hypervalent iodine species with a square planar geometry such as the tetrachloroiodate anion strongly corroborates the associative pathway to be most likely.18
Hypervalent iodine compounds commonly undergo reductive elimination to liberate an iodine(I) species. The exceptionally good leaving group aptitude of a λ3-iodane arises from the facile and energetically favourable generation of the monovalent iodine compound in a reductive process.19 By studying the solvolysis rates of cyclohexenyl derivatives, Ochiai and coworkers20 identified the phenyliodo group as having a remarkably good leaving group capability. It is referred to as a hypernucleofuge (super leaving group) because it was determined to be about 106 times better as a leaving group than the corresponding triflate. Despite the homonymy, reductive elimination in transition metal catalysis differs from that of hypervalent iodine and the same reactivity in the context of hypervalent iodine chemistry should be referred to as ligand coupling, the concerted process retains the stereochemical configuration of the ligands. The actual pathway of the reductive elimination reaction in λ3-iodanes depends strongly on the nature of the ligands on the iodine(III) species, the reaction partner, and the reaction conditions.
Other distinct reaction pathways exhibited by λ3-iodanes are nucleophilic substitution, elimination, or fragmentation. The nucleophilic substitutions are most common and can proceed through either SN2 or SN1-type processes depending on the capability of the ligand to stabilise the positively charged intermediate. In either case, the same product(s) are formed. Base-mediated elimination reactions occur in systems where an acidic proton is present in the α- or β-position of one of the ligands. While β-elimination generates double bonds,21 α-elimination results in the generation of carbenes.22 Less common are processes where the reductive elimination includes a fragmentation.23
Under suitable reaction conditions, iodine(III) compounds may also facilitate transformations which involve the generation of radical species either by homolytic cleavage of the I–L bond (Scheme 3, I) or by Single Electron Transfer (SET) to the λ3-iodane (Scheme 3, II).24
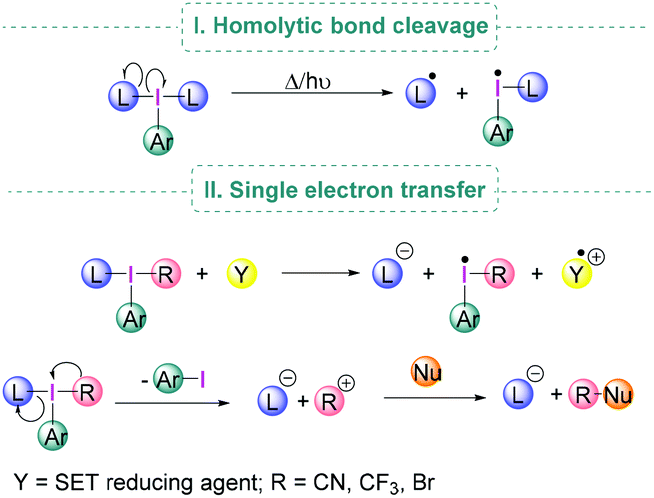 | ||
| Scheme 3 Radical pathways in λ3-iodane chemistry. | ||
Activation of hypervalent iodine compounds
Most reactions employing λ3-iodanes require activation of the hypervalent iodine compound25 which is often achieved by addition of a Lewis or Brønsted acid or base. Lewis bases can be additives, the solvent or a substrate. Acid activation is arguably the most frequently employed method among the above mentioned activation modes. A range of Lewis (e.g. TMSOTf, BF3·OEt2, or metal cations such as Zn, Al, or Mg) or Brønsted (e.g. TfOH, TsOH, AcOH, TFA, or HF) acids that are competent in activating the λ3-iodane have been investigated.26 Although Lewis/Brønsted acid mediated cis-activation or trans-activation of λ3-iodane are common, recently a double-activation mode for hypervalent iodine reagents has also been reported by Liu (Scheme 1).27Tricoordinated boron Lewis acids are a popular choice for the activation of hypervalent iodine compounds, such as BF3, BAr3 and borosilicates. Pervasive use of trivalent boron compounds (typically BF3) to activate hypervalent iodine compounds can be explained due to their high Lewis acidity. The empty p-orbital of the central boron atom can easily be accesed by a lone pair of electrons from a Lewis site in the hypervalent iodine compound enabling activation through increased electrophilicity. This is especially true for iodosylbenzene (15) which is an amorphous, pale yellow solid.28 Its polymeric zigzag structure with O–I⋯O linkages renders it insoluble in most organic solvents.27 In 1982, Ochiai and coworkers29 suggested that BF3·OEt2 activates iodosylbenzene (15) via coordination of the boron Lewis acid to the oxygen atom breaking up the polymeric structure. Analogously, BF3·OEt2 coordination to the acetate oxygen of diacetoxy(m-nitrophenyl)iodane was assumed to activate the λ3-iodane at room temperature to effectively oxidise alcohols to carbonyl compounds (Scheme 4).30
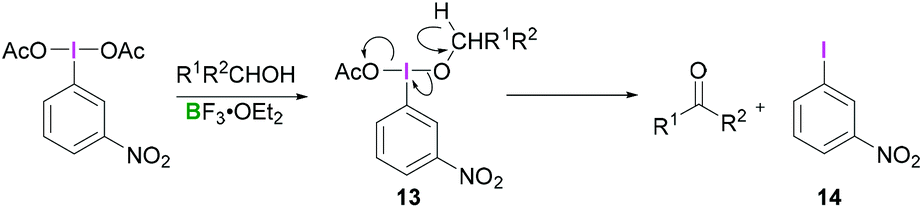 | ||
| Scheme 4 Lewis acid mediated activation of the λ3-iodane. | ||
Presumably, the transformation consists of two steps, both of which are catalysed by BF3. The initial ligand exchange reaction on λ3-iodanes with the alcohol afforded 13 with subsequent β-elimination generating aryliodide 14 along with the respective carbonyl compound. A detailed study of the commercially available PIDA (3)/BF3·OEt2 combination utilised a combined experimental and computational approach for the full investigation of the activation phenomenon.31 Comparison of the 1H NMR spectra of PIDA (3) and a mixture of PIDA/BF3·OEt2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in CDCl3 indicated a downfield shift of the aromatic signals in the latter (ca. 0.1 ppm) which implies the formation of an electron-deficient species. 1H NMR analysis of the titration of PIDA/BF3·OEt2 mixture resulted in the gradual downfield shift of the aromatic and acetyl resonances, confirming the initial finding (Scheme 5). Upon cooling a solution of PIDA/BF3·OEt2 in CDCl3, the single resonance for the acetate ligands splits into two separate signals. This suggests the change of either a fast equilibrium between the BF3 coordinated and the non-coordinated acetate ligand, or an unsymmetrical species with two distinct acetates. X-ray crystallographic analysis31 of the PIDA-BF3 adduct further corroborates the solution phase NMR analysis as the BF3 unit is coordinated to the carbonyl oxygen of one acetate group. Structural comparison of the adduct and PIDA shows a lengthening of d = 0.13 Å for the I–O bond of the BF3·OAc ligand (dI–O = 2.15 Å in PIDA, dI–O = 2.28 Å in the adduct) associated with a contraction of the other I–O bond of d ≈ 0.07 Å (dI–O = 2.076 Å in the adduct) indicative of the existence of the cationic acetoxy(phenyl)iodonium. However, no interaction was observed when PIDA (3) was replaced by PIFA (4) which was attributed to the lower basicity of the latter.
:1) in CDCl3 indicated a downfield shift of the aromatic signals in the latter (ca. 0.1 ppm) which implies the formation of an electron-deficient species. 1H NMR analysis of the titration of PIDA/BF3·OEt2 mixture resulted in the gradual downfield shift of the aromatic and acetyl resonances, confirming the initial finding (Scheme 5). Upon cooling a solution of PIDA/BF3·OEt2 in CDCl3, the single resonance for the acetate ligands splits into two separate signals. This suggests the change of either a fast equilibrium between the BF3 coordinated and the non-coordinated acetate ligand, or an unsymmetrical species with two distinct acetates. X-ray crystallographic analysis31 of the PIDA-BF3 adduct further corroborates the solution phase NMR analysis as the BF3 unit is coordinated to the carbonyl oxygen of one acetate group. Structural comparison of the adduct and PIDA shows a lengthening of d = 0.13 Å for the I–O bond of the BF3·OAc ligand (dI–O = 2.15 Å in PIDA, dI–O = 2.28 Å in the adduct) associated with a contraction of the other I–O bond of d ≈ 0.07 Å (dI–O = 2.076 Å in the adduct) indicative of the existence of the cationic acetoxy(phenyl)iodonium. However, no interaction was observed when PIDA (3) was replaced by PIFA (4) which was attributed to the lower basicity of the latter.
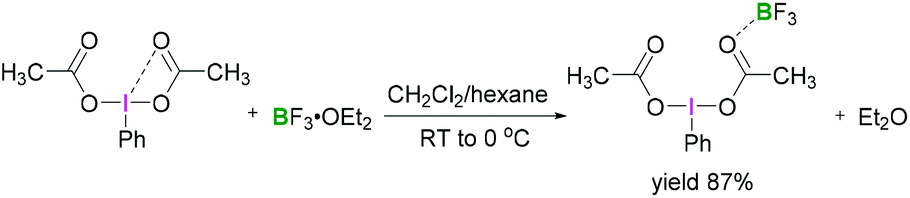 | ||
| Scheme 5 Synthesis and structure of PhI(OAc)2·BF3. | ||
A recent study by Hopkins and Murphy revealed that borosilicates can also be used to activate λ3-iodanes and successfully employed for the gem-difluorination reaction. For these reactions, borosilicate was found to be a better activator compared with BF3·OEt2 (Scheme 6).32
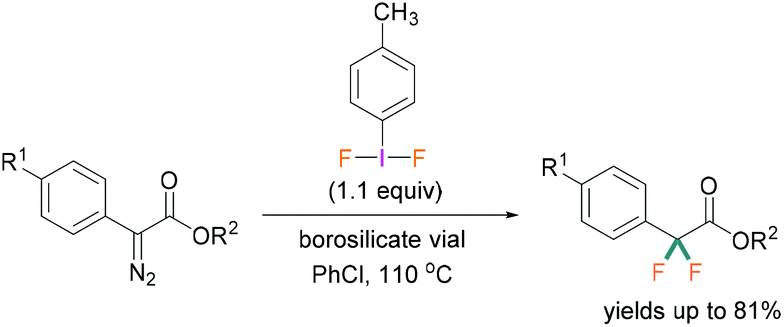 | ||
| Scheme 6 Borosilicate activation of (difluoroiodo)toluene. | ||
BF3-mediated synthesis of iodonium salts
Structurally simple iodine(III) compounds are readily obtained by direct oxidation of the corresponding aryliodide precursor under suitable conditions.33 For example, PIDA (3) can be synthesised by treating iodobenzene with peracetic acid in the presence of acetic acid.34Some λ3-iodanes, most often iodosylbenzene (15) and PIDA (3), can themselves serve as precursors for the synthesis of more complex alkenyl(aryl)iodonium (16),35 alkynyl(aryl)iodonium (17)36 or diaryliodonium salts (18) (Scheme 7).35 Alkenyl(aryl)iodonium compounds represent versatile intermediates providing access to functionalised alkenes upon reaction with a variety of nucleophiles via an addition–elimination process (a formal SN2-type reaction). Additionally, they can undergo α or β-elimination reactions to yield alkylidene carbenes or alkynes, respectively. Reaction of a nucleophilic alkene with 15 or 3 in the presence of BF3·OEt2 followed by anion exchange with NaBF4 yields the corresponding alkenyl(aryl)iodonium tetrafluoroborates 16 in a stereospecific fashion. Synthesis of vinyliodonium salts can be obtained from the reaction between the corresponding β,β-disubstituted vinylsilanes, vinylboronic acid esters and iodine(III) species. It's noteworthy that this electrophilic substitution reaction leads to retention of configuration of the olefin. Using this reaction condition, (E)-alk-1-enylboronates stereoselectively afforded (E)-alk-1-enyliodonium salts.
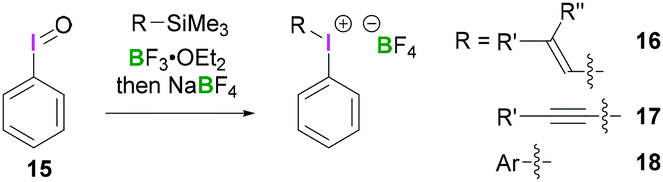 | ||
| Scheme 7 Common strategy for the synthesis of alkenyl-(16) and alkynylaryl iodonium (17) as well as diaryliodonium tetrafluoroborates (18). | ||
Okuyama reported that alkenylboronic esters bearing an acyloxy, alkoxy, or methoxycarbonyl group reacted with PIDA (3) in the presence of BF3·OEt2 to give the alkenyliodonium tetrafluoroborates with complete inversion of configuration.37 Thus (E)- and (Z)-boronates gave (Z)- and (E)-iodonium salts, respectively (Scheme 8).37 A strong solvent effect was evident as selectivity was reversed upon addition of ether to the dichloromethane solution. Neighbouring oxy group participation in the reaction was found to be responsible for the stereoselectivity of these reactions. The anti-addition of an internal oxy group to the electrophilic carbon centre (β-position of the boronic ester) and iodine(III) compound led to the formation of a six membered cyclic intermediate. Concerted anti-elimination of the oxy group and boronic ester afforded the iodonium salts with inversion of configuration of the olefin.
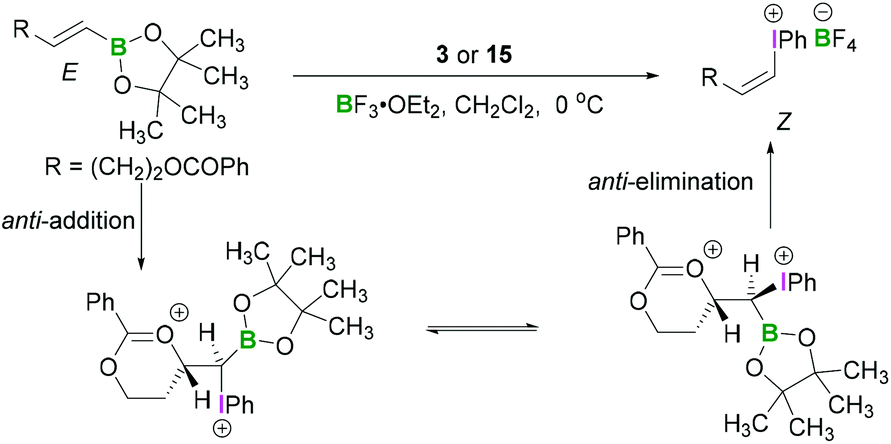 | ||
| Scheme 8 Lewis acid mediated activation of the λ3-iodane. | ||
Compounds 19 and 23 constitute rather unusual examples of vinyl(phenyl)iodonium species. Ochiai and coworkers described the cyclisation of alkynoic acids to iodine(III)-enol species 19 mediated by BF3-activated 15 (Scheme 9, top).38 Stang reported the synthesis of dihydrofuranyl (phenyl)iodonium salt 23 which was obtained from the reaction of allenylphosphonate 20 and PhIF2 in the presence of BF3·OEt2 in CH2Cl2 at −60 °C (Scheme 9, bottom).39 Presumably, the initially formed allyl cation 21, obtained from the regioselective addition of the BF3-activated λ3-iodane to 20, undergoes intramolecular cyclisation (22) followed by loss of the methyl group to afford 23.
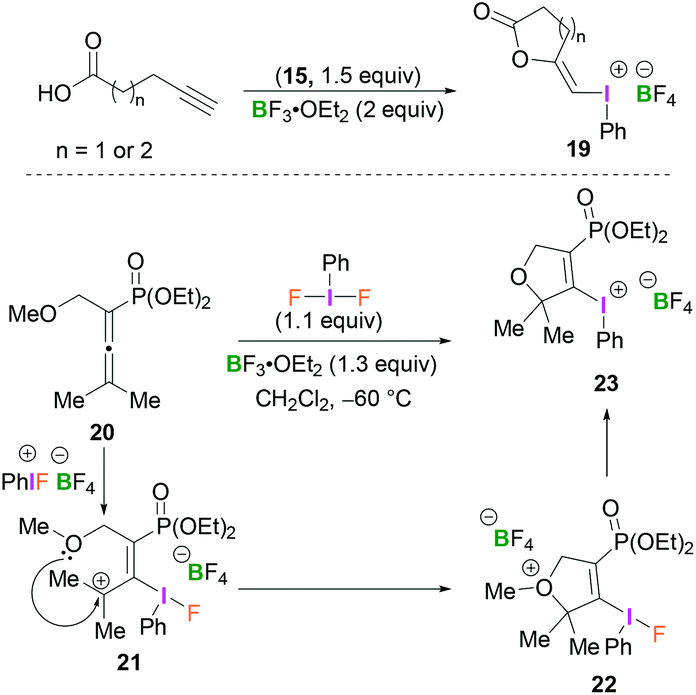 | ||
| Scheme 9 Accessing alkenyliodonium compounds via cyclisation of alkynoic acids (top) or allenylphosponate (bottom). | ||
Furthermore, Ochiai demonstrated a mild reaction protocol for the synthesis of alkynyl(phenyl)iodonium salts.40 Reaction between alkynyltrimethylsilanes and iodosylbenzene (15) in the presence of a Lewis acid led to the formation of iodonium salts 25 and 26 with the trimethylsilyl group replaced by a phenyliodonium group in the product. Initially ethynyltrimethylsilane reacted with 15 and BF3·OEt2 in CH2Cl2 to afford (E)-ethoxy(vinyl)iodonium tetrafluoroborate (24). Protodesilylation of 24 using stoichiometric tetrabutylammonium fluoride in THF at −78 °C produced 25 in 63% yield (Scheme 10, top). The reaction was found to be highly stereoselective and retention of configuration was observed. However, when commercially available bis(trimethylsilyl)ethyne was employed for the reaction with 15 in the presence of BF3·OEt2, phenyl(trimethylsilylethynyl)iodonium tetrafluoroborate (26) was obtained (Scheme 10, bottom).41 Treatment of 26 with hydrogen fluoride afforded the ethynyl(phenyl)iodonium tetrafluoroborate 27 in 83% yield.
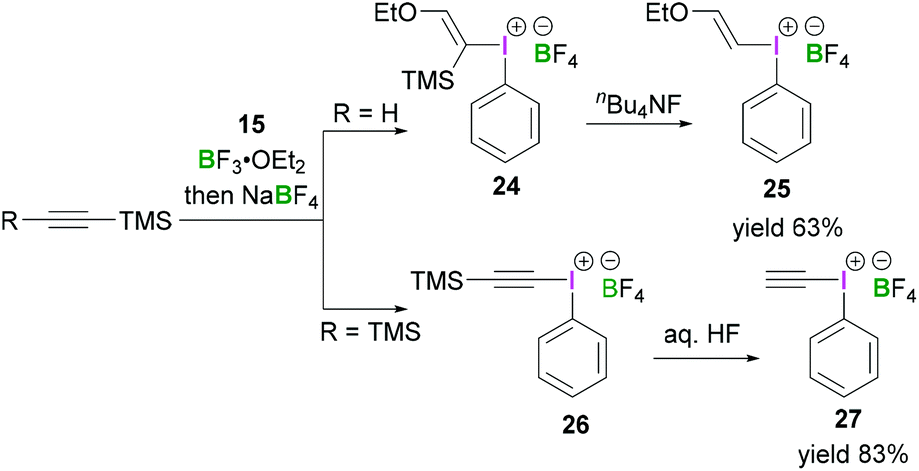 | ||
| Scheme 10 Access to α-silyl substituted (E)-configured vinyliodonium salt 24 and subsequent stereoretentive protodesilylation to give 25 and synthesis of ethynyl(phenyl)iodonium tetrafluoroborate (27). | ||
Employing similar reaction conditions, Ochiai demonstrated the tandem oxidative intramolecular cyclisation of 3-phenylpropanol.42 Formation of 6-chromanyl(phenyl)-λ3-iodanes 28 (Scheme 11, top) originates from the reaction of 15 with either chromane or 3-phenylpropan-1-ol in the presence of BF3·OEt2. The reported work described the tandem oxidative cyclisation and λ3-iodanation of phenylpropanol in which a hypervalent phenyl-λ3-iodanyl group was regioselectively introduced at the C6-position of chromane. A more general approach to access these structures was reported by Olofsson et al. (Scheme 11, bottom).43 After in situ oxidation of an aryl iodide using meta-chloroperoxybenzoic acid (m-CPBA) in the presence of BF3·OEt2, an arylboronic acid was added to the reaction mixture, resulting in the formation of the iodonium borate salt 29.
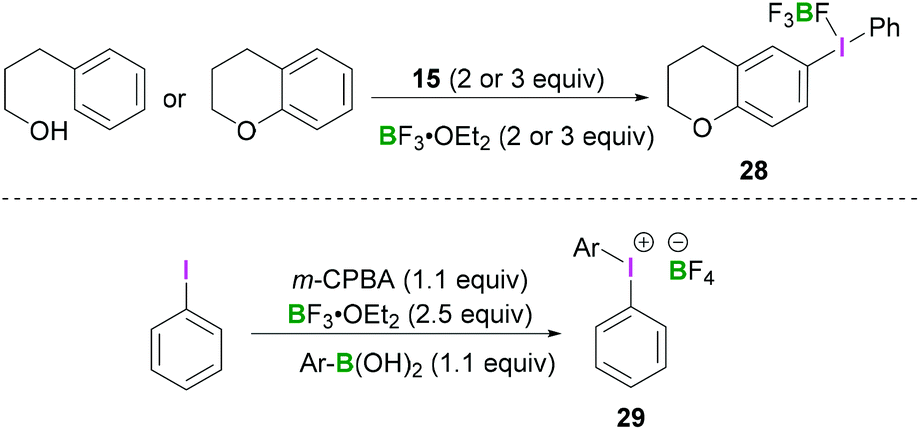 | ||
| Scheme 11 Regioselective synthesis of chromanyl(phenyl)-λ3-iodanes 28 (top) and one-pot synthesis of diaryliodonium tetrafluoroborate 29 (bottom). | ||
This protocol enables the rapid synthesis of a variety of symmetrical and unsymmetrical diaryliodonium compounds with electron-withdrawing or donating substituents in moderate to good yields.
Other noteworthy examples include chiral binaphthyliodonium tetrafluoroborates 31 and 32, both of which can be synthesised from (S)-[1,1′-binaphthalen]-2-yl-λ3-iodanediyl diacetate 30 (Scheme 12).44 Interestingly, the reaction of tetraphenylsilane with 2-(diacetoxyiodo)-1,1′-binaphthyl in the presence of BF3·OEt2 resulted in an intramolecular arylation yielding chiral diaryliodonium salt 31 (intramolecular cyclisation at the C2′ position). However, use of reactive organostannane (tetraphenylstannane), on the other hand, enables Sn–I(III) exchange resulting in binaphthaleneyl(phenyl) iodonium 32 in 76% yield.
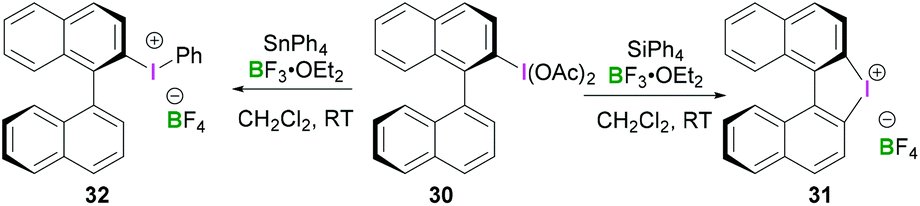 | ||
| Scheme 12 Synthesis of binaphthyl-based iodonium salts 31 and 32. | ||
An extensive study by Ochiai demonstrated the synthesis of different iodinane derivatives. Using 1-hydroxy-1,2-benziodoxol-3(1H)-one as progenitor, tert-butylperoxy iodinane 3345 and alkynyliodinane 3446 were prepared from tert-butylperoxide or alkynylsilane, respectively, upon activation with BF3·OEt2 (Scheme 13).
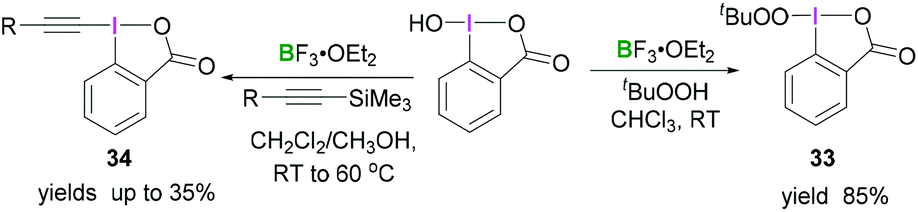 | ||
| Scheme 13 Transformation of benzoiodaoxolone into the corresponding alkylperoxyiodinane 33 and alkynyl derivative 34. | ||
Whilst BF3·OEt2 is the most widely used Lewis acidic borane for the activation of hypervalent iodine compounds, we have demonstrated their activation with the highly Lewis acidic tris(pentafluorophenyl)borane [B(C6F5)3].47 The reaction between triacetoxyiodane, B(C6F5)3 and 2,6-lutidine in CH2Cl2 at −40 °C afforded the ion pair 35. X-Ray crystallographic analysis revealed the formation of a lutidine-stabilised diacetoxyiodonium cation, originating from the abstraction of one acetate unit by B(C6F5)3 generating acetoxytriarylborate as the counterion. The reported structure of 35 showed a bidentate binding mode of one acetate with the second acetate being covalently bound via one oxygen atom (Scheme 14). Reaction of B(C6F5)3 and 2,6-lutidine with the related tris(trifluoroacetoxy)iodane yielded diaryliodonium 36 and perfluorophenyliodane 37 (Fig. 2) by double and single ligand exchange, respectively.
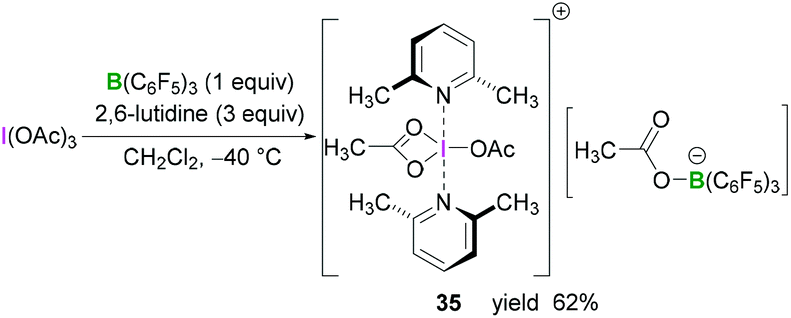 | ||
| Scheme 14 Synthesis of diacetoxy iodonium 35 from I(OAc)3 and B(C6F5). | ||
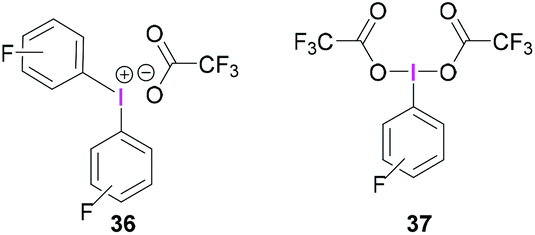 | ||
| Fig. 2 Structure of diaryliodonium 36 and bis(trifluoroacetoxy)iodo pentafluorobenzene (37). | ||
Functionalisation of aryl boronates using trivalent iodine were demonstrated by Kita.48 Lewis acidic boron-substituted aromatic compounds were reacted with PIFA (4) in the presence of acetic acid using hexafluoro-2-propanol (HFIP) and CH2Cl2 (5:1) to afford boron-substituted diaryliodonium salts (Scheme 15). In these reactions the less bulky neopentylglycol boronate ester afforded better yields than BPin. Due to the lower Lewis-acidity of the boronates, it is unlikely that a direct activation of the iodine(III) reagent occurs.
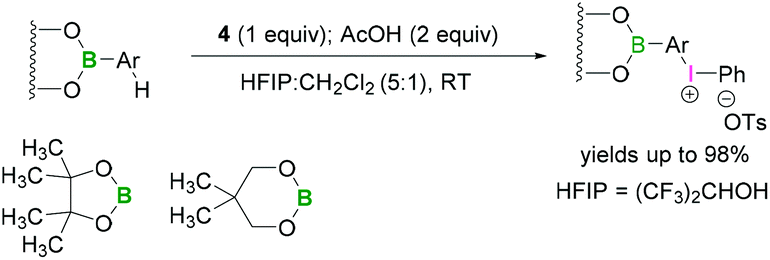 | ||
| Scheme 15 Synthesis of boron-substituted iodonium salts. | ||
Synthetic applications of the BF3·OEt2/iodane system
Stereoselective 1,2-difunctionalisation of alkenes provides the simplest way to introduce useful vicinal bifunctional groups onto a hydrocarbon chain.49 Hypervalent iodine reagents can replace the ubiquitous use of transition metals, commonly utilised as catalysts for such transformations. Lewis acidic boranes, mostly BF3·OEt2, in combination with chiral trivalent iodine compounds have been employed successfully as powerful facilitating tools for enantioselective 1,2-difunctionalisation of alkenes.50The synthesis of a variety of chiral trivalent iodine compounds bearing an α-tetralol was reported recently by Wirth.51 The alcohol was subsequently employed for the synthesis of iodine(III) species 39a–e (Scheme 16, top) in excellent yields (up to 88%). Pyridine containing substituents 39a (N-methyl salicylamide), amide containing substituents 39b, and 5-methoxy-1-tetralone-based substrates 39c–e were introduced at the oxygen centre of α-tetralol to generate the corresponding iodine(III) species. A mild, efficient, and practical reaction protocol has been demonstrated to prepare such chiral trivalent iodine compounds which can be employed as catalysts in combination with boranes for the stereoselective diacetoxylation of styrene (Scheme 16, bottom). Although good to excellent yields of the diacetoxylated compounds were obtained (yields up to 87%), the enantioselectivities (enantiomeric ratio: up to 61:39) were modest. The enantioselective oxyarylation of internal alkenes has also been studied using BF3·OEt2 in combination with a lactate-based chiral iodine(III).
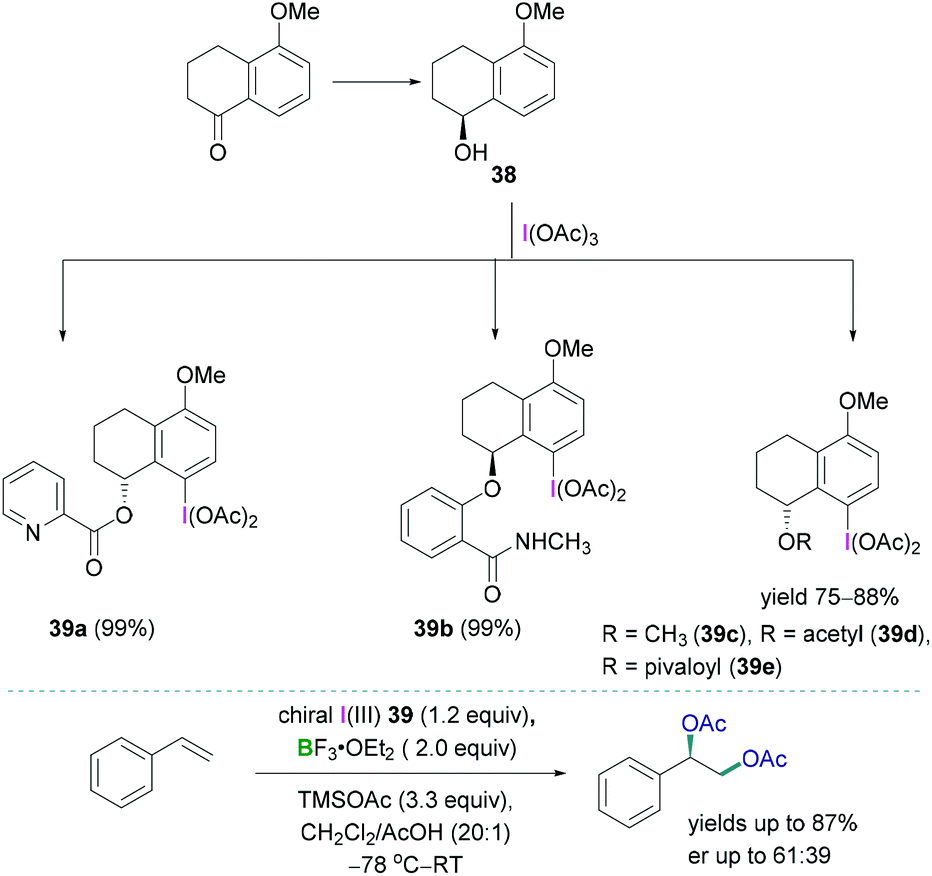 | ||
| Scheme 16 Synthesis of the tetralol-based chiral trivalent iodine compounds (top); stereoselective diacetoxylation using trivalent iodine compounds (bottom). | ||
In 2010, Fujita described the enantioselective oxyarylation of (E)-6-aryl-1-silyloxylhex-3-ene to afford 40 (Scheme 17, top).52 Different Lewis and Brønsted acids were screened for the cyclisation reaction and BF3·OEt2 was found to be the most efficient. To test the effect of the steric bulk, different silyl groups were employed for the cyclisation reaction with slightly better enantioselectivity being observed with the bulky tert-butyldiphenyl silane (TBDPS) compared to the tert-butyldimethylsilyl ether (TBS) group. Several substrates were examined and good yields (up to 88%) of the desired products with high enantioselectivities (up to 93%) were observed. Mechanistic studies (Scheme 18) revealed that the Lewis acidic boranes are required to activate the hypervalent iodine compound which facilitates the electrophilic addition to the olefins. The nucleophilic addition of the internal oxy group afforded 42, and product 40 formed after further nucleophilic addition of the aryl group. The authors determined that the lactate-based hypervalent iodine reagent 41 reacts particularly well with one of the enantiotopic faces of the olefin (which is the rate-determining step) and induces the formation of the cyclised product.
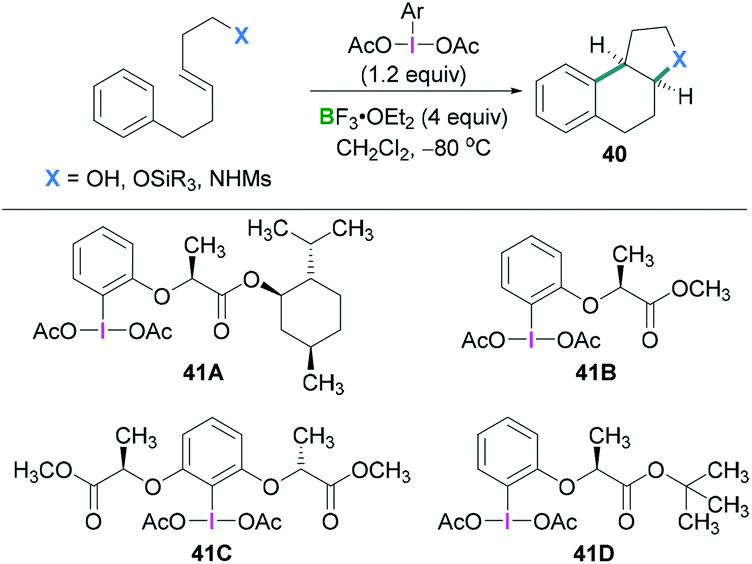 | ||
| Scheme 17 Enantioselective oxyarylation reaction using λ3-iodanes. | ||
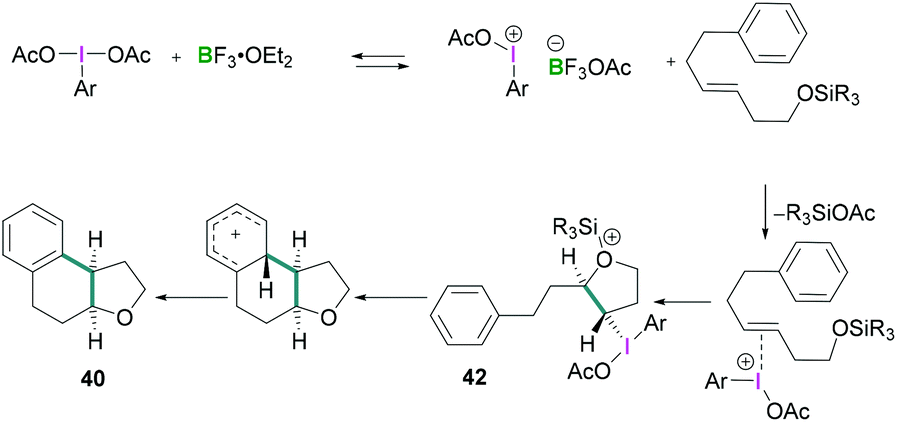 | ||
| Scheme 18 Proposed reaction mechanism for enantioselective oxy arylation reaction. | ||
Preparation of biologically useful isochromanone scaffolds have been carried out using λ3-iodane. In 2010, Fujita demonstrated the regio- and diastereo-selective oxidative lactonisation of 2-vinylbenzoic acid to 4-oxyisochroman-1-one 43a using λ3-iodane as a catalyst (Scheme 19, top).53 BF3·OEt2 was used as the Lewis acidic component to activate the iodine reagent. Vast studies revealed that different oxy groups such as methoxymethyl and hydroxymethyl in the alkenyl moiety does not affect the formation of δ lactone. The reported methodology can be utilised to prepare synthetically useful biologically active compounds 44 (Scheme 19, bottom).
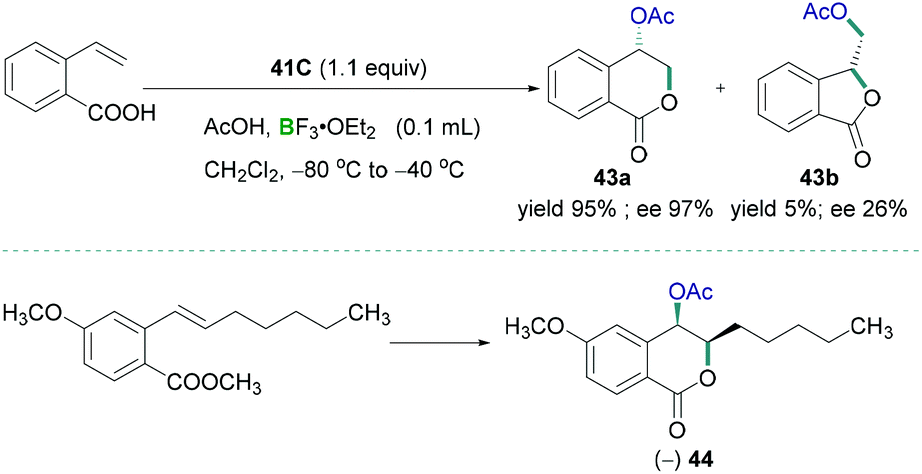 | ||
| Scheme 19 Tosyloxylactonization of 2-ethenylbenzoic acid (top) and synthesis of useful biologically active compound 44 (bottom). | ||
Prévost and Woodward reactions for dioxyacetylation of alkenes using chiral hypervalent iodine compounds have also been carried out by Fujita (Scheme 20). An optically active hypervalent iodine reagent (45) in combination with a Lewis acidic boron BF3·OEt2 and acetic acid were employed for the reaction with different cinnamyl derivative (46) at −80 °C to afford enantioselective dioxyacetylated products (47).54 Diastereoselectivities (syn/anti selectivity) in the dioxyacetylated products showed a dependence on the reaction temperature. When the reaction was carried out at −80 °C to −40 °C, a regioisomeric mixture of monoacetoxy and diacetoxy compounds were obtained. Further treatment with pyridine enabled full conversion to the diacetoxy compounds 47a (Scheme 20, top). High enantioselectivity (up to 96%) of the syn product (1S,2S) and excellent diastereoselectivity between the syn and anti-conformer (98:2) was observed. Intriguingly, the reaction at −80 °C to room temperature afforded the anti-configured diacetoxy compound 47b (1R,2S) as the major product with excellent enantioselectivity (up to 96%) and diastereoselectivity (syn:anti 2:98) (Scheme 20, bottom).
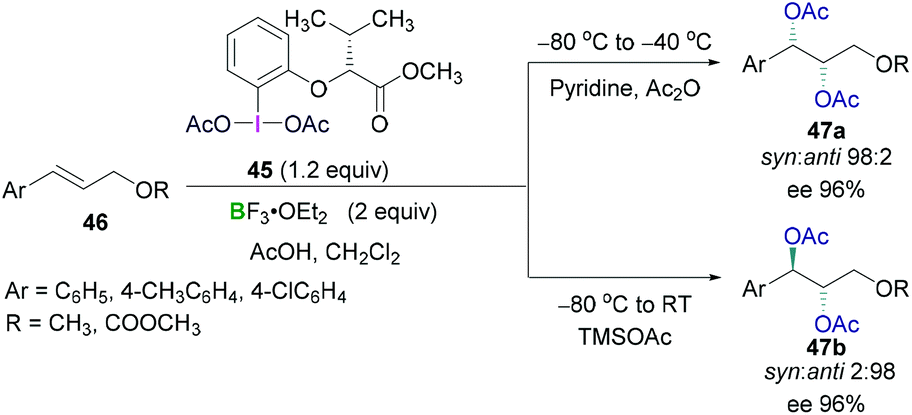 | ||
| Scheme 20 Enantioselective diacetoxylation of olefins using λ3-iodanes. | ||
Wirth and coworkers investigated the enantioselective synthesis of α-acetoxylated ketones using catalytic amounts of chiral λ3-iodanes.55 The λ3-iodanes were generated in situ from the reaction of chiral λ3-iodanes 48 and an oxidising agent in the presence of acetic acid. Lewis acidic BF3·OEt2 was employed to activate the hypervalent λ3-iodanes and then subsequent reaction with the enol ether 49 afforded an intermediate in which the activated λ3-iodane was attached to the α-carbon of the carbonyl functional group. Nucleophilic attack by an acetate anion (SN2 reaction) led to the formation of the desired product 50 (Scheme 21). Excellent yields (up to 97%) of the α-acetoxylated ketones and good enantioselectivities (up to 88%) were obtained. However, cyclic substrates (for example, 3,4-dihydronaphthalen-1-yl acetate) showed poor enantioselectivities.
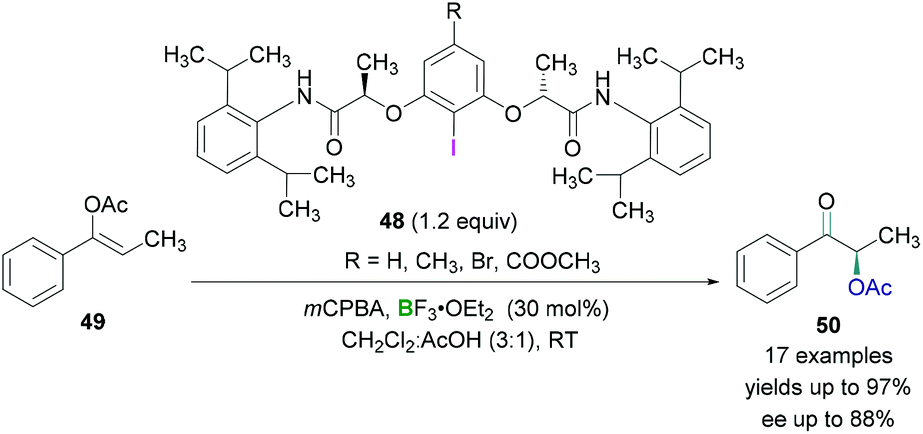 | ||
| Scheme 21 Enantioselective acetoxylation reaction using chiral λ3-iodanes. | ||
Further investigations by Wirth56 on the stereoselective intramolecular diamination reaction of alkenes using a chiral hypervalent iodine reagent 51 revealed that equimolar mixtures of trimethylsilyl triflate (TMSOTf) and BF3·OEt2 afforded BF2OTf·OEt2 which acted as a Lewis acid to activate the λ3-iodanes 51 (Scheme 22). Excellent enantioselectivities (up to 92%) were observed in the products 52.
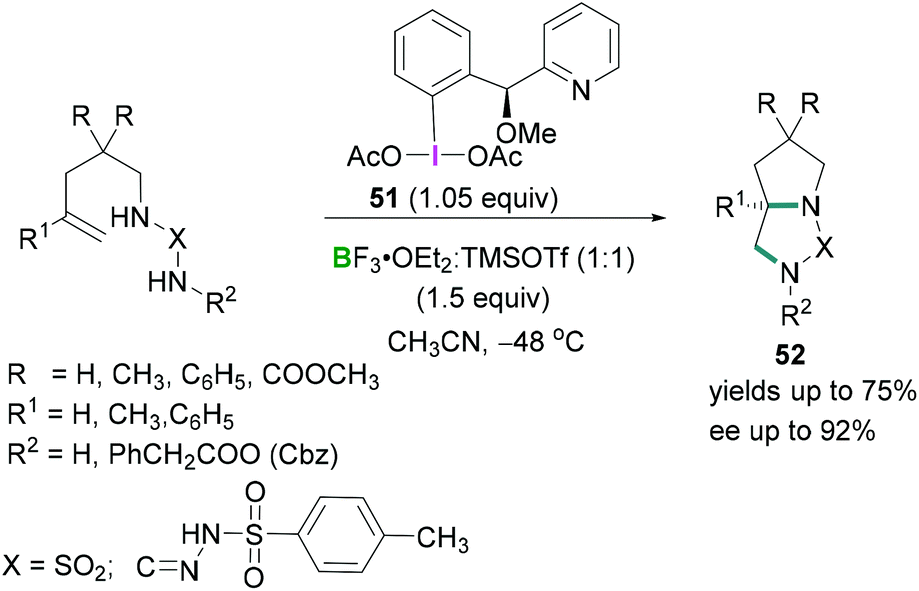 | ||
| Scheme 22 Enantioselective cyclisation reaction using chiral λ3-iodanes. | ||
Hypervalent iodine compounds in combination with Lewis acidic boranes have also been used to introduce the nitrile functionality into an organic molecule. Electrophilic cyanation using iodanes provides an alternate to metal catalysed reactions which are useful for the generation of several biologically active compounds.57 Further investigations revealed that the cyano group can also be introduced enantioselectively into an organic molecule utilising chiral hypervalent iodanes. In 2017, Minakata described the use of 1-cyano-3,3-dimethyl-3-(1H)-1,2-benziodoxole (CDBX) 53 as the source of a CN group which can be activated by the strongly Lewis acidic borane B(C6F5)3 and successively employed for catalytic electrophilic cyanation of silyl enol ethers to afford β-ketonitriles 54 (Scheme 23).58 Commonly used Lewis acidic boranes such as BF3·OEt2 and BEt3 both failed to activate 53 and no product formation was observed, but the more Lewis acidic B(C6F5)3 would activate the CDBX reagent. Mechanistic studies revealed that coordination between the cyano functionality of 53 and B(C6F5)3 (which was confirmed from in situ IR and 13C NMR spectroscopy, see later) resulted in the generation of a highly electrophilic species which facilitates the cyanation reaction. The interaction between the cyano functionality of 53 and B(C6F5)3 was examined spectroscopically. The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch in the IR spectrum was shifted from 2137 cm−1 (starting material-53) to 2216 cm−1 (53 + B(C6F5)3). Moreover, when an equimolar mixture of B(C6F5)3 and 53 in CD2Cl2 was monitored using 13C NMR, a significant downfield shift of the carbon atom of the CN group was seen. Moreover, the tertiary carbon adjacent to the oxygen also exhibits a downfield shift. Based on these observations, it was concluded that the cyano group was coordinated to the Lewis acidic boranes to assist cyano group transfer to silyl enol ethers to generate β-ketonitriles.
N stretch in the IR spectrum was shifted from 2137 cm−1 (starting material-53) to 2216 cm−1 (53 + B(C6F5)3). Moreover, when an equimolar mixture of B(C6F5)3 and 53 in CD2Cl2 was monitored using 13C NMR, a significant downfield shift of the carbon atom of the CN group was seen. Moreover, the tertiary carbon adjacent to the oxygen also exhibits a downfield shift. Based on these observations, it was concluded that the cyano group was coordinated to the Lewis acidic boranes to assist cyano group transfer to silyl enol ethers to generate β-ketonitriles.
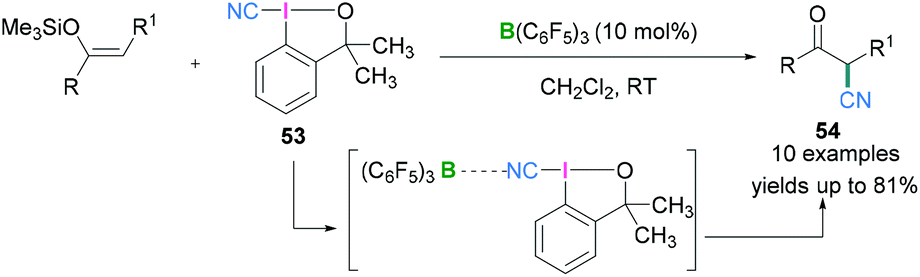 | ||
| Scheme 23 Electrophilic cyanation reaction using Lewis acidic boranes and hypervalent λ3-iodanes. | ||
Kita demonstrated the oxidative cyanation59,60 of electron-rich heterocycles including pyrroles, thiophenes, and indoles. Initial investigations showed that direct oxidative cyanations of electron-rich heterocyclic compounds were possible using a hypervalent iodine(III) reagent (PIFA, 4) and BF3·OEt2. TMSCN was used as the source of the CN group. Premixing of PIFA (4) and TMSCN in situ generated the hypervalent iodine(III)–CN species which was further activated by BF3·OEt2. Cyanation of 1H-pyrrole was investigated but only poor yields of the desired products were obtained. The authors observed that the presence of a strong electron withdrawing group at the nitrogen-position significantly improved the yields of the desired product. Therefore, a tosyl group was introduced and employed for the cyanation reaction. Using the optimised reaction conditions, N-tosylpyrrole was selectively converted to 2-cyano-N-tosylpyrrole in 83% yield but 2 equivalents of PIFA (4) were required. To demonstrate a broad substrate scope, N-tosylpyrroles, substituted thiophenes, and N-tosylindole were tested with satisfactory yields of the products 55 being obtained (Scheme 24).
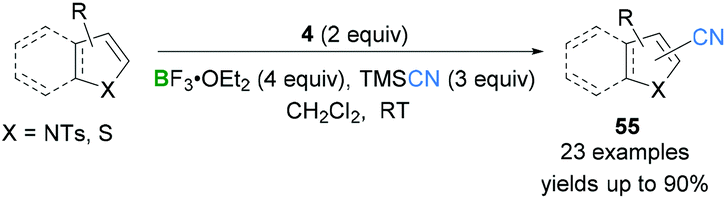 | ||
| Scheme 24 PIFA-mediated oxidative cyanation of heteroarenes. | ||
The reactivity of λ3-iodanes was further investigated and successfully employed for the halogenation reaction.61 Metal-free facile synthesis of organofluorine compounds are desirable as they are ubiquitous structural motifs in pharmaceutical compounds and agrochemicals.62
In 2018, Hu reported the fluorination of aromatic diazonium salts (Scheme 25).63 Catalytic Balz–Schiemann fluorination reactions were investigated using different trivalent iodine compounds and BF3·OEt2. All reactions were carried out in trifluorotoluene and many examples with yields of the products 56 up to 90% were reported using this methodology. Reactive functional groups (iodo, ketone, ester, carboxylic acid, nitrile, and sulfamide) remained intact and formation of undesirable products was not observed. To highlight the mechanistic details, the authors suggested that the Lewis acidic component BF3·OEt2 activated the aryliodonium(III) and generated the aryl cation intermediates.63 Addition of the borane increased the leaving group aptitude of dinitrogen which resulted in the formation of an Ar+BF4− salt and successive nucleophilic attack by a fluoride anion afforded the fluorinated product.
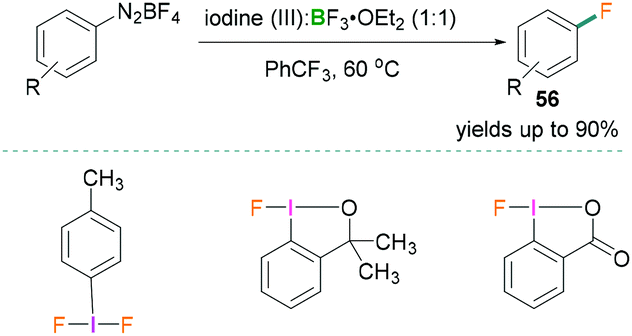 | ||
| Scheme 25 Catalytic Balz–Schiemann fluorination reaction using Lewis acidic boranes and hypervalent λ3-iodanes. | ||
In 2017, Murphy used phenylallene derivatives 57 for the synthesis of α-difluoromethyl styrene compounds 58 which can be employed as fluorinated building blocks.64 Stoichiometric amounts of difluoro(p-tolyl)-λ3-iodane and BF3·OEt2 were reacted with phenylallenes to afford the α-difluoromethyl styrenes 58. The reaction was found to be highly chemoselective with high functional group tolerance and proceeded via a fluorinative rearrangement mechanism. Several substrates such as substituted phenylallenes (phenylallenes bearing phenyl and α-allenyl substituents) and diphenylallenes were investigated.
Moderate to good yields of the desired products (many examples, yields up to 79%) were obtained (Scheme 26, top). To investigate the reaction mechanism, deuterated phenylallene [D2] was used (Scheme 26, bottom) and the crude reaction mixture analysed by multinuclear NMR spectroscopy (1H, 2H, or 19F NMR). As no deuterium scrambling was observed in the product (58a) it was concluded that the terminal double bond of the allene was not reacting (Scheme 26, bottom).
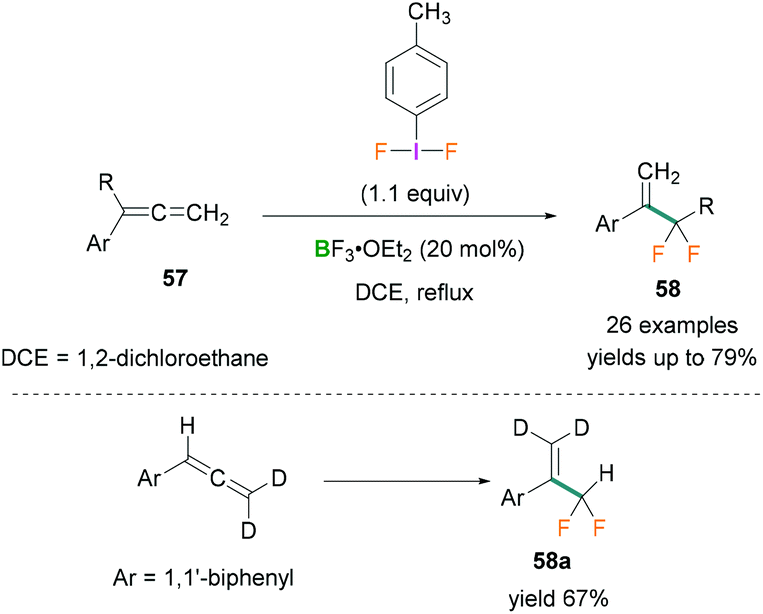 | ||
| Scheme 26 Fluorinative rearrangement of phenylallene using Lewis acidic boranes and λ3-iodanes. | ||
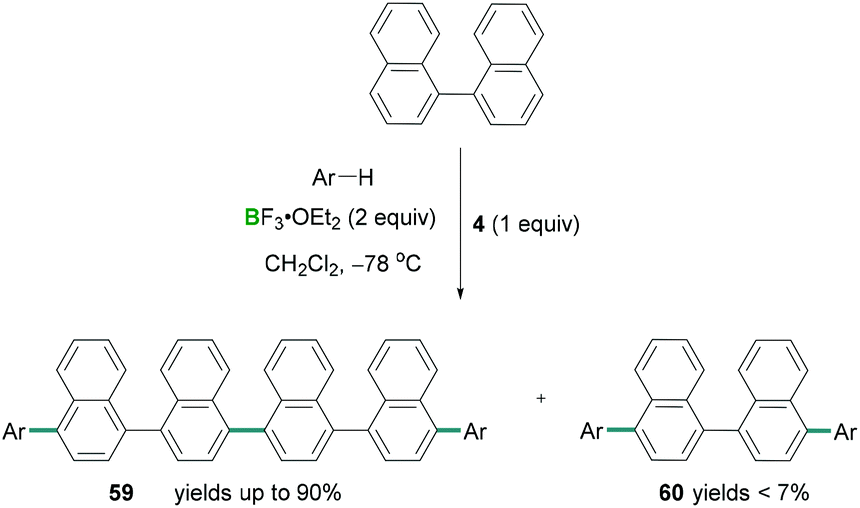
Combinations of hypervalent iodine reagents and Lewis acidic boranes are also capable of promoting C–C cross coupling reactions.65 Following Kita's cyanation reaction conditions, Ramírez de Arellano demonstrated the four-component coupling reaction between naphthalene and substituted benzenes using stoichiometric amounts of PIFA (4) along with an excess of BF3·OEt2.66 Here the functionalisation of C–H bonds using regioselective intermolecular four-component coupling reactions afforded novel C–C cross coupled products (Scheme 27). Direct oxidative coupling between naphthalene and different arenes such as tetramethylbenzene afforded linear tetraarenes with a binaphthalene core. Using this methodology, 1,1′-binaphthalene afforded linear hexaarene (59) chemo-selectively in excellent yields (up to 90%) and formation of tetraarene (60) as a by-product (yields up to 11%). However, when pentamethylbenzene reacted with 1,1′-binaphthalene, the tetraarene 60 was the major (93%) component compared to hexaarene 59 (3%) (Scheme 27).
 | ||
| Scheme 27 Oxidative four-component Kita coupling reaction. | ||
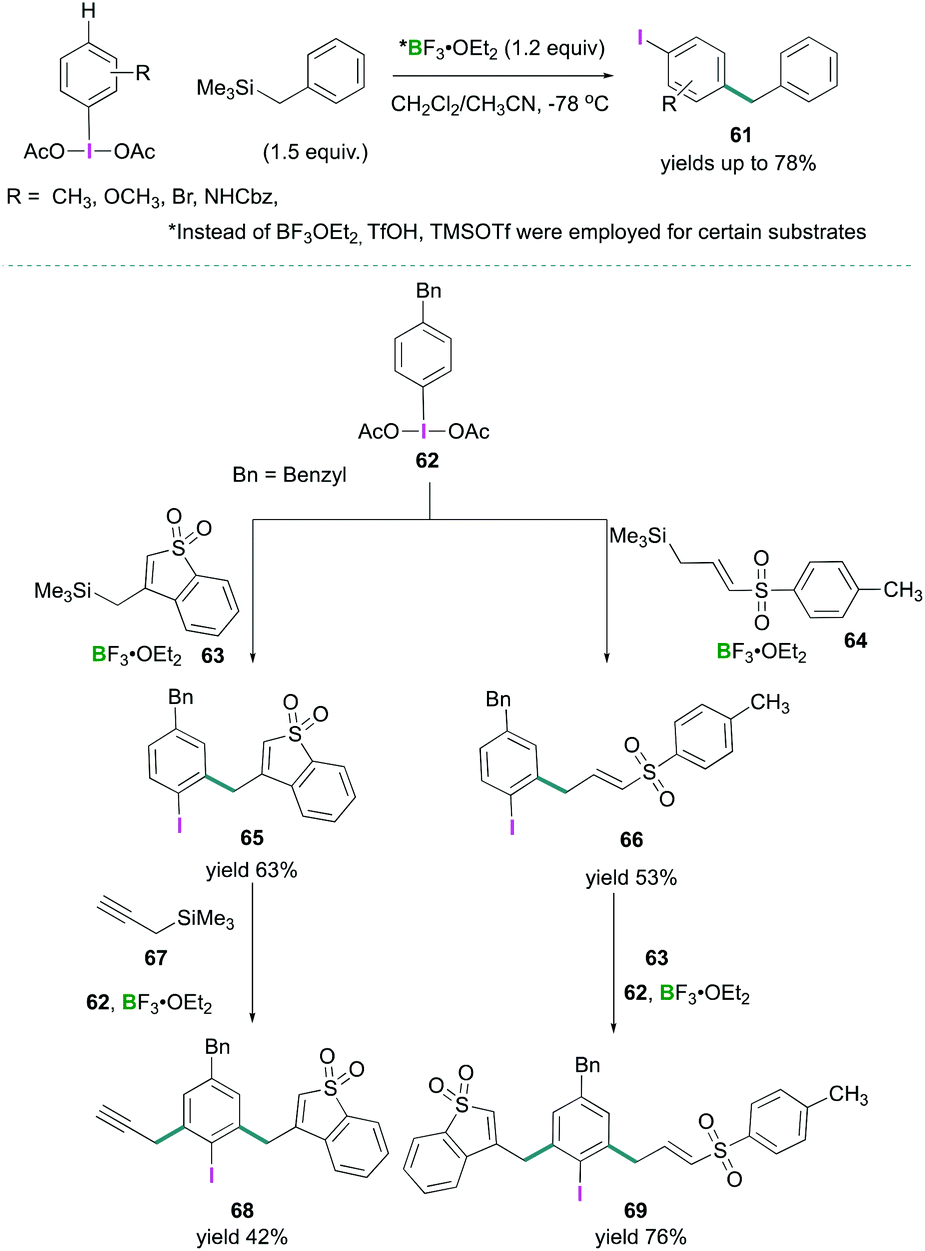
Recently, Shafir investigated the iodine directed ortho and para C–H alkylation of (diacetoxyiodo)benzene (3) to afford poly-substituted arenes.67 A new synthetic strategy for para-selective C–H benzylation, and ortho-selective sulfonylation have been reported (Scheme 28). The reaction between PhI(OAc)2 (3) and ArCH2SiMe3 in the presence of BF3·OEt2 afforded C–C cross-coupled products 61 selectively at the para position to the iodine in good to excellent yields (Scheme 28, top). When para substituted (4-benzylphenyl)-λ3-iodanediyl diacetate (62) was employed for the oxidation reaction with different silane compounds 63 and 64, using the optimised reaction conditions, ortho alkylation (ortho to iodine) compounds 65 and 66 were formed in 63% and 53% yields respectively (Scheme 28, bottom). Further treatment of 66 with cyclic allylsilane 63 led to the formation of 69 (76%), and reaction of 65 with trimethyl(prop-2-yn-1-yl)silane 67 afforded compound 68 in 42% yield. A [3,3] sigmatropic rearrangement mechanism was proposed to account for the formation of these products.
 | ||
| Scheme 28 Iodine directed ortho and para C–H alkylation of PhI(OAc)2. | ||
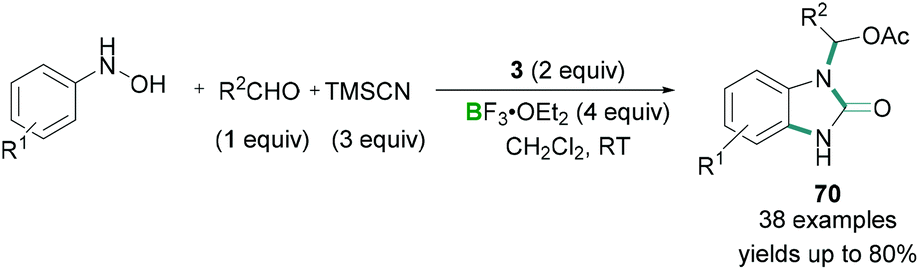
Additionally, hypervalent iodine compounds were tested for multicomponent coupling reactions. A mild reaction protocol was developed by Hu for the synthesis of N-substituted benzimidazolones 70 (Scheme 29).68
 | ||
| Scheme 29 Synthesis of benzimidazolones 70. | ||
Aromatic hydroxylamines, aldehydes, and TMSCN were reacted together in the presence of stoichiometric amounts of PhI(OAc)2 (3) and excess BF3·OEt2 to afford benzimidazolone derivatives in good yields (Scheme 29). Mechanistic details revealed that the aromatic hydroxylamines and aldehydes reacted together to afford a nitrone. Initial activation of the PhI(OAc)2 (3) by BF3·OEt2 and subsequent ligand exchange with TMSCN produced intermediate PhI(OAc)(CN). PhI(OAc)(CN) then underwent a ligand exchange reaction with the nitrone to give a reactive intermediate which undergoes [3,3] rearrangement to afford the final benzimidazolone product. Although high functional group tolerance for these reactions was observed, heterocyclic aldehydes, aliphatic aldehydes, and ketones failed to produce the desired product.
Beyond mere activation: dual role of the borane
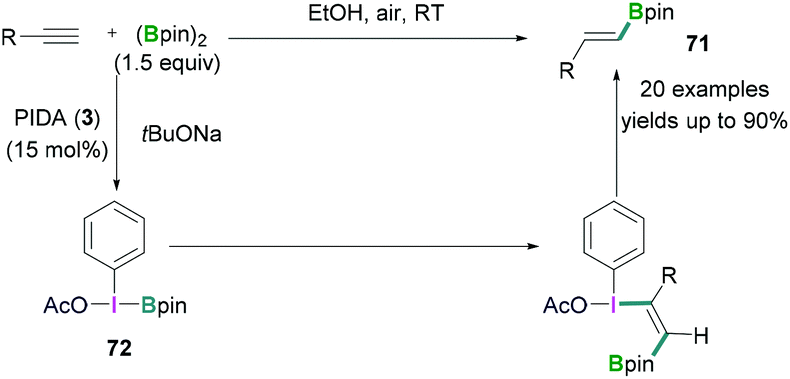
In certain cases, the Lewis acidic boranes do not only activate the hypervalent iodine compounds, but also actively participate in the reaction. Hypervalent iodine compounds in combination with Lewis acidic boranes have successfully been employed for hydroboration and fluorination reactions.In 2017, Wei demonstrated the PhI(OAc)2 (3) mediated hydroboration reaction of terminal alkynes with bis (pinacolato)diboron (B2pin2) using tBuONa (Scheme 30).69 A simple reaction protocol has been discussed for the synthesis of various vinyl boronates 71. It was suggested that PhI(OAc)2 initially reacted with tBuONa to generate PhI(OAc)(OtBu). Likewise, pinacol diborane also reacted with tBuONa to afford tBuOBpin. Subsequent reaction between PhI(OAc)(OtBu) and tBuOBpin afforded PhI(OAc)(Bpin) 72. In the next step, 72 reacted with the terminal alkyne stereoselectively to afford the desired vinyl boronates (71) in good to excellent yields (up to 90%).
 | ||
| Scheme 30 Hydroboration of various terminal alkynes. | ||
Zhang demonstrated the use of BF3·OEt2 as a fluorinating agent with iodosylbenzene for the fluorination of homoallylic amines.70N-(But-3-en-1-yl)-4-methylbenzene sulfonamide 73 exemplifies the intramolecular aminofluorination of homoallylic amines to prepare 3-fluoropyrrolidines 74 (Scheme 31). The reaction proceeds via the activation of PhIO by BF3·OEt2 to afford a λ3-iodane intermediate which reacts further with homoallylic amines to produce an iodonium intermediate 75. This reactive intermediate readily undergoes intramolecular nucleophilic attack by the amino functional group to afford an intermediate iodonium borane pyrrolidine derivative 76 which, upon reductive elimination, formed the cyclic carbocation intermediate 77. This cationic intermediate was readily trapped by the fluoride anion to produce the desired fluoropyrrolidines 74.
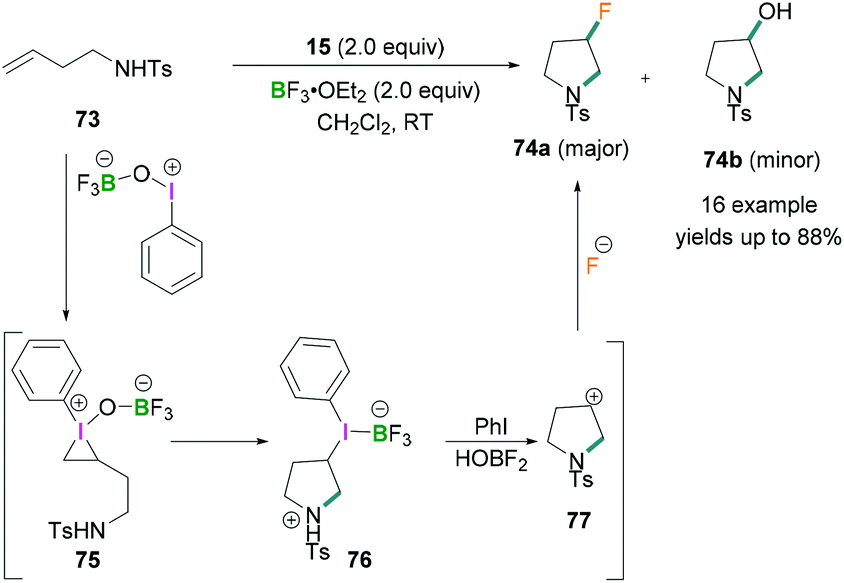 | ||
| Scheme 31 Intramolecular aminofluorination of homoallylic amines. | ||
In 2017, Zhang demonstrated the use of hypervalent iodine compounds towards the ring-contraction monofluorination reaction using BF3 etherate as the fluorine source.71 In the reported work, N-(cyclohex-2-en-1-yl)benzamide 79 was reacted together with iodosobenzene and BF3·OEt2 in CH2Cl2 to afford monofluorinated fused five-membered oxazoline ring products (yields up to 35%). A dramatic change in the yield of the product was observed when 3,5-dichloroiodosobenzene 78 was employed as the oxidant instead of iodosobenzene. Different N-cyclohexenyl amides 79 were tested and good to excellent yields of the desired products were obtained. In contrast with 79a (no substitution at C4 of the cyclohexenyl ring) which afforded 81 in satisfactory yield (up to 72%) (Scheme 32, bottom), when 79b (two methyl groups at C4 of the cyclohexenyl ring) was employed for the fluorination reaction, a very different product (80) with yields up to 97% were obtained (Scheme 32, top). The formation of the different products can be explained based on the stability of the carbocation intermediate. In the reaction, λ3-Iodanes 78 and BF3·OEt2 react together to form an iodine(III) intermediate which activates the double bond of 79 and form the iodonium intermediate.
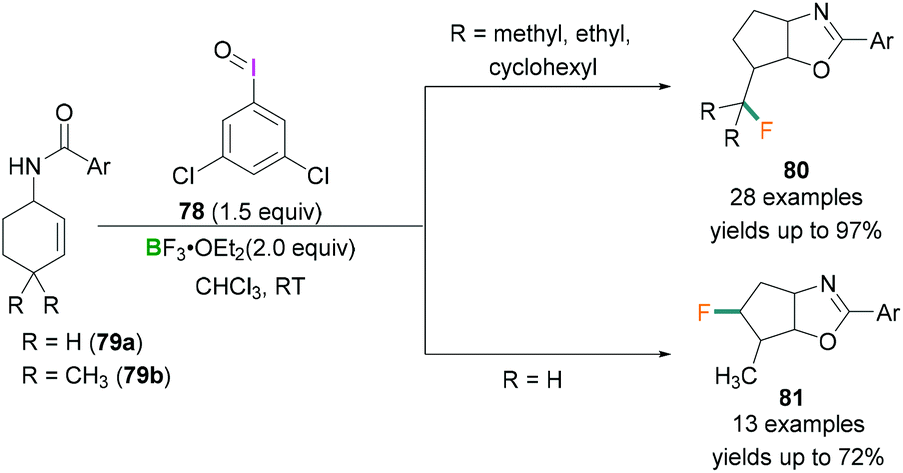 | ||
| Scheme 32 Ring-contraction monofluorination reactions using λ3-iodanes (78) and Lewis acidic boranes. | ||
Intramolecular nucleophilic attack by the oxygen atom of the amide followed by reductive elimination formed the intermediate 82 (Scheme 33). Depending on the nature of the R group, different products were formed. When R = H, alkyl and double hydride shifts generated the carbocation intermediated 83a and successive fluoride attack afforded the product 81. However, when R = CH3, 82 undergoes an alkyl migration to form a more stable tertiary carbocation 83b which reacts with the fluoride ion to afford 80.
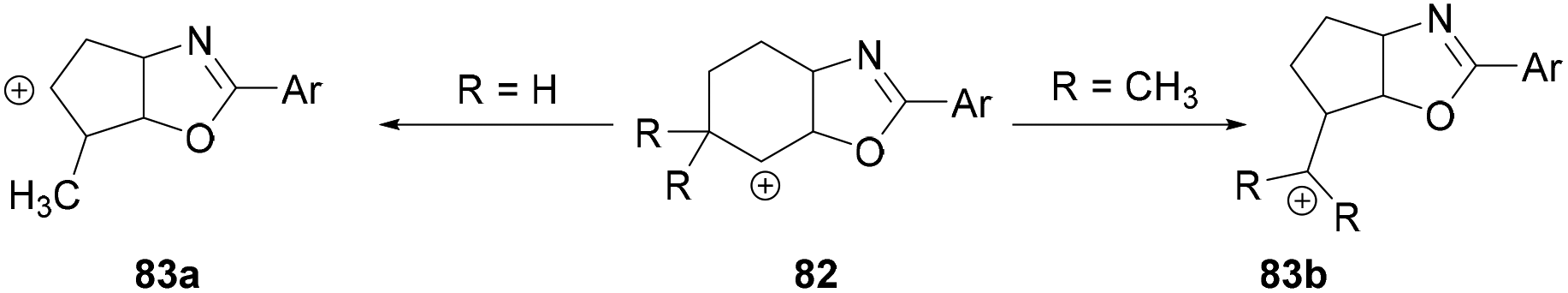 | ||
| Scheme 33 Formation of intermediate 83a and 83b. | ||
The reactions between hydrazones and hydrazides with Lewis acidic triarylfluoroboranes have also been investigated. Recently, we demonstrated the synthesis of different boron dienolates 84 from the iodonium ylides in moderate to good yields (up to 74%).72 Acyclic iodonium ylides reacted with several triarylfluoroboranes to afford the 1,3-carboboration compounds in which aryl group transfer from boron to the iodonium ylide had occurred (Scheme 34). In these reactions, the mechanism of activation was found to be the initial coordination of a boron Lewis acid to the carbonyl group. Cyclic iodonium ylides failed to afford the 1,3-carboboration products.
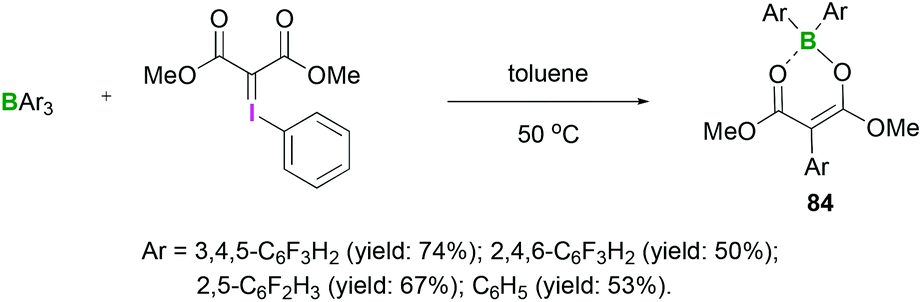 | ||
| Scheme 34 Reaction between iodonium and triarylfluoroboranes to afford the 1,3-carboboration compounds 84. | ||
Conclusions
Expanding the toolbox of the synthetic chemist is crucial for the development of enabling strategies for metal-free synthesis while simultaneously simplifying the formation of ever more complex structures. Hypervalent iodine chemistry is presented as a reliable source to achieve these goals and inspires further development. However, the prerequisite activation of λ3-iodanes by boranes has been limited almost exclusively to BF3·OEt2 with just a few examples with triarylboranes and borosilicate. The few reported examples of other borane-based Lewis acidic activators indicate sufficiently exciting reactivity to promise a great future potential for the further advancement of synthetic protocols. The increased availability of boron reagents and understanding of their Lewis acidities and reactivities will allow the field of hypervalent iodine to expand into novel areas of synthetic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. and R. L. M. are grateful to the EPSRC for funding and the awarding of an EPSRC Early Career Fellowship (R026912/1). We are grateful to the Leverhulme Trust for a research project grant (C. T., P. D. N. and R. L. M. RPG-2016-020).Notes and references
- J. Willgerodt, J. Prakt. Chem., 1886, 33, 154 CrossRef.
- G. F. Koser, A. G. Relenyi, A. N. Kalos, L. Rebrovic and R. H. Wettach, J. Org. Chem., 1982, 47, 2487 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- R. Calvo, A. Le Tellier, T. Nauser, D. Rombach, D. Nater and D. Katayev, Angew. Chem., Int. Ed., 2020, 59, 17162 CrossRef CAS PubMed.
- X. Li, P. Chen and G. Liu, Beilstein J. Org. Chem., 2018, 14, 1813 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- Hypervalent Iodine Chemistry, ed. T. Wirth, Topics in Current Chemistry, 2016, p. 373 Search PubMed.
- M. S. Yusubov, N. S. Soldatova, P. S. Postnikov, R. R. Valiev, A. Yoshimura, T. Wirth, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2019, 55, 7760 RSC.
- M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251 CrossRef CAS PubMed.
- M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086 RSC.
- Z. Wang, New J. Chem., 2021, 45, 509 RSC.
- L. F. Silva, Jr. and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- IUPAC, Compendium of Chemical Terminology, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- R. E. Rundle, J. Am. Chem. Soc., 1947, 69, 1327 CrossRef CAS.
- M. Ochiai, in Top. Curr. Chem, ed. T. Wirth, Springer, Berlin, Heidelberg, 2003, vol. 224, p. 5 Search PubMed.
- B. Ganji and A. Ariafard, Org. Biomol. Chem., 2019, 17, 3521 RSC.
- M. S. Yubusov, V. N. Nemykin and V. V. Zhdankin, Tetrahedron, 2010, 66, 5745 CrossRef.
- A. J. Edwards, J. Chem. Soc., Dalton Trans., 1978, 1723 RSC.
- T. Kitamura, M. Yamane, K. Inoue, M. Todaka, N. Fukatsu, Z. Meng and Y. Fujiwara, J. Am. Chem. Soc., 1999, 121, 11674 CrossRef CAS.
- T. Okuyama, T. Takino, T. Sueda and M. Ochiai, J. Am. Chem. Soc., 1995, 117, 3360 CrossRef CAS.
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS.
- M. Ochiai, Y. Takaoka and Y. Nagao, J. Am. Chem. Soc., 1988, 110, 6565 CrossRef CAS.
- M. Ochiai, T. Ukita, S. Iwaki, Y. Nagao and E. Fujita, J. Org. Chem., 1989, 54, 4832 CrossRef CAS.
- X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef CAS PubMed.
- A. Sreenithya and R. B. Sunoj, Dalton Trans., 2019, 48, 4086 RSC and references cited therein.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2014, 115, 650 CrossRef PubMed.
- S. Shu, Y. Li, J. Jiang, Z. Ke and Y. Liu, J. Org. Chem., 2019, 84, 458 CrossRef CAS PubMed.
- M. Ochiai, Iodosylbenzene-Boron Trifluoride, in Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.ri040.
- M. Ochiai, E. Fujita, M. Arimoto and H. Yamaguchi, J. Chem. Soc., Chem. Commun., 1982, 1108 RSC.
- M. Kida, T. Sueda, S. Goto, T. Okuyama and M. Ochiai, Chem. Commun., 1996, 1933 RSC.
- S. Izquierdo, S. Essafi, I. del Rosal, P. Vidossich, R. Pleixats, A. Vallribera, G. Ujaque, A. Lledós and A. Shafir, J. Am. Chem. Soc., 2016, 138, 12747 CrossRef CAS PubMed.
- G. S. Sinclair, R. Tran, J. Tao, W. S. Hopkins and G. K. Murphy, Eur. J. Org. Chem., 2016, 4603 CrossRef CAS.
- A. Varvoglis, in Best Synthetic Methods, ed. A. B. T.-H. I. in O. S. Varvoglis, Academic Press, London, 1997 Search PubMed.
- R. M. Moriarty, C. J. Chany II, J. W. Kosmeder II and J. Du Bois, in Encyclopedia of Reagents for Organic Synthesis, American Cancer Society, 2006 Search PubMed.
- M. Ochiai, K. Sumi, Y. Takaoka, M. Kunishima, Y. Nagao, M. Shiro and E. Fujita, Tetrahedron, 1988, 44, 4095 CrossRef CAS.
- M. Ochiai, M. Kunishima, K. Sumi, Y. Nagao and E. Fujita, Tetrahedron Lett., 1985, 26, 4501 CrossRef CAS.
- M. Fujita, H. J. Lee and T. Okuyama, Org. Lett., 2006, 8, 1399 CrossRef CAS PubMed.
- M. Ochiai, Y. Takaoka, Y. Masaki, M. Inenaga and Y. Nagao, Tetrahedron Lett., 1989, 30, 6701 CrossRef CAS.
- N. S. Zefirov, A. S. Koźmin, T. Kasumov, K. A. Potekhin, V. D. Sorokin, V. K. Brel, E. V. Abramkin, Y. T. Struckov, V. V. Zhdankin and P. J. Stang, J. Org. Chem., 1992, 57, 2433 CrossRef CAS.
- M. Ochiai, M. Kunishima, K. Fuji, M. Shiro and Y. Nagao, J. Chem. Soc., Chem. Commun., 1988, 1076 RSC.
- M. Ochiai, T. Ito, Y. Takoka, Y. Masaki, M. Kunishima, S. Tani and Y. Nagao, J. Chem. Soc., Chem. Commun., 1990, 118 RSC.
- K. Miyamoto, M. Hirobe, M. Saito, M. Shiro and M. Ochiai, Org. Lett., 2007, 9, 1995 CrossRef CAS PubMed.
- M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 3, 4602 CrossRef PubMed.
- M. Ochiai, Y. Kitagawa, N. Takayama, Y. Takaoka and M. Shiro, J. Am. Chem. Soc., 1999, 121, 9233 CrossRef CAS.
- M. Ochiai, T. Ito and Y. Masaki, J. Am. Chem. Soc., 1992, 114, 6269 CrossRef CAS.
- M. Ochiai, Z. Masaki and M. Shiro, J. Org. Chem., 1991, 56, 5511 CrossRef CAS.
- T. Hokamp, L. Mollari, L. C. Wilkins, R. L. Melen and T. Wirth, Angew. Chem., Int. Ed., 2018, 57, 8306 CrossRef CAS PubMed.
- M. Ito, I. Itani, Y. Toyoda, K. Morimoto, T. Dohi and Y. Kita, Angew. Chem., Int. Ed., 2012, 51, 12555 CrossRef CAS PubMed.
- W. Zhong, J. Yang, X. Meng and Z. Li, J. Org. Chem., 2011, 76, 9997 CrossRef CAS PubMed.
- J. H. Lee, S. Choi and K. B. Hong, Molecules, 2019, 24, 2634 CrossRef PubMed.
- T. Hokamp and T. Wirth, J. Org. Chem., 2019, 84, 8674 CrossRef CAS PubMed.
- M. Shimogaki, M. Fujita and T. Sugimura, Angew. Chem., Int. Ed., 2016, 55, 15797 CrossRef CAS PubMed.
- M. Fujita, Y. Yoshida, K. Miyata, A. Wakisaka and T. Sugimura, Angew. Chem., Int. Ed., 2010, 49, 7068 CrossRef CAS.
- M. Fujita, M. Wakita and T. Sugimura, Chem. Commun., 2011, 47, 3983 RSC.
- T. Hokamp and T. Wirth, Chem. – Eur. J., 2020, 26, 10417 CrossRef CAS PubMed.
- P. Mizar, A. Laverny, M. El-Sherbini, U. Farid, M. Brown, F. Malmedy and T. Wirth, Chem. – Eur. J., 2014, 20, 9910 CrossRef CAS PubMed.
- J. Schörgenhumer and M. Waser, Org. Chem. Front., 2016, 3, 1535 RSC.
- T. Nagata, H. Matsubara, K. Kiyokawa and S. Minakata, Org. Lett., 2017, 19, 4672 CrossRef CAS PubMed.
- T. Dohi, K. Morimoto, Y. Kiyono, H. Tohma and Y. Kita, Org. Lett., 2005, 7, 537 CrossRef CAS.
- T. Dohi, K. Morimoto, N. Takenaga, A. Goto, A. Maruyama, Y. Kiyono, H. Tohma and Y. Kita, J. Org. Chem., 2007, 72, 109 CrossRef CAS PubMed.
- J. Tao, R. Tran and G. K. Murphy, J. Am. Chem. Soc., 2013, 135, 16312 CrossRef CAS.
- X. Zhang, S. Guo and P. Tang, Org. Chem. Front., 2015, 2, 806 RSC.
- B. Xing, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2018, 57, 9896 CrossRef CAS PubMed.
- Z. Zhao, L. Racicot and G. K. Murphy, Angew. Chem., Int. Ed., 2017, 56, 11620 CrossRef CAS PubMed.
- T. Dohi, M. Ito, K. Morimoto, M. Iwata and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 1301 CrossRef CAS PubMed.
- E. Faggi, R. M. Sebastián, R. Pleixats, A. Vallribera, A. Shafir, A. Rodríguez-Gimeno and C. Ramírez de Arellano, J. Am. Chem. Soc., 2010, 132, 17980 CrossRef CAS PubMed.
- Y. Wu, S. Bouvet, S. Izquierdo and A. Shafir, Angew. Chem., Int. Ed., 2019, 58, 2617 CrossRef CAS PubMed.
- H. Zhang, D. Huang, K.-H. Wang, J. Li, Y. Su and Y. Hu, J. Org. Chem., 2017, 82, 1600 CrossRef CAS.
- S. Chen, L. Yang, D. Yi, Q. Fu, Z. Zhang, W. Liang, Q. Zhang, J. Jia and W. Wei, RSC Adv., 2017, 7, 26070 RSC.
- J. Cui, Q. Jia, R.-Z. Feng, S.-S. Liu, T. He and C. Zhang, Org. Lett., 2014, 16, 1442 CrossRef CAS PubMed.
- Y.-C. Han, Y.-D. Zhang, Q. Jia, J. Cui and C. Zhang, Org. Lett., 2017, 19, 5300 CrossRef CAS PubMed.
- T. A. Gazis, B. A. Thaker, D. Willcox, D. M. C. Ould, J. Wenz, J. M. Rawson, M. S. Hill, T. Wirth and R. L. Melen, Chem. Commun., 2020, 56, 3345 RSC.
| This journal is © The Royal Society of Chemistry 2021 |