
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asparaginyl endopeptidases: enzymology, applications and limitations
T. M. Simon
Tang
 a and
Louis Y. P.
Luk
*ab
a and
Louis Y. P.
Luk
*ab
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: lukly@cardiff.ac.uk
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 12th May 2021
Abstract
Asparaginyl endopeptidases (AEP) are cysteine proteases found in mammalian and plant cells. Several AEP isoforms from plant species were found to exhibit transpeptidase activity which is integral for the key head-to-tail cyclisation reaction during the biosynthesis of cyclotides. Since many plant AEPs exhibit excellent enzyme kinetics for peptide ligation via a relatively short substrate recognition sequence, they have become appealing tools for peptide and protein modification. In this review, research focused on the enzymology of AEPs and their applications in polypeptide cyclisation and labelling will be presented. Importantly, the limitations of using AEPs and opportunities for future research and innovation will also be discussed.
 T. M. Simon Tang (left) Louis Y. P. Luk (right) | T. M. Simon Tang (left) studied Natural Sciences at the University of Bath and obtained his MSc in 2017. He obtained his PhD under the supervision of Dr Luk at Cardiff University where he explored the use of asparaginyl endopeptidases for protein bioconjugation. He is currently a research associate working in the group of Professor Jody Mason at the University of Bath. Louis Y. P. Luk (right) is a Lecturer at the School of Chemistry of Cardiff University. He obtained his BSc in Chemistry and Microbiology & Immunology at the University of British Columbia. He obtained his PhD at the same university under the supervisor of Martin E. Tanner, followed by post-doctoral studies in the laboratories of Stephen BH Kent at the University of Chicago and Rudolf K Allemann at Cardiff University. Louis became a University Research Fellow in 2015 and was recently tenured at Cardiff University. Louis' current research combines his training in enzymology and peptide chemistry, focusing on artificial enzyme design and protein bioconjugation chemistry. |
Introduction
Asparaginyl endopeptidase (AEP) are proteases identified from the vacuole of plant cells.1 Also known as legumain,2,3 vacuolar processing enzyme1 or peptide asparaginyl ligase,4 AEPs react selectively at an internal asparagine (Asn) or aspartate (Asp) residue of a peptide backbone via a catalytic cysteine residue. In all cases, the enzymes are folded in the endoplasmic reticulum (ER) as a zymogen with a pro-domain which prevents substrate access to the active site.5–7 Pro-AEPs have been shown to accumulate in ER bodies within the epidermal cells of Arabidopsis thaliana seedlings.5 In response to stress, ER bodies fuse and release zymogenic AEPs into the vacuole.5 AEPs have also been reported to process seed storage proteins, 2S albumins, in prevacuolar multivesicular bodies (MVB) before reaching the protein storage vacuole in A. thaliana embryos.8 The acidic environment within the MVB and vacuole trigger a proteolytic maturation to afford the active AEP (Fig. 1).2,5,7–10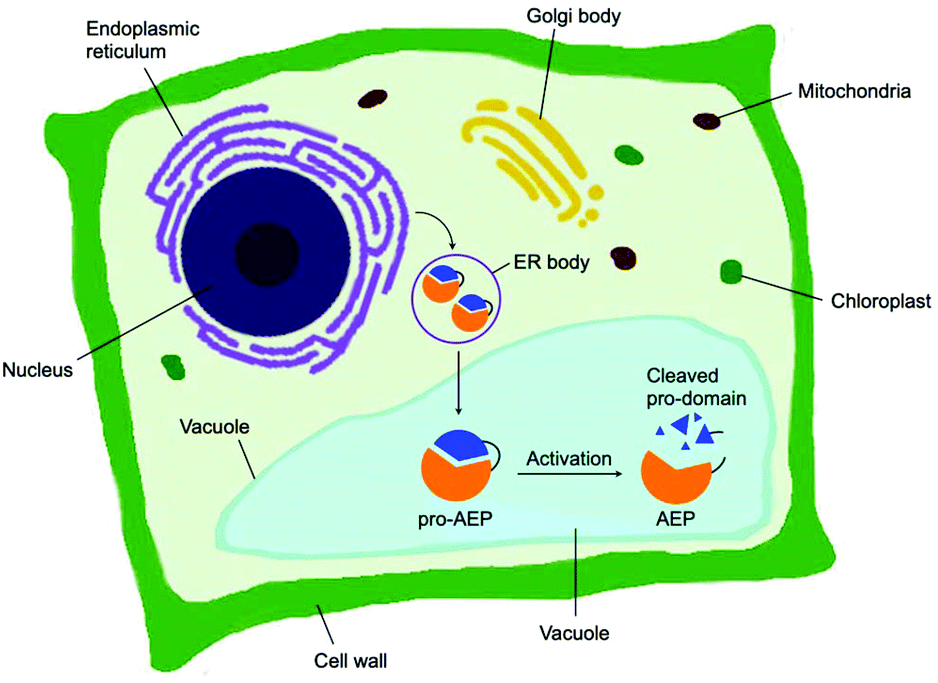 | ||
| Fig. 1 Biosynthesis of AEP in plant cells. Zymogenic AEPs are folded in the ER and stored in ER bodies. The pro-enzyme is trafficked to the vacuole, where the acidic environment enables cleavage of the pro-domain resulting in activation. | ||
Matured AEPs primarily function as peptidases which are associated with several critical cellular processes.2,11 Purified AEP from the seed of castor beans (Ricinus communis) has been shown to mediate the proteolytic processing of pro-proteins to afford mature seed storage proteins including 2S albumin and 11S globulin.1,12 In vegetative organs, AEPs have been shown to facilitate programmed cell death.5,13 When an environmental stress (concentrated salt solution) was applied to healthy seedlings of A. thaliana, ER bodies were found to fuse and deliver AEPs to the vacuole, leading to cell death (Fig. 2).5 While their structures and activity are similar to those of caspases,14–16 AEPs were also identified in mammalians, including humans and mice.2,17 Mammalian orthologues of AEPs, namely legumains, are associated with antigen processing which moderate antigen presentation and T-cell activity.18–20 AEP-mediated proteolytic processing of the microbial tetanus toxin antigen was found to enable antigen presentation by class II major histocompatibility complex molecules.18
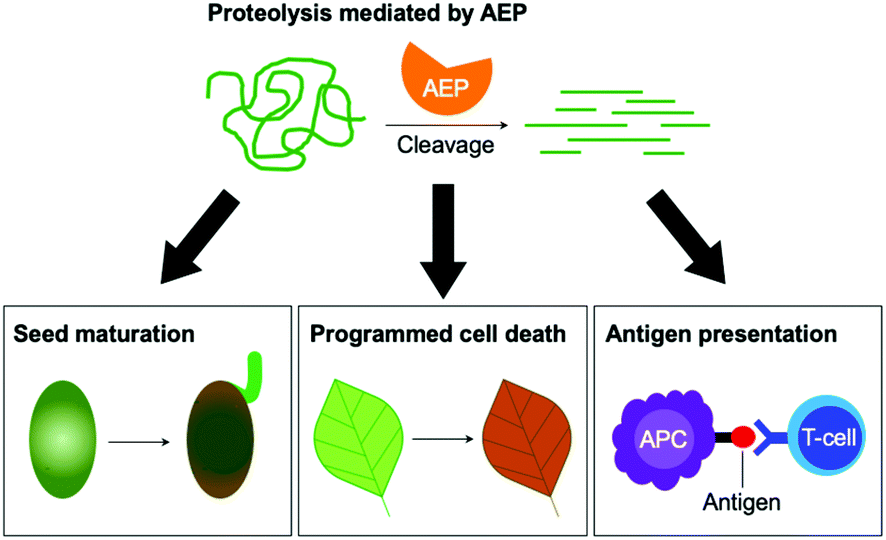 | ||
| Fig. 2 The peptidase activities of AEPs are involved in several critical cellular processes including seed maturation, programmed cell death and the regulation of antigen presentation. | ||
Although most AEPs were shown to facilitate peptide cleavage, some AEPs isolated from plant species were found to exhibit peptide ligase activity.4,21–27 Ligase type AEPs are rare compared to the corresponding peptidases.4,21–23 Various plant species have been found to utilise this peptide ligase activity to facilitate post translational modification to produce backbone cyclised peptides (Fig. 3).4,22–24,26 The cyclisation step during the biosynthesis of kalata B1, a cystine knotted macrocyclic peptide, was shown to be catalysed by an AEP (OaAEP1b) in Oldenlandia affinis.22 Cyclic peptides found in plants exhibit a range of bioactivities,28,29 and demonstrate superior stability compared to their linear counterparts.30 Kalata B1 extracted from O. affinis, a native African plant species, demonstrates insecticidal activities inhibiting the growth of Helicoverpa punctigera larvae at a high concentration.31,32 Furthermore, the cyclic kalata B1 has been shown to display high tolerance towards thermal (370 K, 97 °C) and chemical (8 M urea) denaturation.30 Another example of a cystine knotted cyclic peptide is Cter-M, which is cyclised by an AEP in butterfly pea plants (Clitoria ternatea), dubbed butelase 1.33 Other AEPs such as HaAEP1 from common sunflower (Helianthus annuus) and MCoAEP2 from gac fruit plants (Momordica cochinchinensis) have been reported to facilitate backbone cyclisation during the biosynthesis of cyclic trypsin inhibitor peptides SFTI-1 and MCoTI-II, respectively (Fig. 3).27,34
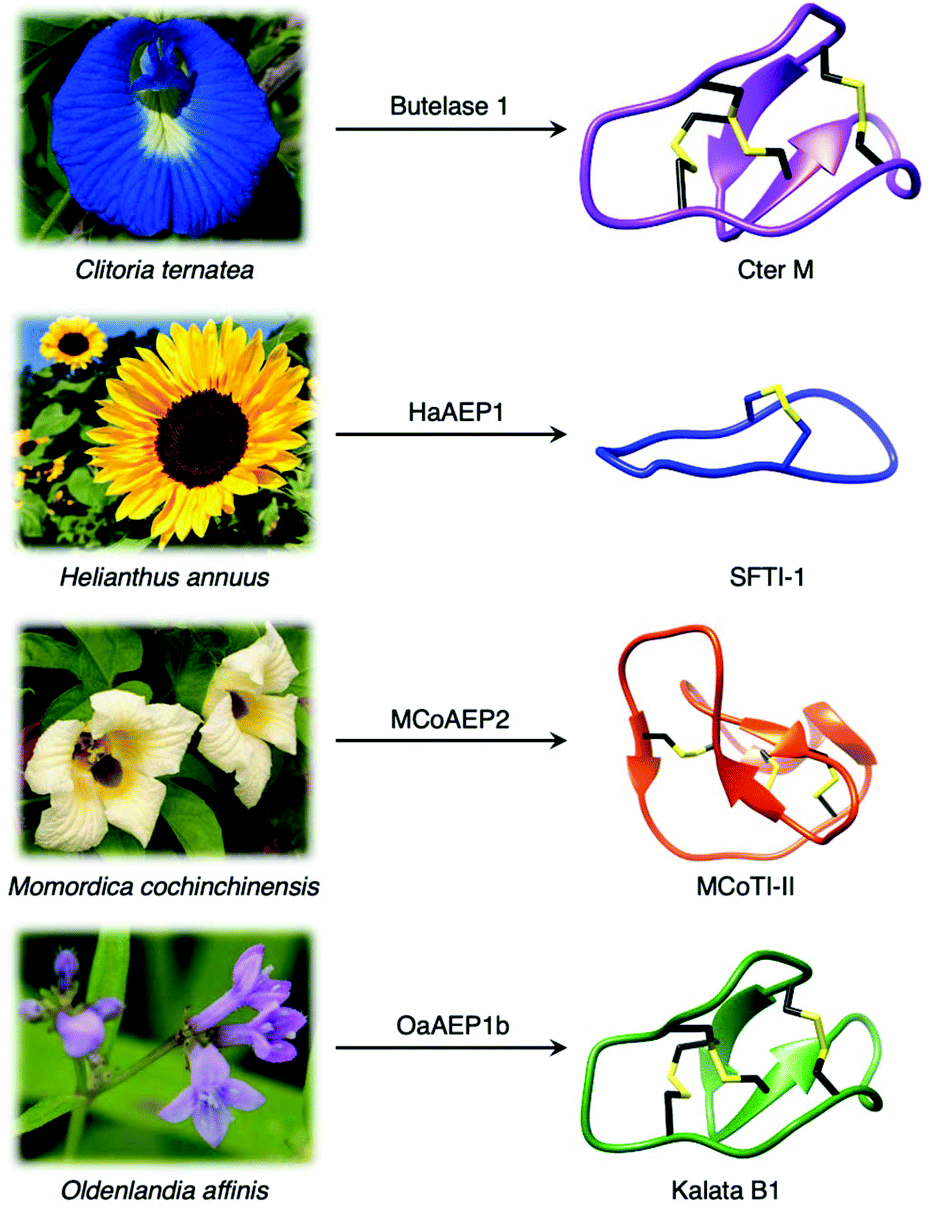 | ||
| Fig. 3 Plant species utilise AEPs with ligase activities to generate backbone cyclised peptides with enhanced stability and bioactivity. Cter M (PDB: 2LAM) from C. ternatea, SFTI-1 (PDB: 1JBL) from H. annuus, MCoTI-II (PDB: 1IB9) from M. cochinchinensis and kalata B1 (PDB: 1NB1) from O. affinis. | ||
Reaction mechanisms of AEPs
Since the initial discovery of the AEP,1 there has been an expansion of research ranging from structural biology, enzymology to chemical biology development. X-ray crystallographic data of various AEPs have provided fundamental knowledge and rationales for the localisation of activity.4,7,10,35,36 Prominent structural features include the catalytic core domain which adopts a protein fold with a six-stranded β-sheet surrounded by five α-helices. In the proenzyme, the catalytic site is covered by the pro-domain which is C-terminal to the core domain (Fig. 4).4,7,10,35,36 Studies performed on an AEP from A. thaliana (AtLEGγ) revealed that pro-AEP adopts a dimeric state at neutral pH which has been shown to prevent enzymatic activation.7 The enzyme adopts a monomeric state at pH 4.0 and the protonation of salt bridges surrounding the active site of the AEP was reported to increase flexibility, enabling the auto-proteolytic cleavage of the pro-domain to generate the active enzyme.5,7,22,35 Taken away from the acidic and reducing environments of the plant vacuole, the cleaved core domain and pro-domain of AtLEGγ have been shown remain bound in a two-chain state.7 The non-covalent association of the cleaved AEP pro-domain was suggested to confer stability to the catalytic core domain at neutral pH.7 | ||
| Fig. 4 Overlaid ribbon representations of human pro-legumain (hLEG, PDB: 4FGU, purple), butelase 1 (PDB: 6DHI, orange), and OaAEP1b (PDB: 5H0I, blue) structures deduced from X-ray crystallography.10,35,36 The black dotted line indicates the separation between the pro-domain and the core domain. After activation, modified butelase 1 (PDB: 6DHI) and OaAEP1b (PDB: 5H0I) structures showing only the catalytic core domains were overlaid with the crystal structure of activated human legumain (PDB: 4AWA, purple). In the box, an enlarged image of the active sites reveals the respective catalytic diads (Cys and His), the conformation adopted by the inhibitor (YVAD-CMK) in complex with activated human legumain, and a plant specific loop region referred to as the poly-proline loop (PPL).26 | ||
The catalytically active core domain of AEPs have been proposed to operate via a mechanism that recruits the catalytic cysteine residue as a nucleophile.14,16,35 The catalytic diad (Cys and His) mediates a nucleophilic attack on the amide carbonyl of an internal Asn residue to form a thioester acyl-enzyme intermediate (Scheme 1).22,23,25,35 For most AEPs, nucleophilic attack from a water molecule completes the peptide bond hydrolysis.35 For ligase-type AEPs, the N-terminal amine of an incoming peptide adopts the role of the nucleophile to resolve the thioester intermediate.22,23 The nucleophilic amine can be either from the same peptide, resulting in an intramolecular cyclisation,22,23 or from a different peptide, resulting in an intermolecular ligation.23,37 A mechanistic study of AEP-mediated peptide ligation employing 18O-labelled water found no isotopic shift in the ligated peptide product.22,24 This finding indicated that water is likely to be excluded from the active site during peptide ligation by AEPs. Peptide ligation proceeds with a direct nucleophilic attack from the N-terminal amine to the thioester intermediate with no participation of solvent water molecules.
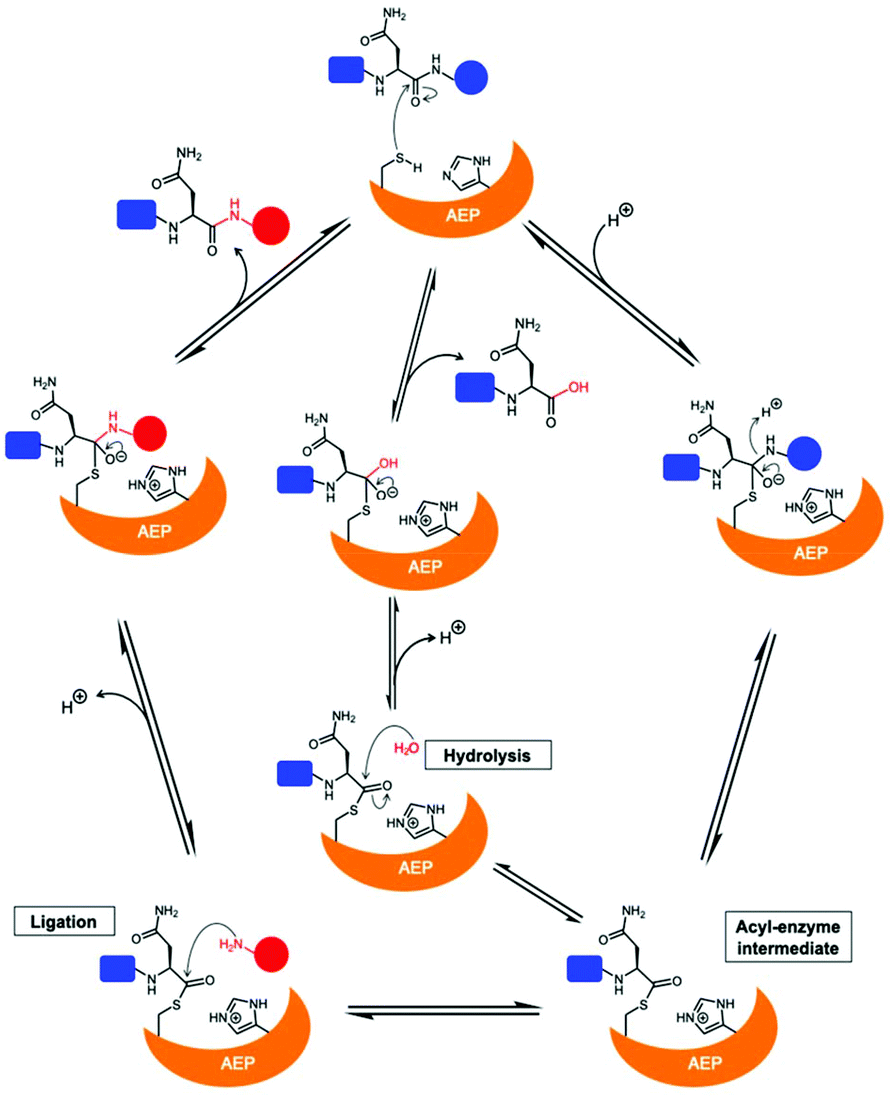 | ||
| Scheme 1 Proposed mechanism of AEPs catalysis with a Cys/His catalytic diad. AEP recognises and cleaves a peptide substrate containing Asn to form an acyl-enzyme intermediate via a tetrahedral intermediate. A nucleophilic attack resolves the thioester intermediate to afford the product peptide and the AEP is regenerated. The nucleophile can be a water molecule or the N-terminus of a peptide, resulting in peptide hydrolysis or ligation, respectively. | ||
A succinimide motif not commonly seen among other enzymes has been observed by X-ray crystallography in active sites of AEP isoforms hLEG, HaAEP1 and AtLEGγ from human, sunflower and A. thaliana, respectively.25,35,38 The metastable structure resulted from the condensation between the sidechain carboxylate of Asp and a backbone amide NH. Nevertheless, Haywood et al. have shown that a mutation of the succinimide forming aspartate residue to an alanine bears no influence on the ligation activity of HaAEP1.25
A comparison of AEPs from the same, or across multiple organisms, reveals that enzymes with ligase activities are uncommon compared to the putative proteases.4,21–23 Various AEP isoforms exhibit a remarkable difference in activity (protease vs. ligase) despite only subtle differences in sequence and structure.4,26,35,36 Furthermore, the activity of AEPs were reported to be influenced by the amino acid sequence, length of substrate peptides, plus reaction conditions such as pH.22,23,39
The growing library of AEPs identified from numerous organisms, accompanied by the increasing availability of sequence and structural information, has enabled comparative studies among isoforms and identification of features that influence the preference towards ligase or protease activity.7,26,27,36,39 Substrate accessibility to the active site of AEPs are governed by a gatekeeper residue (GK), also referred to as ligase-activity determinant 1 (LAD1),4 has been highlighted as a factor that determines activity (Fig. 5).10,26 The identity of this residue, with particular emphasis on the size of its sidechain, was suggested to influence substrate access to the active site. OaAEP1b, isolated from O. affinis, is an AEP variant which exhibits peptide ligase activity.22 Mutagenesis studies of OaAEP1b demonstrated that substitutions of the gatekeeper residue (Cys247) to larger residues such as Ile, Leu and Met abolished ligase activity. In contrast, replacements with smaller Ala and Gly residues at the same position resulted in an increase in peptide cyclisation activity. In addition, the Cys247Gly mutation was shown to enhance peptide hydrolysis when compared to the wild-type and Cys247Ala variants.10 The smaller and less hydrophobic Gly as a gatekeeper residue was proposed to allow water molecules to access the active site, resulting in protease activity. The enhanced ligase activity of OaAEP1-C247A has consequently been adopted to facilitate protein labelling reactions in several studies.40–42
 | ||
| Fig. 5 Key features associated with the determination AEP ligase and protease activity. Multiple sequence alignment of AEPs, from O. affinis (OaAEP1b, 3), P. x hybrida (PxAEP1, 2, 3a, 3b), C. ternatea (butelase 1) and H. sapien (hLEG), centred around amino acid residues reported to influence activity. Catalytic diads (Cys/His) are shown in white letters with black highlight, N-glycosylation site is highlighted in green, the overlaid red boxes show LAD1 (gatekeeper/GK) and LAD2, blue box shows the plant-specific poly-proline loop and the green box highlights the MLA.4,26 In the box, an enlarged overlaid image of OaAEP1 (PDB: 5H0I),10 butelase 1 (PDB: 6DHI)36 and human legumain (PDB: 4AWA)35 active sites. Key features (catalytic diad, LAD1, LAD2, PPL and MLA) are rendered blue, orange and purple for OaAEP1, butelase 1, and human legumain respectively. In black, the conformation adopted by the inhibitor (YVAD-CMK) in complex with human legumain. | ||
Further investigations into the sequence and structures of different AEP isoforms revealed additional polymorphic positions named ligase-activity determinant 2 (LAD2)4 and marker of ligase activity (MLA)26 which are located within, and adjacent to, the substrate binding pocket, respectively. The MLA was identified by a study comparing the sequences of protease and ligase AEPs from garden petunia (Petunia x hybrida) and describes a five amino acid deletion found in AEPs with ligase activity (Fig. 5).26
The impact of the MLA on the peptide cyclisation activity in planta was investigated using tobacco plants (Nicotiana benthamiana) which co-expressed transgenes encoding for AEPs and corresponding peptide substrates. Relative cyclisation efficiencies in planta were measured by matrix-assisted laser desorption ionisation mass spectrometry (MALDI-MS).26 The insertion of five amino acid to the MLA region of OaAEP1b, a putative AEP ligase, was shown to significantly impede its ability to mediate peptide cyclisation.26 However, when the experiments were repeated in vitro, the MLA modification was found to only reduce the rate of peptide cyclisation by OaAEP1b rather than a distinct shift to protease activity.26 The truncation described as the MLA is also not found in the amino acid sequence of butelase 1, an AEP ligase from C. ternatea (Fig. 5). Thus, while the MLA offer an indication to the protease vs. ligase preference of AEPs, it is not a critical determinant of enzyme activity.
LAD2 describes two amino acid residues lining the substrate binding pocket of AEPs. Mutation experiments performed in an AEP isolated from Viola yedoensis (VyPAL3) demonstrated the effect of amino acid substitutions at LAD2. Replacing Tyr with the smaller Gly residue was reported to improve the in vitro ligase activities of VyPAL3.4 Furthermore, a Tyr to Ala substitution at LAD2 of a protease AEP from Viola canadensis (VcAEP) was shown to shift enzyme activity from protease to ligase.4 However, the introduction of protease favouring residues at LAD positions did not switch the activity preference of VyPAL2, an AEP ligase from V. yedoensis, to adapt an increase of protease activity.4
None of the identified structural features were found to provide absolute control over the protease vs. ligase preference of AEPs. Nevertheless, mutations at gatekeeper, MLA and LAD2 have all been shown to significantly impact AEP activity.4,26 These findings suggest that key structural features may operate synergistically to dictate AEP activity, and further investigations are required to fully establish the key determinants of AEP protease and ligase activity. A recent review by Nonis et al. can provide further discussions towards the key determinants of AEP protease and ligase activity.43
Substrate recognition
Amino acid residues surrounding the peptide cleavage site for substrates of AEPs are commonly labelled following the numbering conventions adopted by Schechter and Berger for substrates of proteases (Scheme 2).44 From the cleavage site to the peptide N-terminus, amino acid residues are numbered in ascending order starting from P1. On the other side of the cleavage site, amino acid residues are denoted with a single prime and numbered in ascending order towards the C-terminus starting with P1′. For peptide ligation, the amino acid positions on the incoming peptide are labelled with a double prime and numbered in ascending order from the N-terminus starting with P1′′.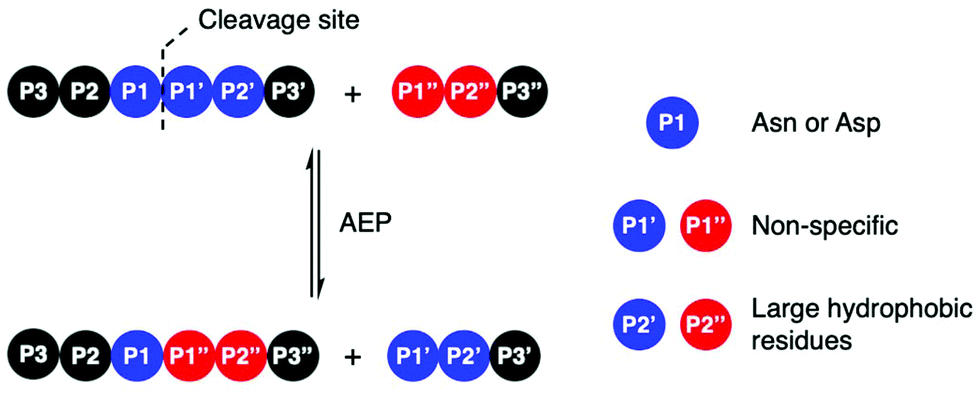 | ||
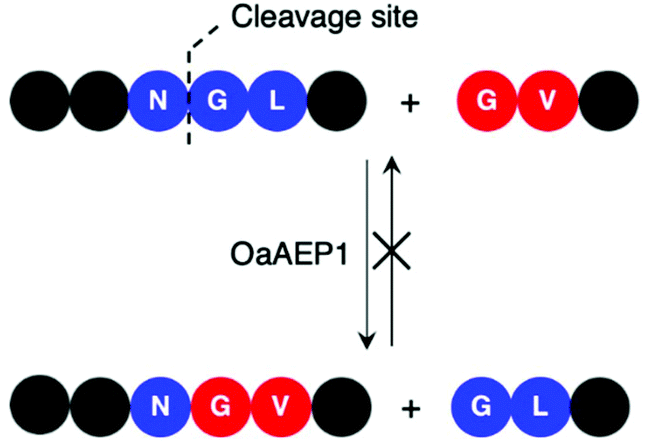
| Scheme 2 AEP-mediated transpeptidation, with amino acid residues numbered according to Schechter and Berger.44 The putative recognition sequences P1–P1′–P2′ and P1′′–P2′′ are coloured in blue and red, respectively. | ||
Initial studies performed with an AEP isolated from castor beans have identified strict substrate specificity towards Asn at the P1 position.1,2,17,18,22,23 The understanding of substrate recognition by AEPs have subsequently expanded with experiments in AEPs isolated from C. ternatea (butelase 1),23,45,46O. affinis (OaAEP1b and OaAEP3-5)22,39 and other plant species.24,27 Substrate recognition by AEPs were shown to be determined mostly by amino acids at positions P1, P1′ and P2′ (Scheme 2). While many AEPs have a strict preference towards Asn at P1, some isoforms such as HaAEP1 and OaAEP1b were found to accept Asp at P1.22,24 The preferences toward P1′ and the ensuing P1′′ from the nucleophilic peptide for ligation are non-specific. A peptide ligation assay which employed a library of peptides, with varying amino acids at the N-terminus, demonstrated that butelase 1 accepts all canonical amino acid residues at P1′′ with the exception of Pro.23 Furthermore, butelase 1 was reported to cyclise peptides with D-amino acids at P1′′ and recognise peptides with a thioester linkage between the P1 and P1′ residues.45,46 AEPs from O. affinis (OaAEP1b, OaAEP3-5) have been shown to process peptides with several different amino acid residues at P1′.22,39,47 In contrast, recognition at P2′ and P2′′ demonstrate greater specificity. While peptides with His or Ala at P2′ were not processed by OaAEP1b, peptide cyclisation was detected when substrates with Leu or Phe at P2′ were used.22,39 Furthermore, butelase 1 was reported to ligate peptides with large, hydrophobic side chains such as Leu, Ile and Val at P2′′.23
Although most investigations towards the substrate specificity of AEPs have focused on the putative recognition sequences at P1–P1′–P2′ and P1′′–P2′′ (Scheme 2), some studies suggested enzyme activity can be affected by other positions in the substrate peptide.22,27,48 A substitution of Arg with Ala at the P2 position was found to significantly reduce the rate of OaAEP1b-mediated peptide cleavage.22 Moreover, the use of Pro at P2 in a short peptide (5–9 amino acids) was shown to favour intermolecular ligation over intramolecular cyclisation by butelase 1. Intermolecular ligation of short peptides followed by head-to-tail cyclisation results in the formation of cyclo-oligomers (Scheme 3A).48 Notably, it was highlighted that butelase 1 was not able to recognise the corresponding cyclo-oligomeric peptide products despite the presence of a suitable recognition sequence.48 Since AEP catalysis is not known to involve co-factors or other external sources of energy, this observation may be rationalised by conformational stabilisation of the cyclised peptide and an entropic gain from the release of the cleaved recognition peptide motif.
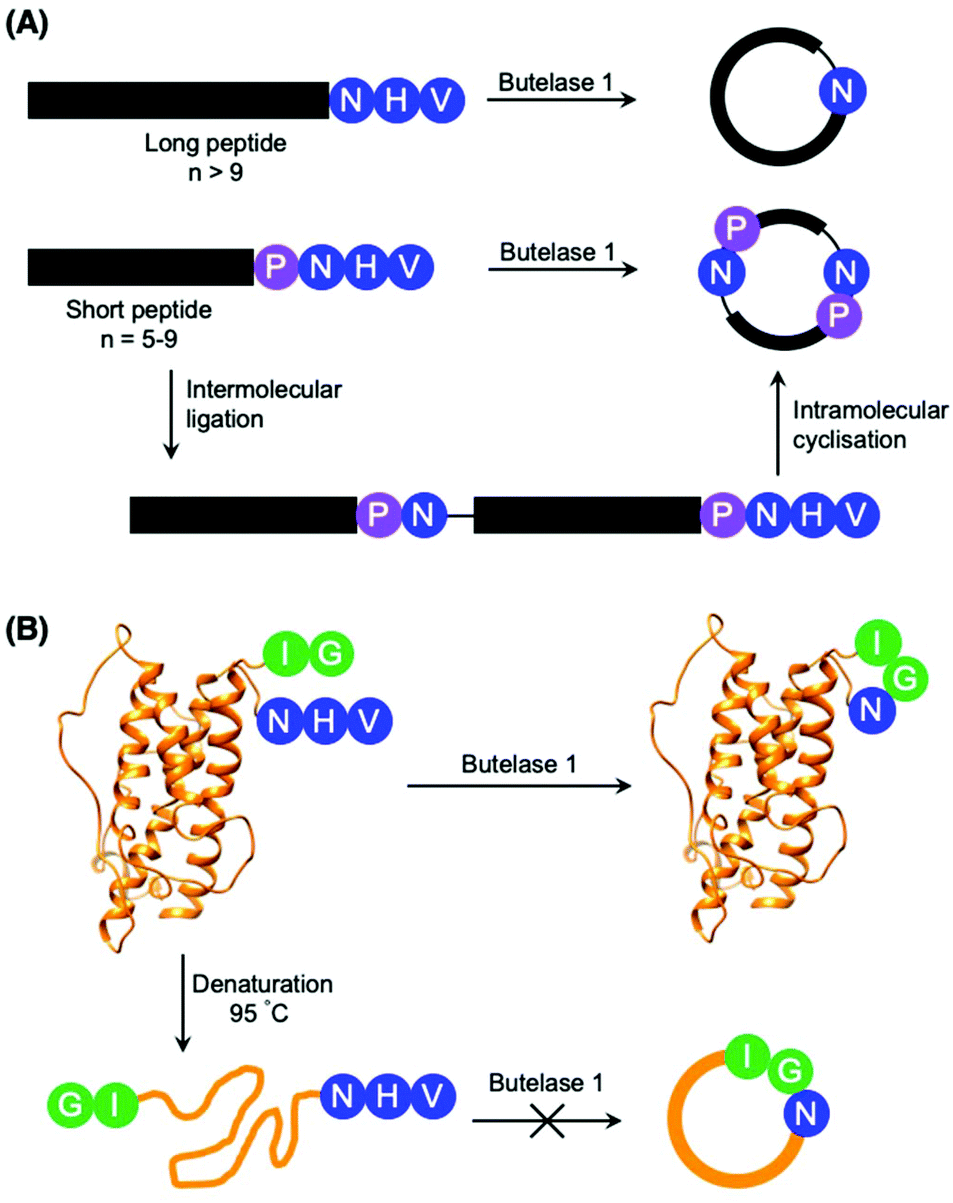 | ||
| Scheme 3 Peptide cyclisation catalysed by butelase 1. (A) Peptides greater than 9 amino acid residues in length and bearing the butelase 1 substrate recognition sequence (NHV) are cyclised by the AEP. Whereas peptides of 5–9 amino acid residues in length, with Pro at P2 and bearing the butelase 1 substrate recognition sequence (NHV) are ligated, then cyclised by AEP to afford cyclic oligomers48 (only cyclo-dimer shown here). (B) Butelase 1 was reported to cyclise the folded somatropin (PDB: 1HGU) but not the denatured protein.49 | ||
The proximity of the N and C-termini was shown to influence the efficiency of AEP-mediated cyclisation of larger peptide and protein substrates. While butelase 1 was reported to effectively cyclise recombinantly prepared interleukin-1 receptor antagonist (IL-1Ra) and human growth hormone (somatropin), no cyclic proteins were detected when IL1-Ra and somatropin were denatured prior to incubation with the enzyme (Scheme 3B).37,49 A similar effect can also be observed in OaAEP1b-mediated protein cyclisation of an intrinsically disordered protein, merozoite surface protein 2 (MSP2), which proceeds in low yield.39
Rate of peptide cyclisation by AEPs
The reported kinetic parameters for peptide cyclisation catalysed by various AEPs have been summarised in Table 1. As investigations have conducted the peptide cyclisation assays under different reaction conditions and substrates, it is difficult to draw direct comparisons. Nevertheless, the catalytic efficiencies of AEPs are relatively high compared to sortase A, a widely used transpeptidase with catalytic efficiency (kcat/KM) values ranging from 0.02 to 2.80 × 104 M−1 s−1 for various engineered variants.50–52| Enzyme | Origin | Recognition motif | k cat/s−1 | k cat/KM/104 M−1 s−1 | Assay conditions | Ref. | |
|---|---|---|---|---|---|---|---|
| Temperature/°C | pH | ||||||
| Butelase 1 | C. ternatea | GI-Xn-NHV | 26.55 | 131 | 42 | 6.0 | 23 and 49 |
| OaAEP1b | O. affinis | GL-Xn-NGL | 0.99 | 0.68 | r.t. | 5.0 | 39 |
| OaAEP1-C247A | O. affinis | GL-Xn-NGL | 13.9 | 3.42 | 37 | 6.0 | 10 |
| OaAEP1-C247A | O. affinis | GL-Xn-NGL | 1.52 | 1.04 | r.t. | 6.0 | 41 |
| OaAEP3 | O. affinis | GL-Xn-NGL | 0.61 | 33.0 | r.t. | 5.0 | 39 |
| OaAEP4 | O. affinis | GL-Xn-NGL | 0.76 | 98.4 | r.t. | 5.0 | 39 |
| OaAEP5 | O. affinis | GL-Xn-NGL | 0.59 | 24.7 | r.t. | 5.0 | 39 |
| Pro-domain-free OaAEP1-C247A | O. affinis | GL-Xn-NGL | 1.54 | 0.79 | r.t. | 6.0 | 41 |
| xCeAEP1 | C. ensiformis | GR-Xn-NGL | N.R. | 0.58 | 37 | 5.0 | 24 |
| MCoAEP2 | M. cochinchinensis | GG-Xn-NAL | 19.86 | 62.1 | 22 | 6.0 | 27 |
With kcat/KM up to 131 × 104 M−1 s−1, butelase 1 extracted from C. ternatea is a highly efficient enzyme for transpeptidation (Table 1).23,37 The enzyme was reported to cyclise >95% of linear substrate peptide within 1 h at 37 °C (0.125 μM butelase 1 and 50 μM peptide).23 On the other hand, OaAEP1b exhibits a lower kcat/KM than butelase 1.22 However, by replacing the gatekeeper (Cys247) near the active site with a smaller Ala residue, the engineered protein was shown to cyclise a model substrate peptide with kcat/KM value of 3.42 × 104 M−1 s−1 at pH 6.0 and 37 °C.10
Subsequently, a suite of AEP ligases from O. affinis (OaAEP3, OaAEP4 and OaAEP5) have been identified.39 OaAEP4 was reported to cyclise >95% of linear substrate peptide within 30 min at room temperature (0.185 μM AEP and 280 μM peptide) with good tolerance to organic co-solvents (acetone, dimethylformamide and methanol). In directly comparable experimental conditions, OaAEP3, 4 and 5 all demonstrated greater kcat/KM than the prototypic OaAEP1b at pH 5.0 and room temperature. Further differences in the activity of AEPs from O. affinis were shown when the peptide cyclisation assay was performed at varying pH values. OaAEP4, the most active among all of the tested AEPs, offered optimal conversion of the linear peptide at pH 4.2 and remained active over a relatively narrow pH range (3.7–5.0). In contrast, OaAEP1b demonstrated optimal activity at pH 5.0 with a broader tolerance to pH compared to OaAEP4 (4.2–7.5). A 50% conversion to the cyclic peptide was detected when linear peptide substrates were incubated with OaAEP1b for 30 min at pH 7.5 (0.528 μM AEP and 280 μM peptide).39
Applications of AEP ligase activity
The growing importance and diverse applications of protein science in contemporary research encompass a range of research topics including protein labelling for the study of post-translational modification;53–56 protein-drug conjugation for the production of biopharmaceuticals;57–59 peptide ligation for the semi-synthesis of small proteins;60 bioconjugation to solid supports for the immobilization of proteins.61,62 The ability to facilitate peptide ligation and backbone cyclisation via a relatively short substrate recognition sequence, with good catalytic efficiency and tolerance to a broad pH range, highlights the significant potential of AEPs in applications.Intramolecular ligation by AEP results in backbone cyclisation
Cyclisation often reduces conformational flexibility and imparts greater thermal and chemical stability.30,39,49,63 The joining of a peptide's termini can provide resistance towards degradation by exopeptidases and improve the oral bioavailability of peptide products.29,30,49,64 Chemical approaches towards backbone cyclisation, such as native chemical ligation, are limited by strict requirements for N-terminal cysteines (or their surrogates), C-terminal thioesters and denaturing conditions.63,65,66 Similarly, the introduction of large split intein domains often impair solubility which limits the versatility of trans-protein splicing strategies.67,68 Cyclisation via small synthetic molecules have been employed to generate diverse libraries of cyclic peptides.69 Chemical linkers with two thiol reactive functional groups were reported to bridge across two cysteine residues to afford cyclic peptide species and allow the insertion of non-native motifs.69 However, this “chemical bridging” approach exploits the thiol reactivity of cysteine residues, which have limited selectivity when applied to cysteine-rich protein constructs.69 Moreover, the termini of bridged cyclic peptides remain unmodified, and thus susceptible to degradation by exopeptidases. The use of peptide ligases offers an alternative approach to peptide cyclisation. With excellent reaction kinetics and a short recognition sequence that is orthogonal to the existing enzymes such as the prolyl oligopeptidase, PCY1,70 and transpeptidase, sortase A,63,71 the addition of AEPs complements existing strategies.Peptide backbone cyclisation has been identified as the native function of AEP ligases isolated from plant species which produce cyclic peptides such as O. affinis (OaAEP1b),21,22 and C. ternatea (butelase 1).23 In nature, AEP ligases process the linear peptide precursors to afford the cyclic product which are typically around 30 amino acids in length.64 Native and engineered AEPs derived from various plants have also been successfully employed to facilitate backbone cyclisation of peptides derived from other plant and non-plant species such as kalata B1;21–23 sunflower trypsin inhibitor 1;23,45,72 histatin-3;23 anti-malarial peptide R1;39 MCoTI-II;27 AS-48,72 and proteins such as GFP;10,40,49 somatropin;49 MSP2;39 p53 binding domain.73 Butelase 1 was reported to generate the cyclic antimicrobial peptide θ-defensin and the cyclic conotoxin MrlA with >95% yield in 1 minute at 42 °C and pH 6.0.45
Cyclotides, backbone cyclised and cysteine knotted peptides, have been touted as a potential scaffold for the development of novel therapeutics.27,29,49 The emergence of convenient and effective peptide cyclisation methods by AEPs have enabled the synthesis of grafted peptides, whereby bioactive peptides are inserted into cyclotides (Fig. 6).74 Exemplified by the cyclisation of MCoSST-01 using MCoAEP2,27,75 this approach employs the highly stable cyclic cysteine knot motif from cyclotides as a scaffold to deliver peptide pharmacophores.
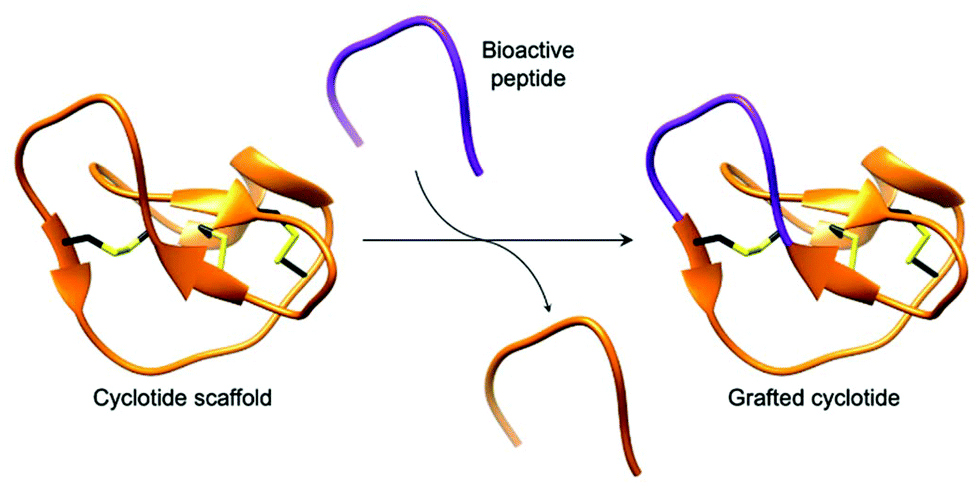 | ||
| Fig. 6 Cartoon representation of a grafted cyclotide. A bioactive peptide (purple) is inserted into a cyclotide backbone, shown here using the structure of MCoTI-II (PDB: 1IB9). | ||
Applications of AEPs in protein research were demonstrated by the butelase 1-mediated cyclisation of the p53 binding domain, a potential target for anti-cancer therapeutics. The cyclic protein was reported to aid structural investigations by NMR and X-ray crystallography, methods that may be hindered by conformational flexibility induced line broadening and poor crystal formation, respectively.73 Furthermore, greater conformational stability in the cyclic protein was suggested to offer improved ligand binding due to a reduction of the associated entropic penalty.73 Protein cyclisation by AEP was also applied to MSP2 from Plasmodium falciparum, a 25 kDa protein for malarial vaccine development.39 When produced recombinantly, the protein is disordered and its antigenic behaviour no longer represents the native protein.76–78 OaAEP1-mediated cyclisation of MSP2 was suggested to reduce disorder and potentially force the protein into adopting a conformation similar to the native antigen.39
Further developments in AEP-based technology can also be found in a recently published article that explored the effects of enzyme immobilisation (Fig. 7).72 The study illustrated that AEPs from C. ternatea (butelase 1) and V. yedoensis (VyPAL2) which were conjugated to a solid support offered greater stability in storage and repeated use for modifications of polypeptide including labelling and cyclisation. Significantly, immobilisation enabled the use of these enzymes at higher concentrations, and to perform peptide cyclisation reactions in continuous flow.72 The findings reported by Hemu et al. represent an advance towards adapting AEP catalysis for large scale and industrial applications.72
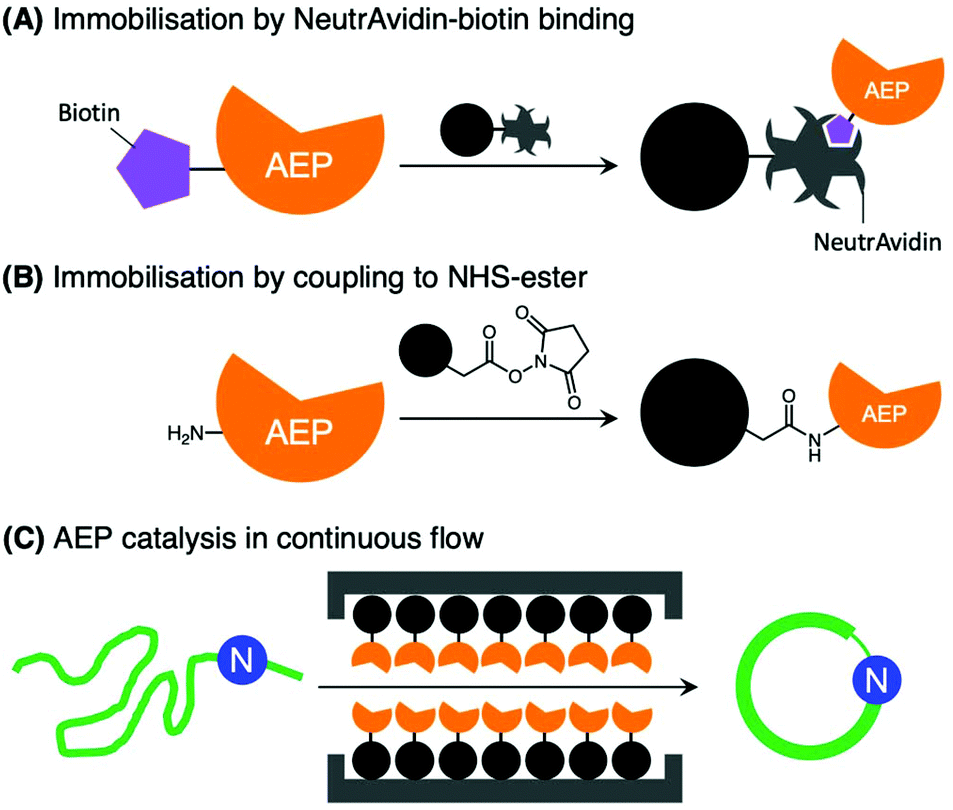 | ||
| Fig. 7 Immobilisation of AEPs on solid support. AEPs, butelase1 and VyPAL2, have been immobilised on agarose beads via (A) NeutrAvidin-biotin affinity binding and (B) direct coupling to N-hydroxysuccinimide (NHS) ester with primary amines presented by lysine residues. (C) The immobilised AEPs were reported to facilitate ligation of proteins and peptides in continuous flow. | ||
Intermolecular ligation by AEP for peptide and protein bioconjugation
Naturally occurring proteins commonly feature non-ribosomal modifications. Glycosylation, methylation, and lipidation can influence protein stability, localisation and interactions with other biological components.54,56,79–81 Protein modifications are routinely employed to study and engineer proteins beyond their native functions for academic research in addition to medicinal and industrial applications.55,82,83 Consequently, the development of novel polypeptide ligation technologies has been a growing theme in biological and organic chemistry research.51,52,58,84–99Currently there is a range of chemical and biological approaches to bioconjugation.51,52,58,84–99 Chemical approaches tend to be more affordable, simpler to perform, but can be hindered by poor selectivity or the availability of specific reactive handles.93,94 Strategies employing enzymes offer an attractive alternative, whereby site-specific reactions can be catalysed under relatively mild conditions.95,96 Transpeptidase from Gram-positive bacteria (sortase A),51,52,97 and other enzymes such as subtiligase,84,98,99 biotin ligase,85,86 transferases87–89 and formyl glycine generating enzymes58,90–92 have been employed for protein bioconjugation. Excellent kinetic parameters and relatively short recognition sequences indicate that AEP methodologies would be a valuable addition to supplement the existing approaches. AEPs have been employed to modify protein substrates including GFP,46,47,100 ubiquitin,37,46,101 ompA,102 DARPin,101 maltose binding proteins,42,47 and nanobodies40 with a range of synthetic labels including click handles, polyethylene glycol, fluorophores and drug molecules.
Introduction of an isotopically labelled segment into protein backbone by OaAEP1b-mediated ligation was shown to enable NMR studies of maltose binding protein.47 The reported method uses a nicked protein, or self-assembling protein domains, to ensure close proximity between the ligating motifs, one of the factors that affects AEP-mediated ligation (Scheme 4).47,100 While preparation of labelled proteins by recombinant gene expression is routinely performed,103–105 this enzymatic approach offers the ability to selectively label specific regions within the protein. In addition, segmental isotopic labelling of proteins may find other applications such as elucidating protein dynamics.106–108
 | ||
| Scheme 4 Ligation of self-assembling protein domains by OaAEP1b. Pre-organisation of the ligating substrates bring the reactive sites into close proximity, which was shown to affect AEP catalysis. | ||
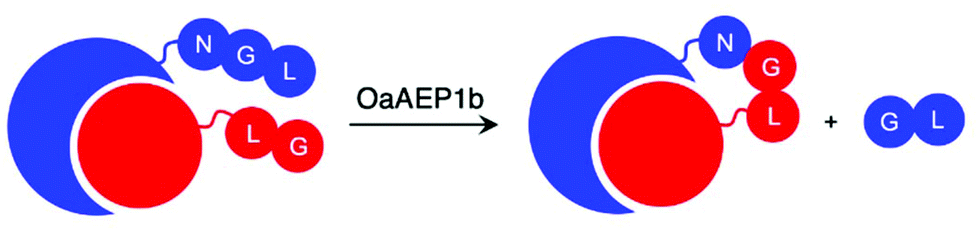
Enzymes commonly target specific substrate recognition motifs defined by the active site. While AEPs have relatively non-specific recognition at certain positions (P1′ and P1′′), changes in the recognition sequence have been shown to influence ligation efficiency.22,23,27,39,40 It was reported that, although Gly-Val was accepted by OaAEP1 at the P1′′–P2′′ positions for ligation, the corresponding product (Asn-Gly-Val at P1–P1′–P2′) was poorly recognised for further enzymatic reactions (Scheme 5).40 This differential recognition of Val at P2′ vs. P2′′ was utilised to construct a protein bearing two different modifications at the N- and C-terminus mediated by a single AEP.40 Butelase 1 has also been employed in conjunction with sortase A to modify immunoglobulin molecules (IgG1) with two fluorescent tags (5,6-carboxyfluorescein and AlexaFluor 647) a one-pot dual labelling reaction.109 These reports highlight the potential use of AEPs in the semi-synthesis of more complex, multiply labelled protein such as antibody–drug conjugates.
 | ||
| Scheme 5 OaAEP1 recognises Val at P2′′, but poorly at P2′. Consequently, this feature in the substrate recognition of OaAEP1 has been exploited to prevent the undesired reverse reaction. | ||
Finally, the reported application of butelase 1 for protein labelling on the surface of live bacteria exemplifies the biocompatibility of enzyme catalysis.102 The outer membrane protein A (OmpA) of Escherichia coli strain BL21(DE3), modified with the butelase 1 recognition sequence at the C-terminus, was successfully labelled with tags such as 5,6-carboxyfluorescein, biotin and the fluorescent protein mCherry.102 The ability to selectively modify proteins on the cell surface can offer an insight towards signalling mechanisms between cells and the extracellular matrix as well as intercellular communications.
Limitations of using AEP as a biocatalytic tool
Preparation of recombinant AEP
While the utility of AEPs have been demonstrated through a number of examples, access to active enzyme has hindered the use of AEPs in biocatalytic applications. Butelase 1 was obtained via an arduous extraction and purification process from the native host, butterfly pea plant, for initial experiments.23,37 Subsequently, procedures for the preparation of recombinant butelase 1 from bacteria (E. coli)36 and yeast (P. pastoris)73 were reported. Similarly, the preparation of recombinant OaAEP1b from E. coli culture has been reported in a number of studies.10,22,39,40Mimicking the production and maturation of AEPs in nature, the recombinant AEPs are produced as a zymogen which can be activated at low pH (pH 3.6–4.0). The acidic environment simulates the plant vacuole, where active AEPs are known to accumulate in cyclotide-producing plant species.7,22,24,25 The gene construct employed to prepare recombinant OaAEP1b encodes for four distinct features: hexahistidine (His6) tag, ubiquitin, AEP core domain and pro-domain, producing a fusion construct. Located at the N-terminus, the His6 tag facilitates purification by immobilised metal affinity chromatography (IMAC). Between the His6 tag and AEP, the ubiquitin sequence has been employed to enhance solubility and expression levels of OaAEP1b and other AEPs.22,27 The pro-domain, a C-terminal section of the zymogenic AEP blocks substrate access to the active site, and thus silences enzyme activity. The pro-enzyme was isolated by IMAC, strong anion exchange (SAX) and size exclusion chromatography (SEC). Incubation at low pH (pH 4.0) was required to trigger the auto-proteolytic activation process (Fig. 8). In some studies, the activated AEP was subjected to a further purification step by ion-exchange chromatography.22,39,42 The reported yield of the activated OaAEP1b ranged between 1.8 and 2.0 mg L−1 from lysogeny-broth (LB) media.22 Most of the procedures for preparing recombinant AEPs involved multiple purification steps and an incubation step at low pH. The time consuming and low yielding aspects of the procedure present a considerable limitation to the uptake of AEPs for bioconjugation applications.
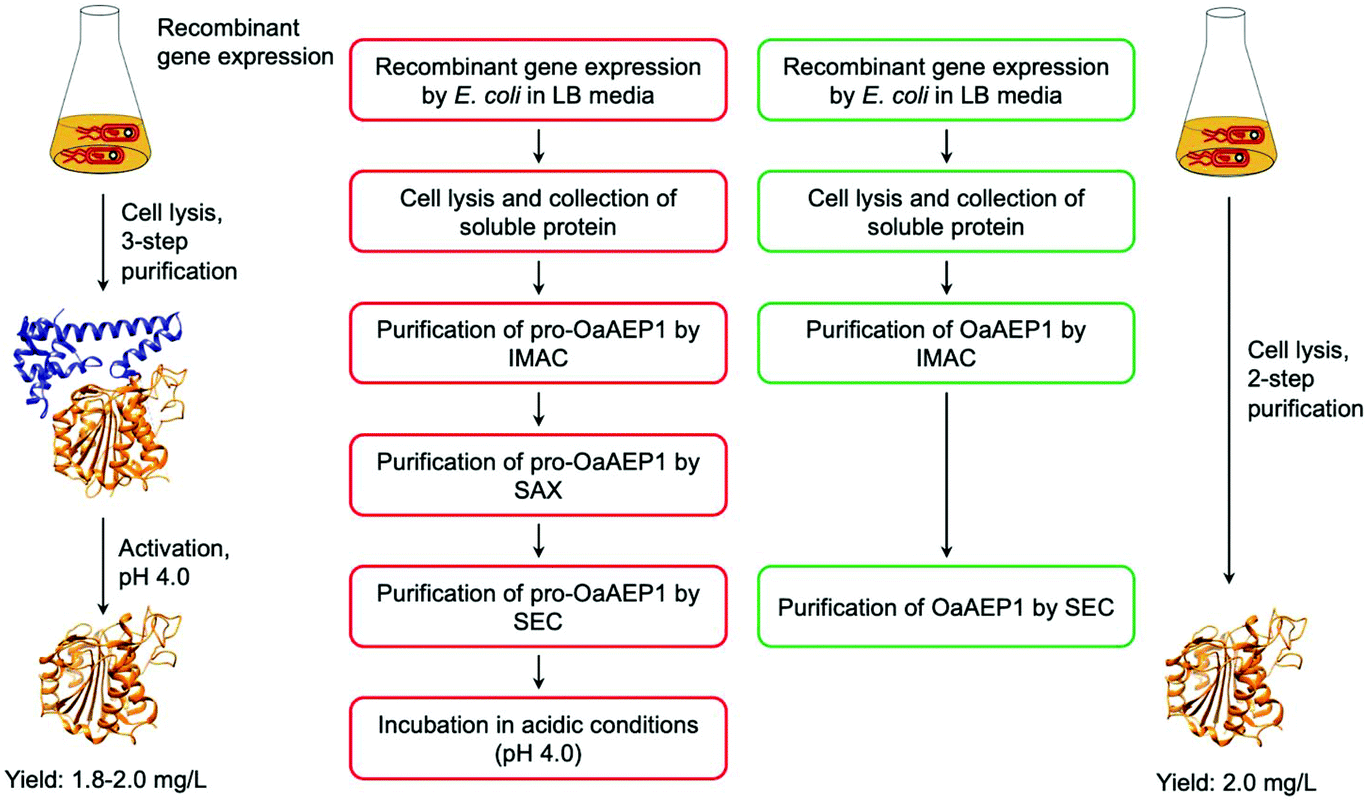 | ||
| Fig. 8 Comparison of procedures for recombinant OaAEP1-C247A. (Red) Recombinant AEP preparation from the pro-enzyme involves multiple purification steps and an incubation period in acidic conditions (pH 4.0). (Green) Recombinant AEP preparation without the pro-domain enabled a streamlined method with less chromatographic steps and does not require activation. (IMAC) Immobilised metal affinity chromatography, (SAX) strong anion exchange and (SEC) size exclusion chromatography. X-ray crystal structure of OaAEP1b (PDB: 5H0I) used here as a representative AEP. | ||
To address this issue, gene expression of different constructs have been tested.41 A construct based on OaAEP1-C247A that can be expressed without the pro-domain was identified. This has enabled the recombinant preparation of active and soluble AEP which can be isolated in only two chromatographic steps (Fig. 8). The purified enzyme demonstrated activities comparable to those of isolated with a pro-domain (Table 1). Nevertheless, the yield is relatively low (2.0 mg L−1) in comparison to most recombinant protein preparation, and hence there remains substantial space for improvement.
Equilibrium in AEP-mediated peptide ligation
The ligation activity of AEPs have been employed to facilitate intermolecular ligation at either the N or C-terminus of proteins and peptides. While AEP mediated intermolecular ligation has been successfully employed in some cases, it operates with limited efficiency. Similar to other transpeptidases such as sortase A and trypsiligase,97,110–112 the challenge in preventing efficient AEP-mediated intermolecular ligation is that the by-product dipeptide (P1′–P2′) acts as a competing nucleophile to reverse the ligation reaction.40,46 Water can also potentially serve as nucleophile, leading to hydrolysis. The efficiency of AEP-mediated ligations is affected by the proximity of the ligation partners and the rate of undesired reverse ligation.37 Unlike intramolecular cyclisation, the ligating termini are unlikely to be in close proximity. Moreover, the entropic penalty from intermolecular ligation negates any entropic benefits from the release of the cleaved recognition peptide motif. Consequently, a large excess of the labelling peptide nucleophile is required to drive the ligation reaction to completion.37,40,46,101,109 The labelling of a nanobody protein using OaAEP1-C247A employed 25 equivalents of a fluorescent peptide label (250 μM), with respect to the protein (10 μM).40 This rendered the transpeptidase-mediated ligations uneconomical, especially when expensive or non-commercially available reagents are used (e.g. radioactive, isotopic or fluorescent labels).47,100,113,114Attempts to address this limitation were made by exploiting the non-specific substrate recognition nature of AEPs.23,40,46,47,100,101 The incorporation of self-assembling protein domains such as nicked-maltose binding protein or designed armadillo repeat protein was the only published approach which enabled effective conjugation without using an excess of labelling reagent.47,100 This method employed protein self-assemblies to bring the ligation partners into close proximity, thus improving ligation efficiency (Scheme 4). However, the insertion of a large protein domain into the substrates limits the versatility of this approach. Therefore, a versatile method to enable effective AEP-mediated intermolecular ligation would unlock the tremendous potential of AEPs to a range of applications in protein sciences.
Butelase 1 have been reported to exhibit considerable reactivity towards synthetic peptide analogues bearing ester and thioester linkages (depsipeptide and thiodepsipeptide, respectively).46,101 This was exploited to develop an irreversible butelase 1-mediated ligation strategy (Scheme 6A).46 While the depsipeptide approach has been employed to ligate a synthetic peptide to the N-terminus of ubiquitin and green fluorescent protein (GFP) with good yields in 2.5 h, the asparaginyl thiodepsipeptide motif has a short half-life of around 45–75 min at pH 6.5 likely because of its susceptibility to rearrangement (Scheme 6B).46,65 Owing to the hydrolysis of thiodepsipeptides, a five-fold excess of this unstable reagent was required to effectively label the target protein substrates.46 The use of an unnatural thioester motif further limits the application of this method to the labelling of protein N-termini.46
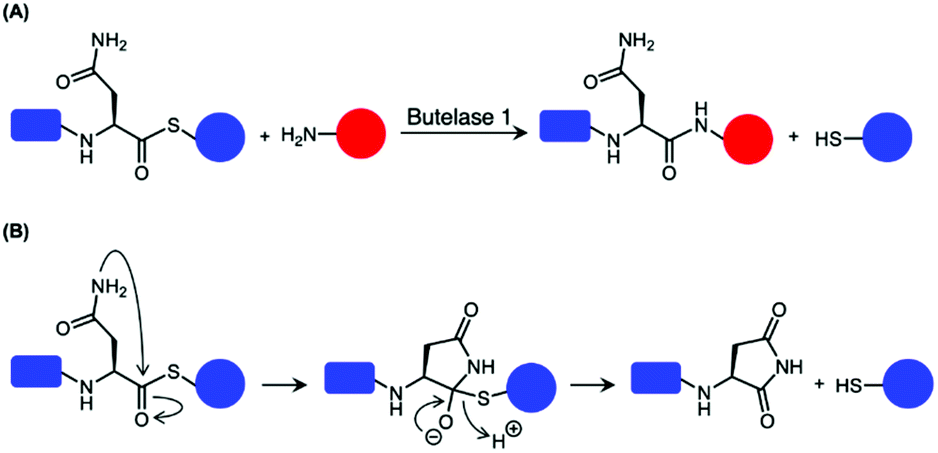 | ||
| Scheme 6 Use of thiodepsipeptide for butelase 1-mediated ligation. (A) Thioester linked Asn is accepted by butelase 1. Peptide ligation generates a peptide by-product with a α-thiol which is not recognised as a nucleophilic peptide substrate. As a result, the reaction was rendered irreversible. (B) Proposed mechanism for the hydrolysis of the unstable thioester. | ||
Without using unnatural (thio)depsipeptide motifs, the reverse reaction mediated by OaAEP1 can be prevented by taking advantage of the subtle difference in reactivity between two recognition sequences. Valine was reported to be an amino acid which is recognised at the P2′′ position, but poorly at P2′. Thus, Gly-Val has been used as a recognition sequence on the nucleophilic peptide to generate a product peptide that is not readily hydrolysed by the enzyme (Scheme 5).40 Nevertheless, a large excess of the nucleophilic peptide (20-fold excess) was required to obtain the ligated product in good yields.40
Recently, reports that coupled AEP catalysis with chemical reactions have been made as an alternative approach to address issues related to reaction equilibrium. In this first report, the recognition Asn-Cys-Leu was recruited to ligate with counterpart carrying a N-terminal Gly-Leu by OaAEP1-C247A. Consequently, the enzymatic reaction can be driven by the addition of 2-formyl phenyl boronic acid (FPBA), which reacts with the released N-terminal cysteine to afford a non-reactive thiazolidine (Scheme 7A).41 In another report, a His residue was introduced to the P3′ position. Consequently, the peptide by-product from the OaAEP1-C247A reaction can be sequestered by chelation with a divalent cation such as Ni2+ (Scheme 7B). Quenching of the nucleophilic by-product prevents the reverse peptide ligation by OaAEP1-C247A, thus driving the reaction equilibrium towards product formation.42 Both of these reports are excellent proof of concepts facilitating protein labelling at the terminus of choice (N or C) with a lowered amount of label. Nevertheless, improvements are still required. Firstly, while the model peptide reactions showed significant improvement by the addition of a quenching agent (+40%), its enhancement was only moderate when applied to protein labelling (+20–30%). Furthermore, the quenching approaches do not address the undesired hydrolysis reaction associated with the protease activity of AEPs. Consequently, the reaction conditions (e.g. pH, enzyme concentration, amount of quenching reagent added, addition of reducing agents, reaction time) need to be carefully screened to minimise unwanted hydrolysis reactions. Finally, the reagents employed to quench the nucleophilic by-product may result in off-target interference. For example, reactive aldehydes are commonly employed to introduce protein glycosylation.92 Therefore, FPBA may not be applicable towards glycosylated peptide labels. Similarly, the use of Ni2+ as a scavenger may also present issues when considering labelling proteins which contain a N-terminal His6 tag or utilise divalent metal co-factors such as Fe2+.
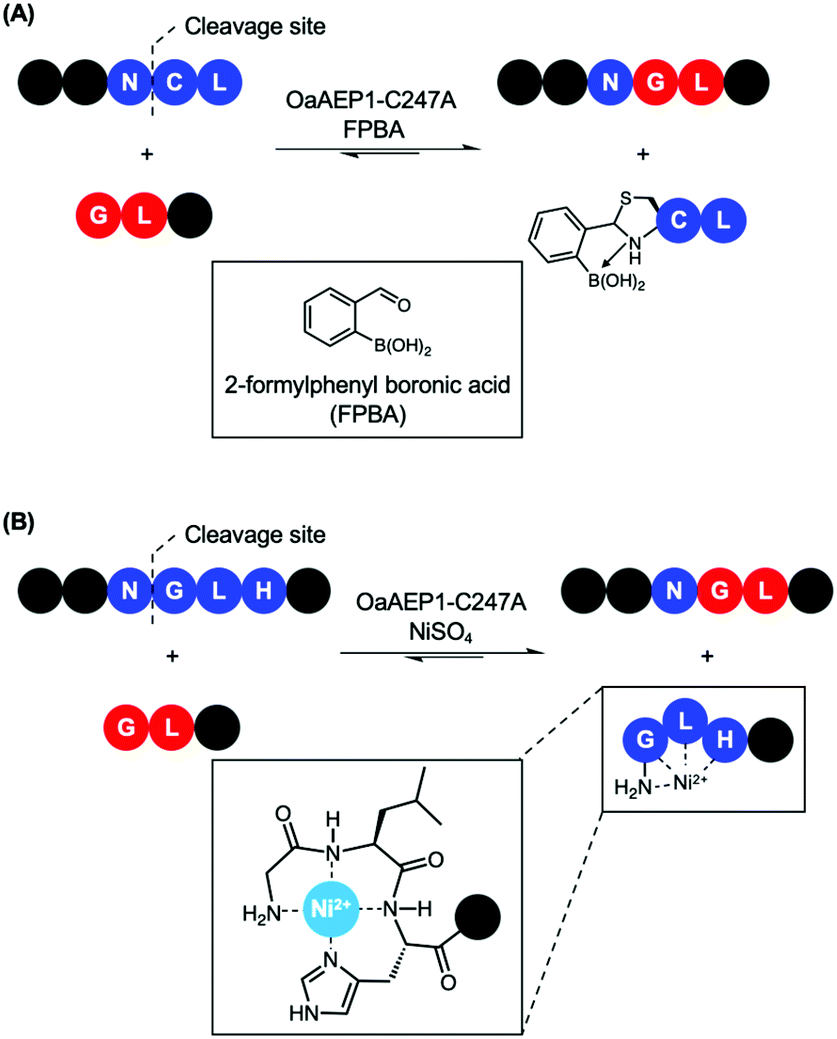 | ||
| Scheme 7 Nucleophile quenching strategies to enable effective peptide ligation by OaAEP1-C247A. (A) FPBA reacts with N-terminal cysteine to thiazolidine derivative to prevent reverse reaction. (B) Divalent Ni2+ quenches the nucleophilic peptide by-product generated by OaAEP1-C247A, thus driving the reaction equilibrium towards product formation. The tripeptide motif Gly-Leu-His at the N-terminus coordinates to Ni2+. | ||
Conclusions
Asparaginyl endopeptidases possess a unique combination of features that draw attention from chemical biologists. They are catalytically efficient, and many can be produced using E. coli as the recombinant host. AEPs have a short recognition site, which is relatively non-specific at the P1′ position and more stringent at the P2′ site. These features together enable development of various polypeptide modification systems. However, most examples reported in the literature remain at the stage of proof-of-concepts, and great strides are needed in order to put AEPs forward into applications. Crucially, a simplified and high-yielding approach to recombinant AEP is essential. The AEP preparation procedure has recently been simplified to only two chromatographic steps with no incubation at low pH needed. Yet, the yield can be further improved by adapting advanced techniques such as use of an appropriate recombinant host and fermentation. Issues surrounding hydrolysis and reversibility also need to be addressed. The addition of scavengers to the AEP reaction appears to be a straightforward solution to prevent the reverse reaction, but its efficacy on protein labelling require further validation. Nevertheless, with significant advances in the structural biology and enzymology research of AEPs, they are anticipated to be widely adopted biocatalysts in the future to generate polypeptide derivatives that cannot be readily created by existing methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank for the financial support provided by BBSRC (BB/T015799/1) for Louis Y. P. Luk as well as the UKRI PhD studentship for T. M. Simon Tang (1928909).Notes and references
- I. Hara-Nishimura, Y. Takeuchi and M. Nishimura, Molecular characterization of a vacuolar processing enzyme related to a putative cysteine proteinase of Schistosoma mansoni, Plant Cell, 1993, 5, 1651 CAS.
- J.-M. Chen, P. M. Dando, R. A. E. Stevens, M. Fortunato and A. J. Barrett, Cloning and expression of mouse legumain, a lysosomal endopeptidase, Biochem. J., 1998, 335, 111–117 CrossRef CAS PubMed.
- E. Dall and H. Brandstetter, Structure and function of legumain in health and disease, Biochimie, 2016, 122, 126–150 CrossRef CAS PubMed.
- X. Hemu, A. El Sahili, S. Hu, K. Wong, Y. Chen, Y. H. Wong, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C.-F. Liu, J. Lescar and J. P. Tam, Structural determinants for peptide-bond formation by asparaginyl ligases, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11737–11746 CAS.
- Y. Hayashi, K. Yamada, T. Shimada, R. Matsushima, N. K. Nishizawa, M. Nishimura and I. Hara-Nishimura, A proteinase-storing body that prepares for cell death or stresses in the epidermal cells of Arabidopsis, Plant Cell Physiol., 2001, 42, 894–899 CrossRef CAS PubMed.
- O. Takeda, Y. Miura, M. Mitta, H. Matsushita, I. Kato, Y. Abe, H. Yokosawa and S. Ishii, Isolation and Analysis of cDNA Encoding a Precursor of Canavalia ensiformis Asparaginyl Endopeptidase (Legumain), J. Biochem., 1994, 116, 541–546 CrossRef CAS PubMed.
- F. B. Zauner, E. Dall, C. Regl, L. Grassi, C. G. Huber, C. Cabrele and H. Brandstetter, Crystal Structure of Plant Legumain Reveals a Unique Two-Chain State with pH-Dependent Activity Regulation, Plant Cell, 2018, 30, 686–699 CrossRef CAS PubMed.
- M. S. Otegui, R. Herder, J. Schulze, R. Jung and L. A. Staehelin, The Proteolytic Processing of Seed Storage Proteins in Arabidopsis Embryo Cells Starts in the Multivesicular Bodies, Plant Cell, 2006, 18, 2567–2581 CrossRef CAS PubMed.
- I. Hara-Nishimura and N. Hatsugai, The role of vacuole in plant cell death, Cell Death Differ., 2011, 18, 1298 CrossRef CAS PubMed.
- R. L. Yang, Y. H. Wong, G. K. T. Nguyen, J. P. Tam, J. Lescar and B. Wu, Engineering a Catalytically Efficient Recombinant Protein Ligase, J. Am. Chem. Soc., 2017, 139, 5351–5358 CrossRef CAS PubMed.
- K. Yamada, T. Shimada, M. Nishimura and I. Hara-Nishimura, A VPE family supporting various vacuolar functions in plants, Physiol. Plant., 2005, 123, 369–375 CrossRef CAS.
- I. Hara-Nishimura, K. Inoue and M. Nishimura, A unique vacuolar processing enzyme responsible for conversion of several proprotein precursors into the mature forms, FEBS Lett., 1991, 294, 89–93 CrossRef CAS PubMed.
- N. Hatsugai, K. Yamada, S. Goto-Yamada and I. Hara-Nishimura, Vacuolar processing enzyme in plant programmed cell death, Front. Plant Sci., 2015, 6, 234 Search PubMed.
- J.-M. Chen, N. D. Rawlings, R. A. E. Stevens and A. J. Barrett, Identification of the active site of legumain links it to caspases, clostripain and gingipains in a new clan of cysteine endopeptidases, FEBS Lett., 1998, 441, 361–365 CrossRef CAS PubMed.
- A. J. Barrett and N. D. Rawlings, Evolutionary Lines of Cysteine Peptidases, Biol. Chem., 2001, 382, 727–734 CAS.
- B. Elsässer, F. B. Zauner, J. Messner, W. T. Soh, E. Dall and H. Brandstetter, Distinct Roles of Catalytic Cysteine and Histidine in the Protease and Ligase Mechanisms of Human Legumain As Revealed by DFT-Based QM/MM Simulations, ACS Catal., 2017, 7, 5585–5593 CrossRef PubMed.
- J.-M. Chen, P. M. Dando, N. D. Rawlings, M. A. Brown, N. E. Young, R. A. Stevens, E. Hewitt, C. Watts and A. J. Barrett, Cloning, Isolation, and Characterization of Mammalian Legumain, an Asparaginyl Endopeptidase, J. Biol. Chem., 1997, 272, 8090–8098 CrossRef CAS PubMed.
- B. Manoury, E. W. Hewitt, N. Morrice, P. M. Dando, A. J. Barrett and C. Watts, An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation, Nature, 1998, 396, 695–699 CrossRef CAS PubMed.
- B. Manoury, D. Mazzeo, L. Fugger, N. Viner, M. Ponsford, H. Streeter, G. Mazza, D. C. Wraith and C. Watts, Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP, Nat. Immunol., 2002, 3, 169–174 CrossRef CAS PubMed.
- F. E. Sepulveda, S. Maschalidi, R. Colisson, L. Heslop, C. Ghirelli, E. Sakka, A.-M. Lennon-Duménil, S. Amigorena, L. Cabanie and B. Manoury, Critical Role for Asparagine Endopeptidase in Endocytic Toll-like Receptor Signaling in Dendritic Cells, Immunity, 2009, 31, 737–748 CrossRef CAS PubMed.
- I. Saska, A. D. Gillon, N. Hatsugai, R. G. Dietzgen, I. Hara-Nishimura, M. A. Anderson and D. J. Craik, An asparaginyl endopeptidase mediates in vivo protein backbone cyclization., J. Biol. Chem., 2007, 282, 29721–29728 CrossRef CAS PubMed.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed.
- G. K. T. Nguyen, S. J. Wang, Y. B. Qiu, X. Hemu, Y. L. Lian and J. P. Tam, Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed.
- K. Bernath-Levin, C. Nelson, A. G. Elliott, A. S. Jayasena, A. H. Millar, D. J. Craik and J. S. Mylne, Peptide Macrocyclization by a Bifunctional Endoprotease, Chem. Biol., 2015, 22, 571–582 CrossRef CAS PubMed.
- J. Haywood, J. W. Schmidberger, A. M. James, S. G. Nonis, K. V. Sukhoverkov, M. Elias, C. S. Bond and J. S. Mylne, Structural basis of ribosomal peptide macrocyclization in plants, eLife, 2018, 7, e32955 CrossRef PubMed.
- M. A. Jackson, E. K. Gilding, T. Shafee, K. S. Harris, Q. Kaas, S. Poon, K. Yap, H. Jia, R. Guarino, L. Y. Chan, T. Durek, M. A. Anderson and D. J. Craik, Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases, Nat. Commun., 2018, 9, 2411 CrossRef CAS PubMed.
- J. Du, K. Yap, L. Y. Chan, F. B. H. Rehm, F. Y. Looi, A. G. Poth, E. K. Gilding, Q. Kaas, T. Durek and D. J. Craik, A bifunctional asparaginyl endopeptidase efficiently catalyzes both cleavage and cyclization of cyclic trypsin inhibitors, Nat. Commun., 2020, 11, 1575 CrossRef CAS PubMed.
- M. R. R. Plan, I. Saska, A. G. Cagauan and D. J. Craik, Backbone Cyclised Peptides from Plants Show Molluscicidal Activity against the Rice Pest Pomacea canaliculata (Golden Apple Snail), J. Agric. Food Chem., 2008, 56, 5237–5241 CrossRef CAS PubMed.
- M. L. Colgrave, A. C. Kotze, Y.-H. Huang, J. O'Grady, S. M. Simonsen and D. J. Craik, Cyclotides: Natural, Circular Plant Peptides that Possess Significant Activity against Gastrointestinal Nematode Parasites of Sheep, Biochemistry, 2008, 47, 5581–5589 CrossRef CAS PubMed.
- M. L. Colgrave and D. J. Craik, Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: the importance of the cyclic cystine knot, Biochemistry, 2004, 43, 5965–5975 CrossRef CAS PubMed.
- C. Jennings, J. West, C. Waine, D. Craik and M. Anderson, Biosynthesis and Insecticidal Properties of Plant Cyclotides: The Cyclic Knotted Proteins from Oldenlandia affinis, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10614–10619 CrossRef CAS PubMed.
- C. V. Jennings, K. J. Rosengren, N. L. Daly, M. Plan, J. Stevens, M. J. Scanlon, C. Waine, D. G. Norman, M. A. Anderson and D. J. Craik, Isolation, Solution Structure, and Insecticidal Activity of Kalata B2, a Circular Protein with a Twist: Do Möbius Strips Exist in Nature?, Biochemistry, 2005, 44, 851–860 CrossRef CAS PubMed.
- A. G. Poth, M. L. Colgrave, R. E. Lyons, N. L. Daly and D. J. Craik, Discovery of an unusual biosynthetic origin for circular proteins in legumes, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10127–10132 CrossRef CAS PubMed.
- J. S. Mylne, M. L. Colgrave, N. L. Daly, A. H. Chanson, A. G. Elliott, E. J. McCallum, A. Jones and D. J. Craik, Albumins and their processing machinery are hijacked for cyclic peptides in sunflower, Nat. Chem. Biol., 2011, 7, 257–259 CrossRef CAS PubMed.
- E. Dall and H. Brandstetter, Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10940–10945 CrossRef CAS PubMed.
- A. M. James, J. Haywood, J. Leroux, K. Ignasiak, A. G. Elliott, J. W. Schmidberger, M. F. Fisher, S. G. Nonis, R. Fenske, C. S. Bond and J. S. Mylne, The macrocyclizing protease butelase 1 remains autocatalytic and reveals the structural basis for ligase activity, Plant J., 2019, 98, 988–999 CAS.
- G. K. T. Nguyen, Y. Qiu, Y. Cao, X. Hemu, C.-F. Liu and J. P. Tam, Butelase-mediated cyclization and ligation of peptides and proteins, Nat. Protoc., 2016, 11, 1977–1988 CrossRef CAS PubMed.
- E. Dall, J. C. Fegg, P. Briza and H. Brandstetter, Structure and Mechanism of an Aspartimide-Dependent Peptide Ligase in Human Legumain, Angew. Chem., Int. Ed., 2015, 54, 2917–2921 CrossRef CAS PubMed.
- K. S. Harris, R. F. Guarino, R. S. Dissanayake, P. Quimbar, O. C. McCorkelle, S. Poon, Q. Kaas, T. Durek, E. K. Gilding, M. A. Jackson, D. J. Craik, N. L. van der Weerden, R. F. Anders and M. A. Anderson, A suite of kinetically superior AEP ligases can cyclise an intrinsically disordered protein, Sci. Rep., 2019, 9, 10820 CrossRef PubMed.
- F. B. H. Rehm, T. J. Harmand, K. Yap, T. Durek, D. J. Craik and H. L. Ploegh, Site-Specific Sequential Protein Labeling Catalyzed by a Single Recombinant Ligase, J. Am. Chem. Soc., 2019, 141, 17388–17393 CrossRef CAS PubMed.
- T. M. S. Tang, D. Cardella, A. J. Lander, X. Li, J. S. Escudero, Y.-H. Tsai and L. Y. P. Luk, Use of an asparaginyl endopeptidase for chemo-enzymatic peptide and protein labeling, Chem. Sci., 2020, 11, 5881–5888 RSC.
- F. B. H. Rehm, T. J. Tyler, K. Yap, T. Durek and D. J. Craik, Improved Asparaginyl-Ligase-Catalyzed Transpeptidation via Selective Nucleophile Quenching, Angew. Chem., 2021, 60, 4004–4008 CrossRef CAS PubMed.
- S. G. Nonis, J. Haywood and J. S. Mylne, Plant asparaginyl endopeptidases and their structural determinants of function, Biochem. Soc. Trans., 2021, 49, 965–976 CrossRef CAS PubMed.
- I. Schechter and A. Berger, On the size of the active site in proteases. I. Papain, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS PubMed.
- G. K. T. Nguyen, X. Hemu, J.-P. Quek and J. P. Tam, Butelase-Mediated Macrocyclization of d-Amino-Acid-Containing Peptides, Angew. Chem., Int. Ed., 2016, 55, 12802–12806 CrossRef CAS PubMed.
- G. K. T. Nguyen, Y. Cao, W. Wang, C. F. Liu and J. P. Tam, Site-Specific N-Terminal Labeling of Peptides and Proteins using Butelase 1 and Thiodepsipeptide, Angew. Chem., Int. Ed., 2015, 54, 15694–15698 CrossRef CAS PubMed.
- K. M. Mikula, I. Tascón, J. J. Tommila and H. Iwaï, Segmental isotopic labeling of a single-domain globular protein without any refolding step by an asparaginyl endopeptidase, FEBS Lett., 2017, 591, 1285–1294 CrossRef CAS PubMed.
- X. Hemu, X. Zhang and J. P. Tam, Ligase-Controlled Cyclo-oligomerization of Peptides, Org. Lett., 2019, 21, 2029–2032 CrossRef CAS PubMed.
- G. K. T. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, Butelase 1: A Versatile Ligase for Peptide and Protein Macrocyclization, J. Am. Chem. Soc., 2015, 137, 15398–15401 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, A general strategy for the evolution of bond-forming enzymes using yeast display, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- L. Chen, J. Cohen, X. Song, A. Zhao, Z. Ye, C. J. Feulner, P. Doonan, W. Somers, L. Lin and P. R. Chen, Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency, Sci. Rep., 2016, 6, 31899 CrossRef CAS PubMed.
- Z. Zou, H. Alibiglou, D. M. Mate, M. D. Davari, F. Jakob and U. Schwaneberg, Directed sortase A evolution for efficient site-specific bioconjugations in organic co-solvents, Chem. Commun., 2018, 54, 11467–11470 RSC.
- B. R. Sabari, D. Zhang, C. D. Allis and Y. Zhao, Metabolic regulation of gene expression through histone acylations, Nat. Rev. Mol. Cell Biol., 2017, 18, 90–101 CrossRef CAS PubMed.
- E. Guccione and S. Richard, The regulation, functions and clinical relevance of arginine methylation, Nat. Rev. Mol. Cell Biol., 2019, 20, 642–657 CrossRef CAS PubMed.
- C. Chatterjee and T. W. Muir, Chemical Approaches for Studying Histone Modifications, J. Biol. Chem., 2010, 285, 11045–11050 CrossRef CAS PubMed.
- K. T.-B. G. Schjoldager, M. B. Vester-Christensen, E. P. Bennett, S. B. Levery, T. Schwientek, W. Yin, O. Blixt and H. Clausen, O-Glycosylation Modulates Proprotein Convertase Activation of Angiopoietin-like Protein 3, J. Biol. Chem., 2010, 285, 36293–36303 CrossRef CAS PubMed.
- T. Hoeg-Jensen, S. Havelund, P. K. Nielsen and J. Markussen, Reversible insulin self-assembly under carbohydrate control., J. Am. Chem. Soc., 2005, 127, 6158–6159 CrossRef CAS PubMed.
- R. Kudirka, R. M. Barfield, J. McFarland, A. E. Albers, G. W. de Hart, P. M. Drake, P. G. Holder, S. Banas, L. C. Jones, A. W. Garofalo and D. Rabuka, Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation, Chem. Biol., 2015, 22, 293–298 CrossRef CAS PubMed.
- C. H. Chau, P. S. Steeg and W. D. Figg, Antibody-drug conjugates for cancer, Lancet, 2019, 394, 793–804 CrossRef CAS.
- R. E. Thompson and T. W. Muir, Chemoenzymatic Semisynthesis of Proteins, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS PubMed.
- L. S. Wong, F. Khan and J. Micklefield, Selective Covalent Protein Immobilization: Strategies and Applications, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS PubMed.
- A. Basso and S. Serban, Industrial applications of immobilized enzymes—A review, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- J. M. Antos, M. W.-L. Popp, R. Ernst, G.-L. Chew, E. Spooner and H. L. Ploegh, A Straight Path to Circular Proteins, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif, J. Mol. Biol., 1999, 294, 1327–1336 CrossRef CAS PubMed.
- B. Dang, T. Kubota, K. Mandal, F. Bezanilla and S. B. H. Kent, Native Chemical Ligation at Asx-Cys, Glx-Cys: Chemical Synthesis and High-Resolution X-ray Structure of ShK Toxin by Racemic Protein Crystallography, J. Am. Chem. Soc., 2013, 135, 11911–11919 CrossRef CAS PubMed.
- J. S. Zheng, S. Tang, Y. K. Qi, Z. P. Wang and L. Liu, Chemical synthesis of proteins using peptide hydrazides as thioester surrogates, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- Y. Minato, T. Ueda, A. Machiyama, I. Shimada and H. Iwaï, Segmental isotopic labeling of a 140 kDa dimeric multi-domain protein CheA from Escherichia coli by expressed protein ligation and protein trans-splicing, J. Biomol. NMR, 2012, 53, 191–207 CrossRef CAS PubMed.
- A. S. Aranko, J. S. Oeemig and H. Iwaï, Structural basis for protein trans -splicing by a bacterial intein-like domain - protein ligation without nucleophilic side chains, FEBS J., 2013, 280, 3256–3269 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zorzi, K. Deyle and C. Heinis, Cyclization of peptides with two chemical bridges affords large scaffold diversities, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- H. Ludewig, C. M. Czekster, E. Oueis, E. S. Munday, M. Arshad, S. A. Synowsky, A. F. Bent and J. H. Naismith, Characterization of the Fast and Promiscuous Macrocyclase from Plant PCY1 Enables the Use of Simple Substrates, ACS Chem. Biol., 2018, 13, 801–811 CrossRef CAS PubMed.
- C. J. S. Barber, P. T. Pujara, D. W. Reed, S. Chiwocha, H. Zhang and P. S. Covello, The Two-step Biosynthesis of Cyclic Peptides from Linear Precursors in a Member of the Plant Family Caryophyllaceae Involves Cyclization by a Serine Protease-like Enzyme, J. Biol. Chem., 2013, 288, 12500–12510 CrossRef CAS PubMed.
- X. Hemu, J. To, X. Zhang and J. P. Tam, Immobilized Peptide Asparaginyl Ligases Enhance Stability and Facilitate Macrocyclization and Site-Specific Ligation, J. Org. Chem., 2020, 85, 1504–1512 CrossRef CAS PubMed.
- N. Pi, M. Gao, X. Cheng, H. Liu, Z. Kuang, Z. Yang, J. Yang, B. Zhang, Y. Chen, S. Liu, Y. Huang and Z. Su, Recombinant Butelase-Mediated Cyclization of the p53-Binding Domain of the Oncoprotein MdmX-Stabilized Protein Conformation as a Promising Model for Structural Investigation, Biochemistry, 2019, 58, 3005–3015 CrossRef CAS PubMed.
- C. K. Wang and D. J. Craik, Designing macrocyclic disulfide-rich peptides for biotechnological applications, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS PubMed.
- L. Y. Chan, D. J. Craik and N. L. Daly, Dual-targeting anti-angiogenic cyclic peptides as potential drug leads for cancer therapy, Sci. Rep., 2016, 6, 35347 CrossRef CAS PubMed.
- C. G. Adda, V. J. Murphy, M. Sunde, L. J. Waddington, J. Schloegel, G. H. Talbo, K. Vingas, V. Kienzle, R. Masciantonio, G. J. Howlett, A. N. Hodder, M. Foley and R. F. Anders, Plasmodium falciparum merozoite surface protein 2 is unstructured and forms amyloid-like fibrils, Mol. Biochem. Parasitol., 2009, 166, 159–171 CrossRef CAS PubMed.
- X. Zhang, M. A. Perugini, S. Yao, C. G. Adda, V. J. Murphy, A. Low, R. F. Anders and R. S. Norton, Solution Conformation, Backbone Dynamics and Lipid Interactions of the Intrinsically Unstructured Malaria Surface Protein MSP2, J. Mol. Biol., 2008, 379, 105–121 CrossRef CAS PubMed.
- C. G. Adda, C. A. MacRaild, L. Reiling, K. Wycherley, M. J. Boyle, V. Kienzle, P. Masendycz, M. Foley, J. G. Beeson, R. S. Norton and R. F. Anders, Antigenic Characterization of an Intrinsically Unstructured Protein, Plasmodium falciparum Merozoite Surface Protein 2, Infect. Immun., 2012, 80, 4177–4185 CrossRef CAS PubMed.
- S. Y. Vakhrushev, C. Steentoft, M. B. Vester-Christensen, E. P. Bennett, H. Clausen and S. B. Levery, Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with simplecells, Mol. Cell. Proteomics, 2013, 12, 932–944 CrossRef CAS PubMed.
- D. T. Tran, L. Zhang, Y. Zhang, E. Tian, L. A. Earl and K. G. Ten Hagen, Multiple members of the UDP-GalNAc: polypeptide N-acetylgalactosaminyltransferase family are essential for viability in Drosophila., J. Biol. Chem., 2012, 287, 5243–5252 CrossRef CAS PubMed.
- M. Rape, Ubiquitylation at the crossroads of development and disease, Nat. Rev. Mol. Cell Biol., 2018, 19, 59–70 CrossRef CAS PubMed.
- J. Choi, L. J. S. Wagner, S. B. P. E. Timmermans, S. A. Malaker, B. Schumann, M. A. Gray, M. F. Debets, M. Takashima, J. Gehring and C. R. Bertozzi, Engineering Orthogonal Polypeptide GalNAc-Transferase and UDP-Sugar Pairs, J. Am. Chem. Soc., 2019, 141, 13442–13453 CrossRef CAS PubMed.
- S. B. Gunnoo and A. Madder, Bioconjugation – using selective chemistry to enhance the properties of proteins and peptides as therapeutics and carriers, Org. Biomol. Chem., 2016, 14, 8002–8013 RSC.
- D. Y. Jackson, J. Burnier, C. Quan, M. Stanley, J. Tom and J. A. Wells, A designed peptide ligase for total synthesis of ribonuclease A with unnatural catalytic residues, Science, 1994, 266, 243–247 CrossRef CAS PubMed.
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- S. A. Slavoff, I. Chen, Y.-A. Choi and A. Y. Ting, Expanding the Substrate Tolerance of Biotin Ligase through Exploration of Enzymes from Diverse Species, J. Am. Chem. Soc., 2008, 130, 1160–1162 CrossRef CAS PubMed.
- C. Kulkarni, M. Lo, J. G. Fraseur, D. A. Tirrell and T. L. Kinzer-Ursem, Bioorthogonal Chemoenzymatic Functionalization of Calmodulin for Bioconjugation Applications, Bioconjugate Chem., 2015, 26, 2153–2160 CrossRef CAS PubMed.
- J. Grünewald, H. E. Klock, S. E. Cellitti, B. Bursulaya, D. McMullan, D. H. Jones, H.-P. Chiu, X. Wang, P. Patterson, H. Zhou, J. Vance, E. Nigoghossian, H. Tong, D. Daniel, W. Mallet, W. Ou, T. Uno, A. Brock, S. A. Lesley and B. H. Geierstanger, Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Phosphopantetheinyl Transferases, Bioconjugate Chem., 2015, 26, 2554–2562 CrossRef PubMed.
- R. Viswanathan, G. R. Labadie and C. D. Poulter, Regioselective covalent immobilization of catalytically active glutathione S-transferase on glass slides, Bioconjugate Chem., 2013, 24, 571–577 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Introducing genetically encoded aldehydes into proteins, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, A Pictet-Spengler ligation for protein chemical modification, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- J. E. Hudak, H. H. Yu and C. R. Bertozzi, Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag., J. Am. Chem. Soc., 2011, 133, 16127–16135 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Selective chemical protein modification, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Choosing an effective protein bioconjugation strategy, Nat. Chem. Biol., 2011, 7, 876 CrossRef CAS PubMed.
- M. Schmidt, A. Toplak, P. Quaedflieg and T. Nuijens, Enzyme-mediated ligation technologies for peptides and proteins, Curr. Opin. Chem. Biol., 2017, 38, 1–7 CrossRef CAS PubMed.
- Y. Zhang, K.-Y. Park, K. F. Suazo and M. D. Distefano, Recent progress in enzymatic protein labelling techniques and their applications, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- J. M. Antos, J. Ingram, T. Fang, N. Pishesha, M. C. Truttmann and H. L. Ploegh, Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation, Curr. Protoc. Protein Sci., 2017, 89, 15.3.1–15.3.19 Search PubMed.
- H. A. I. Yoshihara, S. Mahrus and J. A. Wells, Tags for labeling protein N-termini with subtiligase for proteomics, Bioorg. Med. Chem. Lett., 2008, 18, 6000–6003 CrossRef CAS PubMed.
- T. K. Chang, D. Y. Jackson, J. P. Burnier and J. A. Wells, Subtiligase: a tool for semisynthesis of proteins, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12544–12548 CrossRef CAS PubMed.
- K. M. Mikula, L. Krumwiede, A. Plückthun and H. Iwaï, Segmental isotopic labeling by asparaginyl endopeptidase-mediated protein ligation, J. Biomol. NMR, 2018, 71, 225–235 CrossRef CAS PubMed.
- Y. Cao, G. K. T. Nguyen, J. P. Tam and C.-F. Liu, Butelase-mediated synthesis of protein thioesters and its application for tandem chemoenzymatic ligation, Chem. Commun., 2015, 51, 17289–17292 RSC.
- X. Bi, J. Yin, G. K. T. Nguyen, C. Rao, N. B. A. Halim, X. Hemu, J. P. Tam and C.-F. Liu, Enzymatic Engineering of Live Bacterial Cell Surfaces Using Butelase 1, Angew. Chem., Int. Ed., 2017, 56, 7822–7825 CrossRef CAS PubMed.
- M. T. Lin, L. J. Sperling, H. L. Frericks Schmidt, M. Tang, R. I. Samoilova, T. Kumasaka, T. Iwasaki, S. A. Dikanov, C. M. Rienstra and R. B. Gennis, A rapid and robust method for selective isotope labeling of proteins, Methods, 2011, 55, 370–378 CrossRef CAS PubMed.
- J. Marley, M. Lu and C. Bracken, A method for efficient isotopic labeling of recombinant proteins, J. Biomol. NMR, 2001, 20, 71–75 CrossRef CAS PubMed.
- C. H. Arrowsmith and W. Yu-Sung, NMR of large (s > 25 kDa) proteins and protein complexes, Prog. Nucl. Magn. Reson. Spectrosc., 1998, 32, 277–286 CrossRef CAS.
- A. Ciragan, S. M. Backlund, K. M. Mikula, H. M. Beyer, O. H. Samuli Ollila and H. Iwaï, NMR Structure and Dynamics of TonB Investigated by Scar-Less Segmental Isotopic Labeling Using a Salt-Inducible Split Intein, Front. Chem., 2020, 8, 136 CrossRef CAS PubMed.
- A. Romanelli, A. Shekhtman, D. Cowburn and T. W. Muir, Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein–intein junction, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6397–6402 CrossRef CAS PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernía, A. S. Adesina, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, Chemical Ligation and Isotope Labeling to Locate Dynamic Effects during Catalysis by Dihydrofolate Reductase, Angew. Chem., Int. Ed., 2015, 54, 9016–9020 CrossRef CAS PubMed.
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1, Bioconjugate Chem., 2018, 29, 3245–3249 CrossRef CAS PubMed.
- D. J. Williamson, M. E. Webb and W. B. Turnbull, Depsipeptide substrates for sortase-mediated N-terminal protein ligation, Nat. Protoc., 2014, 9, 253–262 CrossRef CAS PubMed.
- C. Meyer, S. Liebscher and F. Bordusa, Selective Coupling of Click Anchors to Proteins via Trypsiligase, Bioconjugate Chem., 2016, 27, 47–53 CrossRef CAS PubMed.
- S. Liebscher, M. Schöpfel, T. Aumüller, A. Sharkhuukhen, A. Pech, E. Höss, C. Parthier, G. Jahreis, M. T. Stubbs and F. Bordusa, N–Terminal Protein Modification by Substrate–Activated Reverse Proteolysis, Angew. Chem., Int. Ed., 2014, 53, 3024–3028 CrossRef CAS PubMed.
- R. Marega, E. A. Prasetyanto, C. Michiels, L. De Cola and D. Bonifazi, Fast Targeting and Cancer Cell Uptake of Luminescent Antibody-Nanozeolite Bioconjugates, Small, 2016, 12, 5431–5441 CrossRef CAS PubMed.
- L. Karmani, P. Levêque, C. Bouzin, A. Bol, M. Dieu, S. Walrand, T. Vander Borght, O. Feron, V. Grégoire, D. Bonifazi, C. Michiels, S. Lucas and B. Gallez, Biodistribution of 125I-labeled anti-endoglin antibody using SPECT/CT imaging: Impact of in vivo deiodination on tumor accumulation in mice, Nucl. Med. Biol., 2016, 43, 415–423 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |