
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn entry to 2-(cyclobut-1-en-1-yl)-1H-indoles through a cyclobutenylation/deprotection cascade†
Philipp
Natho
,
Zeyu
Yang
 ,
Lewis A. T.
Allen
,
Juliette
Rey
,
Andrew J. P.
White
and
Philip J.
Parsons
*
,
Lewis A. T.
Allen
,
Juliette
Rey
,
Andrew J. P.
White
and
Philip J.
Parsons
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, W12 0BZ London, UK. E-mail: p.parsons@imperial.ac.uk
First published on 19th April 2021
Abstract
A transition-metal-free strategy for the synthesis of 2-(cyclobut-1-en-1-yl)-1H-indoles under mild conditions is described herein. A series of substituted 2-(cyclobut-1-en-1-yl)-1H-indoles are accessed by a one-pot cyclobutenylation/deprotection cascade from N-Boc protected indoles. Preliminary experimental and density functional theory calculations suggest that a Boc-group transfer is involved in the underlying mechanism.
Introduction
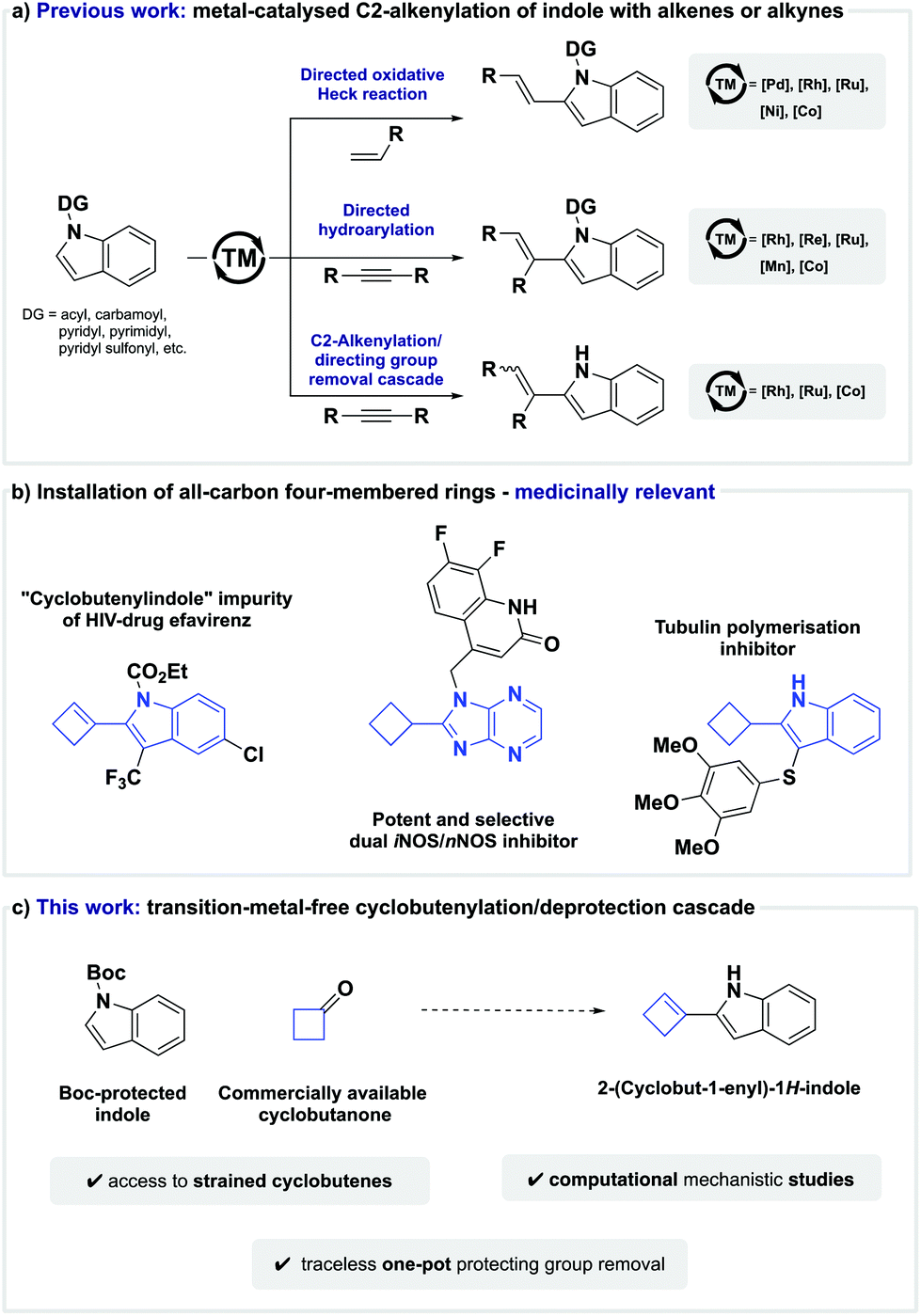
The indole moiety is a privileged structural motif found in a wide range of natural products, active pharmaceutical agents, agrochemical products, and even functional materials.1–3 Based on its versatile applications and properties, the invention of protocols for the regioselective direct functionalisation of indoles has been a long-standing goal in organic synthesis,4–11 and C–H alkenylation is among the effective strategies for the introduction of molecular complexity that received particular attention.12 Classically, alkenylation of indoles has been achieved by the hydroarylation of alkynes or an oxidative Heck reaction between aryl C(sp2)–H bonds and alkenes.13,14In contrast with functionalisation of the electron-rich C3-position of indole, alkenylation of the significantly less activated C2-position is a more challenging task, and as such, new methods to overcome this intrinsic selectivity are highly desirable. Although other tactics have been reported,15–18 the use of metal-chelating directing groups on the indole nitrogen has been established as a general and broadly adopted strategy. To this end, a variety of different directing groups, such as N-carbamoyl,19–22N-acyl,23–25N-pyridyl sulfonyl,26–28N-pyrimidyl,29–35 and others,36–40 have successfully enabled the metal-catalysed C2-selective oxidative Heck reaction between alkenes and indole (Scheme 1a). Similarly, C2-alkenylation of indole can also be effected by a transition-metal-catalysed C–H addition across alkynes when guided by an appropriate directing group, such as N-carbamoyl,41,42N-pyridyl,43,44 or N-pyrimidyl45–51 (Scheme 1a).
 | ||
| Scheme 1 Direct C2-alkenylation of indole and design plan. | ||
Although these elegant C–H functionalisation protocols enable the regioselective C2-alkenylation of indole, removal of the directing group poses significant drawbacks when it is no longer desired in the target molecule. For example, deprotection strategies can limit functional group compatibility, as harsh reaction conditions are often required, or lead to dead ends in multi-step syntheses when the directing group proves to be non-removable. To avoid the aforementioned shortcomings, the development of one-pot C2-alkenylation/directing group-removal cascades is highly desirable, and seminal studies by Kim,52,53 Zeng,54 Matsunaga,55 and Zhao56 have recently established the viability of this approach (Scheme 1a).
Despite these advances, the direct C2-alkenylation of indole is still largely limited to the installation of acyclic alkenes. Strained cyclic alkenes are inaccessible hitherto, and a direct route to 2-(cyclobut-1-en-1-yl)-1H-indoles remains unprecedented.57,58 This is surprising given that 2-(cyclobut-1-en-1-yl)-1H-indole analogues and their saturated counterparts have shown promising biological activity, such as the reduction of pain by selective dual iNOS/nNOS inhibition,59 or anticancer activity by tubulin polymerisation inhibition (Scheme 1b).60
The use of four-membered ring building blocks in medicinal chemistry remains relatively underdeveloped as chemists depend on a small number of viable protocols towards these moieties.61,62 Given our laboratory's interest in the use of four-membered rings to access biologically relevant scaffolds,63–68 we questioned if we could expand the scope of C2-alkenylation protocols to hitherto virtually inaccessible 2-(cyclobut-1-en-1-yl)-1H-indole analogues through a transition-metal-free cyclobutenylation/deprotection cascade (Scheme 1c). Our implementation of these design criteria and computational studies on the mechanistic details are described herein.
Results and discussion
We discovered that treatment of representative N-Boc protected indole 1a with an equimolar amount of n-butyllithium in diethyl ether at −78 °C for one hour, followed by one equivalent of cyclobutanone at the same temperature, furnished cyclobutene 2a in 20% yield (Table 1, entry 1). Investigation into the composition of the crude reaction mixture revealed the presence of trace quantities of indole and approximately 50% starting material.|
|
||
|---|---|---|
| Entry | Variation from above conditionsa | Yieldb (%) |
| a Initial conditions: 1a (1.0 mmol), n-butyllithium (1.1 mmol), Et2O (4 mL), −78 °C, 1 h, then cyclobutanone (1.0 mmol), −78 °C to rt, 17 h. b 1H-NMR yields based on 1,4-dinitrobenzene as internal standard. c Number in parentheses refers to isolated yield. Isolated yields of repeat experiments ranged from 35%–41%. d Reagent stoichiometry was changed to: 1a (1.0 mmol), n-butyllithium (1.1 mmol), cyclobutanone (0.5 mmol). | ||
| 1 | None | 20 |
| 2 | Two equivalents of N-Boc indole and n-BuLi with respect to cyclobutanone | 53 (41)c |
| 3d | sec-BuLi instead of n-BuLi | 23 |
| 4d | tert-BuLi instead of n-BuLi | 15 |
| 5d | LDA instead of n-BuLi | — |
| 6d | TMPMg·LiCl instead of n-BuLi | — |
| 7d | Deprotonation at −40 °C instead of −78 °C | 16 |
| 8d | Deprotonation at rt instead of −78 °C | — |
| 9d | Toluene instead of diethyl ether | — |
| 10d | Pentane instead of diethyl ether | — |
| 11d | Methyl tert-butyl ether instead of diethyl ether | 22 |
| 12d | N-Tosyl instead of N-Boc | — |
| 13d | N-Methyl instead of N-Boc | — |
| 14d | N-Cbz instead of N-Boc | — |
| 15d | N-Ethyloxycarbonyl instead of N-Boc | — |
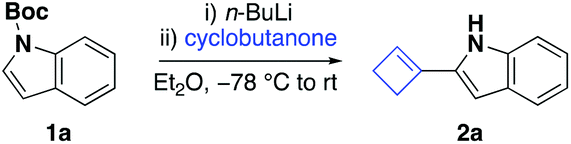
We thus questioned if conversion was limited by incomplete deprotonation of N-Boc protected indole under our reaction conditions and chose to investigate this hypothesis experimentally. To this end, we treated N-Boc protected indole 1a with n-butyllithium in diethyl ether at −78 °C and quenched aliquots of this reaction mixture with deuterium oxide at different time intervals (0.5 h, 1 h, 1.5 h, 2 h). Analysis of the resulting 1H-NMR spectra confirmed our hypothesis as complete C2-deprotonation was not observed even after extended reaction duration (C2–D vs. C2–H ≈ 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), so from the outset of this work we recognised the challenge associated with the deprotonation of N-Boc indole (see ESI†). This observation is also in line with a report by Wu and co-workers.69
:1), so from the outset of this work we recognised the challenge associated with the deprotonation of N-Boc indole (see ESI†). This observation is also in line with a report by Wu and co-workers.69
On the basis of these results, we initially chose to adjust the ratio of 1a and cyclobutanone. Pleasingly, using a two-fold excess of N-Boc protected indole and n-butyllithium with respect to cyclobutanone led to a significant improvement and provided the cyclobutene 2a in 41% yield (Table 1, entry 2).
Encouraged by this result, we next turned our attention to a screen of additional factors influencing the outcome of the reaction. First, the choice of base and reaction temperature were investigated. When n-butyllithium was substituted with sec-butyllithium, tert-butyllithium, lithium diisopropylamide, or a Turbo–Hauser base (TMPMg·LiCl), the reaction proceeded with inferior outcome or was shut down completely (Table 1, entries 3–6). Equally, raising the temperature during deprotonation to −40 °C or room temperature did not improve the outcome of the reaction, which we attributed to the instability of the protecting group under these reaction conditions (Table 1, entries 7 and 8). Next, we decided to screen solvents with different dielectric constants. The use of polar methyl tert-butyl ether led to a significant reduction of the reaction yield, whereas apolar solvents, including toluene and pentane, proved to be unsuitable reaction mediums, leading to complete cessation of the reaction (Table 1, entries 9–11). Finally, we questioned whether other common protecting groups would be compatible with this tandem reaction. Of the additional protecting groups that we screened, including sulfonyl- and alkyl-based moieties, none were suitable for this tandem reaction and no cyclobutene 2a was formed (Table 1, entries 12–15). In the case of N-tosyl indole and N-methyl indole, we obtained the corresponding cyclobutanol addition product, whereas the reaction of N-Cbz indole and N-ethyloxycarbonyl indole mainly afforded starting material. This underscored the unique effectiveness of the Boc-protecting group to engage in this tandem reaction. Full disclosure of our optimisation studies is available in the ESI.†
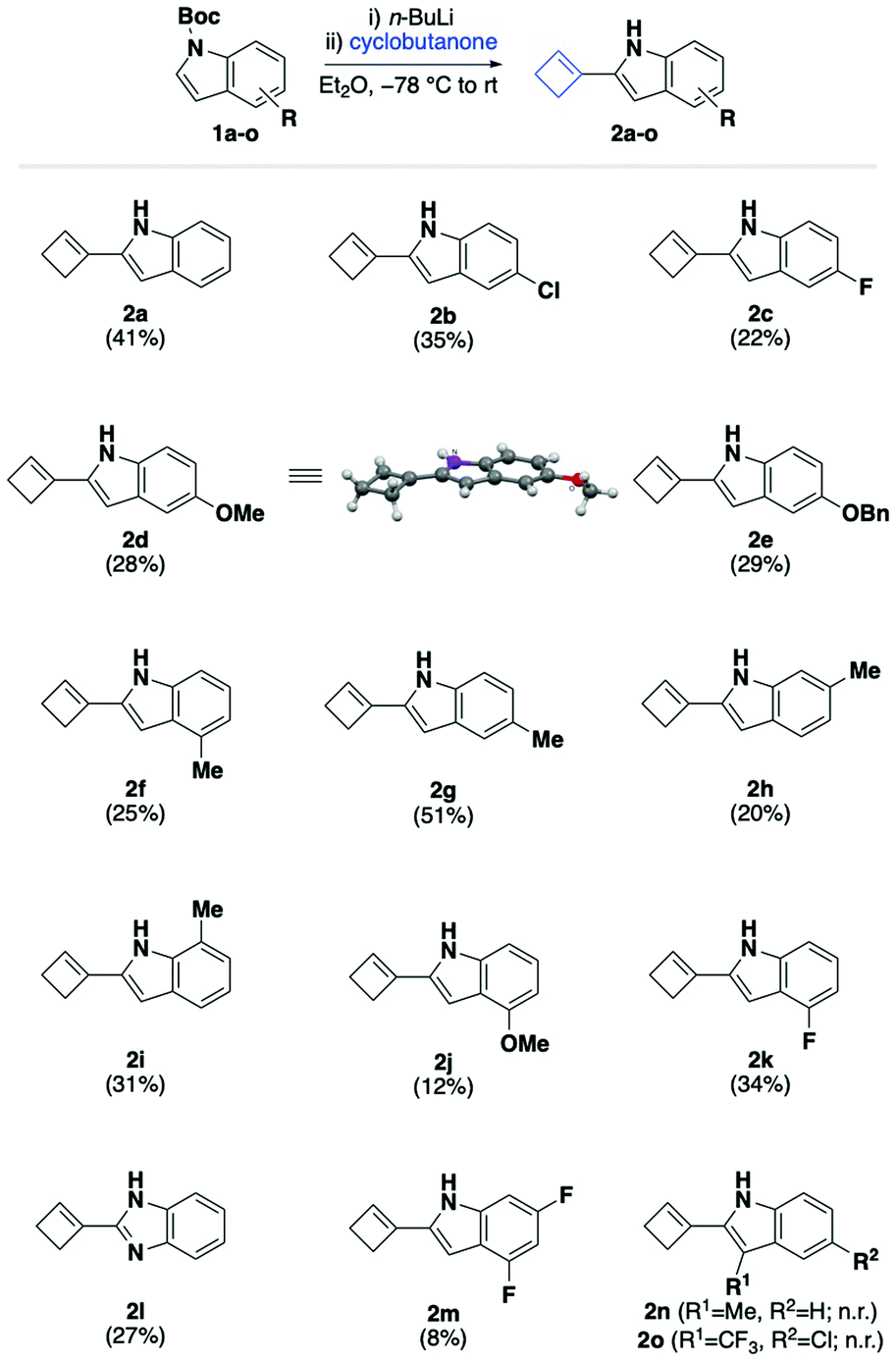
With optimal reaction conditions in hand (Table 1, entry 2), we sought to define the scope and limitations of our cyclobutenylation/deprotection cascade using a variety of differently substituted Boc-protected indoles 1a–o. These substrates were readily synthesised by treatment of commercially available indoles with di-tert-butyl dicarbonate and 4-dimethylaminopyridine in dichloromethane at room temperature (see ESI†). As shown in Scheme 2, our choice of substituted indoles was determined by the ambition to cover a range of electronic properties, while avoiding substrates that are incompatible with n-butyllithium.
 | ||
| Scheme 2 Substrate scope of the cyclobutenylation/deprotection cascade. | ||
In the event, we were pleased to find that weakly activated 5-halogenated indoles 1b and 1c underwent the tandem alkenylation/deprotection sequence in up to 35% isolated yield. Gratifyingly, more strongly activated substances containing methoxy- or benzyloxy substituents 1d and 1e successfully participated in the desired cyclobutenylation/deprotection cascade to afford indoles 2d and 2e in 28% and 29% yield, respectively. The structure of the 5-methoxylated cyclobutenyl indole 2d was confirmed by X-ray crystallography. It is worth noting that in the crystalline form, the cyclobutene moiety resides in-plane with the indole system to extend the conjugated system. Next, we turned our attention to studying the effect of positional isomerism of weakly activated methyl-substituted indole. Indoles containing a methyl substituent in the C4-, C5-, C6-, and C7-position (1f–1i) all afforded the corresponding cyclobutenes 2f–2i in 20–51% yield under our standard reaction conditions. Notably, the best isolated yield was achieved for the 5-methylated substrate 2g, which furnished the corresponding cyclobutene in nearly double the yield compared with the other positional isomers. This positional isomer effect was even more pronounced for strongly electron-donating methoxy-substituted indole, for which the 4-methoxylated analogue 2j was afforded in significantly reduced 12% yield in comparison with 28% yield obtained for the 5-methoxylated analogue 2d. In contrast, electron-deficient 4-fluorinated indole 1k furnished the desired cyclobutene 2k in improved 34% yield compared with the 5-fluorinated derivative 2c. Moreover, we were pleased to find that the reaction of N-Boc protected benzimidazole, a privileged pharmacophore, delivered the expected cyclobutene 2l in 27% yield. Highly electron-deficient Boc-protected 4,6-difluoroindole 1m, on the other hand, afforded the corresponding cyclobutene 2m in measurable, but low 8% yield, which we attributed in part to its rapid decomposition during work-up and isolation. In contrast with other analogues, indole 2m was found to be unstable even at low temperatures under an atmosphere of nitrogen.
Nevertheless, there are limitations to the current cyclobutenylation/deprotection cascade. We discovered that the successful conversion of Boc-protected indoles to cyclobutenes hinged on the presence of an unsubstituted 3-position. For example, when 3-methylated indole 1n was subjected to our standard conditions, a mixture of starting material and 3-methylindole was isolated, which we initially attributed to the facile deprotonation of the methyl group. Consequently, we hypothesised that 3-trifluoromethylated indole 1o would deliver the corresponding cyclobutene product when subjected to our standard conditions. In the event, however, we found that also this analogue was ineffective at participating in the desired cascade. A similar observation was reported for Kim's rhodium-catalysed directed C2-alkenylation, in which C3-substituted indoles also failed to deliver the desired products.53 Moreover, we found that other cycloalkanones or acyclic ketones do not undergo the desired cycloalkenylation/deprotection cascade under these reaction conditions.
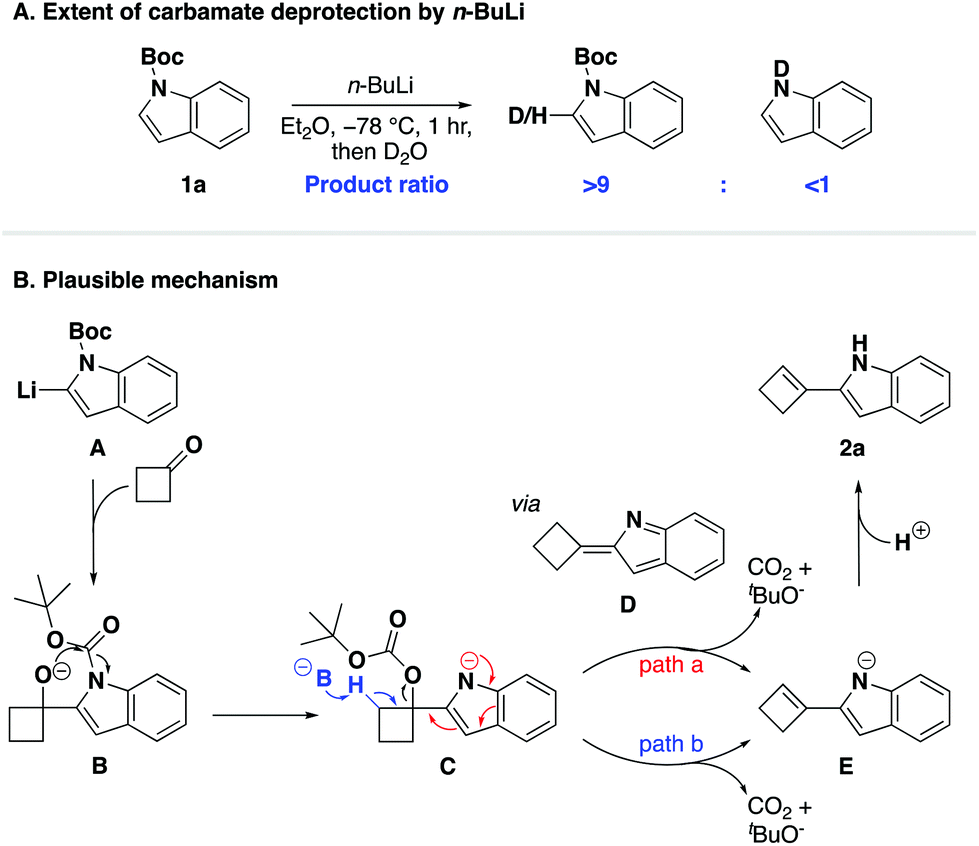
To conclude our study, we sought to elucidate a plausible mechanism for the one-pot cyclobutenylation/deprotection cascade. We found the role of the N-protecting group of particular interest and again turned to deuterium studies to investigate its stability (Scheme 3A and ESI†). To our surprise, these experiments revealed that the N-Boc group was tolerant to treatment with n-butyllithium even after extended reaction durations,70 which to us indicated that the observed deprotection of the Boc-group occurred after the introduction of cyclobutanone to the reaction mixture. We postulated that the unique effectiveness of the Boc-protecting group for this tandem reaction stems from its ideal compromise between stability to tolerate treatment with n-butyllithium and susceptibility to subsequent deprotection. On this basis, a plausible reaction mechanism for the described cascade is presented in Scheme 3B. We hypothesised that in situ generated organolithium A underwent nucleophilic addition to cyclobutanone to reveal alkoxide B which, facilitated by its spatial proximity to the Boc-group, added to the carbamate to furnish carbonate C.71 We postulated that the most likely fate of the carbonate group was then to undergo an elimination, which was thermodynamically driven by the release of carbon dioxide and tert-butanol. Two plausible pathways are thus proposed for the conversion of carbonate C to the final product: (i) an intramolecular elimination pathway via 2-cyclobutylidene-2H-indole D (red, path a); or (ii) an intermolecular process by deprotonation of the neighbouring hydrogen atom (blue, path b).
 | ||
| Scheme 3 Plausible mechanism. | ||
Given the ambiguity of the elimination step, we further probed the underlying mechanism with a computational study. All calculations were performed at the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d,p)72–74 level in diethyl ether with the IEF-PCM solvation model75 and N-Boc protected indole 1a as a representative example (Fig. 1). In line with our experimental results, the C2-deprotonated indole A undergoes nucleophilic addition to cyclobutanone to furnish alkoxide B, in which the anionic charge is mainly located on the oxygen. The calculated transition state energy barrier to TS1 is 11.4 kcal mol−1 and formation of alkoxide B is exothermic by 12.9 kcal mol−1. Next, an intramolecular Boc-group transfer from indole to the alkoxide takes place to yield carbonate Cvia a late transition state TS2 with a relative low energy barrier of 5.4 kcal mol−1. The resulting carbonate C was found to be 11.6 kcal mol−1 lower in energy than carbamate B, which supported our hypothesis that an in situ Boc-group transfer is indeed energetically favourable.
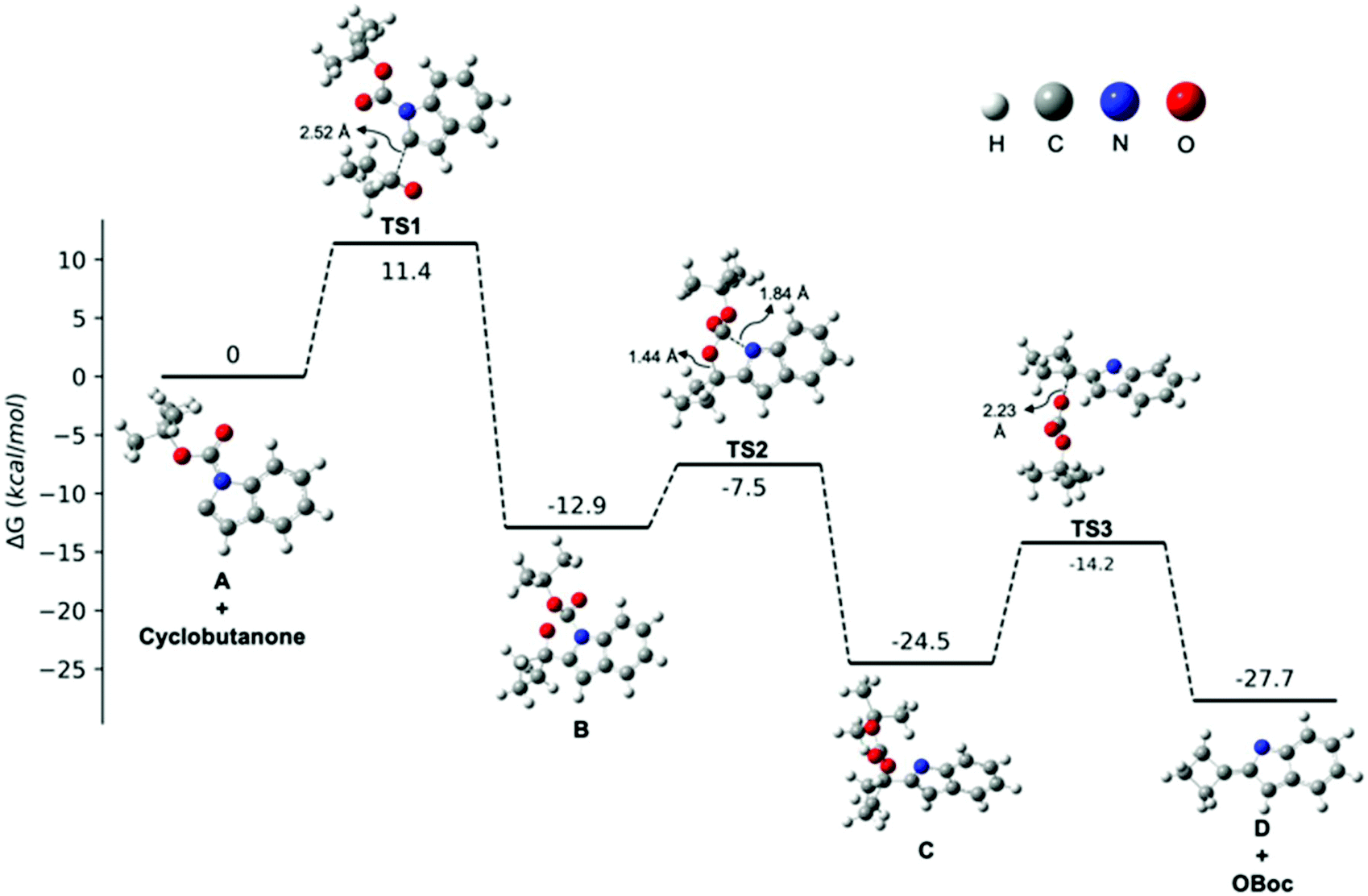 | ||
| Fig. 1 Free energy profile for the transformation from A to 2-cyclobutylidene-2H-indole D (path a in Scheme 3). | ||
Finally, in order to differentiate between the two proposed elimination paths (Scheme 3, path a vs. path b), we compared the relative acidities of intermediates C and D by quantum mechanical methods.76 The pKa values of the β-hydrogen of carbonate C and the allylic hydrogen of D were calculated to be 50.3 and 40.5, respectively (see ESI†). Therefore, the allylic hydrogen of D is more readily deprotonated than C, and as such, we concluded that an intramolecular elimination/isomerisation pathway (path a) is possible. We located the transition state TS3 and our calculations suggest that this elimination step is plausible as it is exothermic overall (3.2 kcal mol−1) and not rate-determining (ΔG‡ = 10.3 kcal mol−1; 1.1 kcal mol−1 lower than the intermolecular nucleophilic addition step). Based on our combined experimental results and computational calculations, we suggest that a mechanism consisting of intramolecular Boc-group transfer followed by intramolecular carbonate elimination/isomerisation (path b) is in operation for the formation of the desired 2-(cyclobut-1-en-1-yl)-1H-indoles.
Conclusions
In summary, we report the first synthesis of 2-(cyclobut-1-en-1-yl)-1H-indoles through a one-pot cyclobutenylation/deprotection cascade. A series of substituted 2-(cyclobut-1-en-1-yl)-1H-indoles was obtained in up to 51% yield in one step from readily accessible N-Boc protected indoles by treatment with n-butyllithium, followed by the addition of cyclobutanone. Furthermore, DFT calculations confirmed that a plausible reaction mechanism involved the transfer of the Boc-group from indole to the intermediate alkoxide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge an EPSRC Imperial College President's Scholarship (to P. N.) and additional funding from Dr Isabel Bader and her late husband Dr Alfred Bader (to P. J. P.) and Oxana Bennett (to P. J. P.). The authors thank Pete Haycock, Corey Fülöp and Dr Lisa Haigh for NMR and mass spectrometric analysis at Imperial College London. The authors also acknowledge the service and support from Imperial College Research Computing Service (DOI: 10.14469/hpc/2232). JChem for Excel was used for producing and handling structure data files (SDF), JChem for Excel 21.3.0.817, ChemAxon (https://www.chemaxon.com).Notes and references
- J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS.
- A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed.
- M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed.
- L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS.
- G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC.
- G. W. Gribble, Pure Appl. Chem., 2003, 75, 1417–1432 CAS.
- S. Agarwal, S. Cammerer, S. Filali, W. Frohner, J. Knoll, M. Krahl, K. Reddy and H.-J. Knolker, Curr. Org. Chem., 2005, 9, 1601–1614 CrossRef CAS.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- I. Moritanl and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef.
- Y. Fujiwara, I. Moritani, M. Matsuda and S. Teranishi, Tetrahedron Lett., 1968, 9, 633–636 CrossRef.
- N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125–3129 CrossRef CAS PubMed.
- Y. Nakao, K. S. Kanyiva, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2006, 128, 8146–8147 CrossRef CAS PubMed.
- A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159–1162 CrossRef CAS PubMed.
- Y.-J. Wang, C.-H. Yuan, D.-Z. Chu and L. Jiao, Chem. Sci., 2020, 11, 11042–11054 RSC.
- B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2013, 19, 11863–11868 CrossRef CAS PubMed.
- L.-Q. Zhang, S. Yang, X. Huang, J. You and F. Song, Chem. Commun., 2013, 49, 8830–8832 RSC.
- B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, J. Org. Chem., 2013, 78, 9345–9353 CrossRef CAS PubMed.
- W. Lin, W. Li, D. Lu, F. Su, T.-B. Wen and H.-J. Zhang, ACS Catal., 2018, 8, 8070–8076 CrossRef CAS.
- F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982–9983 CrossRef CAS PubMed.
- V. Lanke and K. R. Prabhu, Org. Lett., 2013, 15, 2818–2821 CrossRef CAS PubMed.
- W. Chen, H.-J. Li, Q.-Y. Li and Y.-C. Wu, Org. Biomol. Chem., 2020, 18, 500–513 RSC.
- A. García-Rubia, R. G. Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511–6515 CrossRef PubMed.
- A. García-Rubia, B. Urones, R. G. Arrayás and J. C. Carretero, Chem. – Eur. J., 2010, 16, 9676–9685 CrossRef PubMed.
- Z.-L. Yan, W.-L. Chen, Y.-R. Gao, S. Mao, Y.-L. Zhang and Y.-Q. Wang, Adv. Synth. Catal., 2014, 356, 1085–1092 CrossRef CAS.
- L. Yang, G. Zhang and H. Huang, Adv. Synth. Catal., 2014, 356, 1509–1515 CrossRef CAS.
- B. Gong, J. Shi, X. Wang, Y. Yan, Q. Li, Y. Meng, H. E. Xu and W. Yi, Adv. Synth. Catal., 2014, 356, 137–143 CrossRef CAS.
- L. Zhang, R. Qiu, X. Xue, Y. Pan, C. Xu, D. Wang, X. Wang, L. Xu and H. Li, Chem. Commun., 2014, 50, 12385–12388 RSC.
- P. Lu, C. Feng and T.-P. Loh, Org. Lett., 2015, 17, 3210–3213 CrossRef CAS PubMed.
- X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Adv. Synth. Catal., 2016, 358, 573–583 CrossRef CAS.
- S. Nakanowatari, R. Mei, M. Feldt and L. Ackermann, ACS Catal., 2017, 7, 2511–2515 CrossRef CAS.
- Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- L.-J. Li, J.-J. Chen, C.-F. Feng, H.-Y. Li, X. Wang, H. Xu and H.-X. Dai, Org. Lett., 2020, 22, 9169–9173 CrossRef CAS PubMed.
- C.-Y. Tang, Y. Tao, X.-Y. Wu and F. Sha, Adv. Synth. Catal., 2014, 356, 609–615 CrossRef CAS.
- E. Capito, J. M. Brown and A. Ricci, Chem. Commun., 2005, 1854–1856 RSC.
- J. M. Brown, G. Fanton, N. M. Coles, A. R. Cowley and J. P. Flemming, Heterocycles, 2010, 80, 895–901 CrossRef.
- R. A. Jagtap, C. P. Vinod and B. Punji, ACS Catal., 2019, 9, 431–441 CrossRef CAS.
- D. J. Schipper, M. Hutchinson and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6910–6911 CrossRef CAS PubMed.
- H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424–5431 CrossRef CAS.
- L. Liang, S. Fu, D. Lin, X.-Q. Zhang, Y. Deng, H. Jiang and W. Zeng, J. Org. Chem., 2014, 79, 9472–9480 CrossRef CAS PubMed.
- H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15063–15067 CrossRef CAS PubMed.
- Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 4698–4701 CrossRef CAS PubMed.
- L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136–7139 RSC.
- S. Wang, J.-T. Hou, M.-L. Feng, X.-Z. Zhang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2016, 52, 2709–2712 RSC.
- X. Zhou, Y. Luo, L. Kong, Y. Xu, G. Zheng, Y. Lan and X. Li, ACS Catal., 2017, 7, 7296–7304 CrossRef CAS.
- N. Muniraj and K. R. Prabhu, Adv. Synth. Catal., 2018, 360, 1370–1375 CrossRef CAS.
- C. Wang and M. Rueping, ChemCatChem, 2018, 10, 2681–2685 CrossRef CAS.
- C.-L. Duan, X.-Y. Liu, Y.-X. Tan, R. Ding, S. Yang, P. Tian and G.-Q. Lin, Synlett, 2019, 30, 932–938 CrossRef CAS.
- S. Sharma, S. Han, M. Kim, N. K. Mishra, J. Park, Y. Shin, J. Ha, J. H. Kwak, Y. H. Jung and I. S. Kim, Org. Biomol. Chem., 2014, 12, 1703–1706 RSC.
- S. Sharma, S. Han, Y. Shin, N. K. Mishra, H. Oh, J. Park, J. H. Kwak, B. S. Shin, Y. H. Jung and I. S. Kim, Tetrahedron Lett., 2014, 55, 3104–3107 CrossRef CAS.
- W. Zhang, J. Wei, S. Fu, D. Lin, H. Jiang and W. Zeng, Org. Lett., 2015, 17, 1349–1352 CrossRef CAS PubMed.
- H. Ikemoto, R. Tanaka, K. Sakata, M. Kanai, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2017, 56, 7156–7160 CrossRef CAS PubMed.
- X. Wu, Y. Lu, J. Qiao, W. Dai, X. Jia, H. Ni, X. Zhang, H. Liu and F. Zhao, Org. Lett., 2020, 22, 9163–9168 CrossRef CAS PubMed.
- M. Gazvoda, M. Krivec, Z. Časar and J. Košmrlj, J. Org. Chem., 2018, 83, 2486–2493 CrossRef CAS PubMed.
- J. Zhang, J. Shao, J. Xue, Y. Wang and Y. Li, RSC Adv., 2014, 4, 63850–63854 RSC.
- J. E. Payne, C. Bonnefous, K. T. Symons, P. M. Nguyen, M. Sablad, N. Rozenkrants, Y. Zhang, L. Wang, N. Yazdani, A. K. Shiau, S. A. Noble, P. Rix, T. S. Rao, C. A. Hassig and N. D. Smith, J. Med. Chem., 2010, 53, 7739–7755 CrossRef CAS PubMed.
- G. La Regina, R. Bai, W. M. Rensen, E. Di Cesare, A. Coluccia, F. Piscitelli, V. Famiglini, A. Reggio, M. Nalli, S. Pelliccia, E. Da Pozzo, B. Costa, I. Granata, A. Porta, B. Maresca, A. Soriani, M. L. Iannitto, A. Santoni, J. Li, M. M. Cona, F. Chen, Y. Ni, A. Brancale, G. Dondio, S. Vultaggio, M. Varasi, C. Mercurio, C. Martini, E. Hamel, P. Lavia, E. Novellino and R. Silvestri, J. Med. Chem., 2013, 56, 123–149 CrossRef CAS PubMed.
- A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS PubMed.
- E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed.
- P. Natho, M. Kapun, L. A. T. Allen and P. J. Parsons, Org. Lett., 2018, 20, 8030–8034 CrossRef CAS PubMed.
- P. Natho, L. A. T. Allen, A. J. P. White and P. J. Parsons, J. Org. Chem., 2019, 84, 9611–9626 CrossRef CAS PubMed.
- R.-C. Raclea, P. Natho, L. A. T. Allen, A. J. P. White and P. J. Parsons, J. Org. Chem., 2020, 85, 9375–9385 CrossRef CAS PubMed.
- P. Natho, A. B. Rouse, J. L. Greenfield, L. A. T. Allen, A. J. P. White, Z. Yang and P. J. Parsons, Tetrahedron, 2020, 131636 CrossRef CAS.
- A. Petti, P. Natho, K. Lam and P. J. Parsons, Eur. J. Org. Chem., 2021, 854–858 CrossRef CAS.
- P. Natho, L. A. T. Allen and P. J. Parsons, Tetrahedron Lett., 2020, 151695 CrossRef CAS.
- J.-P. Wu, S. Sanyal, Z.-H. Lu and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 5667–5669 CrossRef CAS.
- H. Ila, J. Markiewicz, V. Malakhov and P. Knochel, Synthesis, 2013, 45, 2343–2371 CrossRef CAS.
- C. Agami and F. Couty, Tetrahedron, 2002, 58, 2701–2724 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- Q. Zeng, M. R. Jones and B. R. Brooks, J. Comput.-Aided Mol. Des., 2018, 32, 1179–1189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, NMR spectra and computational methods. CCDC 2057801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob00430a |
| This journal is © The Royal Society of Chemistry 2021 |