
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic trisaccharides reveal discrimination of endo-glycosidic linkages by exo-acting α-1,2-mannosidases in the endoplasmic reticulum†
Kyohei
Nitta
,
Taiki
Kuribara
and
Kiichiro
Totani
 *
*
Department of Materials and Life Science, Seikei University, Musashino-shi, Tokyo, 180-8633, Japan. E-mail: ktotani@st.seikei.ac.jp
First published on 14th April 2021
Abstract
A tri-antennary Man9GlcNAc2 glycan on the surface of endoplasmic reticulum (ER) glycoproteins functions as a glycoprotein secretion or degradation signal after regioselective cleavage of the terminal α-1,2-mannose residue of each branch. Four α-1,2-mannosidases—ER mannosidase I, ER degradation-enhancing α-mannosidase-like protein 1 (EDEM1), EDEM2, and EDEM3—are involved in the production of these signal glycans. Although selective production of signal glycans is important in determining the fate of glycoproteins, the branch-discrimination abilities of the α-1,2-mannosidases are not well understood. A structural feature of the Man9GlcNAc2 glycan is that all terminal glycosidic linkages of the three branches are of the α-1,2 type, while the adjacent inner glycosidic linkages are different. In this study, we examined whether the α-1,2-mannosidases showed branch specificity by discriminating between different inner glycosides. Four trisaccharides with different glycosidic linkages [Manα1-2Manα1-2Man (natural A-branch), Manα1-2Manα1-3Man (natural B-branch), Manα1-2Manα1-6Man (natural C-branch), and Manα1-2Manα1-4Man (unnatural D-branch)] were synthesized and used to evaluate the hypothesis. When synthesizing these oligosaccharides, highly stereoselective glycosylation was achieved with a high yield in each case by adding a weak base or tuning the polarity of the mixed solvent. Enzymatic hydrolysis of the synthetic trisaccharides by a mouse liver ER fraction containing the target enzymes showed that the ER α-1,2-mannosidases had clear specificity for the trisaccharides in the order of A-branch > B-branch > C-branch ≈ D-branch. Various competitive experiments have revealed for the first time that α-1,2-mannosidase with inner glycoside specificity is present in the ER. Our findings suggest that exo-acting ER α-1,2-mannosidases can discriminate between endo-glycosidic linkages.
Introduction
A Glc3Man9GlcNAc2 glycan is transferred to the nascent polypeptide in the side chain of the Asn residue of the consensus Asn-Xaa-Ser/Thr sequence (Xaa is any amino acid other than Pro) by the action of oligosaccharyl transferase1 in the endoplasmic reticulum (ER) (Fig. 1). The Glc3Man9GlcNAc2-glycoprotein is converted to the Glc1Man9GlcNAc2 (G1M9)-glycoprotein by glucosidase I (GI)2 and glucosidase II (GII)3,4 and it interacts with lectin-like molecular chaperones calnexin (CNX)5 and calreticulin (CRT).6 Upon approaching the correct folding state, the binding between the G1M9-glycoprotein and CNX/CRT is weakened, and subsequent cleavage of the terminal glucose by GII produces the Man9GlcNAc2 (M9)-glycoprotein.7 The M9-glycoprotein is recognized by the folding sensor enzyme UDP-glucose:glycoprotein glucosyltransferase 1 (UGGT1),8–10 which checks the folding state. The partially folded glycoprotein is glucosylated by UGGT1 before acceleration of G1M9-glycoprotein folding by CNX/CRT. The folding acceleration/folding check cycle is called the CNX/CRT cycle11 and it plays a pivotal role in the folding mechanism in glycoprotein quality control.12,13 | ||
| Fig. 1 Glycan trimming in ER glycoprotein quality control. | ||
The completely folded M9-glycoprotein and misfolded M9-glycoprotein have their outermost mannose trimmed by ER α-1,2-mannosidases [ER mannosidase I (ERManI),14 ER degradation-enhancing α-mannosidase-like protein 1 (EDEM1),15 EDEM2,16 and EDEM317] to produce glycoproteins with the Man8AGlcNAc2 (M8A), Man8BGlcNAc2 (M8B) or Man8CGlcNAc2 (M8C) glycan.18,19 The M8B-glycoprotein is recognized by the cargo-receptor ER-Golgi intermediate compartment 53 kDa protein (ERGIC53),20 which transports completely folded glycoproteins from the ER to the Golgi apparatus. Thus, the M8B glycan functions as a secretion signal of properly folded glycoproteins. The M8A- or M8C-glycoproteins are further trimmed by ER α-1,2-mannosidases to produce the Man5GlcNAc2 (M5)-glycoprotein. The M5-glycoprotein is excreted from the ER to the cytosol and passed on to the proteasomal degradation system through collaboration with various lectins represented by osteosarcoma-amplified 9 (OS-9).21 Both M8A and M8C are precursors for the degradation signals of misfolded glycoproteins. In this way, ER α-1,2-mannosidases produce different glycan structures that serve as secretion or degradation signals after regioselective cleavage of the terminal mannose of the tri-antennary M9 glycan.
ER α-1,2-mannosidases are exo-acting enzymes belonging to the glycoside hydrolase 47 family. ERManI reportedly cleaves Manα-1,2 linkages at the B-branch of the M9 glycan to produce the M8B glycan in vitro.14 This enzyme preferentially acts on denatured glycoproteins and can cleave all Manα-1,2-linkages of the M9-glycoprotein under high enzyme concentrations.22 According to a comparison of glycan profiles among single knockout cells of various EDEMs, EDEM1, EDEM2, and EDEM3 show mannosidase activity in vivo, but the activity of each EDEM alone has not been detected in vitro.23 Recently, it has been reported that EDEMs show mannosidase activity by forming a complex with ER-resident protein 46, protein disulfide isomerase, or TXNDC11 via their cysteine residue.24–26 It has been suggested that EDEMs recognize the aglycone site of glycoproteins through complex formation.
Most studies on the use of ER α-1,2-mannosidases have focused on discriminating the aglycone state of substrate glycoproteins because these enzymes are responsible for producing secretion and degradation signals. However, the regioselectivity of ER α-1,2-mannosidases according to the structural features of each branch has not been verified. To address this issue, we focused on the differences in the inner glycosidic linkages of each branch (Fig. 2). The A-, B- and C-branches of the M9 glycan have different inner glycosidic linkages of Manα-1,2, Manα-1,3, and Manα-1,6, respectively. We hypothesized that these differences in the inner glycosidic linkages were responsible for the regioselectivity of ER α-1,2-mannosidases.
 | ||
| Fig. 2 Design of trisaccharide substrates for glycan specificity analysis of ER α-1,2-mannosidases. | ||
In this study, we synthesized a series of trisaccharides (A3,27B3,28C3, and D3) with different inner glycosidic linkages (Fig. 2), and conducted glycan specificity analysis and related competitive analysis of ER α-1,2-mannosidases using these trisaccharides. The results clearly showed that the reactivities of α-1,2-mannosidases in the mouse liver ER fraction were in the order A-branch > B-branch > C-branch ≈ D-branch. These findings suggest that the ER fraction contains exo-acting α-1,2-mannosidases with the ability to discriminate endo-glycosidic linkages.
Results and discussion
Synthesis of the trisaccharide substrates
First, we synthesized the monosaccharide units required for the synthesis of the target trisaccharides (Scheme 1). DAST-mediated ring-opening fluorination29 was conducted with the starting material 1 to give mannosyl fluoride 2 as a common glycosyl donor for use in this study. We also used 1 to synthesize glycosyl acceptors 5, 8, 10, and 11 for the formation of α-1,2-, α-1,3-, α-1,6-, and α-1,4-linkages. | ||
| Scheme 1 Synthesis of monosaccharide units. | ||
Next, we examined the synthesis of the target trisaccharides using the common glycosyl donor 2 and the relevant glycosyl acceptor (5, 8, 10, or 11) (Scheme 2 and Table 1). In the synthesis of disaccharide 12, when glycosylation was performed in toluene using AgOTf-Cp2HfCl2 as a promoter, disaccharide 12 was obtained in 98% yield with 3% of the undesired β-product (entry 1). Because purification of the desired α-product from the α/β-mixture was difficult, we explored the reaction conditions to improve the stereoselectivity. The α-selectivity in this glycosylation is attributed to “neighboring group participation”30 in the cation intermediate (major pathway in Fig. 3). In contrast, TfO− derived from AgOTf could coordinate from the α-face to the oxocarbenium cation, and the subsequent SN2-like attack of the glycosyl acceptor should result in the undesired β-product (minor pathway in Fig. 3). Therefore, we investigated the reaction conditions to neutralize TfOH generated in the reaction mixture by adding 2,6-di-tert-butyl-4-methylpyridine (DTBMP)31 as an acid scavenger, and succeeded in synthesizing disaccharide 12 in 93% yield with perfect α-selectivity (entry 2). The DTBMP-H+ ion produced in the reaction mixture might form an ion pair with the triflate anion, enable the suppression of the formation of α-contact ion pairs in the minor pathway, and provide excellent stereoselectivity via the major pathway. Similar reaction conditions were applied for subsequent glycosylation to give trisaccharide 14 in 84% yield with excellent α-selectivity (entry 3). To further improve the product yield, the solvent was changed from toluene to CH2Cl2, which had higher polarity to stabilize the intermediate cation, and a 95% yield was achieved but with lower stereoselectivity (α/β = 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33) (entry 4). The higher reactivity under these reaction conditions might yield more of the β-product by decreasing neighboring group participation. Although we tried to achieve both high yield and high stereoselectivity with a mixture of these two solvents (entries 5 and 6), we could not improve on the results in entry 3. Therefore, we investigated the reaction without DTBMP, which gave a high yield in entry 1 under non-neutralizing acidic glycosylation conditions. We succeeded in synthesizing trisaccharide 14 with a 99% yield and α/β > 95:5 (entry 7). Because 14α and 14β could be separated and purified by silica gel column chromatography, these reaction conditions were identified as the optimum conditions. Through this series of experiments, we found that the yield and stereoselectivity fluctuated with the addition of the base and adjustment of the ratio of solvents with different polarities.
:33) (entry 4). The higher reactivity under these reaction conditions might yield more of the β-product by decreasing neighboring group participation. Although we tried to achieve both high yield and high stereoselectivity with a mixture of these two solvents (entries 5 and 6), we could not improve on the results in entry 3. Therefore, we investigated the reaction without DTBMP, which gave a high yield in entry 1 under non-neutralizing acidic glycosylation conditions. We succeeded in synthesizing trisaccharide 14 with a 99% yield and α/β > 95:5 (entry 7). Because 14α and 14β could be separated and purified by silica gel column chromatography, these reaction conditions were identified as the optimum conditions. Through this series of experiments, we found that the yield and stereoselectivity fluctuated with the addition of the base and adjustment of the ratio of solvents with different polarities.
 | ||
| Scheme 2 Synthesis of the target trisaccharides. Glycosylation conditions are listed in Table 1. (a) NaOMe, MeOH/THF = 1:1, v/v. (b) 20% Pd(OH)2/C, H2, MeOH/THF = 1:1, v/v. (c) LiOH, H2O2 aq., THF. | ||
 | ||
| Fig. 3 Plausible mechanism of the glycosylation reaction. | ||
| Entry | Acceptor | Solvent | Reagents | Time (min) | Product (yield) | α/βd,e |
|---|---|---|---|---|---|---|
| a Reaction performed at −20 to 0 °C. b Reaction performed at −40 °C. c Acceptor 18 was recovered in 27% yield. d The ratio of α/β was determined by 1H NMR spectroscopy. e Stereochemistry was determined by 1JC–H coupling measurements32 using non-decoupling HMQC. f β detected: 3% (1H NMR). g No β detected. h β detected: 1% (1H NMR). | ||||||
| 1a | 5 | Toluene | AgOTf, Cp2HfCl2, MS AW-300 | 40 | 12 (98%) | >95:5f |
| 2a | 5 | Toluene | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 70 | 12 (93%) | α onlyg |
| 3a | 13 | Toluene | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 80 | 14 (84%) | α onlyg |
| 4a | 13 | CH2Cl2 | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 75 | 14 (95%) | 67:33 |
| 5a | 13 | Toluene/CH2Cl2 = 1:1 |
AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 60 | 14 (93%) | 84:16 |
| 6a | 13 | Toluene/CH2Cl2 = 2:1 |
AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 60 | 14 (81%) | 89:11 |
| 7a | 13 | Toluene | AgOTf, Cp2HfCl2, MS AW-300 | 60 | 14 (99%) | >95:5h |
| 8b | 8 | Toluene/CH2Cl2 = 1:1 |
AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 70 | 17 (99%) | α onlyg |
| 9a | 18 | Toluene | AgOTf, Cp2HfCl2, MS AW-300 | 90 | 19 (62%)c | α onlyg |
| 10a | 18 | Toluene/CH2Cl2 = 1:1 |
AgOTf, Cp2HfCl2, MS AW-300 | 50 | 19 (quant.) | >95:5h |
| 11a | 10 | Toluene | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 90 | 22 (90%) | α onlyg |
| 12a | 23 | Toluene | AgOTf, Cp2HfCl2, MS AW-300 | 40 | 24 (92%) | >95:5h |
| 13a | 23 | Toluene | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 40 | 24 (quant.) | α onlyg |
| 14a | 11 | Toluene | AgOTf, Cp2HfCl2, MS AW-300, DTBMP | 70 | 27 (90%) | α onlyg |
| 15a | 28 | Toluene | AgOTf, Cp2HfCl2, MS AW-300 | 50 | 29 (84%) | α onlyg |
For the synthesis of disaccharide 17, we performed glycosylation using a mixture of CH2Cl2 and toluene because acceptor 8 is insoluble in toluene. In this case, we were concerned about β-product formation not because of neighboring group participation but because of the improved reactivity. When the reaction temperature was decreased to −40 °C, disaccharide 17 was obtained in high yield with high α-selectivity (entry 8). In the subsequent synthesis of trisaccharide 19, the optimum conditions for synthesizing trisaccharide 14 (entry 7) were different, and although the resulting stereoselectivity was perfect, the yield was moderate at 62% (entry 9). Therefore, in our experiments investigating the improvement of the yield using a solvent mixture to stabilize the cation intermediate, we succeeded in synthesizing trisaccharide 19 quantitatively with α/β > 95:5 (entry 10). Disaccharides 22 and 27 were both synthesized in high yields with high stereoselectivity (entries 11 and 14) using the same reaction conditions as in the synthesis of disaccharide 12 (entry 2). In the preparation of trisaccharide 24, we synthesized the target product with complete α-selectivity using the reaction conditions with DTBMP (entry 13) because it was difficult to separate and purify trace amounts of the undesired β-product generated under the reaction conditions without DTBMP (entry 12). Trisaccharide 29 was synthesized in high yield with high stereoselectivity (entry 15) using the optimum conditions used for the synthesis of trisaccharide 14 (entry 7). Finally, the protected trisaccharides 14, 19, 24, and 29 were deprotected to convert them to the target trisaccharides A3, B3, C3, and D3, respectively.
Glycan-specificity analysis of ER α-1,2-mannosidases
Next, we analyzed the glycan specificity of ER α-1,2-mannosidases using the four types of synthesized trisaccharides. Because the detection of the activity of the enzyme alone has not been reported in vitro for EDEM1, EDEM2, and EDEM3, it is difficult to analyze individual glycan specificity using the recombinant enzyme. Therefore, we decided to use an ER fraction extracted from biological samples as an enzyme source containing the α-1,2-mannosidases. As a tissue for the extraction of the ER fraction, we selected the senescence-accelerated mouse prone 6 (SAMP6)33 liver, which we have previously used for the analysis of the activity of ER α-1,2-mannosidases using an M9-type synthetic glycan substrate.18,34Extraction of the ER fraction from the SAMP6 liver was performed by centrifugal fractionation.18,34 Because α-mannosidase is also present in the Golgi apparatus,35 the ER/Golgi content of each centrifugal fraction was verified by western blotting using both ER and Golgi marker proteins as indicators. This proved that the extracted ER fraction had no Golgi contamination (Fig. S1†).
Synthetic trisaccharides A3, B3, C3, and D3 were incubated with the ER fractions at 37 °C, and the time courses of the yields of disaccharides A2, B2, C2, and D2 in the reaction mixtures were analyzed quantitatively by HPLC (Fig. 4a). The hydrolysis yields for A3, B3, C3, and D3 at 8 h were 66%, 69%, 35%, and 35%, respectively (Fig. 4b). These results suggest that the reactivities of ER α-1,2-mannosidases change according to differences in the inner glycosidic linkage of the substrate trisaccharide, as we showed in the working hypothesis. D3 has an inner α-1,4-linkage that does not exist in the M9-type glycan, which is a substrate for ER α-1,2-mannosidases in vivo. We proposed two hypotheses for the hydrolysis of D3. The first hypothesis was that ER α-1,2-mannosidase was activated by the recognition of the inner Manα1-4Man linkage, and the second hypothesis was that ER α-1,2-mannosidase had weak recognition of the inner glycosidic linkage. Considering that there is limited evidence of the existence of enzymes that recognize the non-ER type glycosidic linkage, the former hypothesis is unlikely. On the other hand, it has been reported that ERManI primarily cleaves the B-branch in vitro, and also cleaves other branches secondarily.22 Considering these inferences and findings, we conclude that the latter hypothesis is plausible at this stage. Because the cleavage profiles of C3 and D3 were almost identical, it is possible that C3 was also cleaved by ER α-1,2-mannosidases with weak recognition of the inner glycosidic linkage. However, the enzyme involved in the cleavage of C3 may simply be less active than the enzyme involved in the cleavage of A3 and B3, so these issues were re-addressed using the results of competitive experiments with C3 and D3, which are described later in the text.
 | ||
| Fig. 4 Hydrolysis assay of A3, B3, C3 or D3 in the ER fraction. (a) Typical HPLC profiles of the glycan trimming reaction. Conditions for HPLC: a TSK-GEL Amide-80 column (25 cm × 4.6 mm i.d.), mobile phase CH3CN/H2O, gradient from 98:2 v/v to 90:10 v/v over 10 min followed by that from 90:10 v/v to 65:35 v/v over 10 min, and a mobile phase flow rate of 1.0 mL min−1 at 40 °C. (b) Hydrolysis yields of branch trisaccharides A3, B3, C3, and D3 (250 μM) treated with the ER fraction (3 mg protein per mL). Each data point represents the mean value with the standard deviation (n = 9). | ||
The hydrolysis profiles of A3 and B3 were similar (Fig. 4b). However, considering that the cleavage of the terminal Man at the A and B branches is responsible for the production of glycoprotein secretion and degradation signals, respectively,12,13 it is unlikely that the cleavage at each branch is caused by the same ER α-1,2-mannosidase. In this experiment, the possibility of further cleavage of the product A2 was not considered because it was undetectable as the HPLC retention time of A1 coincided with those of components derived from the ER fraction. Therefore, differences in the reactivity of ER α-1,2-mannosidases with A3 and B3 were evaluated using further experiments, which are described later in the text. At this stage, the cleavage specificity of ER α-1,2-mannosidases inferred from the results in Fig. 4 was A3 ≈ B3 > C3 ≈ D3.
Because each branch trisaccharide is present in the M9-type glycan structure, we then examined the competition in mannose trimming when the branch trisaccharides A3, B3, and C3 were added to the same reaction mixture. For the competitive experiment, an internal standard was necessary to calculate the hydrolysis yields from the HPLC chromatograms because the retention times of the mannose trimming products A2, B2, and C2 were identical (Fig. S2†). Because D2 had a different retention time from the other mannose trimming products, we decided to use this compound as an internal standard after confirming that the addition of D2 did not affect the hydrolysis reactions of A3, B3, and C3 (Fig. S3†).
The competitive mannose trimming experiments were conducted in the ER fraction with branch trisaccharides A3, B3, and C3 and the internal standard D2 (Fig. 5). In the competitive experiments, remarkable decreases in the activity were observed for B3 and C3 compared with the individual experiments (Fig. 4), which gave a glycan specificity order of A3 > B3 > C3. We compared the resulting glycan specificity with the glycan trimming profile of ER α-1,2-mannosidases for M9-Gly-BODIPY reported in our previous study,18 which was A-branch ≈ B-branch > C-branch (Fig. 6). The reported specificity (Fig. 6c) was similar to the specificity obtained in the individual experiments (Fig. 4b). These results indicate that the branches of the M9 glycan do not interact with each other. In fact, a foldback conformation in which the B-branch interacts with the core structure (Manβ1-4GlcNAcβ1-4GlcNAc) has been reported for the M9 glycan.36,37 In the competitive experiment shown in Fig. 5, free diffusion of each branch trisaccharide substrate may have caused competition among A3, B3, and C3, leading to a decrease in the cleavage activity for B3 and C3.
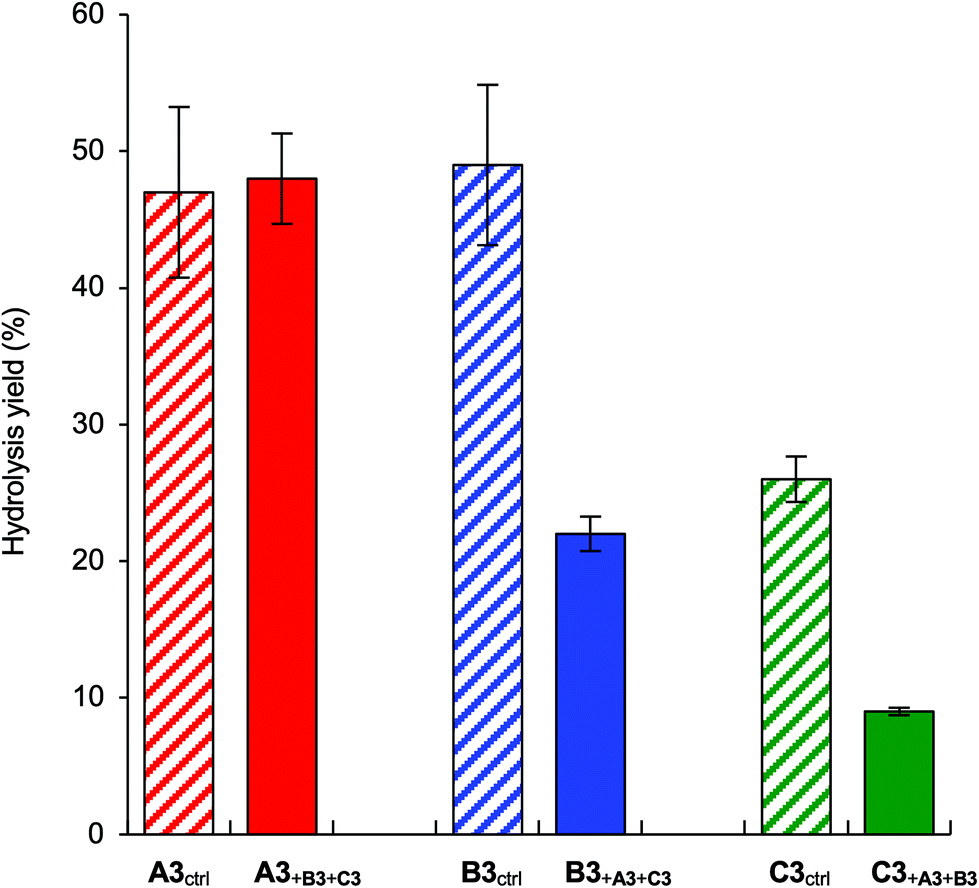 | ||
| Fig. 5 Hydrolysis yields after 6 h from the competitive assay of A3/B3/C3 mixtures (250 μM each) in the ER fraction (3 mg protein per mL). The diagonal bars represent the individual hydrolysis yields of A3, B3, and C3. Each data point represents the mean value with the standard deviation (n = 3). | ||
 | ||
| Fig. 6 Mannose trimming reaction of M9-Gly-BODIPY in the ER fraction. (a) Assay conditions. (b) Structure of M9-Gly-BODIPY. (c) HPLC chromatogram of the reaction mixture showing the mannose profile obtained from ref. 18. | ||
The results in Fig. 5 suggest that the A-branch structure with the inner α-1,2-linkage would be most reactive for ER α-1,2-mannosidases. To confirm this, it is necessary to discuss the reactivity of ER α-1,2-mannosidases with the product A2. As mentioned above, the hydrolysis product A2 can be further cleaved by the enzymes. In fact, ERManI reportedly finally trims Manα1-2Manα1-2Man to a monosaccharide.22 If A2 is recognized by the ER α-1,2-mannosidases responsible for the cleavage of B3 and C3, the apparent cleavage activity of B3 and C3 will be lower than the original activity. These phenomena were not consistent with our working hypothesis. Therefore, we investigated the effect of A2 on the production of disaccharides from each branch trisaccharide.
In a system with D2 added as an internal standard, the hydrolysis yield of A2 can be calculated even if the retention time of the product A1 and the retention time of the component derived from the ER fraction are identical in the HPLC analysis. Using this experimental procedure, we examined whether A2 was cleaved into A1 by ER α-1,2-mannosidases, and found that the trimming activity for A2 was similar to those for A3 and B3 (Fig. 7). Next, the time courses of the hydrolysis yields of A2 and A1 from A3 were analyzed (Fig. 8). In the hydrolysis using A2 as a substrate, the product A1 was observed immediately after the reaction started (Fig. 7). In contrast, when A3 was used as a substrate, the formation of A1 was not observed until 4 h after the reaction started, and approximately 10% was formed after 8 h (Fig. 8). Thus, the cleavage of B3 and C3 shown in Fig. 5 would not be affected by A2. Although the specificities of the ER α-1,2-mannosidases were in the order A3 ≈ B3 > C3 ≈ D3 (Fig. 4), the true specificity seemed to be A3 > B3 > C3 ≈ D3 considering the further cleavage of generated A2.
 | ||
| Fig. 7 Hydrolysis yield of A2 (250 μM) in the ER fraction (3 mg protein per mL). The dashed lines represent the individual hydrolysis profiles of A3, B3, and C3. Each data point represents the mean value with the standard deviation (n = 3). | ||
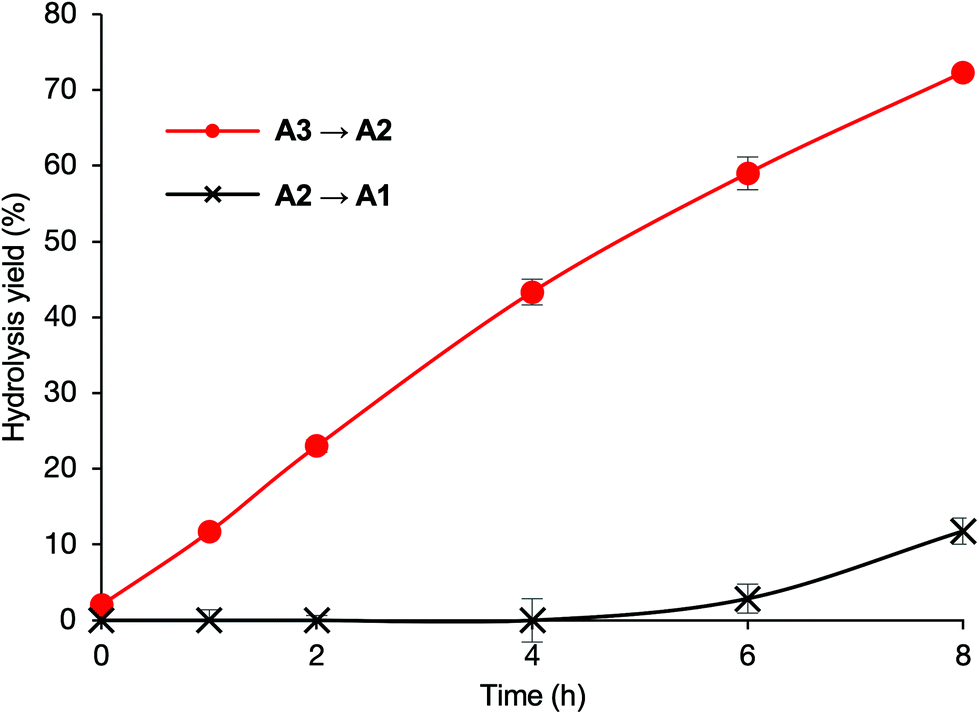 | ||
| Fig. 8 Time course of hydrolysis yields from A3 (250 μM) to A2 and A2 to A1 in the ER fraction (3 mg protein per mL). Each data point represents the mean value with the standard deviation (n = 3). | ||
Next, to clarify the competitive relationship between each branch, we conducted competitive experiments in which two types of synthetic trisaccharides were systematically added to the reaction mixture (Fig. 9). A competitive experiment with A3 and B3 showed relatively reduced activity for B3 (Fig. 9a), and a competitive experiment with A3 and C3 showed reduced activity for C3 (Fig. 9b). In addition, a competitive experiment with B3 and C3 showed relatively reduced activity for C3 (Fig. 9c). According to these results, we concluded that the branch specificities of the ER α-1,2-mannosidases under competitive conditions were in the order A3 > B3 > C3. We further examined the possibility that product inhibition by A2 affected the cleavage activity for B3 and C3 (Fig. 5). When A2 was added to the reaction mixture as shown in Fig. 9c, the cleavage activity for B3 and C3 was not significantly affected (Fig. S4†). This finding shows that the difference in the cleavage activity for each branch observed in this study is not caused by the effect of product inhibition by A2 but by differences in the inner glycosidic linkages. This series of experiments revealed that ER α-1,2-mannosidases could discriminate between endo-glycosidic linkages.
 | ||
| Fig. 9 Hydrolysis yields after 6 h from competitive assays with two-branch trisaccharides. The diagonal bars represent the individual hydrolysis yields of the corresponding trisaccharides. Results of competitive assays with A3 (250 μM) and B3 (250 μM) (a), A3 (250 μM) and C3 (250 μM) (b), and B3 (250 μM) and C3 (250 μM) (c) in the ER fraction (3 mg protein per mL). Each data point represents the mean value with the standard deviation (n = 3). | ||
Finally, we examined the competitive experiments of natural trisaccharides A3, B3, and C3 with unnatural trisaccharide D3 (Fig. 10). On addition of D3, the cleavage activity for A3 and B3 greatly improved compared with that for A3 and B3 without D3 (Fig. 10a and b). This phenomenon can be explained by the fact that D3 acts as a pharmacological chaperone38 to contribute to the restoration of the activity of the partially unfolded ER α-1,2-mannosidases. Pharmacological chaperone is a general term for compounds that weakly interact with enzymes that have been denatured by diseases to promote their refolding and restore enzyme activity. As mentioned above, the ER fraction used in this study was extracted from the senescence-accelerated mouse liver. Because the mouse model showed aging symptoms such as osteoporosis, the activity of the corresponding ER α-1,2-mannosidases might be somewhat reduced. Specifically, these results indicate that D3 might be a lead compound in the development of pharmacological chaperones for ER α-1,2-mannosidases. Conversely, no significant changes in activity were detected in the competitive assay of C3 with D3. This suggests that the enzymes responsible for the cleavage of C3 and D3 are different. Although C3 and D3 may have been cleaved by the same ER α-1,2-mannosidase, as shown in Fig. 4, the result in Fig. 10c revealed that both trisaccharides were cleaved by different enzymes. Overall, we speculate that there are two categories of α-1,2-mannosidases in the ER: (a) the enzymes responsible for the cleavage of A3 and B3 are sensitive to the enhancement by D3 and (b) the enzyme responsible for the cleavage of C3 is barely affected by D3.
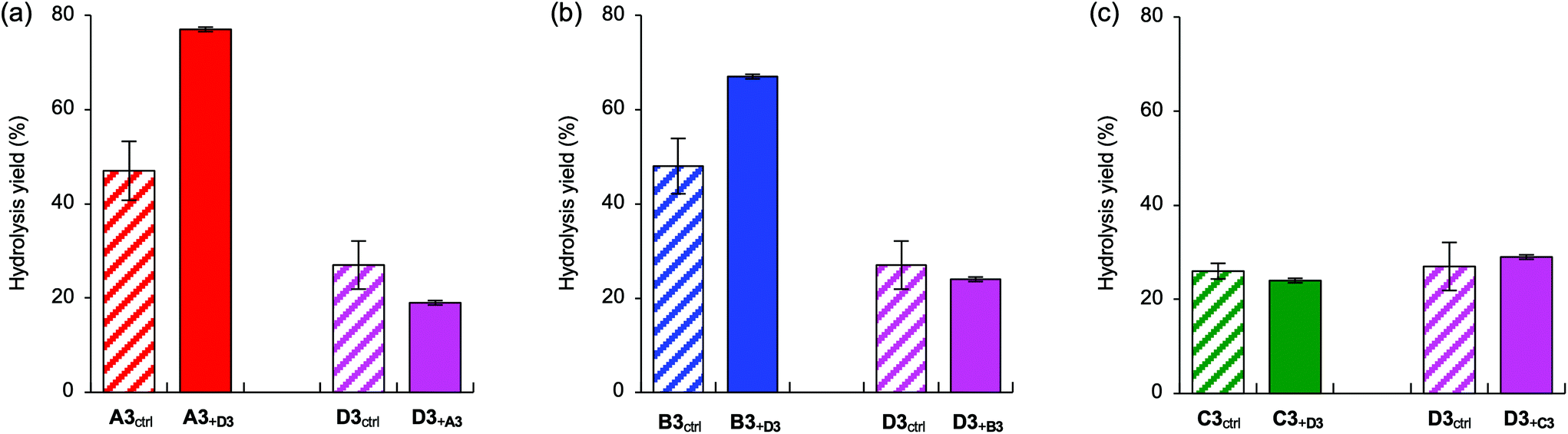 | ||
| Fig. 10 Hydrolysis yields after 6 h from competitive assays of natural A3, B3, or C3 branches (250 μM) with the unnatural D3 branch (250 μM) in the ER faction (3 mg protein per mL). The diagonal bars represent the individual hydrolysis yields of the corresponding trisaccharides. (a) Competitive assay of A3 with D3. (b) Competitive assay of B3 with D3. (c) Competitive assay of C3 with D3. Each data point represents the mean value with the standard deviation (n = 3). | ||
Conclusions
In this study, we synthesized a series of trimannosides with different inner glycosidic linkages, which corresponded to the three branched chains of a high-mannose type glycan. The resulting trimannosides were applied to analyze the branch discrimination abilities of ER α-1,2-mannosidases. Various competitive experiments using synthetic trimannosides and the mouse liver ER fraction revealed that some ER α-1,2-mannosidases could discriminate endo-glycosidic linkages. In the synthesis of trimannoside substrates A3, B3, C3, and D3, glycosyl donors and acceptors were efficiently prepared from a common starting material. Moreover, the stereoselectivities of glycosylation reactions were optimized by tuning the solvent polarity and/or adding a mild base. In glycan specificity analysis of the ER α-1,2-mannosidases, a branch specificity order of A3 > B3 > C3 was found by a combination of the results from individual cleavage assays of A3, B3, and C3, competitive cleavage assays of a mixture of A3 and B3 with C3, and competitive cleavage assays of A3 with B3, A3 with C3, and B3 with C3. This is the first experimental demonstration that ER α-1,2-mannosidases can discriminate inner glycosidic linkages in the cleaved outer Manα1-2Man linkage. Competitive assays of D3 with A3, B3, or C3 provided preliminary insight into the pharmacological chaperone activity of D3 trisaccharide.Although the enzyme responsible for the cleavage of each branch has not been identified in this study, the discovery of the ability of ER α-1,2-mannonsidases to discriminate inner glycosidic linkages will enable the identification of the responsible enzyme in future research. For instance, the responsible enzyme may be identified by comparing the mannose trimming of trisaccharides synthesized in this study by ER fractions extracted from single knockout cells of each ER α-1,2-mannosidase. Our findings will also open the door for the manipulation of glycoprotein secretion/degradation signals with selective inhibitors based on the synthetic trisaccharides A3, B3 and C3 as lead compounds. Thus, the results of this study are expected to lead to advanced research to obtain a precise understanding of the complementary functions of ER α-1,2-mannosidases in glycoprotein quality control and the related protein-folding diseases.39 Further studies are in progress along this line and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (JSPS KAKENHI) (grant numbers JP16H06290 and JP16K01938). We thank Dr Yukishige Ito for helpful discussions.Notes and references
- R. E. Dempski and B. Imperiali, Curr. Opin. Struct. Biol., 2002, 6, 844–850 CrossRef CAS.
- P. A. Romero, G. J. Dijlgraaf, S. Shahinian, A. Herscovics and H. Bussey, Glycobiology, 1997, 7, 997–1004 CrossRef CAS PubMed.
- E. S. Trombetta, J. F. Simons and A. Helenius, J. Biol. Chem., 1996, 271, 27509–27516 CrossRef CAS PubMed.
- K. Totani, Y. Ihara, I. Matsuo and Y. Ito, J. Biol. Chem., 2006, 281, 31502–31508 CAS.
- F. E. Ware, A. Vassilakos, P. A. Peterson, M. R. Jackson, M. A. Lehrman and D. B. Williams, J. Biol. Chem., 1995, 270, 4697–4704 CrossRef CAS PubMed.
- K. H. Krause and M. Michalak, Cell, 1997, 88, 439–443 CrossRef CAS PubMed.
- M. Hirano, Y. Adachi, Y. Ito and K. Totani, Biochem. Biophys. Res. Commun., 2015, 466, 350–355 CrossRef CAS PubMed.
- J. J. Caramelo, O. A. Castro, L. G. Alonso, G. de Prat-Gay and A. J. Parodi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 86–91 CrossRef CAS PubMed.
- K. Totani, Y. Ihara, I. Matsuo, H. Koshino and Y. Ito, Angew. Chem., Int. Ed., 2005, 44, 7950–7954 CrossRef CAS PubMed.
- T. Kudo, M. Hirano, T. Ishihara, S. Shimura and K. Totani, Bioorg. Med. Chem. Lett., 2014, 24, 5563–5567 CrossRef CAS PubMed.
- J. J. Caramelo and A. J. Parodi, J. Biol. Chem., 2008, 283, 10221–10225 CrossRef CAS PubMed.
- T. Kuribara and K. Totani, Curr. Opin. Struct. Biol., 2021, 68, 41–47 CrossRef CAS PubMed.
- T. Suzuki and H. Fujihira, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2020, B978-0-12-409547-2.14947-9 Search PubMed.
- D. S. Gonzalez, K. Karaveg, A. S. Vandersall-Nairn, A. Lal and K. W. Moremen, J. Biol. Chem., 1999, 274, 21375–21386 CrossRef CAS PubMed.
- N. Hosokawa, I. Wada, K. Hasegawa, T. Yorihuzi, L. O. Tremblay, A. Herscovics and K. Nagata, EMBO Rep., 2001, 2, 415–422 CrossRef CAS PubMed.
- S. Olivari, G. Carmela, H. Alanen, L. Ruddock and M. Molinari, J. Biol. Chem., 2005, 280, 2424–2428 CrossRef CAS PubMed.
- K. Hirao, Y. Natsuka, T. Tamura, I. Wada, D. Morito, S. Natsuka, P. Romero, B. Sleno, L. O. Tremblay, A. Herscovics and K. Nagata, J. Biol. Chem., 2006, 281, 9650–9658 CrossRef CAS PubMed.
- T. Kuribara, M. Hirano, G. Speciale, S. J. Williams, Y. Ito and K. Totani, ChemBioChem, 2017, 18, 1027–1035 CrossRef CAS PubMed.
- T. Kuribara, Trends Glycosci. Glycotechnol., 2020, 32, E31–E34 CrossRef.
- Y. Kamiya, D. Kamiya, K. Yamamoto, B. Nyfeler, H.-P. Hauri and K. Kato, J. Biol. Chem., 2008, 283, 1857–1861 CrossRef CAS PubMed.
- N. Hosokawa, Y. Kamiya, D. Kamiya, K. Kato and K. Nagata, J. Biol. Chem., 2009, 284, 17061–17068 CrossRef CAS PubMed.
- J. Aikawa, I. Matsuo and Y. Ito, Glycoconjugate J., 2012, 29, 35–45 CrossRef CAS PubMed.
- S. Ninagawa, T. Okada, Y. Sumitomo, Y. Kamiya, K. Kato, S. Horimoto, T. Ishikawa, S. Takeda, T. Sakuma, T. Yamamoto and K. Mori, J. Cell Biol., 2014, 206, 347–356 CrossRef CAS PubMed.
- S. Yu, S. Ito, I. Wada and N. Hosokawa, J. Biol. Chem., 2018, 293, 10663–10674 CrossRef CAS PubMed.
- M. Shenkman, E. Ron, R. Yehuda, R. Benyair, I. Khalaila and G. Z. Lederkremer, Commun. Biol., 2018, 1, 172 CrossRef PubMed.
- G. George, S. Ninagawa, H. Yagi, T. Saito, T. Ishikawa, T. Sakuma, T. Yamamoto, K. Imami, Y. Ishihama, K. Kato, T. Okada and K. Mori, eLife, 2020, 9, e53455 CrossRef PubMed.
- T. Ogawa and H. Yamamoto, Carbohydr. Res., 1982, 104, 271–283 CrossRef CAS.
- N. Bovin and A. Chinarev, US Pat, US20080076137A1, 2008 Search PubMed.
- I. Cumpstey, A. J. Fairbanks and A. J. Redgrave, Org. Lett., 2001, 3, 2371–2374 CrossRef CAS PubMed.
- A. A. Hettikankanamalage, R. Lassfolk, F. S. Ekholm, R. Leino and D. Crich, Chem. Rev., 2020, 120, 7104–7151 CrossRef CAS PubMed.
- H. M. Christensen, S. Oscarson and H. H. Jensen, Carbohydr. Res., 2015, 408, 51–95 CrossRef CAS PubMed.
- K. Bock and C. A. Pedersen, Acta Chem. Scand., Ser. B, 1975, 29, 258–264 CrossRef.
- K. Takahashi, T. Tsuboyama, M. Matsushita, R. Kasai, H. Okumura, T. Yamamuro, Y. Okamoto, K. Toriyama, K. Kitagawa and T. Takeda, Bone Miner., 1994, 24, 245–255 CrossRef CAS.
- S. Iwamoto, M. Isoyama, M. Hirano, K. Yamaya, Y. Ito, I. Matsuo and K. Totani, Glycobiology, 2013, 23, 121–131 CrossRef CAS PubMed.
- A. Lal, P. Pang, S. Kalelkar, P. A. Romero, A. Herscovics and K. W. Moremen, Glycobiology, 1998, 8, 981–995 CrossRef CAS PubMed.
- Y. Kamiya, K. Yanagi, T. Kitajima, T. Yamaguchi, Y. Chiba and K. Kato, Biomolecules, 2013, 3, 108–123 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Sakae, Y. Zhang, S. Yamamoto, Y. Okamoto and K. Kato, Angew. Chem., Int. Ed., 2014, 53, 10941–10944 CrossRef CAS PubMed.
- V. Bernier, M. Lagacé, D. G. Bichet and M. Bouvier, Trends Endocrinol. Metab., 2004, 15, 222–228 CrossRef CAS PubMed.
- M. Wang and R. J. Kaufman, Nature, 2016, 529, 326–335 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures (Fig. S1–S3), details of synthesis, characterization of new compounds, and methods for enzyme assays. See DOI: 10.1039/d1ob00428j |
| This journal is © The Royal Society of Chemistry 2021 |