
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the binding properties of phosphorylated glycoluril-derived molecular tweezers and selective nanomolar binding of natural polyamines in aqueous solution†
Michael
Heilmann‡
 a,
Melina
Knezevic
a,
GiovanniMaria
Piccini
b and
Konrad
Tiefenbacher
*ac
a,
Melina
Knezevic
a,
GiovanniMaria
Piccini
b and
Konrad
Tiefenbacher
*ac
aUniversity of Basel, Department of Chemistry, Mattenstrasse 24a, 4058 Basel, Switzerland. E-mail: konrad.tiefenbacher@unibas.ch; tkonrad@ethz.ch
bETH Zurich, Department of Chemistry and Applied Biosciences, c/o USI campus, Via Guiseppe Buffi 13, 6900 Lugano, Switzerland
cETH Zurich, Department of Biosystems Science and Engineering, Mattenstrasse 26, 4058 Basel, Switzerland
First published on 30th March 2021
Abstract
A modular synthetic platform for the construction of flexible glycoluril-derived molecular tweezers was developed. The binding properties of four exemplary supramolecular hosts obtained via this approach towards 16 organic amines were investigated by means of 1H NMR titration. In this work, we compare the Ka values obtained this way with those of three structurally related molecular tweezers and provide a computational approach towards an explanation of the observed behavior of those novel hosts. The results showcase that certain structural modifications lead to very potent and selective binders of natural polyamines, with observed binding of spermine below 10 nM.
Introduction
Over the past decades, the growing understanding of intermolecular processes has set the foundation for studying molecular recognition, both in biological and artificial systems, in an increasingly exact fashion.1 Numerous synthetic supramolecular host structures with different sizes, shapes and functionalization patterns have been shown to interact with a multitude of classes of guest molecules.2 Among those are biologically highly relevant targets, including proteins, drugs or drug-like molecules or biogenic small molecules.3 One class of such constructs called molecular tweezers is defined as U-shaped molecules with aromatic panels that are connected with rigid linkers.4 Several different frameworks for such tweezers have been shown suitable to accommodate a guest molecule within the pre-organized cavity.2b,4h,k,5 Importantly, by deliberate decoration of a hydrophobic core structure with solubilizing groups like phosphates, phosphonates, or carboxylates, such tweezers can be adapted to bind organic guests in aqueous solution.6 For instance, the phosphate derivative of tweezer 1 (Fig. 1A, R = O−) has been shown to be remarkably selective for lysine and arginine derivatives and was even successfully employed to tightly bind these moieties in oligopeptides and on protein surfaces.7 However, its structural design limits the possibilities of facile derivatization. On the other hand, the glycoluril building block is a well-established subunit for an entire family of acyclic tweezers of varying size, shape and flexibility that have been utilized as hosts for a broad range of viable guest molecules.3d One example is host 2, which is very similar to 1 in size and shape of its cavity, but has a higher level of flexibility due to the lack of rigid bicyclic linkers.6b,9 Inspired by these contributions and motivated by our group's interest in catalysis inside supramolecular containers and within molecular tweezers,10 we sought to develop a modular and derivatization-friendly platform for the synthesis of molecular tweezers. Ultimately, we envision this to enable the selective binding and modification of biologically relevant targets. We recently showed that the conformationally flexible phosphorylated host 3 binds very tightly to numerous sterically undemanding diamine guests.8 Guided by these results, we have designed a streamlined synthetic procedure for tweezers of the general structure 4 with the goal of understanding host–guest interactions of these hosts in more detail. Here, we report a comparative study of the binding properties of four derivatives of the general structure 4 and the previously described tweezers 1, 2 and 3.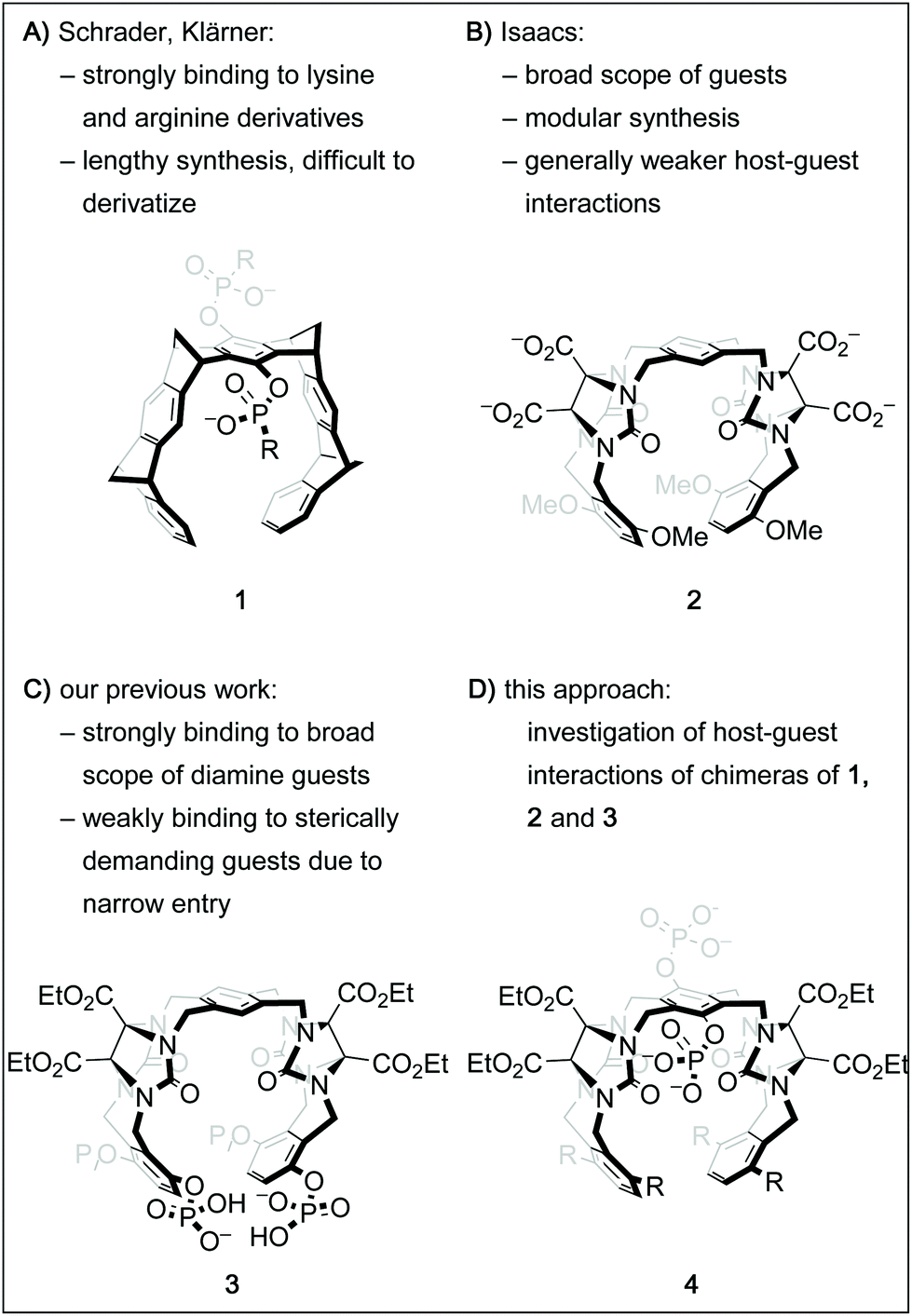 | ||
| Fig. 1 Chemical structures of molecular tweezer 16a and the similarly sized tweezer 2.6b Tweezer 38 shares the framework with 2 but the phosphate groups attached to it provide unique binding properties. Tweezers of the general structure 4 were synthesized with the goal of having access to a broad range of chimeras via a modular platform. | ||
Results and discussion
For this study, we chose to synthesize and investigate the following derivatives (Scheme 1): benzo-tipped tweezer 4a which closely resembles 1; naphtho-tipped tweezer 4b which is related to 4a but features a larger cavity due to increased angular distortion resulting from repulsion of the larger tips; dimethoxybenzo-tipped tweezer 4c which shares features of both 1 and 2; and hexaphosphorylated tweezer 4d which serves as a chimera of 3 and 4a. The synthetic strategy of tweezers 4a–d is based on the convergent alkylation of the respective glycoluril derivatives 7a–d′ with tetrabromide 6 (Scheme 1). Tetrabromide 6 is accessible from commercially available duroquinone (5) in three steps. After a zinc mediated reduction11 and phosphorylation employing sodium hydride and diethyl chlorophosphate, fourfold benzylic bromination delivered 6 in good yield (55% over three steps). While the preparation of the partially substituted glycoluril derivatives 7a–d′ was easily accomplished by alkylation of the parent unsubstituted glycoluril with the corresponding o-xylylene dibromides (see ESI† for details), the direct construction of the final frameworks 8a–d′ proved to be a highly challenging transformation. Numerous attempts of employing different inorganic and organic bases in a variety of solvents led to virtually no conversion at room temperature, likely due to the steric congestion of electrophile 6, and resulted in decomposition without formation of 8 when heated. The use of differently protected derivatives of 6, including several alkyl, acetal, or acyl protecting groups, did not resolve the issue. Attempts to use different leaving groups instead of bromides as in 6, including readily accessible chlorides and acetates, were not successful either.12 Gratifyingly, trace product formation was observed when we subjected 6 and 7a to the weak inorganic base potassium carbonate and sodium iodide at 40 °C. Further optimization showed that using catalytic amounts of tetramethylammonium iodide and cesium carbonate smoothly gave the desired compounds in good yields. Unlike in our previously presented synthesis of 38 but also observed in the synthesis of 2,6b mixtures of the “C-shaped” 8a–d′ and their undesired “S-shaped” diastereomers were obtained that necessitated separation via HPLC (see ESI†). Subsequent deprotection of the obtained diastereopure diethylphosphates by addition of bromotrimethylsilane followed by ethanol furnished the desired phosphate tweezers 4a–d in excellent yields.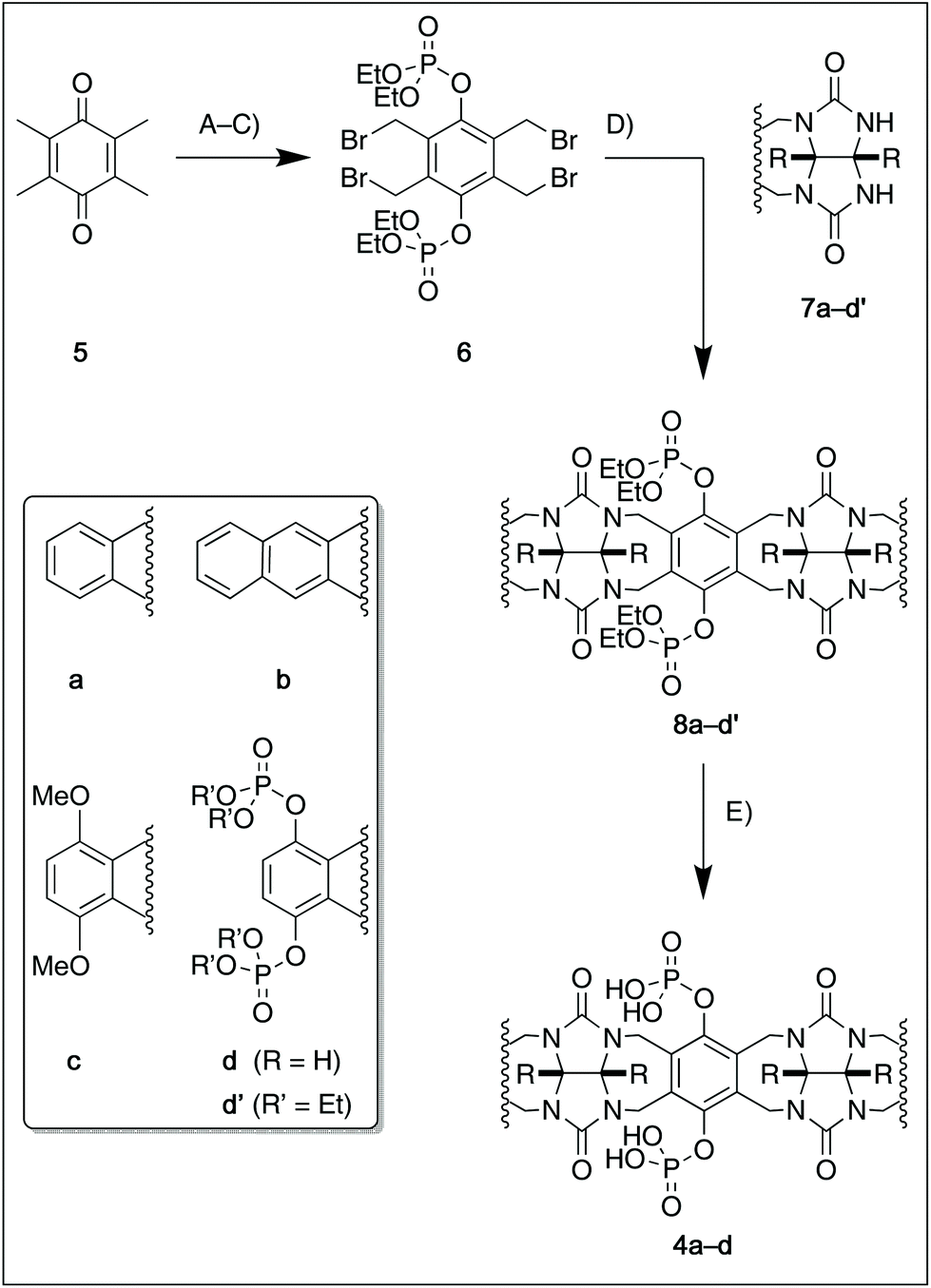 | ||
| Scheme 1 Synthesis of tweezers 4a–d. R = CO2Et. (A) Zn, AcOH, 130 °C, 60 min. (B) NaH, (EtO)2P(O)Cl, DMF, 0 °C, 18 h, 64% (2 steps). (C) NBS, AIBN, CCl4, 95 °C, 16 h, 87%. (D) 6, 7 (2.0 equiv.), Cs2CO3, TMAI, 35–58% (dr = 1.3/1 … 1.9/1). (E) TMSBr, 0 °C, 24 h then EtOH, rt, 60 min, 82–88%. NBS: N-Bromosuccinimide, AIBN: azobis-iso-butyronitrile, TMAI: tetramethylammonium iodide, TMS: trimethylsilyl. | ||
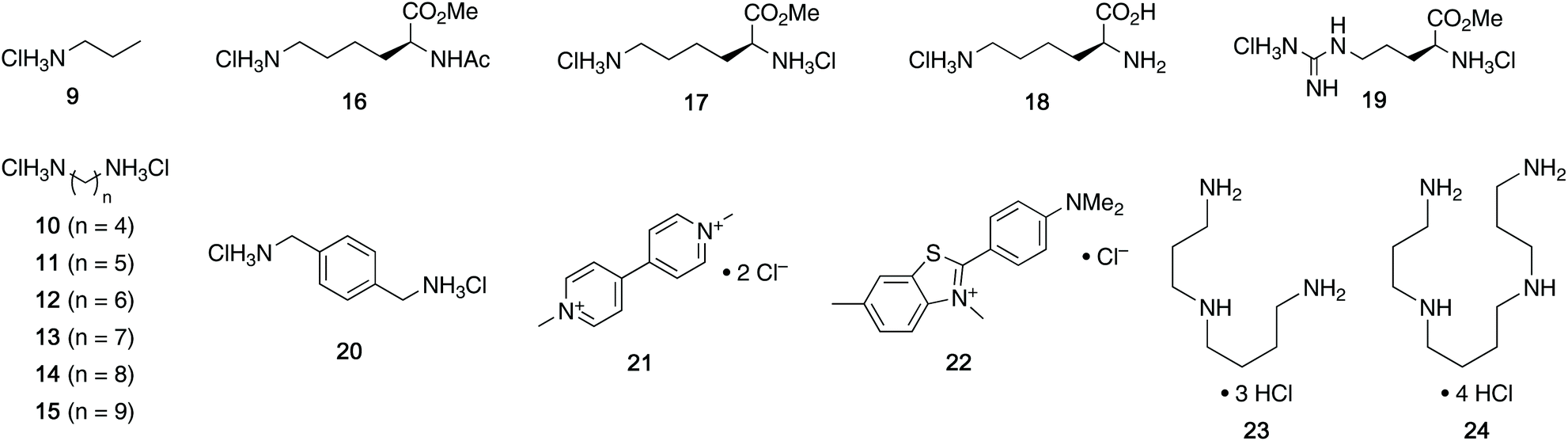
With the desired tweezers in hand, we started investigating their binding properties by means of 1H NMR titration in buffered D2O (70 mM phosphate buffer, pD = 7.2), and determining the equilibrium constants via non-linear regression.13 We found that 4a and 4d undergo weak dimerization (Kdim = 32.7 M−1 and 35.0 M−1, respectively), while this process is considerably more pronounced in 4b (773 M−1) and 4c (624 M−1), with the orders of magnitude in good agreement with comparable systems.6a,7a,9b Job plots indicated that these tweezers bind their guests in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 fashion (see ESI†). Next, the guest scope was explored in detail. Table 1 summarizes the binding constants (Ka) of complexes of 16 guests and the four tweezers 4a–d as well as available literature values of tweezers 1–3 for comparison. Similar to previous findings with 2 and 3, 4a and 4c bind short aliphatic monoamine compounds like propylammonium chloride (9) relatively weakly, and 4b did not show significant uptake of such short-chained diammonium guests. The stronger affinity of 4d to 9 is in agreement with the stronger ionic interactions facilitated by the six phosphate moieties in 4d as compared to two in 1 and 4a-c. Interestingly, the binding properties of tweezers 3 and 4a–d did not change substantially when we investigated the same equilibrium in unbuffered D2O, although we observed diminished solubility of 4a–d, likely due to stronger self-aggregation in this solvent.6b As expected, we were not able to observe any binding in control titration experiments of the exemplary S-shaped deprotected construct dia-4c; in agreement with the exclusive accommodation of the aliphatic chains of the guests in the tweezers’ C-shaped cavity.
:1 fashion (see ESI†). Next, the guest scope was explored in detail. Table 1 summarizes the binding constants (Ka) of complexes of 16 guests and the four tweezers 4a–d as well as available literature values of tweezers 1–3 for comparison. Similar to previous findings with 2 and 3, 4a and 4c bind short aliphatic monoamine compounds like propylammonium chloride (9) relatively weakly, and 4b did not show significant uptake of such short-chained diammonium guests. The stronger affinity of 4d to 9 is in agreement with the stronger ionic interactions facilitated by the six phosphate moieties in 4d as compared to two in 1 and 4a-c. Interestingly, the binding properties of tweezers 3 and 4a–d did not change substantially when we investigated the same equilibrium in unbuffered D2O, although we observed diminished solubility of 4a–d, likely due to stronger self-aggregation in this solvent.6b As expected, we were not able to observe any binding in control titration experiments of the exemplary S-shaped deprotected construct dia-4c; in agreement with the exclusive accommodation of the aliphatic chains of the guests in the tweezers’ C-shaped cavity.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Guest |
|
|
|
|
Related tweezers | ||
| Schrader 1 (Ka/M−1)c | Isaacs 2 (Ka/M−1)e | Our prev. work 3 (Ka/M−1)f | |||||
| a Titration performed in unbuffered D2O. b Determined via competitive displacement titration at 100 μM 4d and 10 mM or 200 mM 21 as a competitor. c Values for the phosphate derivative of tweezer 1 as determined by Klärner & Schrader.6a d Value for the methylphosphonate derivative of tweezer 1 as determined by Klärner & Schrader.14 e Values for tweezer 2 as determined by Isaacs.6b f Values for tweezer 3 as determined in our previous work.8 | |||||||
| 9 | 138 | <10 | 88.7 | 3310 | 105 | 345 | |
| 9a | 208 | <10 | 275 | 4260 | 890d | 295 | |
| 10 | 35.8 | <10 | <10 | 26100 |
724 | 10700 |
|
| 11 | 247 | 83.5 | 377 | 287000 |
5960 | 122000 |
|
| 12 | 569 | 354 | 1170 | 148000 |
15200 |
133000 |
|
| 13 | 838 | 870 | 2580 | 49600 |
6460 | 155000 |
|
| 14 | 517 | 1200 | 1900 | 14300 |
7060 | 179000 |
|
| 15 | 240 | 831 | 861 | 3150 | 36300 |
||
| 16 | <10 | <10 | <10 | 28.3 | 58800 |
<10 | |
| 17 | 88.0 | <10 | 23.0 | 1430 | 4220 | ||
| 18 | 32.1 | <10 | <10 | 30.8 | 47600 |
57.1 | |
| 19 | 14.3 | <10.0 | 14.8 | 1260 | 253 | ||
| 20 | 26.2 | 103 | 99.2 | 18800 |
496 | 33800 |
|
| 21 | 144 | 1750 | 130.4 | 27100 |
2060 | 21600 |
|
| 22 | 147 | 585 | 175 | <10 | 54.0 | ||
| 23 | 200 | 45 | 94.4 | 2370000b |
388000 |
||
| 24 | 815 | <10 | 325 | 148000000b |
7920000 |
||
We next investigated the dependency of aliphatic diamine guest binding on the length of their methylene linkers. As expected, we found moderate-length guests to be optimal, the size of which suit the tweezers’ cavities most closely (C7-diammonium chloride 13 for 4a and 4c, C8-diammonium chloride 14 for the larger host 4b). However, the binding of diammonium guests in 4a–c is weak compared to the derivatives 4d and 3.8 Interestingly, the values for 10–14@4c and 10–14@2 suggest that the phosphates present in 4c but missing in 2 do not increase but even decrease binding.6b This observation was surprising as the added phosphates were expected to increase binding substantially due to strong ion–ion interactions. We interpret these results as an indication that the central phosphate moieties destabilize the desired tweezer conformation, potentially favouring more open conformations. The conformational flexibility of the related host 2 was described by Isaacs and co-workers previously.6b While 4d proved to be a much stronger binder than 4a–c, a similar observation was made: the additional phosphate moieties in 4d compared to 3 appeared to offer a limited contribution to the binding of diamine guests 10–15, although a stronger preference for shorter methylene linkers and reduced affinity for longer-chained guests was observed (maximum with C5-diammonium chloride 11 for 4d and maximum with C8-diammonium chloride 14 for 3).8
We were also interested in the formation of complexes of the novel tweezers and basic amino acid derivatives. Very little interaction was observed between tweezer 4a and the investigated lysine derivatives Ac-Lys-OMe (16, Ka < 10 M−1), H-Lys-OMe (17, 88.0 M−1) and H-Lys-OH (18, 32.1 M−1) as well as arginine derivative H-Arg-OMe (19, 14.3 M−1). These results were surprising, since the size and shape of the cavity of 4a is very similar to that of 1, which binds lysine derivatives very tightly,6a but are in good agreement with the previous observation of comparably poor binding in 3. It is important to note that 4a has a much more flexible framework than 1 and that it can populate different non-active conformations besides the desired “tweezer-like” conformation by means of rotation around the methylene bridges (vide infra). Similar to 4a, interactions between amino acid-derived guests 16–19 and hosts 4b and 4c are weak. The binding properties of 4d towards these guests are very similar to those of 3, which is in good agreement with the finding that the central phosphate moieties do not contribute to binding.
When we investigated (hetero-)aromatic guests, we found that the association constants for the complexes of 4d with p-xylylenediammonium chloride (20, Ka = 1.88 × 104 M−1) and methyl viologen chloride (21, 2.71 × 104 M−1) are similar to the values for 20@3 and 21@3 (3.88 × 104 M−1 and 2.16 × 104 M−1, respectively). Additionally, both 4d and 3 show no or very weak interaction with the sterically more demanding Thioflavin T (22). This further showcases that the installation of additional phosphate moieties at the central aromatic unit does not generally increase binding. Given this observation, we were not surprised to see relatively weak binding of 20 and 21 with 4a–c. Interestingly, especially 20 and 21, both comparably large aromatic cations, showed stronger binding to naphtho-tipped host 4b (Ka = 1.75 × 103 M−1 and 5.85 × 102 M−1, respectively) which displays larger aromatic surfaces than 4a and 4c.
We did, however, observe significantly stronger binding of the natural polyamines spermidine (23) and spermine (24) by the hexaphosphorylated host 4d (Ka = 2.37 × 106 M−1 for 23@4d and 1.48 × 108 M−1 for 24@4) than the tetraphosphorylated host 3 whereas 4a–c interact only weakly with those guests. This particularly strong interaction is consistent with our observation that the additional phosphate moieties in 4d compared to 3 were most advantageous for binding of diamines with relatively short aliphatic linkers. Given that the polyamine metabolism and homeostasis is strongly regulated in humans and elevated levels are linked to excessive cell proliferation and also neurodegenerative diseases, these are viewed as potential handle for future treatments.15 Therefore, the nanomolar binding of these guests (422 nM for spermidine and 6.76 nM for spermine) in aqueous buffer is remarkable. Furthermore, while comparable affinities to spermidine and spermine have been reported for synthetic supramolecular hosts before,9a,16 the level of selectivity for spermidine (23) and especially spermine (24) over their parent biosynthetic precursor putrescine (10) is substantially higher for 4d compared to previously reported hosts.
In order to better understand the guest binding capabilities of the new hosts 4a–d, we performed MD simulations of the conformational space of hosts 2 and 4c in water (Fig. 2B and C, for detailed methodology see ESI†). The conformational changes in molecular tweezers are similar to those encountered in the folding process of small proteins.17 However, the symmetric structure of the tweezers as well as their well-defined local conformations allows a simpler yet more efficient definition of their overall conformational state. In this case, ten different conformers are possible with regard to the four pairs of methylene bridges in these hosts, defining the correlation between the units that are linked by the methylene bridges either as syn (s, when the substituent at the glycoluril backbone and the aromatic unit are oriented towards each other) or anti (a, when they are oriented away from each other).18 We found that while in both 2 and 4c the s,a,a,s conformation is the most favored one, the free energy difference between this state and the “active” a,a,a,a conformation is lower in 2 (≈−9 kJ mol−1) than in 4c (≈−12 kJ mol−1). This indicates that the active tweezer conformation is more accessible in solution for tweezer 2 than for 4c. Furthermore, the energy barriers between the different conformations are much higher in 4c than in 2, indicating that interconversion between different conformers happens at lower rates in 4c than in 2. As a result, and in agreement with our observations in the binding studies with hosts 4a–d, installation of phosphate groups at the central aromatic unit apparently destabilizes the desired tweezer conformation in favor of more open conformations that do not bind guest molecules efficiently. These results clarify the surprising finding that central phosphate groups do not generally increase binding although additional ion–ion interactions with cationic guests would become available. The binding of spermidine (23) and particularly spermine (24) to the hexaphosphate tweezer 4d is the exception identified. We assume that the increased ion–ion interactions between the triply/quadruply charged guest and the hexaphosphate tweezer 4d are overriding other factors in these two cases.
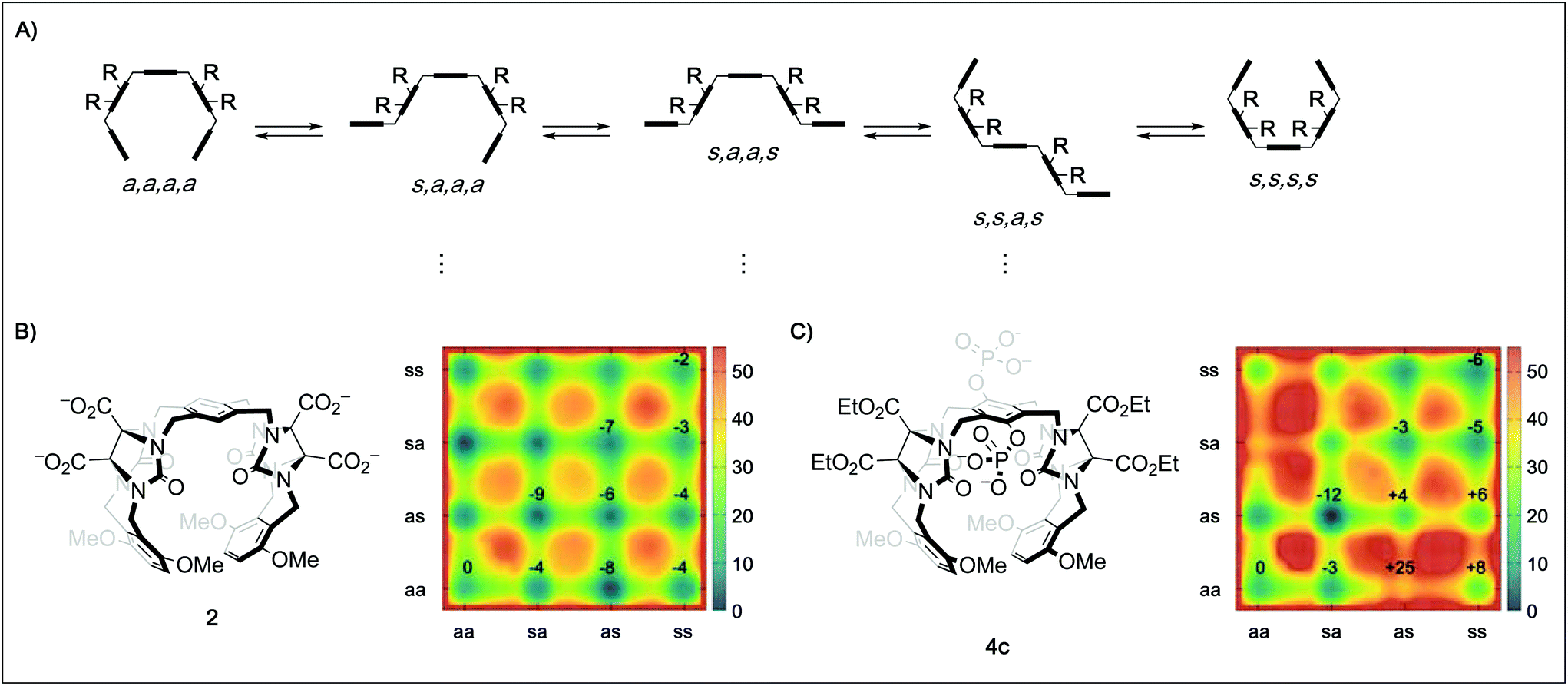 | ||
| Fig. 2 (A) Schematic depiction of five of the ten possible conformers of a tweezer with the general framework of 2, 3, and 4 by rotation around the four pairs of methylene bridges using the nomenclature defined by Nolte.18 (B) Metadynamics estimation of the free energy surface (kJ mol−1) for the conformers of 2 in water reveal a global minimum for the s,a,a,s state. (C) Metadynamics estimation of the free energy surface (kJ mol−1) for the conformers of 4c in water also reveal a global minimum for the s,a,a,s state, but a much less favored “active” a,a,a,a state as well as higher energy barriers between the different conformations. Values given in the surface plots are relative to the a,a,a,a conformer. | ||
Conclusions
In summary, we have developed a modular synthetic platform for phosphorylated molecular tweezers based on glycoluril linker units of the general structure 4 and were able to showcase the synthetic versatility by synthesizing the four derivatives 4a–d with diverse tips. The comparative study of those and related tweezers shows that the installation of phosphate groups at the central aromatic unit of the tweezers did not generally improve binding of ammonium guests. Comparison of the binding properties of 4a and 1, 4c and 2 as well as 4d and 3 together with MD simulations of the conformational space of 4c and 2 suggest that these central phosphate groups substantially reduce the stability of the active tweezer conformation and result in more open conformations in which the hosts cannot interact with guest efficiently in most cases. Nevertheless, in the case of host 4d, the central phosphate groups appear to strongly contribute to the binding of natural polyamines in particular, resulting in nanomolar affinity (422 nM for spermidine and 6.76 nM for spermine) in aqueous solution. Together, these results grant an understanding of the impact of substituents on the binding properties depending on their location in the framework of this class of molecular tweezers. This combination of insights will guide efforts towards further generations of optimized flexible molecular tweezers. The short and highly modular synthetic route will enable the facile construction of future customized tweezer hosts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation as part of the NCCR Molecular Systems Engineering program. M. H. acknowledges Dr Michael Pfeffer for HR-MS analysis and Dr Daniel Häussinger for support in NMR titration experiments. Dr GiovanniMaria Piccini performed the MD simulation on the ETH Euler cluster.Notes and references
- (a) E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed; (b) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- (a) J. Rebek, Science, 1987, 235, 1478 CrossRef CAS PubMed; (b) A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Acc. Chem. Res., 1999, 32, 995–1006 CrossRef CAS; (c) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed; (d) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC; (e) H.-J. Schneider and A. K. Yatsimirsky, Chem. Soc. Rev., 2008, 37, 263–277 RSC; (f) D. Ajami, L. Liu and J. Rebek Jr., Chem. Soc. Rev., 2015, 44, 490–499 RSC.
- (a) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC; (b) X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed; (c) S. van Dun, C. Ottmann, L.-G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960–13968 CrossRef CAS PubMed; (d) S. Ganapati and L. Isaacs, Isr. J. Chem., 2018, 58, 250–263 CrossRef CAS PubMed.
- (a) S. C. Zimmerman, in Supramolecular Chemistry I—Directed Synthesis and Molecular Recognition, Springer Berlin Heidelberg, Berlin, Heidelberg, 1993 Search PubMed; (b) F.-G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef PubMed; (c) M. Hardouin-Lerouge, P. Hudhomme and M. Sallé, Chem. Soc. Rev., 2011, 40, 30–43 RSC; (d) M. Hardouin-Lerouge, P. Hudhomme and M. Salle, Chem. Soc. Rev., 2011, 40, 30–43 RSC; (e) F.-G. Klärner and T. Schrader, Acc. Chem. Res., 2013, 46, 967–978 CrossRef PubMed; (f) V. Valderrey, G. Aragay and P. Ballester, Coord. Chem. Rev., 2014, 258–259, 137–156 CrossRef CAS; (g) S. C. Zimmerman, Beilstein J. Org. Chem., 2016, 12, 125–138 CrossRef CAS PubMed; (h) C. W. Chen and H. W. Whitlock, J. Am. Chem. Soc., 1978, 100, 4921–4922 CrossRef CAS; (i) J. Rebek, B. Askew, N. Islam, M. Killoran, D. Nemeth and R. Wolak, J. Am. Chem. Soc., 1985, 107, 6736–6738 CrossRef CAS; (j) C. S. Wilcox, L. M. Greer and V. Lynch, J. Am. Chem. Soc., 1987, 109, 1865–1867 CrossRef CAS; (k) S. C. Zimmerman and C. M. VanZyl, J. Am. Chem. Soc., 1987, 109, 7894–7896 CrossRef CAS.
- (a) M. Harmata and C. L. Barnes, J. Am. Chem. Soc., 1990, 112, 5655–5657 CrossRef CAS; (b) H. Kurebayashi, T. Haino, S. Usui and Y. Fukazawa, Tetrahedron, 2001, 57, 8667–8674 CrossRef CAS.
- (a) S. Dutt, C. Wilch, T. Gersthagen, P. Talbiersky, K. Bravo-Rodriguez, M. Hanni, E. Sánchez-García, C. Ochsenfeld, F.-G. Klärner and T. Schrader, J. Org. Chem., 2013, 78, 6721–6734 CrossRef CAS PubMed; (b) C. A. Burnett, D. Witt, J. C. Fettinger and L. Isaacs, J. Org. Chem., 2003, 68, 6184–6191 CrossRef CAS PubMed.
- (a) F.-G. Klärner, B. Kahlert, A. Nellesen, J. Zienau, C. Ochsenfeld and T. Schrader, J. Am. Chem. Soc., 2006, 128, 4831–4841 CrossRef PubMed; (b) P. Talbiersky, F. Bastkowski, F.-G. Klärner and T. Schrader, J. Am. Chem. Soc., 2008, 130, 9824–9828 CrossRef CAS PubMed; (c) D. Bier, R. Rose, K. Bravo-Rodriguez, M. Bartel, J. M. Ramirez-Anguita, S. Dutt, C. Wilch, F.-G. Klärner, E. Sanchez-Garcia, T. Schrader and C. Ottmann, Nat. Chem., 2013, 5, 234 CrossRef CAS PubMed.
- M. Heilmann and K. Tiefenbacher, Chem. – Eur. J., 2019, 25, 12900–12904 CrossRef CAS PubMed.
- (a) D. Ma, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2010, 75, 4786–4795 CrossRef CAS PubMed; (b) N. She, D. Moncelet, L. Gilberg, X. Lu, V. Sindelar, V. Briken and L. Isaacs, Chem. – Eur. J., 2016, 22, 15270–15279 CrossRef CAS PubMed.
- (a) Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed; (b) M. Knezevic, M. Heilmann, G. M. Piccini and K. Tiefenbacher, Angew. Chem., Int. Ed., 2020, 59, 12387–12391 CrossRef CAS PubMed.
- M. Fuchs, Y. Simeo, B. T. Ueberbacher, B. Mautner, T. Netscher and K. Faber, Eur. J. Org. Chem., 2009, 833–840 CrossRef CAS.
- (a) W. J. Burke, J. L. Bishop and J. A. Warburton, J. Org. Chem., 1962, 27, 4003–4005 CrossRef CAS; (b) D. L. Fields, J. B. Miller and D. D. Reynolds, J. Org. Chem., 1964, 29, 2640–2647 CrossRef CAS.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) B. D. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- M. Fokkens, T. Schrader and F.-G. Klärner, J. Am. Chem. Soc., 2005, 127, 14415–14421 CrossRef CAS PubMed.
- (a) R. A. Casero Jr., T. Murray Stewart and A. E. Pegg, Nat. Rev. Cancer, 2018, 18, 681–695 CrossRef PubMed; (b) A. K. Handa, T. Fatima and A. K. Mattoo, Front. Chem., 2018, 6, 10 CrossRef PubMed.
- M. V. Rekharsky, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Supramol. Chem., 2007, 19, 39–46 CrossRef CAS.
- (a) K. Lindorff-Larsen, S. Piana, R. O. Dror and D. E. Shaw, Science, 2011, 334, 517 CrossRef CAS PubMed; (b) D. Mendels, G. Piccini, Z. F. Brotzakis, Y. I. Yang and M. Parrinello, J. Chem. Phys., 2018, 149, 194113 CrossRef PubMed.
- R. P. Sijbesma and R. J. M. Nolte, J. Am. Chem. Soc., 1991, 113, 6695–6696 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00379h |
| ‡ Current address: Merck Center for Catalysis at Princeton University, Princeton, NJ 08544, USA. |
| This journal is © The Royal Society of Chemistry 2021 |