
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-Oxazolone complexes conjugated to an engineered enzyme: improving fluorescence and catalytic properties†
Carla
Garcia-Sanz‡
a,
Alicia
Andreu‡
a,
Blanca
de las Rivas
b,
Ana I.
Jimenez
 c,
Alexandra
Pop
d,
Cristian
Silvestru
d,
Esteban P.
Urriolabeitia
*c and
Jose M.
Palomo
*a
c,
Alexandra
Pop
d,
Cristian
Silvestru
d,
Esteban P.
Urriolabeitia
*c and
Jose M.
Palomo
*a
aDepartment of Biocatalysis, Institute of Catalysis (ICP-CSIC), Marie Curie 2, 28049 Madrid, Spain. E-mail: josempalomo@icp.csic.es
bDepartment of Microbial Biotechnology, Institute of Food Science, Technology and Nutrition (ICTAN-CSIC), José Antonio Novais 10, 28040 Madrid, Spain
cInstituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: esteban@unizar.es
dSupramolecular Organic and Organometallic Chemistry Centre, Department of Chemistry, Babeş-Bolyai University, Str. Arany Janos 11, RO-400028 Cluj-Napoca, Romania
First published on 2nd March 2021
Abstract
Different Pd-complexes containing orthometallated push–pull oxazolones were inserted by supramolecular Pd-amino acid coordination on two genetically engineered modified variants of the thermoalkalophilic Geobacillus thermocatenolatus lipase (GTL). Pd-lipase conjugation was performed on the solid phase in the previously immobilized form of GTL under mild conditions, and soluble conjugated Pd-GTL complexes were obtained by simply desorbing by washing with an acetonitrile aqueous solution. Three different Pd complexes were incorporated into two different genetically modified enzyme variants, one containing all the natural cysteine residues changed to serine residues, and another variant including an additional Cys mutation directly in the catalytic serine (Ser114Cys). The new Pd–enzyme conjugates were fluorescent even at ppm concentrations, while under the same conditions free Pd complexes did not show fluorescence at all. The Pd conjugation with the enzyme extremely increases the catalytic profile of the corresponding Pd complex from 200 to almost 1000-fold in the hydrogenation of arenes in aqueous media, achieving in the case of GTL conjugated with orthopalladated 4a an outstanding TOF value of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 428 min−1. Also the applicability of GTL-C114 conjugated with orthopalladated 4b in a site-selective C–H activation reaction under mild conditions has been demonstrated. Therefore, the Pd incorporation into the enzyme produces a highly stable conjugate, and improves remarkably the catalytic activity and selectivity, as well as the fluorescence intensity, of the Pd complexes.
428 min−1. Also the applicability of GTL-C114 conjugated with orthopalladated 4b in a site-selective C–H activation reaction under mild conditions has been demonstrated. Therefore, the Pd incorporation into the enzyme produces a highly stable conjugate, and improves remarkably the catalytic activity and selectivity, as well as the fluorescence intensity, of the Pd complexes.
Introduction
Metalloenzymes are enzymes that contain at least one metal incorporated into their structure, with the metal being necessary to develop their function correctly.1 It is well known that the binding sites at the cavities of the enzymes, usually N or S atoms or carboxylate groups of the amino acids of the protein, located at very specific positions in tight environments and used as second coordination spheres, confer remarkable properties to the metals housed in these positions. The conjugation of Pd complexes with proteins and enzymes, generating new types of artificial metalloenzymes, has shown to be an interesting and efficient tool to obtain materials with improved properties such as, among others, better stability, luminescence, catalytic activity and selectivity.2–9 Additional advantages are based on the applicability of chemical processes in aqueous media, which represents more sustainable conditions for Pd-chemistry.Here we describe for the first time the conjugation of Pd complexes, containing orthometallated 5(4H)-oxazolones as ancillary ligands, with an enzyme in an aqueous media system at room temperature. The aim of the work is the design and development of stable but highly active Pd-catalysts, as well as organometallic Pd-based fluorophores. Following our previous interest in highly efficient Pd-catalyzed processes4 and in luminescent complexes,10 we have selected oxazolones as the polyfunctional ligands and a lipase as the enzyme. Oxazolones are strong C^N-chelates structurally analogous to naturally occurring chromophores; therefore they impart fluorescent properties and chemical stability.11 More specifically, we have employed (Z)-4-arylidene-5(4H)-oxazolones, which are precursors of (Z)-4-arylidene-5(4H)-imidazolones, chromophores of the Green Fluorescent Proteins (GFPs).12 We are also interested in the thermoalkalophilic lipase from Geobacillus thermocatenulatus (GTL), which was chosen as representing an enzyme. This enzyme presents an interesting characteristic as a lipase, namely that the enzymatic mechanism is based on the movement of an oligopeptide chain from a closed conformation, which makes the active site inaccessible to the solvent, to an open conformation where this active site is accessible (Fig. 1).13 During this opening-closing mechanism two oligopeptides move at the same time, this fact being a particular feature of this enzyme compared to others. In addition, this enzyme also possesses extremely high hydrophobic active site surroundings.14 This property allows the selective adsorption of the enzyme in a hydrophobic material, thus fixing the open conformation.15 This process involves the whole area surrounding the active site. In this protein the N-terminal (the most reactive NH2 group) and C-terminus group (the most reactive COOH moiety) are on the opposite side of this hydrophobic phase, leaving it completely accessible to the solvent in the immobilized form (Fig. 1), bringing thus advantages for metal coordination.
 | ||
| Fig. 1 Three-dimensional structure of the G. thermocatenulatus lipase in open conformation. The protein structure was obtained from the Protein Data Bank (pdb code: 2W22) and the picture was created using Pymol v. 0.99. | ||
The novel Pd–enzyme conjugates demonstrated an improvement in fluorescence and catalytic capacity compared to the free Pd complexes. This strategy allowed the successful use of these unstable Pd complexes in aqueous media, making it possible to work with these organometallic molecules in conditions not possible in their free form.
Results and discussion
Synthesis and characterization of different Pd-oxazolones complexes (4a, 4b, 5a) and their stability in aqueous media
Three Pd-complexes containing orthometallated oxazolones (4a, 4b, 5a) were synthesized as shown in Scheme 1. The oxazolones 1a and 1b were synthesized by the Erlenmeyer–Plöchl method as reported.16–25 They were selected as complex scaffolds due to their push–pull electronic nature, which enhances charge separation and luminescence properties, and also because of their different steric requirements (Fig. S1†). The characterization of 1a and 1b showed that they have Z-configuration for the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and for 1b, the styryl fragment had E-configuration. The incorporation of the Pd atom at the ortho-position of the 4-arylidene ring of the oxazolone skeleton was performed by C–H bond activation processes, following methods developed in our group.10,11,26–29
C bond and for 1b, the styryl fragment had E-configuration. The incorporation of the Pd atom at the ortho-position of the 4-arylidene ring of the oxazolone skeleton was performed by C–H bond activation processes, following methods developed in our group.10,11,26–29
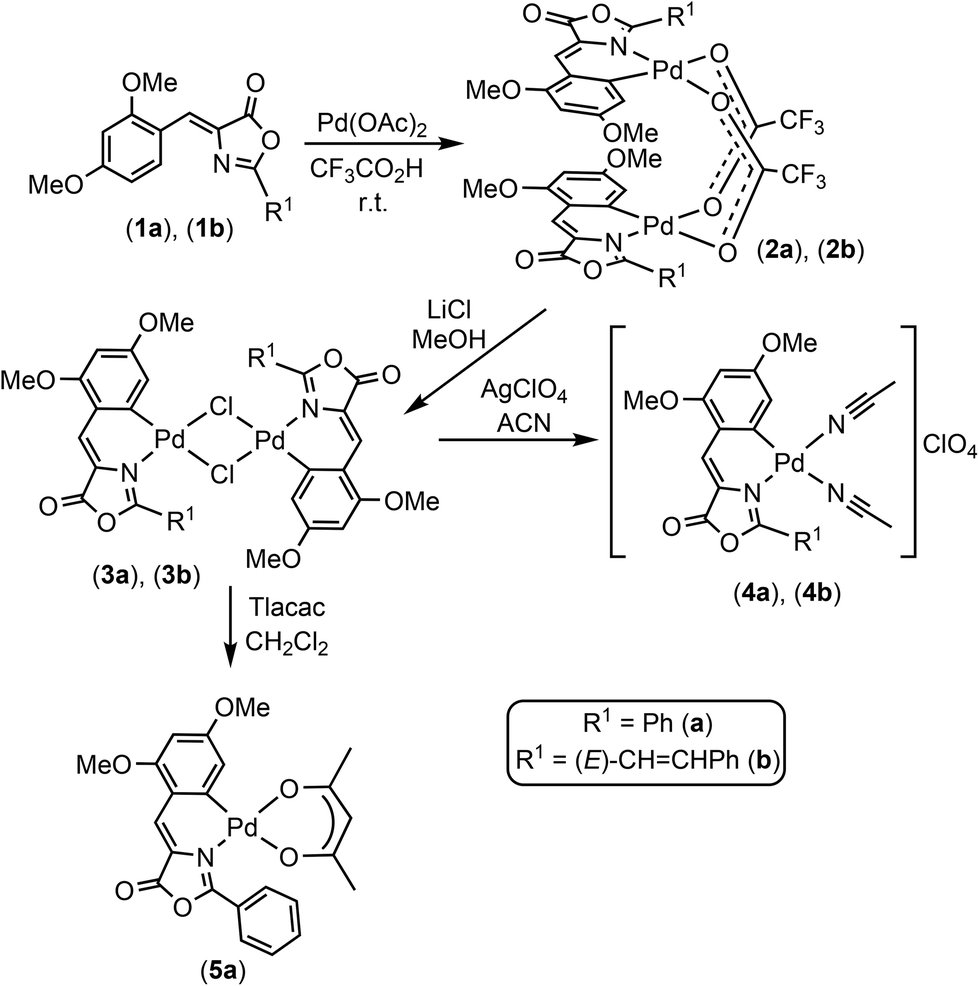 | ||
| Scheme 1 Synthesis of Pd-oxazolones complexes. | ||
The treatment of 1a (or 1b) with Pd(OAc)2 (1:1 molar ratio) in CF3CO2H at room temperature for 40 min afforded 2a (or 2b) as deep red solids in good isolated yields. Further treatment of 2a (2b) with excess of LiCl in MeOH at room temperature promoted the metathesis of the carboxylate bridges by chloride bridges and the formation of dinuclear 3a (3b) (Scheme 1) as very insoluble deep-red solids. Complexes 3a (3b) react with a halide scavenger such as AgClO4 (1:2 molar ratio) in a coordinating solvent (acetonitrile, ACN), allowing the isolation of the bis-solvato derivatives 4a (4b) as bright red solids, which are stable towards the oxygen and moisture even in solution. The presence of labile ACN ligands in the coordination sphere of the Pd center is very convenient for our purposes because it provides two masked vacant coordination sites that are able to react with donor atoms present in the cavity of the protein.
The reaction of 3a with Tlacac (1:2 molar ratio, acac = acetylacetonate) in CH2Cl2 takes place with the precipitation of TlCl and formation of 5a, which contains the acac ligand O,O′-chelated. The molecular structure of 5a has been determined by single-crystal X-ray diffraction methods. Fig. 2 shows a molecular drawing of the complex, while crystallographic parameters and most relevant bond distances (Å) and angles (°) are included in the ESI (Tables S1 and S2†).
 | ||
| Fig. 2 Molecular structure of 5a. | ||
The Pd atom is located in a square-planar environment, slightly distorted with respect to the ideal geometry expected for a Pd(II) complex. The acetylacetonate ligand (defined as the best least-square plane containing the O5–C21–C20–C19–O6 atoms) is almost coplanar with respect to the best coordination plane defined by Pd–C1–N1–O5–O6 (dihedral angle = 0.44(4)°). However, the oxazolone ligand is severely distorted and too far away to be coplanar with the coordination plane, with the dihedral angle between the coordination plane and the best plane defined by the C1–C6–C9–C10–N1 atoms being 143.54(4)°. The intramolecular repulsions between the Ph ring at the 2-position of the oxazolone heterocycle and the fragment O6–C19–C22 of the acac ligand seems to be responsible for this severe deformation and loss of planarity. Internal bond distances and angles show values similar to those found in other complexes reported in the literature.27
Considering that conjugation of Pd complexes with the enzymes must be performed in aqueous solution, the stability of the complexes was evaluated in different aqueous solutions by monitoring their UV absorbance under these conditions in the range of 200–600 nm (Fig. 3). The study was performed using compound 4a. The compound was dissolved in pure acetonitrile at a concentration of 1 mg mL−1 and then 200 μL of this solution was diluted in 2 mL of the different aqueous solutions.
 | ||
| Fig. 3 Stability of 4a in different aqueous solutions at room temperature. | ||
Complex 4a was stable in distilled water, distilled water adjusted at pH 7 and also in an aqueous solution of acetonitrile. However, the color was rapidly lost in aqueous solutions containing different buffers (phosphate buffer, MES, Tris) or directly in DMSO. Considering these results, we selected the distilled water adjusted at pH 7 as the best condition for continuing our studies.
Preparation and characterization of the new Pd-GTL enzymes
Two genetically modified variants of the native G. thermocatenulaus lipase were used, the first one where the two natural cysteines were removed and substituted by serines in a conservative change (GTL), and another one where the catalytic serine114 was modified by a Cys residue (GTL-C114), generating a lipase containing a unique cysteine. These two enzyme variants present as a unique difference the possible coordination of Pd in the active site (Fig. S2†).Firstly, enzyme variants were previously immobilized on a C4-support (butyl-Sepharose) for a solid-phase modification. This procedure allows the lipase to be selectively adsorbed against other proteins in the extract.15 Thus, in addition to having it fixed in a heterogeneous way, it allowed purification, ensuring that we only have our protein on the solid support before incorporating the complex (Fig. S3†).
Previous to the conjugation step, experimental conditions to avoid any undesired unspecific adsorption of Pd complex on the solid C4-support were studied. For this purpose, 4a was dissolved in solutions containing different concentrations of acetonitrile or surfactant (Triton X-100) in water adjusted at pH 7. Solid butyl-Sepharose was added to the 4a solutions and the adsorbance of the supernantant was measured at different times (Fig. S4†). In this study, the addition of 20% ACN resulted as the optimal conditions because after 30 min 85% of 4a still was remaining on the supernatant. In this way, the stability of the three complexes 4a, 4b and 5a was tested in these optimized conditions, i.e. 20% ACN in water adjusted at pH 7. In the three cases, more than 90% of the initial UV absorption was preserved after 4 h of incubation. Thus, the enzyme–metal bioconjugation was performed following a strategy based on two steps (Fig. 4). The first step was the direct supramolecular coordination of the Pd complexes to the protein.
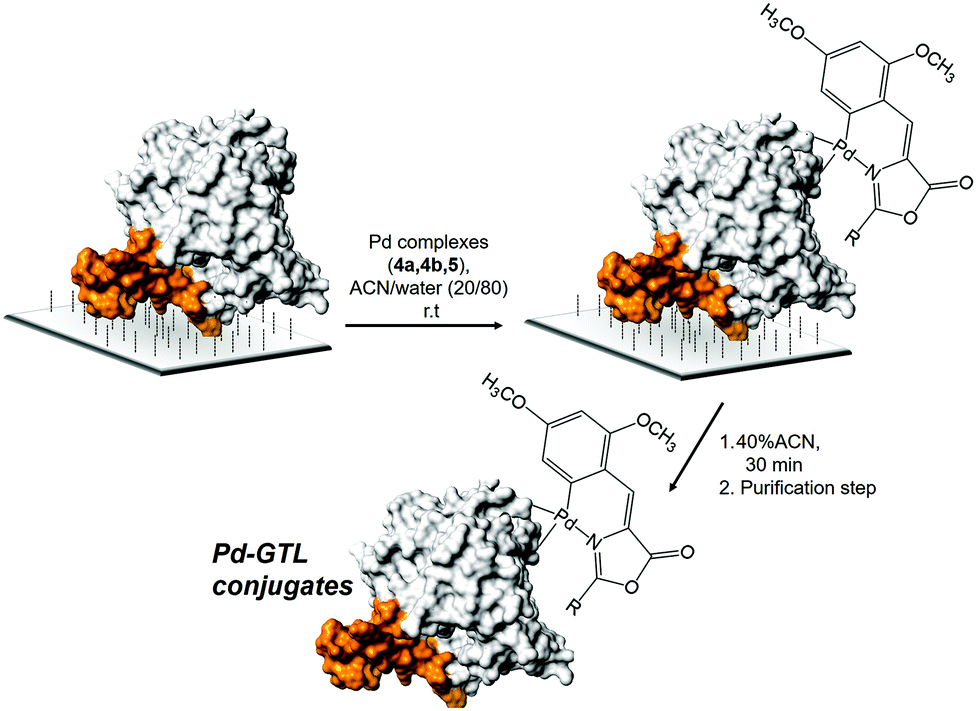 | ||
| Fig. 4 Strategy for the site-specific Pd-complex incorporation on GTL. Enzyme lid oligopeptide (orange). | ||
The incorporation of the Pd complexes was performed in the two different enzymes immobilized on the C4-support in these reaction conditions at room temperature (Fig. 4). The chemical modification was followed by the disappearance of the absorbance signal in the UV spectrum in the supernatant. 100 eq. of Pd complexes with respect to the protein was added in all cases. The adsorption speed was slightly faster for 4b and 5a although in all cases around 50% of the compound offered was adsorbed after 4 h (Fig. 5). Longer incubation times did not improve the results. Although the adsorption effect was evaluated, the high amount of Pd offered indicated that free Pd was also absorbed into the solid material. However, the reduction in the amount of the compound produced worse results. The second step is the recovery of the enzyme–Pd conjugate in solution. To achieve that, the modified immobilized enzymes were desorbed from the solid support by incubation with water containing 40% ACN (Fig. 4). Under these conditions, lipases can be desorbed from these hydrophobic supports.15
 | ||
| Fig. 5 Adsorption of the Pd complexes on the immobilized GTL enzymes. | ||
After desorption, the sample was purified in order to eliminate all the free Pd compound desorbed also from the resin. After the purification, yellow or red solutions from the different conjugates were obtained (Fig. S5†). This strategy afforded much better performance compared to the preparation of the conjugate in solution. The content of protein per ml of solution in the conjugates was determined by the Bradford Assay (Table 1). The Pd content in each solution was determined by ICP-OES mass analysis (Table 1). The results have shown a protein concentration around 0.1 mg mL−1 with a Pd content of 0.3–0.5 ppm (Table 1). The determination of nmols of protein and Pd, for example in the case of GTL-4a and GTL-C114-4a, showed that in the former the ratio nmolsGTL/nmolsPd was 1, whereas in the latter the ratio was 2 (Table 1).
| Sample | Protein concentration (mg ml−1) | Pd content (mg ml−1) | Proteinb (nmol) | Pdb (nmol) |
|---|---|---|---|---|
| a The conjugates were prepared using 0.4 g of immobilized enzymes. b Amount of protein or Pd per mL of solution. | ||||
| GTL-4a | 0.125 | 0.31 × 10−3 | 2.90 | 2.92 |
| GTL-C114-4a | 0.08 | 0.35 × 10−3 | 1.86 | 3.3 |
| GTL-4b | 0.125 | 0.44 × 10−3 | 2.90 | 4.1 |
| GTL-C114-4b | 0.16 | 0.50 × 10−3 | 3.71 | 4.7 |
| GTL-5aa | 0.10 | 0.17 × 10−3 | 2.32 | 1.6 |
| GTL-C114-5aa | 0.06 | 0.21 × 10−3 | 1.39 | 2.0 |
This could indicate that coordination of the Pd complexes took place, probably, in a selective way at the N-terminus of the protein (the most reactive group at this pH) in GTL-4a, whereas in GTL-C114-4a both the N-terminus and the S atom of the cysteine in the active site are involved in the coordination, and two metallic centers can be captured by the same protein. The 3D-analysis of the environments and neighbouring residues surrounding both groups (NH2 at N-terminus and S atom from the cysteine) on the protein structure (Fig. 6) clearly suggests that in the case of GTL-C114-4a, the possible coordination of the Pd complex in the active site would occur between SH and some of the His around that group (His365), which is one of the three amino acids of the catalytic triad and therefore is more activated from its nucleophilic nature (Fig. 6A).
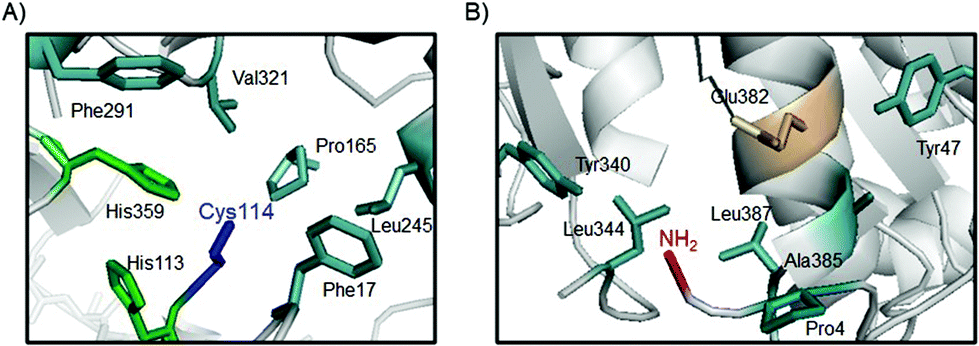 | ||
| Fig. 6 Core sites in the GTL protein for Pd coordination. (A) Active site of GTL-C114. (B) Structural environment around the N-terminus of GTL enzymes. The protein structure was obtained from the Protein Data Bank (pdb code: 2W22) and the pictures were created using Pymol v. 0.99. | ||
However, analyzing the environment of the N-terminal region we can observe that there is a proline (Pro4), and a carboxyl group. These could intervene together with the NH2 group of the N-terminal in the coordination to the Pd center, the interaction being different from that observed in the active site (Fig. 6B). In this case, different hydrophobic groups could stabilize the structure of the complex (Fig. 6).
In order to demonstrate this hypothesis, a chemical modification in the N-terminus30 of the two variants GTL and GTL-C114 on the solid phase was performed following a strategy recently reported31 by us using N-acetyl-tyrosine (Ac-Tyr) as molecule (Fig. S6†). The carboxylic group of the molecule was previously activated by the EDC/NHS activation step. Then, the Pd orthopalladated 4a incorporation protocol was repeated in the modified Ac-Tyr-variants (GTL and GTL-C114) and in both cases no fluorescence emission signal was observed, with a purified protein solution completely transparent (Fig. S7†), demonstrating that the Pd coordination is performed by the N-terminus in GTL and N-terminus and SH active site in GTL-114.
Evaluation of Pd–GTL conjugates as mimics of fluorescent proteins
The fluorescence properties of the Pd–enzyme conjugates were studied (Table 2 and Fig. S5†), and the comparison of the obtained results with those of the free species 4a, 4b, and 5a was performed. The free organometallic complexes 4a, 4b and 5a showed strong fluorescent emission in pure organic solvents such as ACN (Table S3 and Fig. S8†). They were also tested in the previous specific aqueous conditions (water at pH 7 with 20% ACN) (Table S4 and Fig. S9†). In all of the studied cases, the excitation wavelength was fixed at 450 or 475 nm, and strong emissions were observed in the green region of the spectrum (526–579 nm). Evaluation at concentrations of 0.1 and 0.25 mg ml−1 (100 or 250 ppm, respectively) showed the expected differences of fluorescence intensity depending on the type of complex and their concentration: compound 4a showed the strongest fluorescence intensity under these conditions followed by 5a, and 4b being the weakest fluorescent molecule (Table S4†).| Sample | Pd content (ppm) | Excitation λ (nm) | Emission λ (nm) | Intensity (counts) |
|---|---|---|---|---|
| GTL | — | 450 | — | 0 |
| GTL-C114 | — | 450 | — | 0 |
| 4a | 0.50 | 450 | — | 0 |
| GTL-4a | 0.3109 | 450 | 523.19 | 50.94 |
| GTL-4a | 0.3109 | 475 | 523.19 | 56.89 |
| 4b | 0.50 | 450 | — | 0 |
| GTL-4b | 0.44 | 450 | 550 | 105 |
| GTL-C114-4b | 0.50 | 450 | 550 | 195 |
| 5a | 0.50 | 450 | — | 0 |
| GTL-5a | 0.17 | 450 | 524.83 | 67.18 |
| GTL-C114-5a | 0.21 | 450 | 523.12 | 53.12 |
| GTL-5a | 0.17 | 475 | 524.83 | 71.53 |
| GTL-C114-5a | 0.21 | 475 | 523.12 | 58.23 |
In the case of artificial Pd-proteins, samples with a concentration of Pd in the range 0.17–0.50 ppm were evaluated. That is, the content of Pd in the samples of the metalloenzymes is 200 times smaller than in the free organometallic complexes 4a, 4b, or 5a described in the last paragraph (100–250 ppm). The fluorescence of the samples of the metalloenzymes was compared with that observed for 4a, 4b, and 5a, prepared in the same concentration (0.50 ppm) and in larger concentrations (100–250 ppm). In the case of the free complexes 4a, 4b, or 5a, no fluorescence signal at all was found at 0.50 ppm concentration (Table 2). The proteins without the Pd complex did not show any fluorescence intensity. However, the new Pd–GTL enzymes showed strong fluorescence signals under these conditions at the lowest concentrations (Fig. S10†).
The conjugation of Pd complexes with the enzyme improved the fluorescence of the compounds to a different extent. For example, the highest fluorescence intensity was achieved using the bioconjugate enzyme-4b. In particular, the insertion of 4b in the active site of GTL-C114 allows to obtain the conjugate GTL-C114-4b as the representative showing the strongest fluorescence, almost twice compared with that of the conjugate GTL-4b (Table 2). This is quite interesting because complex 4b gave the weakest fluorescence emission in free form in aqueous solution (Table S4†).
In the other two Pd complexes (4a and 5a), the insertion in the proteins also gave a fluorescence signal at these extremely low concentrations, showing better results at both excitation wavelengths (450 and 475 nm) using the conjugation with GTL-C114 (Table 2). Besides the huge qualitative increase of the intensity of the fluorescence of the Pd(oxazolone) fragment after bioconjugate formation, it seems that there are not clear correlations between such increases and other molecular parameters, such as the type of substituents and/or the nature of the enzyme involved in each particular case. The lower intensity in the case of GTL-C114-5a could be explained considering that 5a presents a more rigid structure in the Pd coordination in free form; may be the modification of the active site is causing a conformational change in the whole protein structure affecting the final fluorescence signal of the bioconjugate.
Evaluation of the new Pd–GTL conjugates catalysis in water
The strong change in the environment around the metal center from free complexes 4a, 4b, and 5a to the artificial metalloenzymes most likely will also modify the catalytic properties of the palladium.32 In this respect, we have studied the selective reduction of an aromatic derivative such as p-nitrophenol (pNP) in aqueous media at room temperature as the model reaction for evaluating the catalytic capacity of these new Pd bioconjugates in comparison with free molecules (Table 3 and Fig. S11–S14†).| Catalyst | Pd content (μg) | time (min) | TOF valueb (min−1) | pNP conversionc |
|---|---|---|---|---|
| a Reaction conditions: 1.0 mM (3 mg) nitroarene, 40 mM (3 mg) NaBH4, 2 mL of distilled water, air and room temperature. b The TOF value was defined as the μmols of pAP per μmol of Pd and per minute. c pNP conversion was determined by UV spectrophotometer after 10 min reaction. | ||||
| GTL | — | 1 | — | 0 |
| GTL-C114 | — | 1 | — | 0 |
| 4a | 0.75 | 1 | 137 | 95 |
| GTL-4a | 0.0047 | 1 | 27428 |
80 |
| GTL-C114-4a | 0.0052 | 1 | 22857 |
>95 |
| 4b | 1 | 3 | 27 | 71 |
| GTL-4b | 0.0067 | 1 | 15873 |
>95 |
| GTL-C114-4b | 0.0075 | 1 | 15714 |
>95 |
| 5a | 7.5 | 1 | 12 | >95 |
| GTL-5a | 0.0026 | 3 | 13605 |
65 |
| GTL-C114-5a | 0.0031 | 7 | 4892 | 63 |
From the free Pd complexes, 4a showed the best catalytic activity in the reduction of pNP with a TOF value of 137 min−1, 5 and 10 times faster than 4b and 5a, respectively. As for the hybrids, the results showed how the conjugation of the three Pd complexes on the protein extremely improved their catalytic performance, exhibiting an increase in the TOF value of 200 times for GTL-4a conjugates compared with the free Pd molecule. The best TOF value was achieved with GTL-4a, 27428 min−1, which is, as far as we know, the highest TOF value ever achieved by a transition metal catalyst in this process.33–36 The improvement was even higher for the other two compounds, with a TOF value enhanced almost 600 times for 4b in GTL-4b, and almost 1000 times for 5a in GTL-5a (Table 3). These results showed for the first time an extraordinary improvement of a metal organometallic complex by enzyme coordination.
Finally, GTL-114-4b was applied in a site-selective C–H activation reaction, by the C-2 aryl modification of N-acetyl-l-tryptophan methyl ester37 under mild conditions (methanol/water (1/1) as the solvent and at room temperature). These conditions were recently reported by us as excellent for this modification by using other Pd–enzyme hybrids.38 The reaction was performed using 4-methoxybenzenediazonium tetrafluoroborate, as an electrophilic arylating coupling partner, and >99% of C-2′ arylated tryptophan was obtained after 16 h incubation (Fig. S15†), showing the potential application of these bioconjugates in selective complex reactions.
Experimental
General information
Butyl-Sepharose® 4 Fast Flow was from GE Healthcare (Uppsala, Sweden). Sodium borohydride, Triton® X-100, dimethylsulfoxide (DMSO), 4-methoxybenzenediazonium tetrafluoroborate (pOMe-BDTFB), p-nitrophenol (pNP), p-nitrophenylpropionate (pNPP), p-aminophenol (pAP), N-acetyl-tyrosine, N-hydroxysuccinimide (NHS), 2-dipyridyldisulfide (2-PDS) and dithiothreitol (DTT) were from Sigma-Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) was purchased from Tokyo Kasei (Japan). N-Acetyl-L-tryptophan methyl ester (AcTrpOMe) was bought from Alfa Aesar. Inductively coupled plasmaoptical emission spectrometry (ICP-OES) was performed on a PerkinElmer OPTIMA 2100 DV equipment. C, H, N and S elemental microanalyses were carried out on a PerkinElmer 2400-B Series II Analyser. Electrospray Ionisation (ESI) mass spectra were recorded using a Bruker Esquire 3000 plus™ ion-trap mass spectrometer equipped with a standard ESI source. High-resolution mass spectra-ESI (HRMS-ESI) were recorded using a Bruker MicroToF-Q™ equipped with an API-ESI source and a Q-ToF mass analyzer, which allows a maximum error in the measurement of 5 ppm. Acetonitrile and methanol were used as solvents. For all types of MS measurements, samples were introduced in a continuous flow of 0.2 mL min−1 and nitrogen served both as the nebulizer gas and the dry gas. Infrared spectra were recorded on a Spectrum 100 PerkinElmer FTIR Spectrophotometer, with a Universal Attenuated Total Reflectance (UATR) accessory (4000–250 cm−1). The 1H, 13C{1H} and 19F NMR spectra were recorded on a Bruker Avance-300 spectrometer (δ in ppm; J in Hz). All experiments were recorded on solution at room temperature using CD2Cl2 or CDCl3 as the deuterated solvents. Other conditions were specified on each particular case. The 1H and 13C{1H} spectra were referenced using the residual solvent signal as internal standard, while 19F spectra were referenced to CFCl3. The N-cinnamoylglycine used for the synthesis of 1b was prepared by the Schotten–Baumann method.39 Oxazolone 1b appears in Scifinder as commercially available, but there are no references associated to its synthesis nor to its characterization, so a complete description is provided here.Site-directed mutagenesis, cloning and expression of Geobacillus thermocatenulatus lipase (GTL)
The gene corresponding to the mature lipase from G. thermocatenulatus (GTL) was cloned into pT1GTL expression vector as previously described.40,41 All site-directed mutagenesis experiments were carried out by PCR using mutagenic primers. To introduce the amino acid change, the corresponding pair of primers was used as homologous primer pair in a PCR reaction using a specific plasmid as the template and Prime Start HS Takara DNA polymerase. The product of the PCR was digested with endonuclease DpnI that exclusively restricts methylated DNA.40E. coli DH10B cells were transformed directly with the digested product. The plasmid with mutated GTL were identified by sequencing and then transformed into E. coli BL21 (DE3) cells to express the corresponding proteins. Firstly, C65S was created, and the resulting plasmid was used as the template to create the double mutant C65S/C296S-GTL (GTL).41 This plasmid (pT1BGTLmutCys) was used as the template to construct additional mutations (S114C), creating the mutants GTL-C114, using the mutagenic primers S114C-for CATCGCTCATTGCCAAGGAGGAC and S114C-rev GTCCTCCTTGGCATGAGCGATG. The gene corresponding to the mature lipase from G. thermocatenulatus (GTL) was cloned into a pT1 expression vector as previously described.40,41 Cells carrying the recombinant plasmid pT1GTL were grown at 30 °C and overexpression was induced by raising the temperature to 42 °C for 20 h.Enzymatic activity assay
The activities of the soluble and immobilized GTL variants were analyzed spectrophotometrically measuring the increment in absorbance at 348 nm produced by the release of p-nitrophenol (pNP) (∈ = 5150 M−1 cm−1) in the hydrolysis of 0.4 mM pNPP in 25 mM sodium phosphate at pH 7 and 25 °C. To initialize the reaction, 0.05–0.2 mL of lipase solution or suspension was added to 2.5 mL of the substrate solution. Enzymatic activity is given as micromoles of hydrolyzed pNPP per minute per milligram of enzyme (IU) under the conditions described above.Synthesis and characterization of compounds 1a–5a
N), 164.00 (q, 2JCF = 38.5 Hz, CO2 bridging), 161.95 (s, C2–O, C6H2), 160.39 (s, CO), 159.18 (s, C4–O, C6H2), 139.39 (s, C, C6–Pd, C6H2), 133.98 (s, CH, Cp, C6H5), 132.45 (s, CH, Cvinyl), 130.14 (s, CH, Co, C6H5), 128.32 (s, CH, Cm, C6H5), 123.22 (s, Ci, C6H5), 119.06 (s, C), 114.67 (q, 1JCF = 288 Hz, CF3), 113.81 (s, C, C1, C6H2), 109.47 (s, CH, C5, C6H2), 96.40 (s, CH, C3, C6H2), 55.93 (s, OCH3), 55.45 (s, OCH3). 19F NMR (CD2Cl2, 282.40 MHz): δ = −75.31 (s, CF3). IR (ν, cm−1): 1783 (vs, CO), 1671, 1644 (vs, O2CCF3), 1572, 1534 (vs, O–CN).
O), 1620, 1562, 1534 (O–CN), 314 (Pd–Cl).
CHvinyl), 7.80–7.68 (m, 3H, Hm, Hp, C6H5), 6.56 (d, 4JHH = 2.1 Hz, 1H, C6H2, H5), 6.29 (d, 4JHH = 2.1 Hz, 1H, C6H2, H3), 3.92 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 2.44 (s, very broad, 3H, CH3CN), 2.26 (s, very broad, 3H, CH3CN). 13C{1H} NMR (CD2Cl2, 75.47 MHz): δ = 165.81 (s, CN), 162.85 (s, C2–O, C6H2), 160.57 (s, CO), 160.19 (s, C4–O, C6H2), 134.97 (s, Cp, C6H5), 130.34 (s, CH, Cvinyl), 129.17 (2s, overlapped, Co, Cm, C6H5), 123.44 (s, Ci, C6H5), 119.46 (s, C), 116.15 (s, CH, C5, C6H2), 114.90 (s, C, C1, C6H2), 95.53 (s, CH, C3, C6H2), 56.06 (s, OCH3), 55.99 (s, OCH3), 3.48 (s, very broad, CH3, CH3CN). Signals due to the C6 carbon of the orthometallated C6H2 fragment, as well as signals due to the 13C nuclei of CH3CN ligands, were not observed, probably hidden in the baseline due to their involvement in dynamic processes.
CHvinyl), 7.61 (m, 1H, Hp, C6H5), 7.51 (m, 2H, Hm, C6H5), 7.00 (d, 1H, H5, C6H2, 4JHH = 2.4 Hz,), 6.24 (d, 1H, H3, C6H2, 4JHH = 2.4 Hz), 5.24 (s, 1H, CH-acac), 3.94 (s, 3H, OMe), 3.91 (s, 3H, OMe), 2.02 (s, 3H, CH3-acac), 1.21 (s, 3H, CH3-acac). 13C{1H} NMR (CD2Cl2, 75.47 MHz): δ = 187.39 (s, CO-acac), 185.66 (s, CO-acac), 164.19 (s, CN), 162.38 (s, C2–O, C6H2), 162.24 (s, CO), 160.08 (s, C4–O, C6H2), 151.54 (s, C, C6–Pd, C6H2), 133.95 (s, CH), 133.09 (s, Cp, C6H5), 130.10 (s, Co, C6H5), 127.82 (s, Cm, C6H5), 125.03 (s, Ci, C6H5), 119.77 (s, C), 116.35 (s, C, C1, C6H2), 111.28 (s, CH, C5, C6H2), 99.63 (s, CH, C3H, acac), 95.23 (s, CH, C3, C6H2), 55.69 (s, OMe), 55.42 (s, OMe), 27.06 (s, CH3-acac), 26.15 (s, CH3-acac).
CHvinyl), 7.62 (d, J = 16.5 Hz, 1H, CHolef), 7.57 (m, 2H, Ho, C6H5), 7.44–7.40 (m, 3H, Hm, Hp, C6H5), 6.79 (d, J = 16.2 Hz, 1H, CHolef), 6.63 (dd, J = 8.7, 1.8 Hz, 1H, H5, C6H3), 6.43 (d, J = 2.1 Hz, 1H, H3, C6H3), 3.88 (s, 6H, OCH3). 13C{1H} NMR (CDCl3, 75.47 MHz): δ = 167.82 (s, CO), 164.10 (s, C2–O, C6H3), 161.78 (s, C4–O, C6H3), 161.06 (s, CN), 142.31 (s, CH, Colef), 134.85 (s, C), 134.35 (s, CH, C6, C6H3), 130.49 (s, Ci, C6H5), 130.36 (s, CH, Cp, C6H5), 129.04 (s, CH, Co, C6H5), 127.97 (s, CH, Cm, C6H5), 125.75 (s, CH, Cvinyl), 116.21 (s, C1, C6H3), 113.69 (s, CH, Colef), 106.32 (s, CH, C5, C6H3), 97.68 (s, CH, C3, C6H3), 55.66 (s, OCH3), 55.55 (s, OCH3).
N), 161.58 (s, C–O, C6H2), 160.46 (s, CO), 158.82 (s, C–O, C6H2), 145.95 (s, CH, Colef), 139.08 (s, C), 134.01 (s, Ci, C6H5), 131.57 (s, CH, Cp, C6H5), 130.87 (s, CH, Cvinyl), 129.04 (s, CH, Co, C6H5), 128.81 (s, CH, Cm, C6H5), 118.73 (s, C, C6H2), 113.68 (s, C–Pd, C6H2), 110.69 (s, CH, Colef), 108.97 (s, CH, C6H2), 96.66 (s, CH, C6H2), 55.86 (s, OCH3), 55.21 (s, OCH3). Signals due to the presence of the bridging ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) F3OO ligand were not found in this spectrum, despite the use of long accumulation trials, probably due to the low solubility of this compound. 19F NMR (282.40 MHz, CDCl3): δ = −74.49 (s, CF3). IR (ν, cm−1): 1790 (CO), 1662, 1571 (O–CN), 1642 (CF3COO bridging).
O), 1637, 1560 (O–CN), 314 (Pd–Cl).
CHvinyl), 7.75 (d, J = 16.2 Hz, 1H, CHolef), 7.72 (m, 2H, Ho, C6H5), 7.52–7.50 (m, 3H, Hm, Hp, C6H5), 7.12 (d, J = 15.9 Hz, 1H, CHolef), 6.49 (s, broad, 1H, C6H2, H5), 6.26 (d, J = 1.8 Hz, 1H, C6H2, H3), 3.91 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 2.51–1.98 (very broad, 6H, CH3CN). 13C{1H} NMR (75.47 MHz, CH2Cl2): δ = 164.90 (s, C2–O, C6H2), 163.14 (s, CN), 160.81 (s, CO), 160.78 (s, C4–O, C6H2), 147.73 (s, CH, Colef), 135.02 (2C overlapped, C + Ci, C6H5), 132.87 (s, CH, Cvinyl), 132.61 (s, Cp, C6H5), 130.29 (s, Co, C6H5), 129.91 (s, Cm, C6H5), 120.45 (s, Pd–C6, C6H2), 115.69 (s, C
F3OO ligand were not found in this spectrum, despite the use of long accumulation trials, probably due to the low solubility of this compound. 19F NMR (282.40 MHz, CDCl3): δ = −74.49 (s, CF3). IR (ν, cm−1): 1790 (CO), 1662, 1571 (O–CN), 1642 (CF3COO bridging).
O), 1637, 1560 (O–CN), 314 (Pd–Cl).
CHvinyl), 7.75 (d, J = 16.2 Hz, 1H, CHolef), 7.72 (m, 2H, Ho, C6H5), 7.52–7.50 (m, 3H, Hm, Hp, C6H5), 7.12 (d, J = 15.9 Hz, 1H, CHolef), 6.49 (s, broad, 1H, C6H2, H5), 6.26 (d, J = 1.8 Hz, 1H, C6H2, H3), 3.91 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 2.51–1.98 (very broad, 6H, CH3CN). 13C{1H} NMR (75.47 MHz, CH2Cl2): δ = 164.90 (s, C2–O, C6H2), 163.14 (s, CN), 160.81 (s, CO), 160.78 (s, C4–O, C6H2), 147.73 (s, CH, Colef), 135.02 (2C overlapped, C + Ci, C6H5), 132.87 (s, CH, Cvinyl), 132.61 (s, Cp, C6H5), 130.29 (s, Co, C6H5), 129.91 (s, Cm, C6H5), 120.45 (s, Pd–C6, C6H2), 115.69 (s, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, CH3CN), 112.05 (s, CH, Colef), 96.45 (s, CH, C3, C6H2), 56.95 (s, OCH3), 56.75 (s, OCH3), 23.59 (s, CH3, CH3CN). Signals due to the C1 and C5 carbons of the orthometallated C6H2 fragment, as well as signals due to the 13C nuclei of one NCMe ligand, were not observed, probably being hidden in the baseline.
N, CH3CN), 112.05 (s, CH, Colef), 96.45 (s, CH, C3, C6H2), 56.95 (s, OCH3), 56.75 (s, OCH3), 23.59 (s, CH3, CH3CN). Signals due to the C1 and C5 carbons of the orthometallated C6H2 fragment, as well as signals due to the 13C nuclei of one NCMe ligand, were not observed, probably being hidden in the baseline.
X-ray crystallography
Single crystals of 5a of suitable quality for X-ray diffraction were grown by slow evaporation of a saturated solution of 5a in Et2O at 25 °C. One selected single crystal was mounted at the end of a quartz fiber in a random orientation, covered with magic oil and placed under a cold stream of N2 gas. Crystallographic measurements were carried out at 100 K on a Bruker Smart APEX CCD diffractometer, using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). A hemisphere of data was collected in each case based on ω-scan or ϕ-scan runs. The diffraction frames were integrated using the program SAINT42 and the integrated intensities were corrected for absorption with SADABS.43 The structures were solved and developed by Patterson and Fourier methods.44 All non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms were placed at idealized positions and treated as riding atoms. Each H atom was assigned an isotropic displacement parameter equal to 1.2–1.5 times the equivalent isotropic displacement parameter of its parent atom. For structure solving and refinement, SHELX-97 Software Package was used. The structure was refined to Fo2, and all reflections were used in the least-squares calculations.45 CCDC 2045156 (5a)† contains the supplementary crystallographic data for this paper.Stability evaluation of Pd complexes
The different Pd complexes 4a, 4b and 5a were dissolved at 10 mg mL−1 in different solvents and aqueous solutions (phosphate buffer, water containing Triton X-100, SDS or acetonitrile as co-solvent) and their UV absorbance was measured in a UV spectrophotometer JASCO in the range of 340 to 600 nm. Stability of the complex in aqueous solution was determined by measuring the conservation of the absorbance (ΔAbs) intensity.Preparation of the Pd–enzyme conjugates
The GTL variant (GTL-NC or GTL-C114) crude extract from E. coli (5 mL) was added to 15 mL of 25 mM sodium phosphate at pH 7.0 (obtaining a final enzyme concentration of 8 mg ml−1 determined by the Bradford assay.46 Then, butyl-Sepharose was added in a 1/10 (v/v) proportion and gently stirred for 3 h at 25 °C. Periodically, the activity of the suspensions and supernatants was measured by the pNPP assay described above. After that, the adsorbed lipase preparation was abundantly washed with distilled water. More than 95% of the enzyme was adsorbed as confirmed by SDS-PAGE analysis in both cases (Fig. S2†). Then, 0.5 g of the immobilized GTL variant was incubated with 10 mL of a solution of acetonitrile/water adjusted at pH 7 (2/8) with Pd complex, previously dissolving 10 mg of Pd complex in 1.9 mL of acetonitrile and this was added to 8.1 mL of water adjusted at pH 7. The mixture was maintained in a roller-stirrer at room temperature for 4 h. Then, the solid was transferred to a 50 mL syringe reactor containing a filter, and the liquid was removed. The solid was washed with acetonitrile/water adjusted at pH 7 (2/8) (3 × 50 mL) and then 5 mL of a solution acetonitrile/water adjusted at pH 7 (4/6) was added to the solid. The mixture was incubated and maintained in the roller stirrer for 1 h. After that, the mixture was filtered and the liquid (containing the desorbed modified proteins) was recovered. Finally, in order to remove any amount of Pd complex that is free, the solution was purified by centrifugation using Centrifugation filters (Amicon® Ultra-4 mL, 10 K cut-off), previously diluting the initial solution of 4 ml with distilled water (4 ml) and centrifuging at 8000 rpm for 10 min several times. Finally, 2 mL of the pure Pd–enzyme conjugate was obtained in each case. The content of Pd in each case was determined by ICP-OES analysis.Chemical modification of N-terminus of GTL variants
5 mg of N-acetyl-L-tyrosine (Ac-Tyr), 10 eq. of EDC (42.17 mg) and 15 eq. of NHS (37.46 mg) were added in 5 mL of H2O adjusted to pH 4.8–5.2.31 This solution was allowed to react for 1 h. Then, 0.5 mL (50 eq.) of the previous solution was added to 3.5 mL of H2O adjusted to pH = 7 and 0.5 g of GTL or GTL-C114 immobilized on butyl-Sepharose protein at r.t., and the mixture was left to react overnight. Then, the mixture was filtered and the solid was washed several times with distilled water.Then this solid was used as previously described in the protocol of modification with Pd complex (4a).
Protection of thiol in GTL-C114 by 2-PDS
In order to test if the free cysteine in GTL-C114 was also modified by this protocol, 0.2 g of the modified immobilized protein was incubated in the presence of 2 ml of DTT solution (50 mM in 25 mM sodium phosphate at pH 8) for 30. After that, the solid was washed with distilled water until the DTT smell disappeared. Then the solid was added to 3 ml of 2-PDS solution (1.5 mM substrate in a mixture of DMSO (5%, v/v)-25 mM phosphate buffer (95%, v/v) at pH 8.0) for 1 h. The cysteine PDS activation was followed spectrophotometrically by measuring the increase of the absorbance at 343 nm of the solution. No modification was found in the GTL variant.Fluorescence spectroscopy measurements
The steady-state excitation–emission spectra of 4a, 4b and 5a and the corresponding Pd–enzyme conjugates in pure acetonitrile or aqueous solution containing 20% acetonitrile was determined in a PerkinElmer Fluorophotometer. The measurements were carried out at room temperature using quartz cuvettes of 1 cm path length. An excitation wavelength of 450 or 475 nm was used, and emission wavelengths were monitored, registering the counts value in each case. A filter of 3.5% was used.Catalytic reduction of nitroarenes
p-Nitrophenol (pNP) was dissolved in 2 mL of distilled water to 1 mM concentration. Then, solid NaBH4 (3.2 mg) was added to the solution. After this addition, the light yellow solution changes to a strong yellow colour, generating the formation of 4-nitrophenolate ions (the substrate UV-peak undergoes an immediate shift from 317 to 400 nm). After 30 s, 15 to 20 μl of the complex free or bioconjugated was added under gentle stirring at room temperature in an orbital shaker. The reaction progress was monitored by taking out an aliquot of the solution (0.1 mL) at different times, diluting it with distilled water (2 mL) and measuring the absorption spectrum between 500 and 300 nm in a quartz cuvette.General procedure for C–H activation of AcTrpOMe37,38
0.0192 mmol (5 mg) of AcTrpOMe and 0.0192 mmol (4.2 mg) of ionic liquid pOMe-BDTFB were added to a glass flask containing 1.2 mL of pure GTL-114-4b (0.6 μg of pd) in 10% ACN/90% water. Then 0.5 mL of methanol was added to the solution. The mixture was kept at room temperature for 18 h. Then the conversion was monitored by TLC analysis of the reaction's sample. The eluent was 60% ethyl acetate and 40% hexane and the product has a Rf of 0.35. The product was confirmed using a pure standard sample.Conclusions
In conclusion, we have prepared a new type of enzyme–Pd bio-conjugate by the selective coordination of different palladium complexes containing orthometallated oxazolones to genetically produced thermoalkalophilic lipases (GTL and GTL-C114). The resulting Pd–metalloenzymes show outstanding fluorescence properties and extraordinary catalytic capacity. The amplification of the fluorescence is up to 600 times higher in the metalloenzyme with respect to the free orthopalladated complex, in such a way that the metalloenzymes show strong emissions under conditions (0.2–0.5 ppm) where free Pd complexes were not fluorescent at all. In addition, the metalloenzymes showed catalytic activity from 200 to 1000 times higher than the corresponding free Pd complexes, being able to obtain in one bioconjugate (GTL-4a), the highest TOF value ever described for this reaction. The insertion of these complexes on the protein—as we have demonstrated—could open the applicability of these bioconjugate systems in different kinds of chemical reactions (formation of C–C and/or C-heteroatom bonds through C–H bond activation, for instance) in aqueous solution, conditions where in many cases free Pd complexes cannot be used.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish National Research Council (CSIC, Spain), the Aragón Government (Spain, Project LMP144_18: Programa Operativo FEDER Aragón 2014-2020, “Construyendo Europa desde Aragón” and research group Aminoácidos y Péptidos E19_20R) and the Spanish Government (Ministerio de Ciencia e Innovación) (Projects No. AGL2017-84614-C2-2-R and PID2019-106394GB-I00) for funding. Authors thank the European Cooperation in Science and Technology (COST) program under CA15106 grant (CHAOS: CH Activation in Organic Synthesis). A. P. is grateful for the financial support of the Romanian Ministry of Education and Research through the grant PN-III-P1-1.1-MC-2018-2580. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- M. Hoppert, in Metalloenzymes, ed. J. Reitner and V. Thiel, Encyclopedia of Geobiology. Encyclopedia of Earth Sciences Series, Springer, Dordrecht, 2011. DOI:10.1007/978-1-4020-9212-1_134.
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576 CrossRef CAS PubMed.
- M. Dieguez, J.-E. Bäckvall and O. Pamiés, Artificial Metalloenzymes and MetalloDNAzymes in Catalysis: From Design to Applications, 2018, pp. 1–1200 Search PubMed.
- M. Filice, O. Romero, A. Aires, J. M. Guisan, A. Rumbero and J. M. Palomo, Adv. Synth. Catal., 2015, 357, 2687 CrossRef CAS.
- A. Chatterjee, H. Mallin, J. Klehr, J. Vallapurackal, A. D. Finke, L. Vera, M. Marsh and T. R. Ward, Chem. Sci., 2016, 7, 673 RSC.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512 CrossRef PubMed.
- P. Pocquet, N. Vologdin, G. F. Mangiatordi, I. Ciofini, O. Nicolotti, S. Thorimbert and M. Salmain, Eur. J. Inorg. Chem., 2017, 3622 CrossRef.
- P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083 RSC.
- Y. Yao, C.-L. Hou, Z.-S. Yang, G. Ran, L. Kang, C. Li, W. Zhang, J. Zhang and J.-L. Zhang, Chem. Sci., 2019, 10, 10170 RSC.
- L. Bischoff, C. Baudequin, C. Hoarau and E. P. Urriolabeitia, Adv. Organomet. Chem., 2018, 69, 73 CrossRef CAS.
- S. Collado, A. Pueyo, C. Baudequin, L. Bischoff, A. I. Jiménez, C. Cativiela, C. Hoarau and E. P. Urriolabeitia, Eur. J. Org. Chem., 2018, 6158 CrossRef CAS.
- S. J. Remington, Protein Sci., 2011, 20, 1509 CrossRef CAS PubMed.
- L. Brady, A. M. Brzozowski, Z. S. Derewenda, E. Dodson, G. Dodson, S. Tolley, J. P. Turkenburg, L. Christiansen, B. Huge-Jensen, L. Norskov, L. Thim and U. Menge, Nature, 1990, 343, 767 CrossRef CAS PubMed.
- C. Carrasco-Lopez, C. Godoy, B. de Las Rivas, G. Fernandez-Lorente, J. M. Palomo, J. M. Guisan, R. Fernandez-Lafuente, M. Martinez-Ripoll and J. A. Hermoso, J. Biol. Chem., 2009, 284, 4365 CrossRef CAS PubMed.
- G. Fernandez-Lorente, Z. Cabrera, C. Godoy, R. Fernandez-Lafuente, J. M. Palomo and J. M. Guisan, Process Biochem., 2008, 43, 1061 CrossRef CAS.
- J. Plöchl, Chem. Ber., 1883, 16, 2815 CrossRef.
- J. Plöchl, Chem. Ber., 1884, 17, 1623 CrossRef.
- E. Erlenmeyer, Justus Liebigs Ann. Chem., 1893, 275, 1 CrossRef.
- H. E. Carter, Org. React., 1946, 3, 198 Search PubMed.
- R. Filler, Advances in Heterocyclic Chemistry, ed. A. R. Katrizky, Academic Press, New York, 1954, ch. 4, p. 75 Search PubMed.
- Y. S. Rao and R. Filler, Synthesis, 1975, 749 CrossRef CAS.
- Y. S. Rao and R. Filler, Oxazolones, in Chemistry of Heterocyclic Compounds, ed. I. J. Turchi, John Wiley and Sons, New York, 1986, vol. 45, ch. 3, p. 361 Search PubMed.
- J. P. Lambooy, J. Am. Chem. Soc., 1954, 76, 133 CrossRef CAS.
- A. K. Barua, M. Chakrabarty, P. K. Datta and S. Ray, Phytochemistry, 1988, 27, 3259 CrossRef CAS.
- A. R. Khosropour, M. M. Khodaei and S. J. H. Jomor, J. Heterocycl. Chem., 2008, 45, 683 CrossRef CAS.
- D. Roiban, E. Serrano, T. Soler, I. Grosu, C. Cativiela and E. P. Urriolabeitia, Chem. Commun., 2009, 4681 RSC.
- G.-D. Roiban, E. Serrano, T. Soler, G. Aullon, I. Grosu, C. Cativiela, M. Martinez and E. P. Urriolabeitia, Inorg. Chem., 2011, 50, 8132 CrossRef CAS PubMed.
- E. Serrano, A. Juan, A. García-Montero, T. Soler, F. Jiménez-Márquez, C. Cativiela, M. V. Gomez and E. P. Urriolabeitia, Chem. – Eur. J., 2016, 22, 144 CrossRef CAS PubMed.
- E. P. Urriolabeitia, P. Sánchez, A. Pop, C. Silvestru, E. Laga, A. I. Jiménez and C. Cativiela, Beilstein J. Org. Chem., 2020, 16, 1111 CrossRef CAS PubMed.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697 CrossRef CAS PubMed.
- J. Jaramillo, I. Rodriguez-Oliva, O. Abian and J. M. Palomo, SN Appl. Sci., 2020, 2, 1731 CrossRef CAS.
- J. M. Palomo and E. P. Urriolabeitia, Gen. Chem., 2020, 6, 190030 Search PubMed.
- E. Menumerov, R. A. Hughes and S. Neretin, Catal. Sci. Technol., 2017, 7, 1460 RSC.
- L. Gregor, A. K. Reilly, T. A. Dickstein, S. Mazhar, S. Bram, D. G. Morgan, Y. Losovyj, M. Pink, B. D. Stein, V. G. Matveeva and L. M. Bronstein, ACS Omega, 2018, 3, 14717 CrossRef CAS PubMed.
- A. Fernández-Lodeiro, J. Djafari, D. Lopez-Tejedor, C. Perez-Rizquez, B. Rodríguez-González, J. L. Capelo, J. M. Palomo, C. Lodeiro and J. Fernández-Lodeiro, Nano Res., 2019, 12, 1083 CrossRef.
- M. Ródenas, J. El Haskouri, J. V. Ros-Lis, M. D. Marcos, P. Amorós, M. Á. Úbeda and F. Pérez-Pla, Catalysts, 2020, 10, 449 CrossRef.
- T. J. Williams, A. J. Reay, A. C. Whitwood and I. J. S. Fairlamb, Chem. Commun., 2014, 50, 3052 RSC.
- C. Perez-Rizquez, O. Abian and J. M. Palomo, Chem. Commun., 2019, 55, 12928 RSC.
- Y. Kim, Y. H. Ko, M. Jung, N. Selvapalam and K. Kim, Photochem. Photobiol. Sci., 2011, 10, 1415 CrossRef CAS PubMed.
- C. Schmidt-Dannert, M. L. Rua, H. Atomi and R. D. Schmid, Biochim. Biophys. Acta, 1996, 1301, 105 CrossRef.
- C. A. Godoy, B. de las Rivas, V. Grazu, T. Montes, J. M. Guisan and F. Lopez-Gallego, Biomacromolecules, 2011, 12, 1800 CrossRef CAS PubMed.
- SAINT, Version 5.0 ed, Bruker Analytical X-Ray Systems, Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS, Program for absorption and other corrections, Göttingen University, 1996 Search PubMed.
- G. M. Sheldrick, SHELXS-86, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-97, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra of all prepared compounds; IR spectra of selected compounds, fluorescence spectra, additional experiment information, etc. See DOI: 10.1039/d1ob00305d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |