
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Horeau amplification in the sequential acylative kinetic resolution of (±)-1,2-diols and (±)-1,3-diols in flow†
Arianna
Brandolese
ab,
Mark D.
Greenhalgh
ac,
Titouan
Desrues
d,
Xueyang
Liu
d,
Shen
Qu
a,
Cyril
Bressy
 *d and
Andrew D.
Smith
*a
*d and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
bDepartment of Chemical and Pharmaceutical Sciences, University of Ferrara, Via L. Borsari, 46, 44121 Ferrara, Italy
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
dAix Marseille Univ, CNRS, Centrale Marseille, iSm2 Marseille, France. E-mail: cyril.bressy@univ-amu.fr
First published on 8th March 2021
Abstract
The sequential acylative kinetic resolution (KR) of C2-symmetric (±)-1,2-syn and (±)-1,3-anti-diols using a packed bed microreactor loaded with the polystyrene-supported isothiourea, HyperBTM, is demonstrated in flow. The sequential KRs of C2-symmetric (±)-1,2-syn and (±)-1,3-anti-diols exploits Horeau amplification, with each composed of two successive KR processes, with each substrate class significantly differing in the relative rate constants for each KR process. Optimisation of the continuous flow set-up for both C2-symmetric (±)-1,2-syn and (±)-1,3-anti-diol substrate classes allowed isolation of reaction products in both high enantiopurity and yield. In addition to the successful KR of C2-symmetric (±)-1,2-syn and (±)-1,3-anti-diols, the application of this process to the more conceptually-complex scenario involving the sequential KR of C1-symmetric (±)-1,3-anti-diols was demonstrated, which involves eight independent rate constants.
Introduction
The Horeau principle (often referred to as Horeau amplification) is a powerful strategy that can be used within a synthetic process to generate material of high enantiopurity. These terms can be applied to synthetic reaction processes in which material of high enantiopurity is delivered either (i) from starting material of lower enantiopurity through conversion into diastereomeric products, subsequent separation and recovery; or (ii) through carrying out successive enantioselective transformations on polyfunctional starting materials.1 Initially demonstrated synthetically and formalised in a landmark manuscript by Horeau in 1973,2 the basic principles of this phenomena were expanded upon mathematically by Rautenstrauch and Kagan.3 Over the following decades the power of this process has been extended to a range of catalytic enantioselective sequences with an elegant review from Harned demonstrating the power of this process in a range of synthetic sequences.4A representative example of this powerful amplification process is the acylative double catalytic kinetic resolution (DoCKR) of C2-symmetric (±)-1,2-syn and (±)-1,3-anti-diols (Fig. 1).5–8 Due to the bis-alcohol functionality within the substrates, two KR processes can be in operation and are represented by the rate constants k1 and k2 for the first KR, and k3 and k4 for the second KR. If k1 > k2 and k3 > k4, the enantiomer of monoester preferentially generated in the 1st KR is also preferentially acylated in the 2nd KR (so called additive Horeau amplification). In principle, this allows the generation of both the diester (from two successive KRs) and diol in high enantiopurity and with opposite absolute configuration (shown in the blue and red boxes in Fig. 1). The enantiopurity and absolute configuration of the monoester product will be highly dependent upon reaction conversion, with its configuration matching that of the diester at low conversion, and the diol at high conversion. By necessity, there will be an inflection point between these two extremes where the monoester is racemic. In addition, the product distribution and hence relative yields of diester, diol and monoester obtained in such a sequential KR process will be dependent upon the relative magnitude of the combined rate constants for the 1st KR process relative to the 2nd KR process. If both KR processes are reasonably selective, then this can be simplified as the relationship between the largest rate constant for each KR step (i.e. in the example in Fig. 1, k1vs. k3).
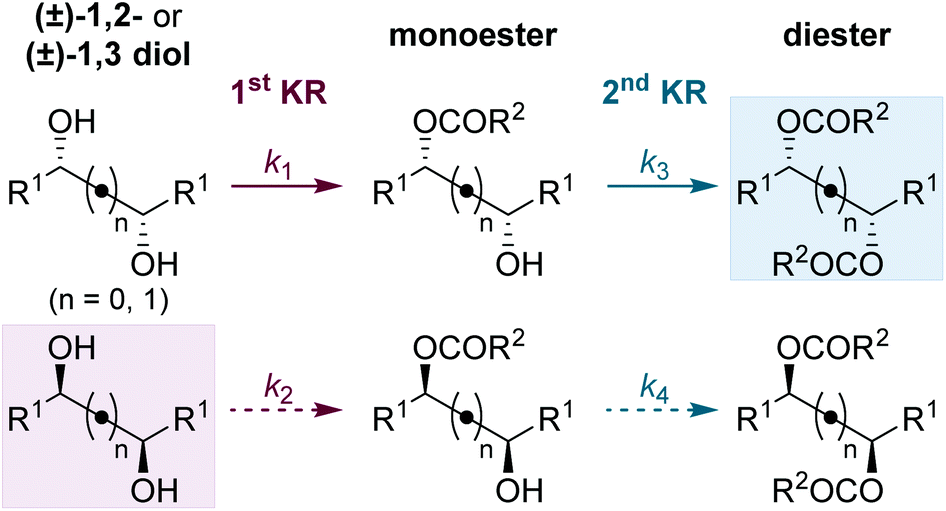 | ||
| Fig. 1 Horeau amplification in the sequential double acylative KR of C2-symmetric (±)-diols, where k1 > k2 and k3 > k4. | ||
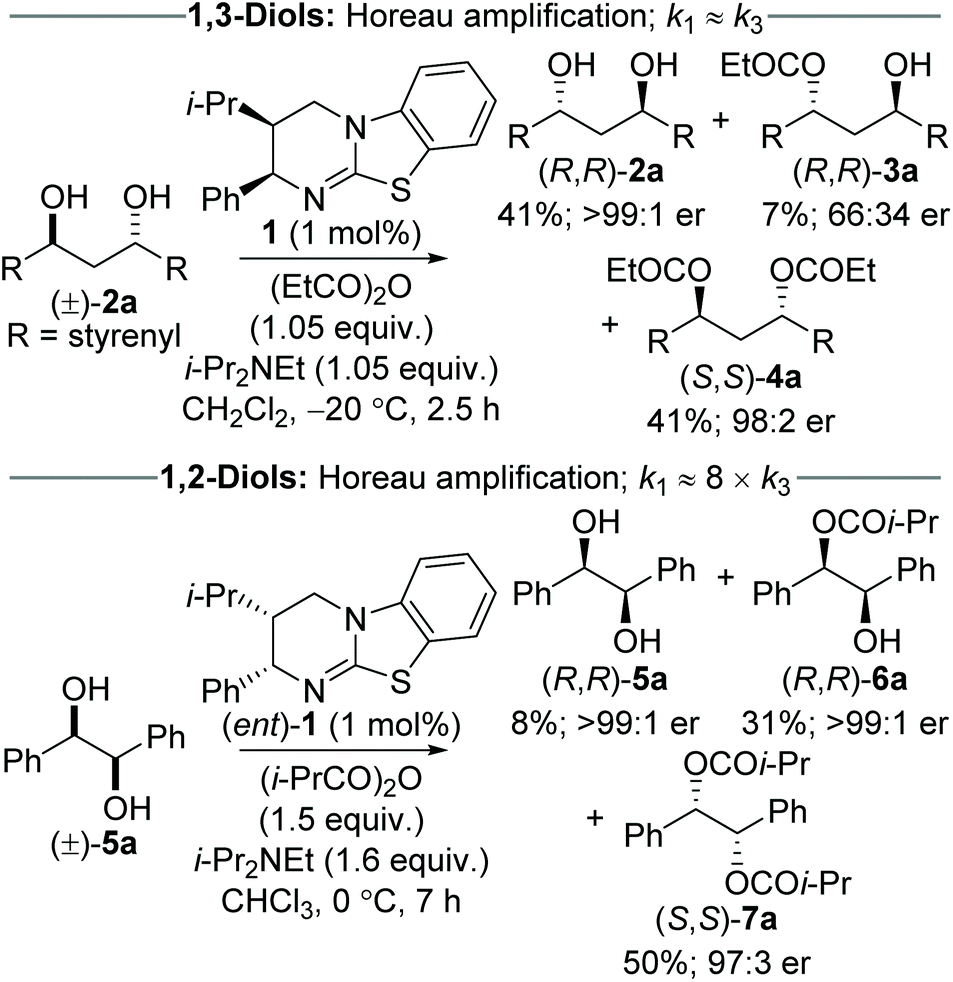
Despite the potential to harness additive Horeau amplification in these processes, the majority of examples of catalytic acylative KR of diols are selective for formation of the monoester product,5,6 and hence do not benefit from amplification of product enantiopurity imparted by the 2nd KR process. To date, only work from our research groups has demonstrated this concept, through the use of isothiourea catalysis in the sequential KR of (±)-1,3-anti and (±)-1,2-syn diols (Scheme 1).7,8 The sequential KR of (±)-1,3-anti-diol 2a was achieved using (2R,3S)-HyperBTM 1 as catalyst (1 mol%) and propionic anhydride as acylating agent (1.05 equiv.), to give (R,R)-diol 2a (41% yield, >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er), (R,R)-monoester 3a (7% yield, 66:34 er) and (S,S)-diester 4a (41% yield, 98:2 er). This process was shown to exhibit Horeau amplification, where both KR steps displayed the same sense of enantiodiscrimination.7 Based on the product distribution, it can be estimated that the relative rates of each KR process are close to parity (i.e. k1 ≈ k3). The sequential KR of (±)-1,2-syn-diol 5a was also shown to satisfy the conditions of Horeau amplification; however the relative rates of the two KR steps were significantly different, with the 1st KR being ∼8 times that of the 2nd KR.8 The significantly smaller rate constant for the second acylation can presumably be attributed to the increased steric hindrance associated with the α-ester substituent introduced following the first acylation event. This scenario makes it impossible to isolate diol and diester in both high yield and enantiopurity, and necessitates the sequential KR being driven to higher conversion to access highly enantiomerically enriched material. As such, treatment of (±)-1,2-syn-diol 5a with (2S,3R)-HyperBTM (ent)-1 (1 mol%) and isobutyric anhydride (1.5 equiv.) in CHCl3 gave (R,R)-diol 5a (8% yield, >99:1 er), (R,R)-monoester 6a (31% yield, >99:1 er) and (S,S)-diester 7a (50% yield, 97:3 er).8 In this case, the diol and monoester can be combined to give the (R,R)-enantiomer in 39% yield and >99:1 er. These two sequential KR processes, although related, demonstrate the complexity of these systems, and how modulation of reaction conversion can be used to maximise both the yield and enantiopurity of the various reaction products.
:1 er), (R,R)-monoester 3a (7% yield, 66:34 er) and (S,S)-diester 4a (41% yield, 98:2 er). This process was shown to exhibit Horeau amplification, where both KR steps displayed the same sense of enantiodiscrimination.7 Based on the product distribution, it can be estimated that the relative rates of each KR process are close to parity (i.e. k1 ≈ k3). The sequential KR of (±)-1,2-syn-diol 5a was also shown to satisfy the conditions of Horeau amplification; however the relative rates of the two KR steps were significantly different, with the 1st KR being ∼8 times that of the 2nd KR.8 The significantly smaller rate constant for the second acylation can presumably be attributed to the increased steric hindrance associated with the α-ester substituent introduced following the first acylation event. This scenario makes it impossible to isolate diol and diester in both high yield and enantiopurity, and necessitates the sequential KR being driven to higher conversion to access highly enantiomerically enriched material. As such, treatment of (±)-1,2-syn-diol 5a with (2S,3R)-HyperBTM (ent)-1 (1 mol%) and isobutyric anhydride (1.5 equiv.) in CHCl3 gave (R,R)-diol 5a (8% yield, >99:1 er), (R,R)-monoester 6a (31% yield, >99:1 er) and (S,S)-diester 7a (50% yield, 97:3 er).8 In this case, the diol and monoester can be combined to give the (R,R)-enantiomer in 39% yield and >99:1 er. These two sequential KR processes, although related, demonstrate the complexity of these systems, and how modulation of reaction conversion can be used to maximise both the yield and enantiopurity of the various reaction products.
 | ||
| Scheme 1 Acylative DoCKR of (±)-1,3- and (±)-1,2-diols using isothiourea catalysis. | ||
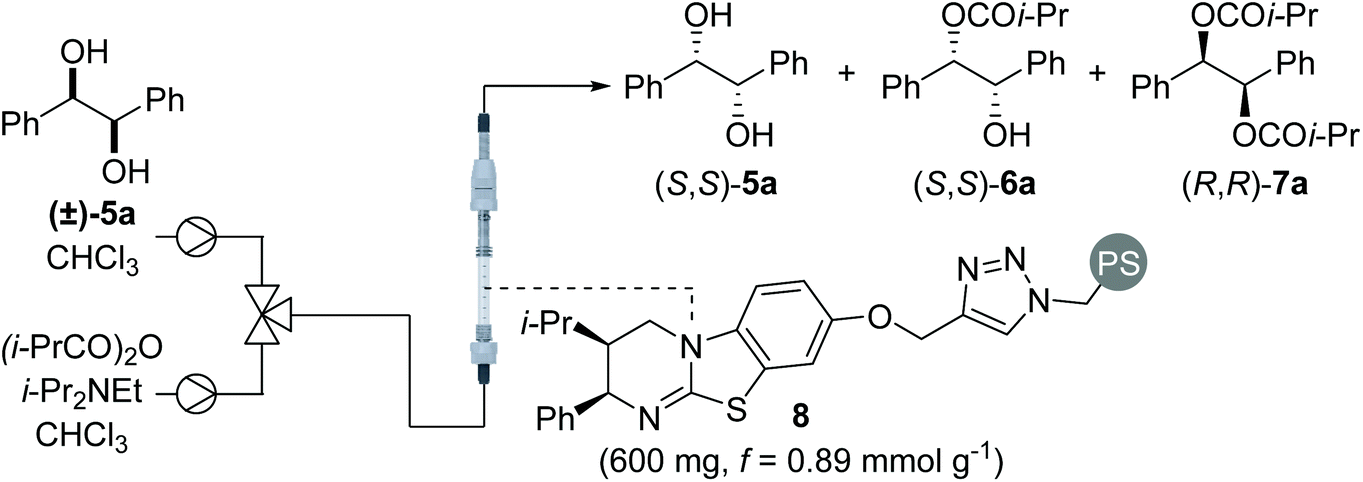
One approach to improve the sustainability of organocatalytic transformations is the immobilisation of the organocatalyst on a heterogeneous polymer support.9,10 In order to off-set the additional time and expense associated with synthesis, the supported catalyst must be readily recovered and recycled multiple times without loss in activity. The use of a packed bed reactor and application in a continuous flow set-up is a particularly-attractive technological solution that significantly simplifies catalyst recovery.9e–h The use of polymer-supported isothioureas as recyclable enantioselective catalysts was first demonstrated by Pericàs in formal cycloaddition processes.11 Further collaborative work between Pericàs and Smith has established the use of polymer-supported isothioureas for the KR of secondary alcohols, tertiary alcohols and BINOL derivatives in batch and flow.12 Building on these precedents, this manuscript investigates the possibility of performing the sequential KR of C2-symmetric (±)-1,2-syn- and (±)-1,3-anti-diols, as well as C1-symmetric (±)-1,3-anti-diols, in flow13 using a polymer-supported variant of HyperBTM.
Results and discussion
Sequential KR of C2-symmetric (±)-1,2-syn-diols in flow
Using a packed bed microreactor loaded with polystyrene-supported (2R,3S)-HyperBTM 8,12a initial studies focused on the KR of (±)-1,2-diphenylethane-1,2-diol 5a using isobutyric anhydride as the acylating agent and i-Pr2NEt as the base (Table 1). Unlike simple KRs where the selectivity factor (s) can be easily calculated using substrate or product enantiopurity and reaction conversion,14 interrogation of the selectivity of a sequential KR is complicated by the operation of two enantioselective processes.15 As such, reaction optimisation was assessed by targeting the isolation of all reaction components in high enantiopurity. Initially replicating the optimal conditions used in our previously-reported homogenous system,8 the use of 1.5 equivalents of isobutyric anhydride allowed isolation of diester (R,R)-7a and recovered diol (S,S)-5a with high enantioenrichment, however monoester (R,R)-6a was obtained with only moderate enantioenrichment (Table 1, entry 1). Encouraged by this initial result, increasing the enantioenrichment of monoester (1R,2R)-6a was targeted by increasing reaction conversion and aiming for ∼50% conversion to diester. Increasing the concentration of reagents was hampered by the low solubility of diol 5a in CHCl3 above 0.2 M, whilst decreasing the concentration of all the three reagents provided both the monoester 6a and diester 7a in lower enantiopurity (entry 2). Subsequent studies focused on increasing the equivalents of anhydride and base.16 Using 2 equivalents of anhydride gave almost full consumption of diol 5a and 59% conversion to the diester 7a (entry 3). This high conversion had a negative impact on the enantiopurity of diester 7a, demonstrating the need for a subtle balance to obtain all three products in high enantiopurity. Using 1.75 equivalents of anhydride provided the ideal compromise, with all three products obtained in high enantiopurity (entry 4). Finally, the method for delivering the reagents was varied, with the combination of diol and anhydride in a single syringe proving more optimal that the initial set-up, in which the anhydride and base were delivered from the same syringe. Increasing the scale of this process to 2 mmol allowed isolation of diester (R,R)-7a in 48% yield and 94:6 er, along with the (S,S)-enantiomer of both diol 5a and monoester 6a in >99:1 er and 98:2 er, respectively, and a combined yield of 38% (entry 5).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | (±)-5a [M] | (i-PrCO)2O [M] | i-Pr2Net [M] | Product ratio (5a:6a:7a)b |
5a er (S,S):(R,R)c |
6a er (S,S):(R,R)c |
7a er (R,R):(S,S)c |
| a Conditions: (±)-5a (0.5 mmol), r.t., flow rate 0.1 mL min−1. b Determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. c Determined by CSP-HPLC analysis. d Diol and anhydride in one syringe, i-Pr2NEt in the other. e 2.0 mmol scale. f Isolated yield. | |||||||
| 1 | 0.2 | 0.3 | 0.32 | 17:39:44 |
99:1 |
85:15 |
95:5 |
| 2 | 0.1 | 0.15 | 0.16 | 10:44:46 |
>99:1 |
75:25 |
94:6 |
| 3 | 0.2 | 0.4 | 0.44 | 1:40:59 |
— | >99:1 |
75:25 |
| 4 | 0.2 | 0.35 | 0.35 | 7:41:52 |
99:1 |
97:3 |
90:10 |
| 5 , | 0.2 | 0.35 | 0.35 |
7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :40:53 :40:53
|
>99:1 (3%)
|
98:2 (35%)
|
94:6 (48%)
|
Using the conditions optimised for diol 5a, the generality of this sequential KR process in flow was investigated using a selection of electronically- and sterically-differentiated (±)-1,2-diols (Scheme 2). (±)-1,2-Diarylethane-1,2-diol derivatives 5b–5d were all resolved with excellent selectivity with results comparable with those obtained with the homogeneous catalyst.8 The sequential KR of p-methoxy-substituted diol 5d provided products with higher enantioenrichment than substrates 5b and 5c bearing electron-withdrawing p-chloro or p-trifluoromethyl substituents. These results are consistent with the KR of simple benzylic alcohols using isothiourea catalysts, where the presence of an electron-rich aromatic substituent provides enhanced stabilisation of the acylation transition state through π–cation interactions.17 The method was also extended to naphthyl-substituted diol 5e, however low solubility of 5e in CHCl3 required the use of a different solvent system (1:1 THF:CHCl3). Under these conditions, diol 5e and diester 7e were recovered with high enantioenrichment (97:3 er and 94:6 er, respectively); while monoester 6e was obtained with only moderate enantioenrichment (70:30 er). This can be attributed to slightly lower conversion being observed, presumably reflecting the reduced swelling of the catalyst resin in 1:1 THF:CHCl3 and consequently leading to a lower residence time. Finally, the KR of (±)-1,2-diols 5f and 5g bearing alternative π-donor systems (alkene and alkyne) was also performed. Only moderate selectivity, coupled with higher conversions were obtained in both cases, consistent with the sequential KR of these substrates using the homogenous catalyst.8 These results are also consistent with the observation that the isothiourea-catalysed KR of simple allylic and propargylic alcohols is generally less selective than the KR of benzylic alcohols.17
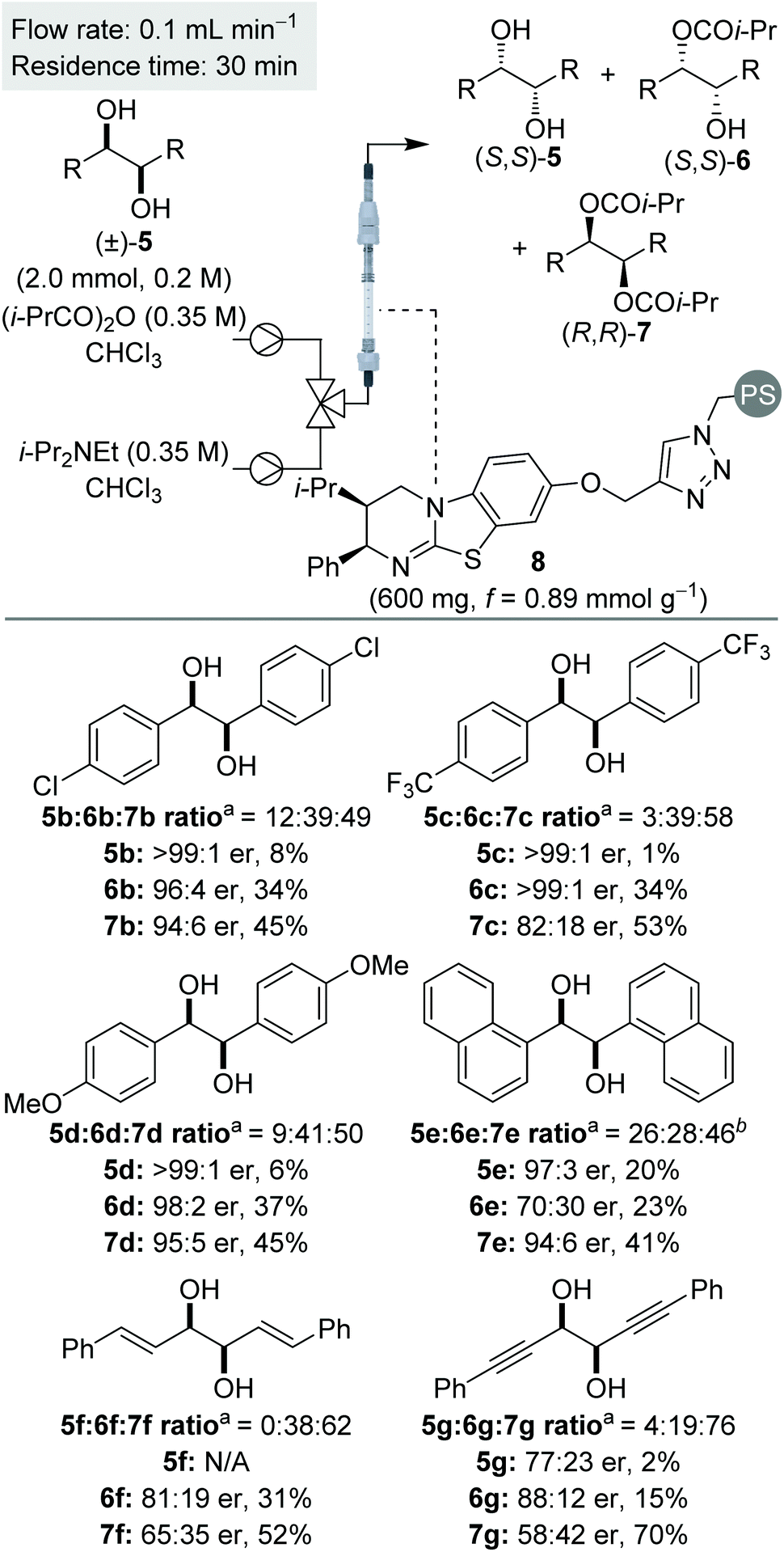 | ||
| Scheme 2 Reaction scope for C2-symmetric (±)-syn-1,2-diols. aRatio determined by 1H NMR spectroscopic analysis of the crude reaction product. bSolvent 1:1 THF:CHCl3. | ||
Sequential KR of C2-symmetric (±)-1,3-anti-diols in flow
Considering the positive outcome of the sequential KR of 1,2-diols in continuous flow, we sought to extend this process to (±)-anti-1,3-diols. In our previous work on the sequential KR of 1,3-diols using a homogenous isothiourea catalyst, the KRs were performed at −20 °C in CH2Cl2,7 however low substrate solubility under these conditions prohibited use of these conditions in flow. Low diol solubility was also observed when using CHCl3, however using 1:1 THF:CHCl3 at room temperature provided good solubility. To test the suitability of this solvent system for KR selectivity, control experiments were conducted using the homogenous catalyst, HyperBTM 1, in both CH2Cl2 and 1:1 THF:CHCl3 for the KR of 1,3-diol 2a at room temperature.16 Comparable conversion and product enantioenrichment was obtained and therefore process optimisation in flow was conducted using this mixed solvent system. A brief optimisation was conducted using 1,3-diol 2a, with the use of fewer equivalents of anhydride, lower reagent concentration and a higher flow rate, relative to the KR of 1,2-diols, found to be optimal.16
Using the optimised conditions, the sequential KR of (±)-anti-1,3-diols was explored (Scheme 3). In all cases, the (±)-anti-1,3-diols substrates were synthesised in >95:5 dr, with only trace amounts of the syn-diastereomer present. In keeping with the KR of these substrates using the homogenous catalyst,7 the diol and diester products were obtained in consistently good yield and enantioenrichment, whilst the monoester was isolated as a minor component with low enantioenrichment in each case. Also in keeping with the previous work,7 the monoester was generally isolated as a mixture of anti- and syn-diastereomers, whilst both diol and diester were obtained as essentially single diastereomers. This phenomenon results in the sequential KR enhancing the diastereopurity of the products, in addition to the anticipated separation of enantiomers, and can presumably be attributed to a small rate constant for the 2nd acylation for the syn-diastereomer.
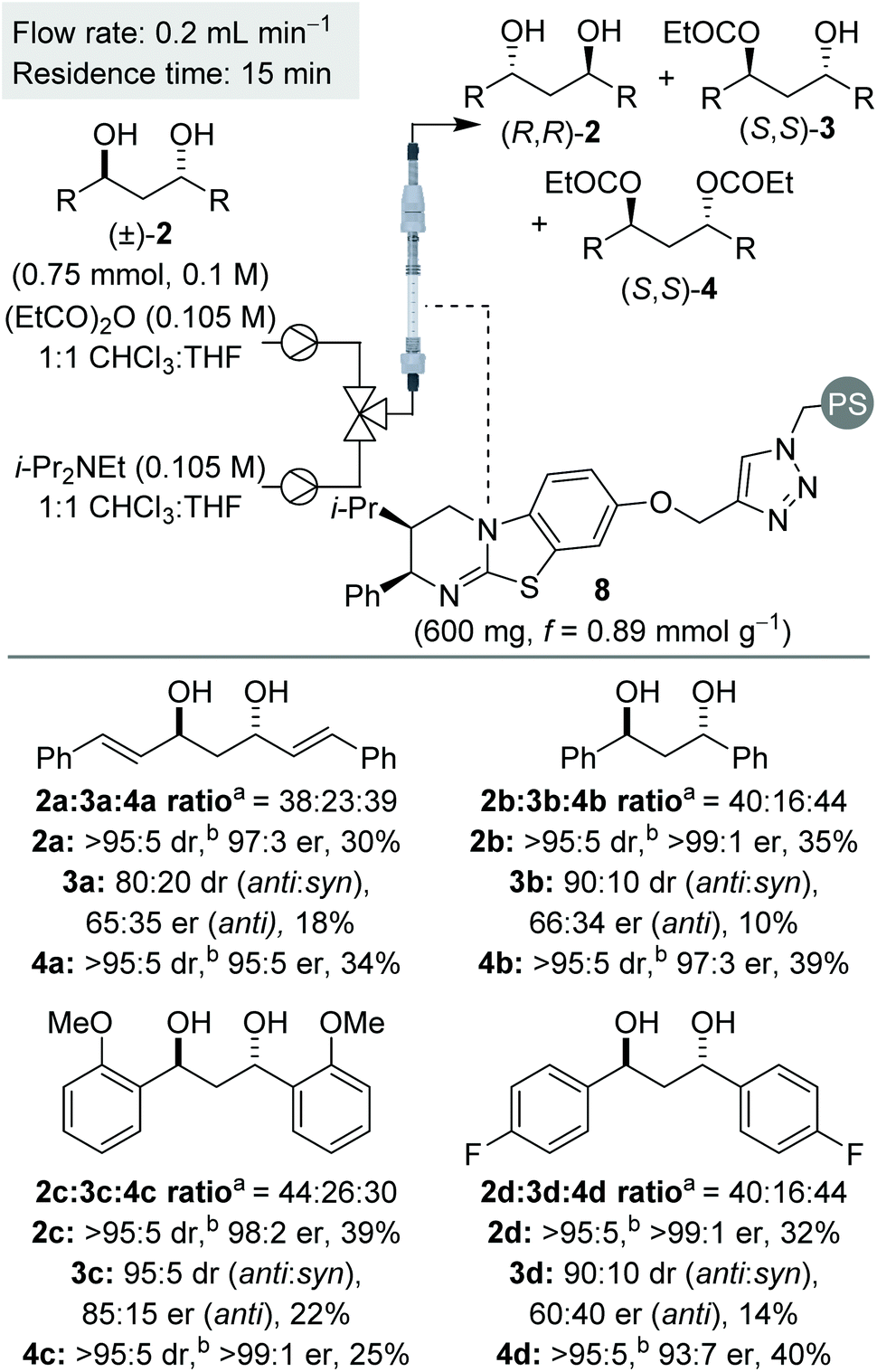 | ||
| Scheme 3 Reaction scope for C2-symmetric (±)-anti-1,3-diols. aRatio determined by 1H NMR spectroscopic analysis of the crude reaction product. bdr given is anti:syn ratio of diastereomers. er shown corresponds to anti diastereomer. | ||
Initially, the sequential KR process was applied to C2-symmetric 1,3-diols 2a–d, bearing a π-system adjacent to the reactive carbinol centres (Scheme 3). Allylic diol 2a was resolved with both diol and diester obtained with excellent enantioenrichment. This result provides some contrast to the KR of 1,2-diols, where the resolution of allylic and propargylic diols was significantly less selective. The KR of benzylic 1,3-diol 2b was achieved with even higher selectivities, again consistent with the usual observation that benzylic alcohols are resolved more selectively than allylic alcohols.17 Variation of the electronic nature of the aromatic substituents was next probed, with the introduction of either electron-donating or electron-withdrawing substituents (2c and 2d) providing diol and diester products with excellent enantioenrichment.
Sequential KR of C1-symmetric (±)-anti-1,3-diols in flow
Next, we were intrigued to investigate the KR of C1-symmetric (±)-anti-1,3-diols in flow. For the KR of these substrates, the kinetic scenario is further complicated by the intermediacy of four isomeric monoesters: two sets of enantiomers arising from acylation of either hydroxyl group (Scheme 4). Overall this leads to a sequential KR composed of 8 independent rate constants, where both the formation and consumption of each isomeric monoester is controlled by the chiral isothiourea catalyst.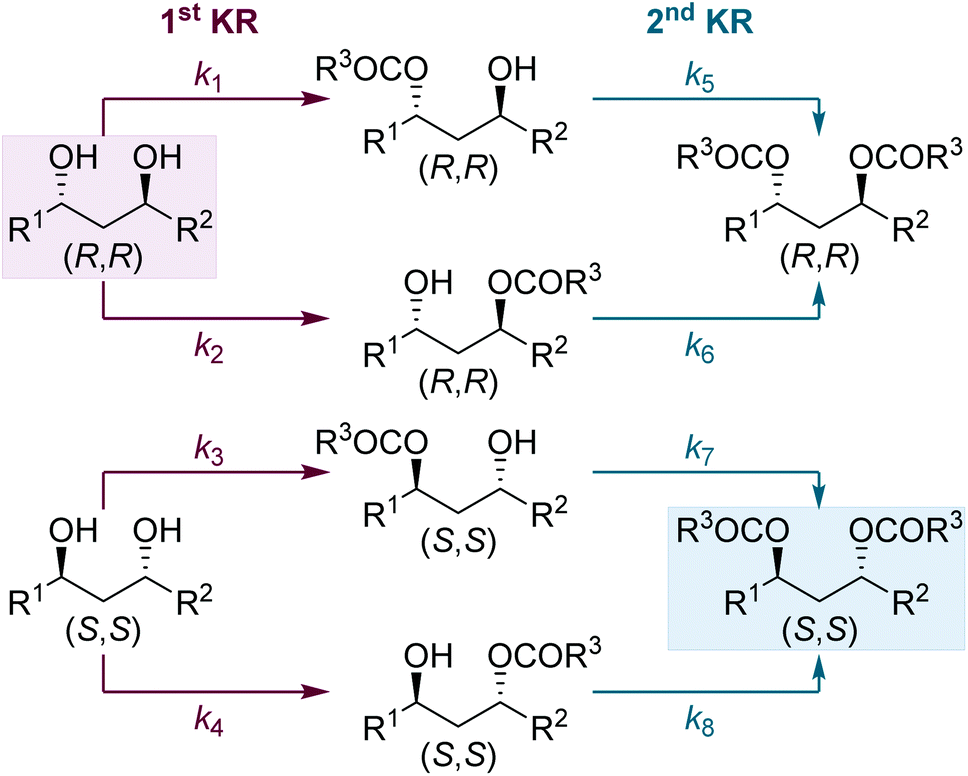 | ||
| Scheme 4 Double organocatalytic kinetic resolution of C1-symmetric (±)-anti-1,3-diols. | ||
To probe this scenario, two C1-symmetric (±)-anti-1,3-diols, 2e and 2f, were applied in the sequential KR process with excellent selectivity. Diols 2e and 2f, and their corresponding diesters, 4e and 4f, were recovered in good yield and with high enantioenrichment. The corresponding monoesters 3e′ and 3e′′ and 3f′ and 3f′′ were all isolated with low er and with lower dr than the starting anti-1,3-diols. Of particular note, the two constitutional isomers of monoester obtained from each reaction (for example e′ and e′′) were determined to have opposite configuration: whilst monoester e′/f′ was enriched in the same enantiomeric series as the (R,R)-diol, the monoester e′′/f′′ was enriched in the same enantiomeric series as the (S,S)-diester. At low conversion both constitutional isomers of monoester would be enriched in the (S,S)-enantiomer, whilst at high conversion both constitutional isomers of monoester would be enriched in the (R,R)-enantiomer. The intriguing results displayed in Scheme 5 can therefore be explained as a product of the 8 independent rate constants outlined above and the reaction conversion, which is controlled by the concentration of reagents, catalyst bed loading and flow-rate. Overall these results conveniently highlight the kinetic complexity of this system, and embody many of the principles popularised by Horeau.
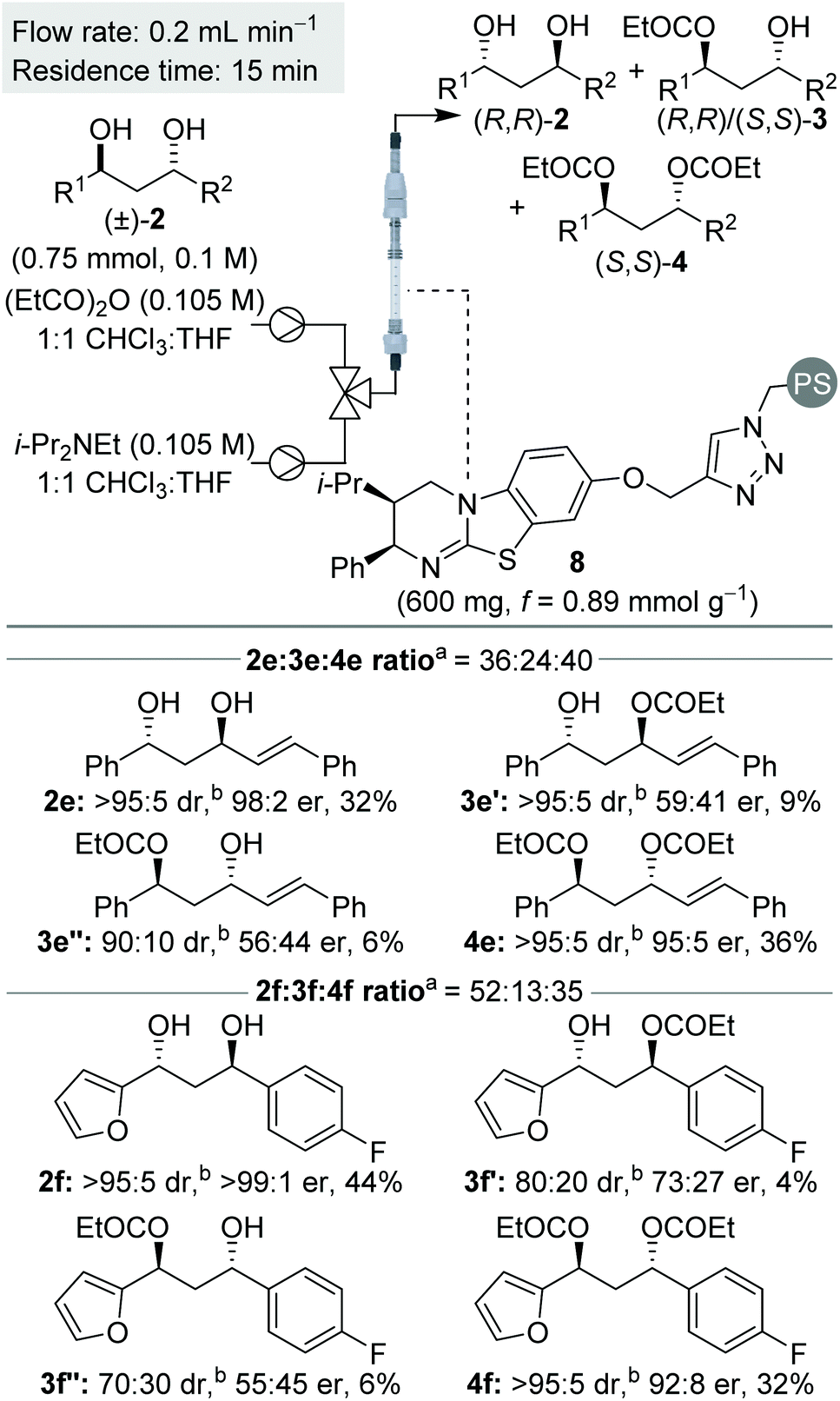 | ||
| Scheme 5 Reaction scope for C1-symmetric (±)-anti-1,3-diols. aRatio determined by 1H NMR spectroscopic analysis of the crude reaction product. bdr given is anti:syn ratio of diastereomers. er shown corresponds to anti diastereomer. | ||
Finally, to provide further insight into these processes, and contrast the sequential KR of (±)-syn-1,2- and (±)-anti-1,3-diols, selectivity values (s)14 for the individual KR steps were calculated for each process (Scheme 6). The s value for the first KR step was evaluated in each case by using the conversion and er of the recovered diol. This calculation assumes the diol only participates in the first step, and that its er is unaffected by the KR of the monoester. Due to the high conversion required for the sequential KR of (±)-syn-1,2-diols, the s value calculated for the 1st KR would be very inaccurate and therefore the sequential KR was repeated using 1 equiv. of anhydride.16 The s value for the 2nd KR is more conveniently assessed by performing a simple KR of a racemic sample of monoester. For the sequential KR of (±)-syn-1,2-diols, the s values for the 1st and 2nd KR steps were calculated as 13 and 38, respectively, demonstrating the 2nd KR step is significantly more selective than the 1st KR. This trend is in keeping with the sequential KR process using the homogenous catalyst,8,12a albeit the magnitude of the s values in flow are slightly lower than observed in batch. For the KR of (±)-anti-1,3-diols, the opposite relationship between the two KR steps was observed, with the s values for the 1st and 2nd KR steps calculated as 12 and 7, respectively, demonstrating the 1st KR step is more selective. This subtle difference between the KR of 1,2- and 1,3-diols, in addition to the difference in product distribution obtained due to different overall rate constants for diol and monoester acylation, highlights how appreciation of Horeau's pioneering work and insight is essential for understanding these sequential KR processes. It is also notable that although none of the individual s values are particularly high (e.g. >50), the products are still obtained in highly enantioenriched form due to the operation of the additive Horeau amplification.
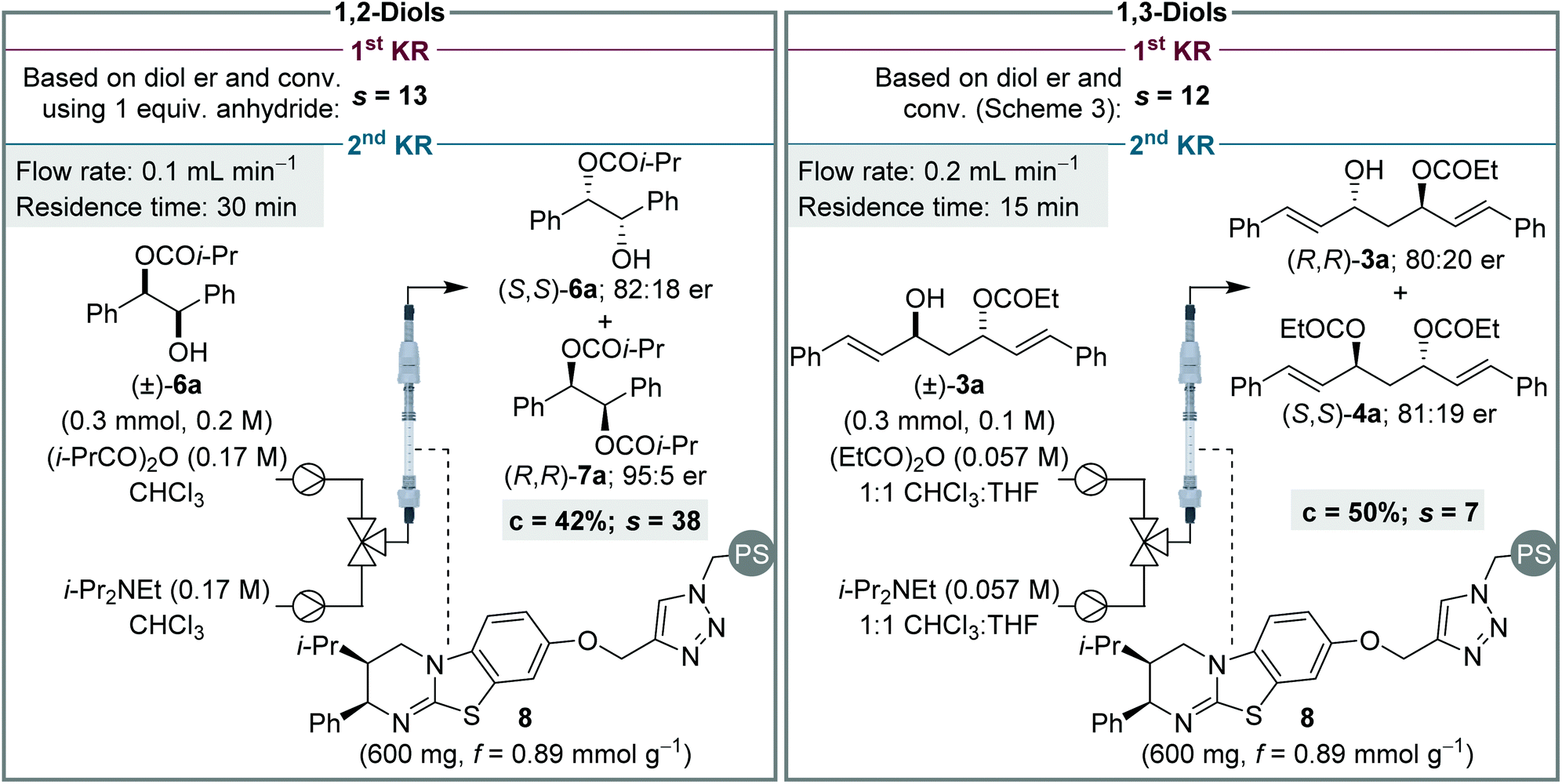 | ||
| Scheme 6 Assessment and comparison of s values for individual KR steps for the sequential KR of both (±)-1,2- and (±)-1,3-diols. | ||
Conclusions
In conclusion an isothiourea-catalysed sequential kinetic resolution of (±)-1,2-syn-diols and (±)-1,3-anti-diols in continuous flow has been presented. Excellent results have been obtained for both processes, allowing isolation of diol, diester, and in some cases monoester products in excellent enantiopurity and yield. It is notable that the developed procedure has been conducted at room temperature, in place of the low temperatures typically used when using the homogeneous version of the catalyst.7,8,17 The use of room temperature makes the process operationally simple to perform, whilst the use of a continuous flow system simplifies catalyst recovery and recycling to improve the overall sustainability of the process. Moreover, the same packed bed reactor loaded with immobilised HyperBTM resin was used for all optimisation and reaction scope experiments reported in this manuscript, without loss in efficiency.18 This equates to more than 100 h operation time using the same catalyst and demonstrates the highly recyclable nature of this immobilised organocatalyst.19Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the University of Ferrara (Bando Giovani Ricercatori 2019) for financial support (AB). The Chinese Scholarship Scheme and University of St Andrews are thanked for a CSC Scholarship (S. Q.). Aix-Marseille University, CNRS, and ANR (Agence Nationale de la Recherche) (AMPLI project ANR-18-CE07-0036) are also gratefully acknowledged for funding (TD, XL and CB).Notes and references
- For selected examples of this principle see: (a) M. Suginome, T. Iwanami, Y. Ohmori, A. Matsumoto and Y. Ito, Chem. – Eur. J., 2005, 11, 2954–2965 CrossRef CAS; (b) C. Yu, Y. Zhang, A. Song, Y. Ji and W. Wang, Chem. – Eur. J., 2011, 17, 770–774 CrossRef CAS PubMed; (c) P. K. Dornan, P. L. Leung and V. Y. Dong, Tetrahedron, 2011, 67, 4378–4384 CrossRef CAS; (d) L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344–16347 CrossRef CAS PubMed; (e) M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed; (f) J. Merad, P. Borkar, T. Bouyon Yenda, C. Roux, J.-M. Pons, J.-L. Parrain, O. Chuzel and C. Bressy, Org. Lett., 2015, 17, 2118–2121 CrossRef CAS PubMed; (g) Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900–8903 CrossRef CAS PubMed; (h) D. A. Tickell, E. V. Lampard, J. P. Lowe, T. D. James and S. D. Bull, J. Org. Chem., 2016, 81, 6795–6799 CrossRef CAS PubMed; (i) S. Tong, A. Limouni, Q. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 14192–14196 CrossRef CAS PubMed; (j) X. Bao, J. Rodriguez and D. Bonne, Chem. Sci., 2020, 11, 403–408 RSC; (k) Q.-Y. Feng, J. Zhu, M.-Z. Wang and S. Tong, Org. Lett., 2020, 22, 483–487 CrossRef CAS PubMed.
- J. P. Vigneron, M. Dhaenes and A. Horeau, Tetrahedron, 1973, 29, 1055–1059 CrossRef CAS.
- (a) V. Rautenstrauch, Bull. Soc. Chim. Fr., 1994, 131, 515–524 CAS; (b) S. El Baba, K. Sartor, J.-C. Poulin and H. B. Kagan, Bull. Soc. Chim. Fr., 1994, 131, 525–533 CAS For an historical perspective see: (c) G. Heller, Angew. Chem., Int. Ed., 2000, 39, 495–499 CrossRef For selected other related review topics see: (d) P. L. Polavarapu, Org. Biomol. Chem., 2020, 18, 6801–6806 RSC.
- For an excellent overview see: (a) A. M. Harned, Tetrahedron, 2018, 74, 3797–3841 CrossRef CAS For selected other reviews see: (b) L. Frasson and C. Moberg, ChemCatChem, 2010, 2, 1523–1532 CrossRef; (c) D. S. Glueck, Catal. Sci. Technol., 2011, 1, 1099–1108 RSC.
- For Lewis base-catalysed acylative KRs of (±)-1,2-diols, that in each case result in only minimal formation of the diester, see: (a) S. Mizuta, Y. Ohtsubo, T. Tsuzuki, T. Fujimoto and I. Yamamoto, Tetrahedron Lett., 2006, 47, 8227–8229 CrossRef CAS; (b) C. E. Müller, L. Wanka, K. Jewell and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 6180–6183 CrossRef PubMed; (c) R. Hrdina, C. E. Müller and P. R. Schreiner, Chem. Commun., 2010, 46, 2689–2690 RSC; (d) R. Hrdina, C. E. Müller, R. C. Wende, L. Wanka and P. R. Schreiner, Chem. Commun., 2012, 48, 2498–2500 RSC; (e) C. E. Müller, D. Zell, R. Hrdina, R. Wende, L. Wanka, S. M. Schuler and P. R. Schreiner, J. Org. Chem., 2013, 78, 8465–8484 CrossRef PubMed; (f) C. Hofmann, S. M. M. Schuler, R. C. Wende and P. R. Schreiner, Chem. Commun., 2014, 50, 1221–1223 RSC; (g) C. Hofmann, J. M. Schümann and P. R. Schreiner, J. Org. Chem., 2015, 80, 1972–1978 CrossRef CAS PubMed; (h) S. Kuwano, S. Harada, B. Kang, R. Oriez, Y. Yamaoka, K. Takasu and K.-I. Yamada, J. Am. Chem. Soc., 2013, 135, 11485–11488 CrossRef CAS PubMed; (i) S. Iwahana, H. Iida and E. Yashima, Chem. – Eur. J., 2011, 17, 8009–8013 CrossRef CAS PubMed; (j) K. Fujii, K. Mitsudo, H. Mandai and S. Suga, Adv. Synth. Catal., 2017, 359, 2778–2788 CrossRef CAS; (k) H. Mandai, K. Fujii, H. Yasuhara, K. Abe, K. Mitsudo, T. Korenaga and S. Suga, Nat. Commun., 2016, 7, 11297 CrossRef PubMed.
- For other non-enzymatic catalytic acylative KRs of (±)-1,2-diols see: (a) Y. Matsumura, T. Maki, S. Murakami and O. Onomura, J. Am. Chem. Soc., 2003, 125, 2052–2053 CrossRef CAS PubMed; (b) C. Mazet, S. Roseblade, V. Kohler and A. Pfaltz, Org. Lett., 2006, 8, 1879–1882 CrossRef CAS PubMed; (c) S. Jammi, L. Rout, P. Saha, V. K. Akkilagunta, S. Sanyasi and T. Punniyamurthy, Inorg. Chem., 2008, 47, 5093–5098 CrossRef CAS PubMed; (d) R. Rasappan, T. Olbrich and O. Reiser, Adv. Synth. Catal., 2009, 351, 1961–1967 CrossRef CAS; (e) M. Durini, E. Russotto, L. Pignataro, O. Reiser and U. Piarulli, Eur. J. Org. Chem., 2012, 5451–5461 CrossRef CAS.
- J. Merad, P. Borkar, F. Caijo, J.-M. Pons, J.-L. Parrain, O. Chuzel and C. Bressy, Angew. Chem., Int. Ed., 2017, 56, 16052–16056 CrossRef CAS PubMed.
- S. Harrer, M. D. Greenhalgh, R. M. Neyyappadath and A. D. Smith, Synlett, 2019, 30, 1555–1560 CrossRef CAS.
- (a) M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401–3429 CrossRef CAS PubMed; (b) A. F. Trindade, P. M. P. Gois and C. A. M. Alfonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed; (c) T. E. Kristensen and T. Hansen, Eur. J. Org. Chem., 2010, 3179–3204 CrossRef CAS; (d) B. Altava, M. I. Burguete, E. Garcia-Verdugo and S. V. Luis, Chem. Soc. Rev., 2018, 47, 2722–2771 RSC; (e) C. Rodríguez-Escrich and M. A. Pericàs, Eur. J. Org. Chem., 2015, 1173–1188 CrossRef; (f) C. Rodríguez-Escrich and M. A. Pericàs, Chem. Rec., 2019, 19, 1872–1890 CrossRef PubMed; (g) T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada and P. Li, Chem. – Eur. J., 2020, 26, 5729–5747 CrossRef CAS PubMed; (h) C. De Risi, O. Bortolini, A. Brandolese, G. Di Carmine, D. Ragno and A. Massi, React. Chem. Eng., 2020, 5, 1017–1052 RSC.
- For KRs of alcohols using solid-supported catalysts see the following: immobilized peptides and transition metal catalysts: (a) G. T. Copeland and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 6496–6502 CrossRef CAS PubMed; (b) A. Gissibl, M. G. Finn and O. Reiser, Org. Lett., 2005, 7, 2325–2328 CrossRef CAS PubMed; (c) A. Gissibl, C. Padié, M. Hager, F. Jaroschik, R. Rasappan, E. Cuevas-Yañez, C.-O. Turrin, A.-M. Caminade, J.-P. Majoral and O. Reiser, Org. Lett., 2007, 9, 2895–2898 CrossRef CAS PubMed; (d) A. Schätz, M. Hager and O. Reiser, Adv. Funct. Mater., 2009, 19, 2109–2115 CrossRef For supported Lewis base catalysts for the KR of alcohols see: (e) B. Clapham, C.-W. Cho and K. D. Janda, J. Org. Chem., 2001, 66, 868–873 CrossRef CAS PubMed; (f) B. Pelotier, G. Priem, I. B. Campbell, S. J. F. Macdonald and M. S. Anson, Synlett, 2003, 679–683 CAS; (g) K. Ishihara, Y. Kosugi and M. Akakura, J. Am. Chem. Soc., 2004, 126, 12212–12213 CrossRef CAS PubMed; (h) Y. Kosugi, M. Akakura and K. Ishihara, Tetrahedron, 2007, 63, 6191–6203 CrossRef CAS; (i) O. Gleeson, R. Tekoriute, Y. K. Gun'ko and S. J. Connon, Chem. – Eur. J., 2009, 15, 5669–5673 CrossRef CAS PubMed.
- (a) J. Izquierdo and M. A. Pericàs, ACS Catal., 2016, 6, 348–356 CrossRef CAS; (b) S. Wang, J. Izquierdo, C. Rodríguez-Escrich and M. A. Pericàs, ACS Catal., 2017, 7, 2780–2785 CrossRef CAS; (c) S. Wang, C. Rodríguez-Escrich and M. A. Pericàs, Angew. Chem., Int. Ed., 2017, 56, 15068–15072 CrossRef CAS PubMed.
- (a) R. M. Neyyappadath, R. Chisholm, M. D. Greenhalgh, C. Rodríguez-Escrich, M. A. Pericàs, G. Hähner and A. D. Smith, ACS Catal., 2018, 8, 1067–1075 CrossRef CAS; (b) N. R. Guha, R. M. Neyyappadath, M. D. Greenhalgh, R. Chisholm, S. M. Smith, M. L. McEvoy, C. M. Young, C. Rodríguez-Escrich, M. A. Pericàs, G. Hähner and A. D. Smith, Green Chem., 2018, 20, 4537–4546 RSC; (c) J. Lai, R. M. Neyyappadath, A. D. Smith and M. A. Pericàs, Adv. Synth. Catal., 2020, 362, 1370–1377 CrossRef CAS; (d) S. Qu, S. M. Smith, V. Laina-Martín, R. M. Neyyappadath, M. D. Greenhalgh and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 16572–16578 CrossRef CAS PubMed.
- The KR of a 1,2-diol using a Cu(II)-azabis(oxazoline) catalyst supported on a Co/C nanoparticle has been reported in continuous flow, see: (a) A. Schätz, R. N. Grass, Q. Kainz, W. J. Stark and O. Reiser, Chem. Mater., 2010, 22, 305–310 CrossRef For the non-catalytic acylative parallel KR of amines in continuous flow see: (b) I. Kreituss and J. W. Bode, Nat. Chem., 2017, 9, 446–452 CrossRef CAS.
- (a) H. B. Kagan and J. C. Fiaud, Top. Stereochem., 1988, 18, 249–330 CAS; (b) M. D. Greenhalgh, J. E. Taylor and A. D. Smith, Tetrahedron, 2018, 74, 5554–5560 CrossRef CAS.
- W. Kroutil, A. Kleewein and K. Faber, Tetrahedron: Asymmetry, 1997, 8, 3263–3274 CrossRef CAS.
- See ESI† for full details.
- For representative examples see: (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed; (b) V. B. Birman and L. Guo, Org. Lett., 2006, 8, 4859–4861 CrossRef CAS PubMed; (c) I. Shiina and K. Nakata, Tetrahedron Lett., 2007, 48, 8314–8317 CrossRef CAS; (d) V. B. Birman and X. Li, Org. Lett., 2008, 10, 1115–1118 CrossRef CAS PubMed; (e) Q. Hu, H. Zhou, X. Geng and P. Chen, Tetrahedron, 2009, 65, 2232–2238 CrossRef; (f) I. Shiina, K. Nakata, K. Ono, M. Sugimoto and A. Sekiguchi, Chem. – Eur. J., 2010, 16, 167–172 CrossRef CAS PubMed; (g) D. Belmessieri, C. Joannesse, P. A. Woods, C. MacGregor, C. Jones, C. D. Campbell, C. P. Johnston, N. Duguet, C. Concellón, R. A. Bragg and A. D. Smith, Org. Biomol. Chem., 2011, 9, 559–570 RSC; (h) X. Li, H. Jiang, E. W. Uffman, L. Guo, Y. Zhang, X. Yang and V. B. Birman, J. Org. Chem., 2012, 77, 1722–1737 CrossRef CAS PubMed; (i) K. Nakata, K. Gotoh, K. Ono, K. Futami and I. Shiina, Org. Lett., 2013, 15, 1170–1173 CrossRef CAS PubMed; (j) I. Shiina, K. Ono and T. Nakahara, Chem. Commun., 2013, 49, 10700–10702 RSC; (k) S. F. Musolino, O. S. Ojo, N. J. Westwood, J. E. Taylor and A. D. Smith, Chem. – Eur. J., 2016, 22, 18916–18922 CrossRef CAS PubMed For review, see: ; (l) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589–5610 CrossRef CAS.
- The efficiency of the packed bed catalyst was evaluated prior to starting this study and at the end of the work, by conducting a KR of (±)-1-phenyethanol as reported in ref. 12a.
- The research data underpinning this publication can be found at: https://doi.org/10.17630/5476cf74-a501-4121-b3ec-e2271fed0bcc.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation and HPLC analyses. See DOI: 10.1039/d1ob00304f |
| This journal is © The Royal Society of Chemistry 2021 |