
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eosin: a versatile organic dye whose synthetic uses keep expanding
Artemis
Bosveli
,
Tamsyn
Montagnon
,
Dimitris
Kalaitzakis
 and
Georgios
Vassilikogiannakis
*
and
Georgios
Vassilikogiannakis
*
Department of Chemistry, University of Crete, Vasilika Vouton, 71003 Iraklion, Crete, Greece. E-mail: vasil@uoc.gr
First published on 19th March 2021
Abstract
Organic dyes, which absorb light in the visible region of the electromagnetic spectrum, offer a lower cost, greener alternative to precious metals in photocatalysis. In this context, the organic dye eosin's uses are currently expanding at a significant rate. For a long time, its action as an energy transfer agent dominated, more recently, however, there has been a growing interest in its potential as an electron transfer agent. In this short review, we highlight some recent (from 2016 onwards) contributions to the field with a focus on the breadth of the reactions eosin can catalyse.
 From left to right in the photo: Artemis Bosveli, Dr Tamsyn Montagnon, Dr Dimitris Kalaitzakis, Prof. Georgios Vassilikogiannakis | Artemis Bosveli obtained her M.Sc. (2019) with Prof. Trikalitis from the University of Crete. She is currently working towards her Ph.D. in Prof. Vassilikogiannakis’ group. Tamsyn Montagnon obtained her Ph.D. (2000) with Prof. Parsons from the University of Sussex. From 2001–2004, she was a postdoctoral fellow at the Scripps Research Institute with Prof. Nicolaou where she co-authored the book “Molecules That Changed the World”. She is currently working as a Senior Researcher at the University of Crete. Dimitris Kalaitzakis obtained his Ph.D. (2006) with Prof. Smonou from the University of Crete. In 2011 he joined the group of Prof. Vassilikogiannakis as a postdoctoral fellow. He is currently working as a Senior Researcher at the University of Crete. Georgios Vassilikogiannakis obtained his Ph.D. (1998) with Prof. Orfanopoulos from the University of Crete. From 1999–2002, he was a postdoctoral fellow at the Scripps Research Institute with Prof. Nicolaou. His independent career started (2002) at the University of Crete, where he was promoted to full Professor (2013). He is a member of the advisory board of OBC and the editorial board of ChemPhotoChem. He is the recipient of ERC starting and PoC grants. The research interests of the authors are in the field of sustainable synthetic organic chemistry with a particular focus on the development and application of new photocatalytic synthetic methodologies. |
1 Introduction
The synthetic organic dye eosin was named -albeit, indirectly- after the Titaness Eos who was the bringer of light at Dawn and it has now graced the shelves of chemical laboratories for almost 150 years.1 Van Gogh was an early user of eosin; indeed, his famous irises were originally a unique purple colour due to the red eosin he had included in his paint.2 Eosin was invented in a time when dyes could make chemists rich and were driving the growth of the nascent chemical industry,3 but, for over a century eosin languished in obscurity being used in synthetic chemistry only sporadically as a photosensitizer for generating singlet oxygen4 (an energy transfer process). However, with the explosive resurgence of interest in photocatalysis that has occurred over the last decade and a half,5 eosin too has seen a very rapid expansion in its uses (now including both energy and electron transfer processes). There have been a number of excellent reviews focusing on eosin in synthetic chemistry published already6 (particularly worthy of note are those from Hari and König in 20146a and from Srivastava and Singh in 20176b), but, the true extent of its versatility is, nonetheless, only just beginning to come to light, and, with this review of some of the most recent contributions to the field (after 2016), we hope to highlight this fact.Before we begin to review recently published eosin-catalysed reactions, some of eosin's basic features and characteristic behaviours need to be established. These issues are very comprehensively dealt with in the superb review on Organic Photoredox Catalysis by Romero and Nicewicz.5b Instead of regurgitating all the material here, we will try, to highlight some of the most important take home messages. Firstly, it is eosin Y, the most commonly used photocatalyst from the eosin family, which will be discussed throughout this review. Secondly, eosin Y commonly comes in either its neutral form or as the disodium salt, and, unfortunately, it is not always clearly indicated which of the two has been employed in the experimental protocols that have been published (Scheme 1). Thirdly, understanding of the whole fluorescein family of dyes5b is complicated by both the complex tautomermism and the acid–base equilibria which exist with these compounds (Scheme 1). For example, the closed lactone ring tautomer 1b does not absorb visible light. A corollary of the existence of these equilibria is that the absorption wavelength and intensity are both pH and solvent dependent for eosin. In contrast, the redox potentials, however, do not vary significantly.5b Eosin undergoes very fast intersystem crossing (ISC, 1[EY] − hν → 1[EY]* − ISC → 3[EY]*), so the initially formed singlet excited state(1[EY]*) has a very short lifetime and it is, therefore, the triplet excited state (3[EY]*) that is considered to be the most relevant. Triplet excited state eosin is both a moderate oxidant and reductant in single electron transfer (SET) pathways (redox potentials: for 3EY* → EY˙− = +0.83 V vs. SCE and for 3EY* → EY˙+ = +1.15 V vs. SCE),5b but it is also an adept energy transfer agent (including; for the generation of singlet oxygen = its historical use4). It is of note that Romero and Nicewicz5b assert that eosin along with the other fluorescein dyes (fluorescein itself, rose Bengal and erythrosine) are the most analogous of all the organic dyes in their photoredox activity to the commonly used precious transition metal photocatalysts. They should, therefore, always be considered as alternatives because they are not only lower in cost, but their use is also more sustainable.
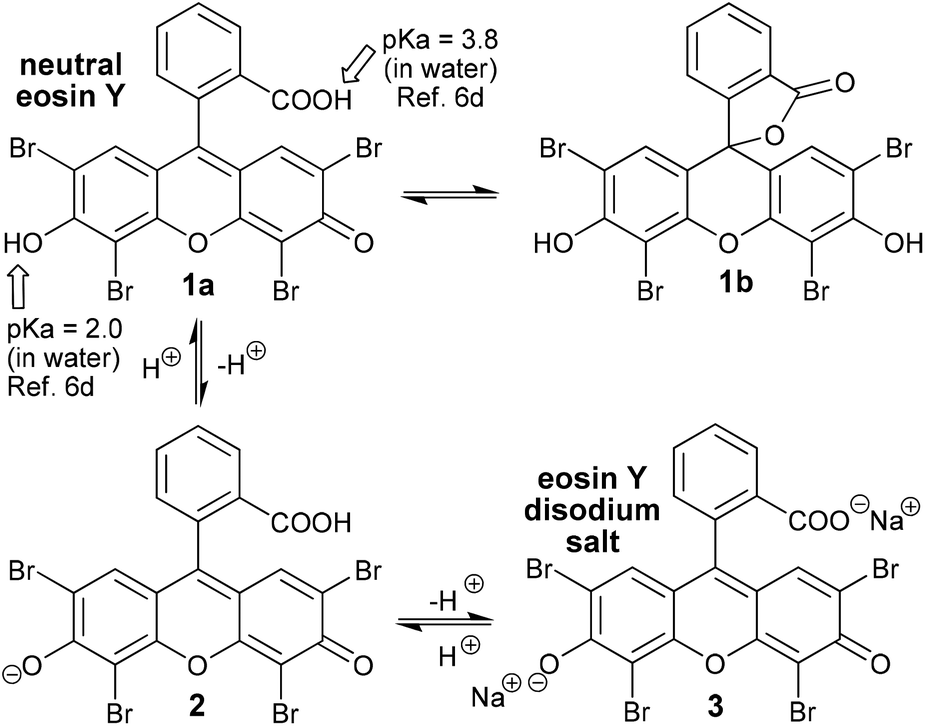 | ||
| Scheme 1 Relevant tautomeric and acid–base equilibria for eosin Y. | ||
2 Eosin-catalysed reactions
We have categorised the examples of eosin-catalysed reactions that will be presented in this review, firstly, by the type of bond(s) formed, and, then, within these categories, by the role eosin is proposed (by each of the work's authors) to have played mechanistically. In the schemes, the newly formed bonds are highlighted in bold (this does not indicate relative stereochemistry).2.1 Eosin-catalyzed C–C bond forming reactions (no ring formed)
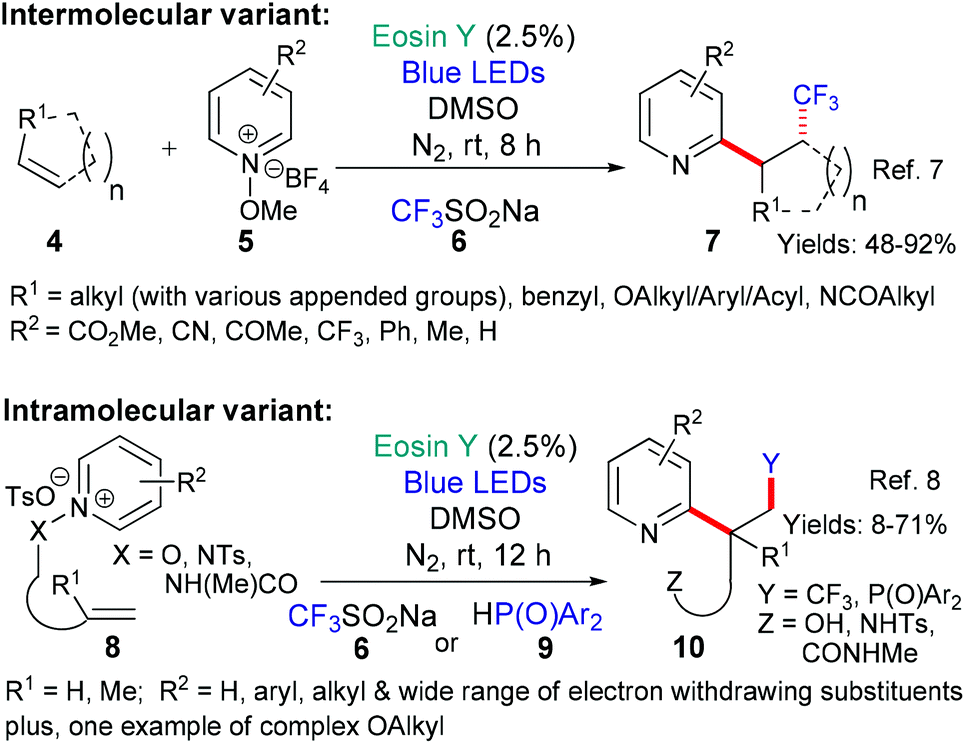 | ||
| Scheme 2 Synthesis of substituted pyridines (7 and 10) using an eosin-catalyzed domino sequence. | ||
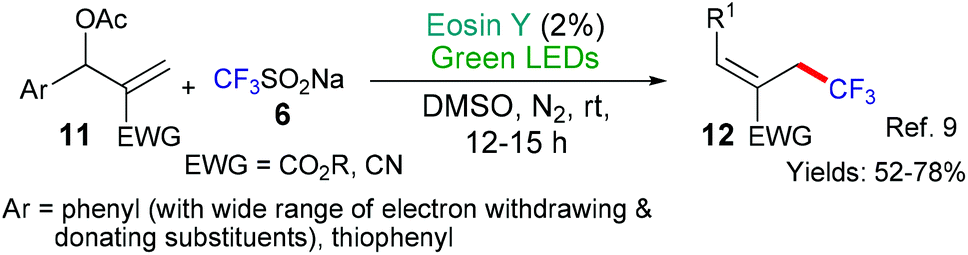 | ||
| Scheme 3 Eosin-light catalyzed addition of trifluoromethyl radicals to allylic acetates 11. | ||
This general strategy is not limited to trifluromethyl radical additions; alongside Hong's work there are two reports wherein various fluorinated alkyl radicals are added to aromatic heterocycles from families that exhibit a broad range of biological activities. In 2018, Zhang, Deng and coworkers presented the difluoromethylation of courmarins in pursuit of new antifungal agents (Scheme 4A).10a This work was followed by a report from Wei et al. showcasing the addition, using the same conditions, of a variety of fluorinated alkyl groups to quinoxalinones and xanthines (Scheme 4B and C, respectively).11 Difluoromethylation (or monofluoromethylation) of a different type was investigated by Feng, Xu and coworkers (Scheme 5).12 The authors propose that DIPEA acts as a sacrificial electron donor reducing excited state eosin to the corresponding radical anion (*EY → EY˙−); this species, in turn, donates an electron to the bromoacetate substrate 22 which fragments to give the halide anion (Br−) and the corresponding fluorinated alkyl radical. The fluorinated radical adds regioselectivity to a cinnamic acid derivative (formed in situ by reaction of the cinnamic acid 21 with BI-OH). This intermediate then fragments to yield the desired product 23.
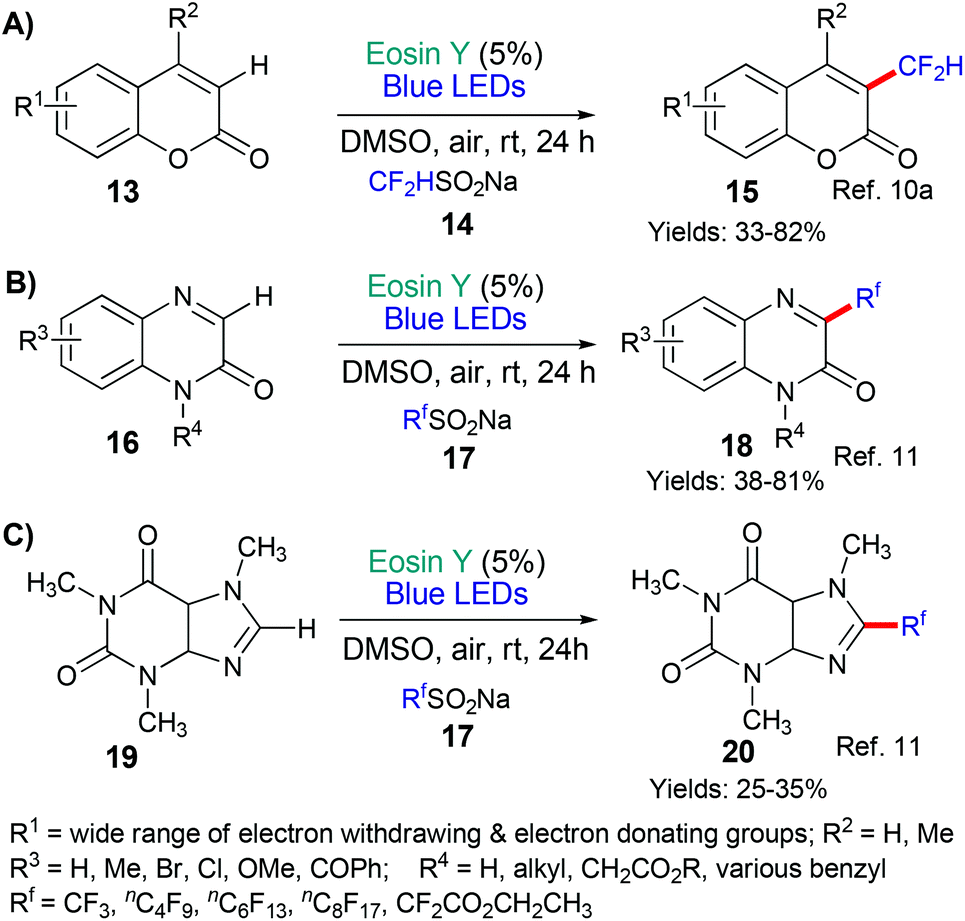 | ||
| Scheme 4 Addition of various fluorinated alkyl radicals to aromatic heterocycles of interest. | ||
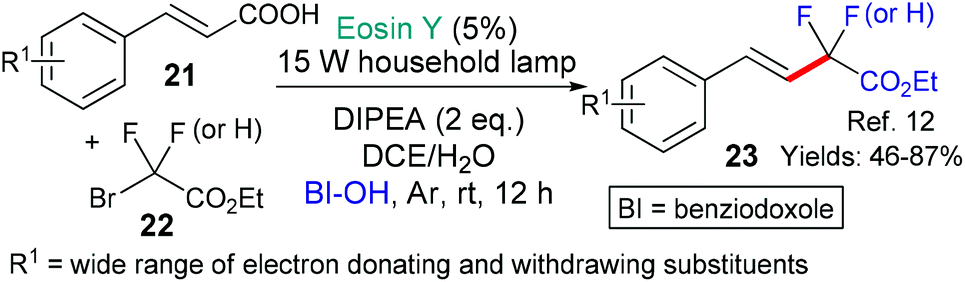 | ||
| Scheme 5 (Di)Fluoromethylation of various cinnamic acids 21. | ||
 | ||
| Scheme 6 Examples where heterolytic cleavage of a carbon-halide bond generates a C-centered radical for addition to a suitable substrate. | ||
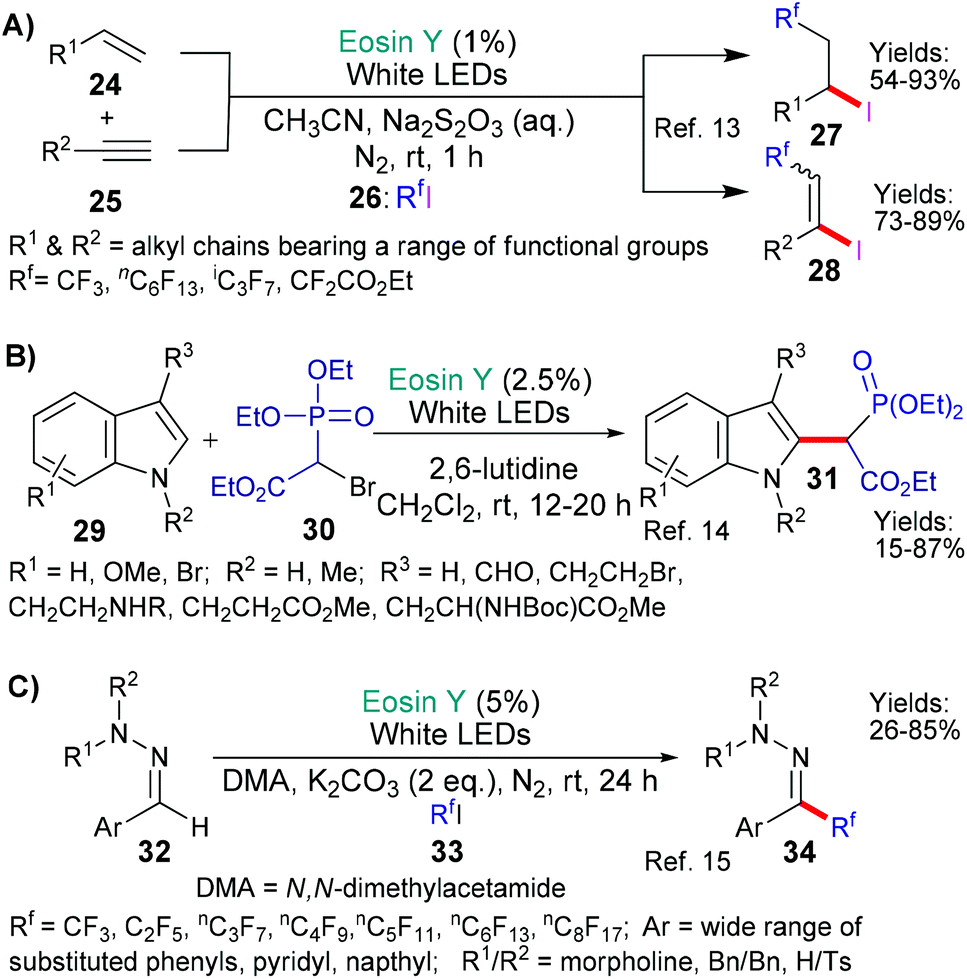
In the first of these examples from Yajima and Ikegami (Scheme 6A),13 two generalised iodoperfluoroalkylation reactions are presented wherein an iodide and a perfluoroalkyl group have been regioselectively added across either a terminal unactivated double or triple bond (24 or 25 → 27 or 28, respectively). The developed methodology showed good functional group tolerance and may be used to provide important fluorinated building blocks for use in synthesis. In the second example (Scheme 6B), from the group of Opatz,14 indoles 31 bearing the necessary functionality for onward elaboration via a Horner–Wadsworth–Emmons reaction were successfully targeted. Once again wide functional group tolerance was seen as a big advantage of the metal-free method. In the final and most recent example of the three from Zhou, Li and coworkers (Scheme 6C),15 fluorinated alkyl groups were successfully added to aromatic hydrazone substrates (32 → 34, Scheme 6C).
Earlier, we saw excited state eosin operating as an oxidant to generate trifluoromethyl radicals (Schemes 2 and 3); in Scheme 7, an alternative means to generate the same trifluoromethyl radical is shown by the group of Balaraman that uses excited state eosin as a reductant (*EY → EY˙+) and CF3SO2Cl.16 The susbstrates are aromatic nitroalkenes and after addition of the trifluoromethyl radical it is proposed that NO2 is eliminated to yield the trans 1-trifluoromethylalkenes, selectively.
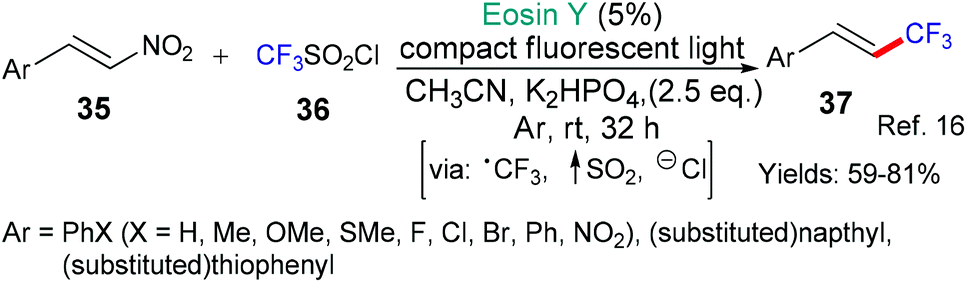 | ||
| Scheme 7 Addition of trifluoromethyl radical to aromatic nitroalkenes 35. | ||
In the next examples,17,18 the inherent weakness and reducibility of N–O bonds has been exploited (Scheme 8). In 2018, Chen, Guo and Yu presented a new method for synthesizing δ-alkenyl amides (40, Scheme 8A).17a The reaction sequence was initiated by the reduction of the N–O bond (by *EY → EY˙+) in substrates 38 to yield the corresponding aromatic carboxylate anion and an amidyl radical. This radical then undergoes 1,5-hydrogen atom transfer (HAT) to shift the position of the radical to the carbon at the γ-position (relative to the carbonyl group) on the side chain. This new carbon-centred radical then reacts with an alkenyl boronic acid 39 to eventually yield the desired δ-alkenyl amides 40. Increasing steric hindrance (increasing substitution) at the carbon centre γ to the amide led to better trans-selectivity when the product's (40) double bond was formed. In the second example from the group of Liu and Zhou,18 a strained oxime additive (43) was used to generate the desired radicals and initiate the sequence (Scheme 8B). Specifically, excited state eosin is proposed to reduce the N–O bond of 43 generating the corresponding alkoxide anion and N-centred imine radical (I), which, in turn, fragments opening the cyclobutyl ring to form a cyano-functionality and a primary alkyl radical (II). It is proposed that this radical abstracts an H atom from 42 to generate yet another radical intermediate (III) which adds to the double bond of 41. Next after ipso-addition of the resulting homobenzylic radical to an adjacent aromatic residue, a 1,2-migration-oxidation sequence is completed to afford the final product 44.
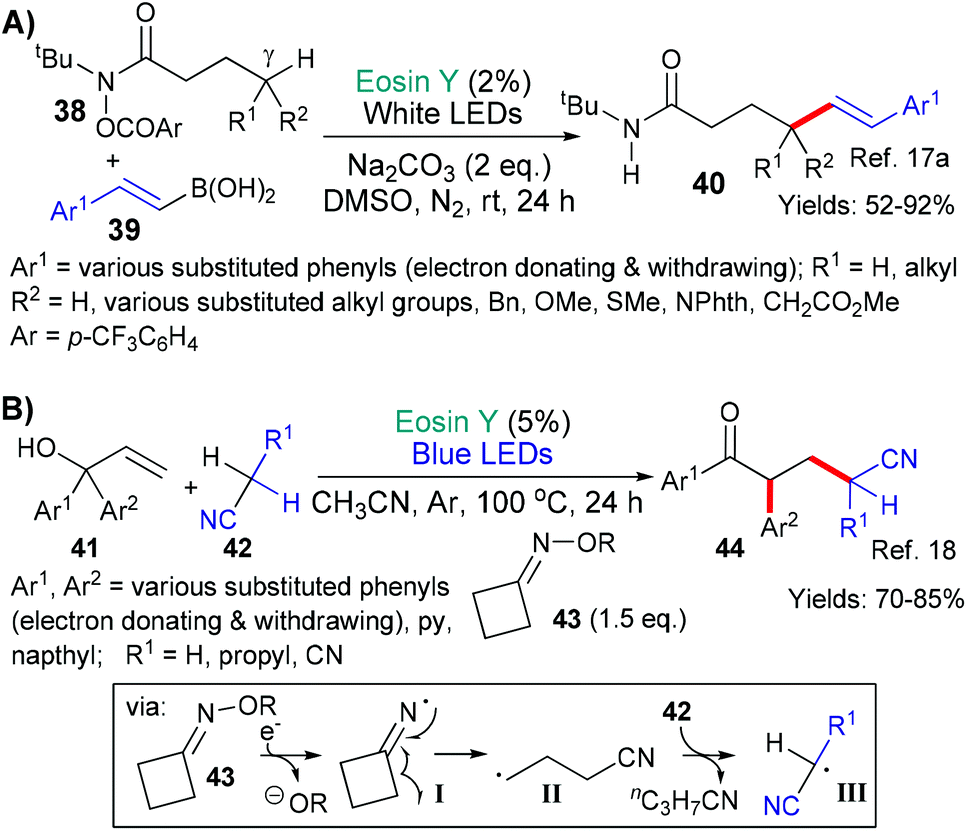 | ||
| Scheme 8 Exploiting the weakness of N–O bonds to generate radicals for further reaction. | ||
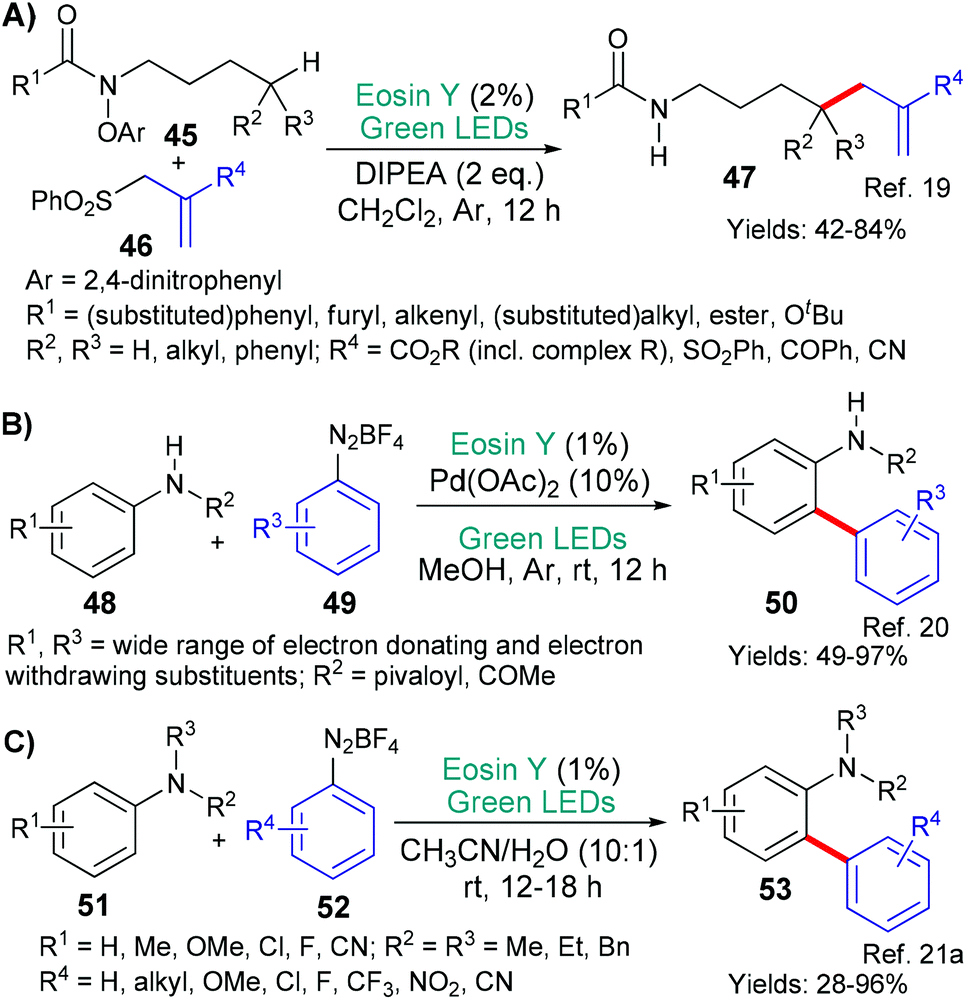
Our next example shares some characteristics with the one shown in Scheme 8A, but, in this case, it is proposed by the group of Flechsig and Wang that the aromatic functionality (of N-OAr in 45) first accepts an electron from excited state eosin (*EY → EY˙+, Scheme 9A).19 This initial reduction of the aromatic ring is followed by cleavage of the N–O bond to yield the corresponding phenoxide anion and amidyl radical. The fate of the latter mirrors the previous example (Scheme 8A); thus, a 1,5-HAT relocates the radical to the δ-position of the side chain and the resulting carbon-centred radical reacts with allyl sulfones 46 to form the adducts 47.
 | ||
| Scheme 9 Examples where excited state eosin reduces an aromatic functionality to initiate a reaction sequence. | ||
Aromatic diazonium salts have also proven to be easily reducible substrates (Scheme 9B and C).20,21 In both instances, excited state eosin donates an electron (*EY → EY˙+) to the aromatic diazonium salt (49 or 52) which then fragments to release nitrogen gas and the corresponding nucleophilic aromatic carbon-centred radical (an aryl radical). In the older of the two examples from group of Balaraman (2017),20 this aryl radical reacts with a complex formed between the palladium catalyst and substrate 48. After several steps standard to palladium chemistry, the biaryl products 50 are produced. In the later (2019), metal-free, example from Kapoor, Chawla and Yadav,21a the aryl radical produced adds directly to the ortho-position of the aniline substrate 51. Only, an oxidation and deprotonation step are then required to afford the biaryl products 53.
In our final example of this section from the groups of An and Li, we return again to the generation of trifluoromethyl radicals (Scheme 10).22 In this example, a third way of generating these radicals is presented (for the two previous methods; see, Schemes 2, 3 and 7). In this instance, the authors propose that excited state eosin reduces the peroxydisulfate anion (S2O82−) to form a sulfate anion (SO42−) and a SO4˙− radical. This radical, in turn, reacts with sodium triflinate (CF3SO2Na also known as Langois’ reagent) to generate the trifluoromethyl radical which then adds regioselectively to the 8-amino quinoline substrate 54.
 | ||
| Scheme 10 Addition of trifluoromethyl radical to 8-amino quinolines 54. | ||
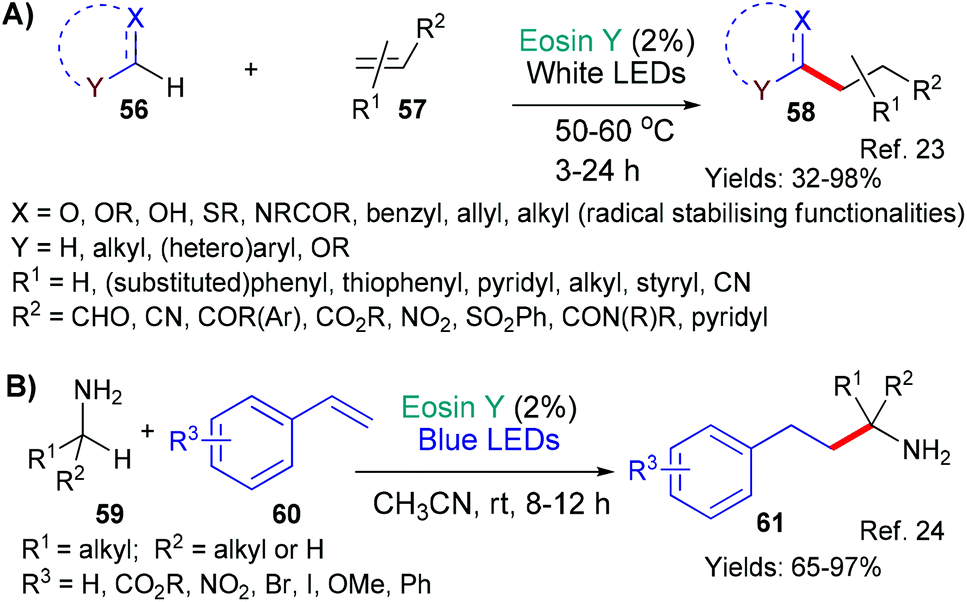
The first example, from the group of Wu in 2018,23 represents a very wide ranging study into the successful addition of a variety of radicals (derived from 56), formed through the mechanism described above, to electron deficient double bonds (56 + 57 → 58, Scheme 11A). This methodology is an effective and sustainable way of generating important building blocks (58) for synthetic applications.
 | ||
| Scheme 11 Examples where excited state eosin abstracts a hydrogen atom. | ||
The second contribution comes from Srivastava, Singh and Singh who showed that a similar strategy could be used to add a variety of α-amino alkyl radicals (derived from 59, Scheme 11B) to styrenes 60.24
2.2 Eosin-catalyzed cyclization reactions (C–C and/or C–X bond forming reactions)
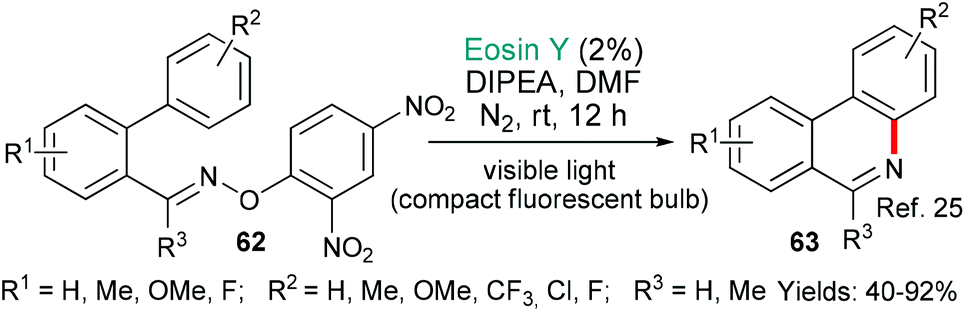
This section of the review contains really diverse examples because it encompasses not only C–C bond forming reactions, but also C–X (X = a heteroatom) bond forming reactions. Additionally, the number of bonds formed in each example varies from one to three. As such, the section forms a bridge between the preceding one, where a single C–C bond was formed in each case, and the next, where C–X bonds become the focus.In the first example, phenanthridines are targeted. These alkaloids exhibit a very wide range of biological activities. They were made by the group of Xie in 2016 using an eosin- mediated cyclization (Scheme 12).25 More specifically, DIPEA acts as a sacrificial electron donor to generate EY˙−, which, in turn, donates an electron to the 2,4-dinitrophenyl oxime group of the substrates 62. A fragmentation then occurs to generate an iminyl radical, which cyclizes onto the proximal pendent phenyl group to yield the products 63.
 | ||
| Scheme 12 Synthesis of phenanthridines 63 using eosin-mediated cyclization. | ||
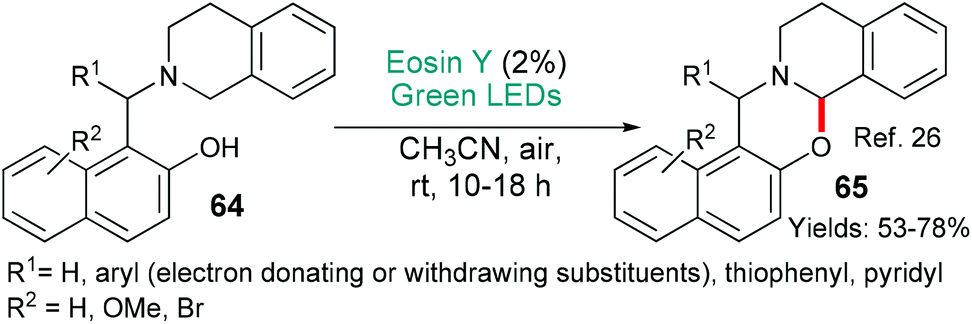
1,3-Oxazines also exhibit a wide variety of activities and can be made using an eosin-mediated cyclization (Scheme 13).26 In this investigation, from the Borpatra, Deb and Baruah, the substrates 64 contain an internal electron donor (the tertiary amine) which cedes an electron to excited state eosin (*EY → EY˙−). H-Atom abstraction from the intermediate then yields an imminium ion which is attacked by the nucleophilic phenol to give the products 65. This methodology may be said to be greener than previous variants that used copper salts to achieve the same overall transformation.
 | ||
| Scheme 13 Synthesis of 1,3-oxazines 65 using eosin-mediated cyclization. | ||
Oxazoles make up a large and very popular class of synthetic targets often having interesting biological activities. In 2019, a new eosin-mediated method for their synthesis was published by the group of Gu and Li starting from α-bromoketones 66 and primary amines (67, Scheme 14).27 In this instance, two new bonds are formed in the key eosin-mediated reaction. A complex multi-component mechanism is proposed for this new synthesis of oxazoles.
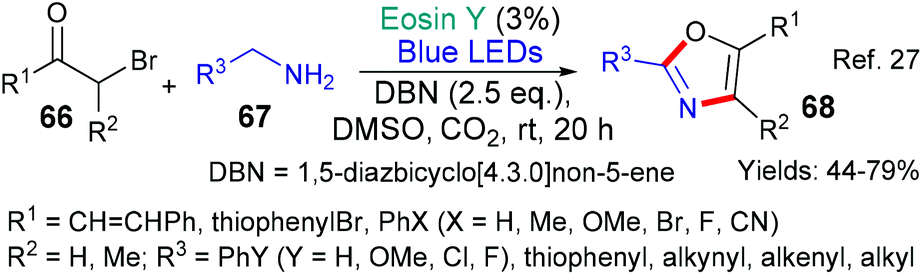 | ||
| Scheme 14 Synthesis of oxazoles 68 using eosin-mediated cyclization. | ||
Another reaction that forms two new bonds was reported in 2018 by Kshirsagar's group (Scheme 15).28a In this case, sulfenylindoles 71 are formed from 2-alkynyl-azidoarenes 69. It is proposed that the thiol partner initiates the process by donating an electron to excited state eosin (*EY → EY˙−). After a proton transfer from the thiol radical cation, a thiol radical (R3S˙) is generated that adds to the alkyne forming a vinyl radical, which, in turn, adds onto the azide ejecting nitrogen gas to form the products 71. This work builds on an earlier example from Gu et al.28b that made 2,3-disubstituted indoles from similar substrates using eosin Y.
 | ||
| Scheme 15 Synthesis of sulfenylindoles 71 using eosin-mediated cyclization. | ||
The next example from the groups of Chen and Xiao (2018) is particularly interesting mechanistically (Scheme 16).29 In this synthesis of 3-hydroxyisoindolinones 73, it was proposed, following mechanistic investigations supported by DFT calculations, that ground state eosin donates an electron to Selectfluor initiating the first stages of the sequence which involve abstraction of a benzylic hydrogen atom from the substrate 72. In the latter stages of the sequence, eosin fulfills a second more classical role when in its excited state it accepts an electron (*EY → EY˙−) to participate in the final benzylic oxidation.
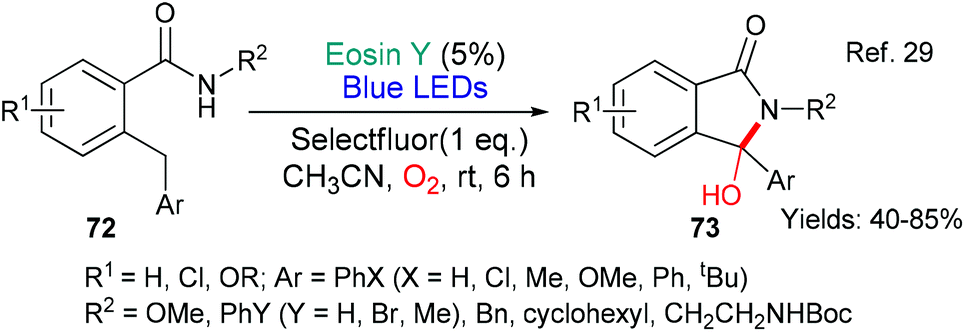 | ||
| Scheme 16 Synthesis of 3-hydroxyisoindolinones 73 using dual role eosin-mediated reactions. | ||
Huang's group published an eosin-mediated synthesis of dihydroisoquinolinones 75 in 2019 (Scheme 17).30 These compounds were targeted due to previously reported biological activities. The authors propose that DBU deprotonates the amide in the starting benzamide 74. The so-formed anion then donates an electron to excited state eosin (*EY → EY˙−) generating the N-centred amidyl radical which cyclises onto the proximal double bond to give the products 75.
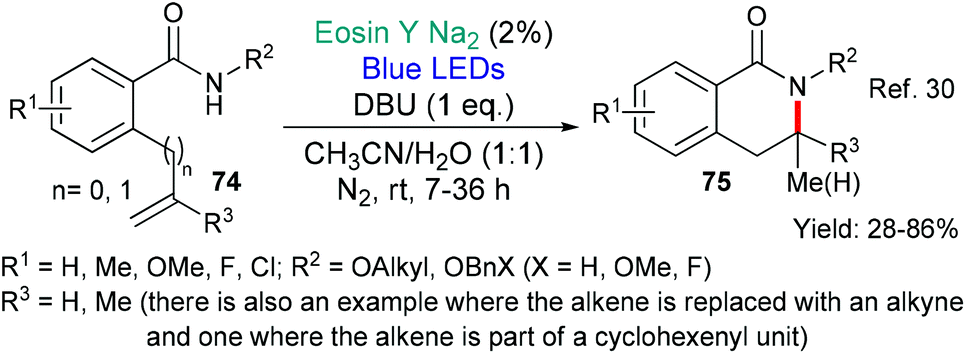 | ||
| Scheme 17 Synthesis of dihydroisoquinolinones 75 using eosin-mediated reactions. | ||
The first example in this section, from Leonori's group, uses classic concepts to give versatile access to a wide range of high value N-containing 5-membered ring products (Scheme 18).31 Excited state eosin donates an electron (*EY → EY˙+) to the 2,4-dinitrophenylhydroxylamine moiety which fragments to generate the amidyl radical (compare with other examples that also form an amidyl radical, Schemes 9A and 17). This radical then undergoes 5-exo cyclization with 1,4-cyclohexadiene acting as the requisite hydrogen atom source to complete the transformation into the product 77 (Scheme 18).
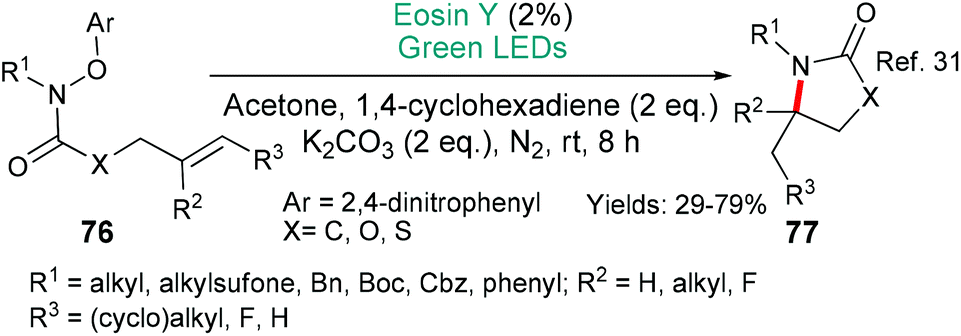 | ||
| Scheme 18 Synthesis of high value heterocycles 77 using eosin-mediated cyclization. | ||
In 2018, Sun, Yin and Zhang published an eosin-mediated synthesis of 3-arylsulfonylquinolines 80 (Scheme 19).32 Here it is proposed that excited state eosin donates an electron (*EY → EY˙+) to the diaryliodonium salt 79 which then fragments to generate the aromatic radical (Ar˙). This radical reacts with DABSO (DABCO(SO2)2) to form the arylsulfonyl radical (ArSO2˙) which adds to the alkyne (in 78) giving a vinyl radical. The vinyl radical cyclizes onto the aromatic ring to afford the products 80.
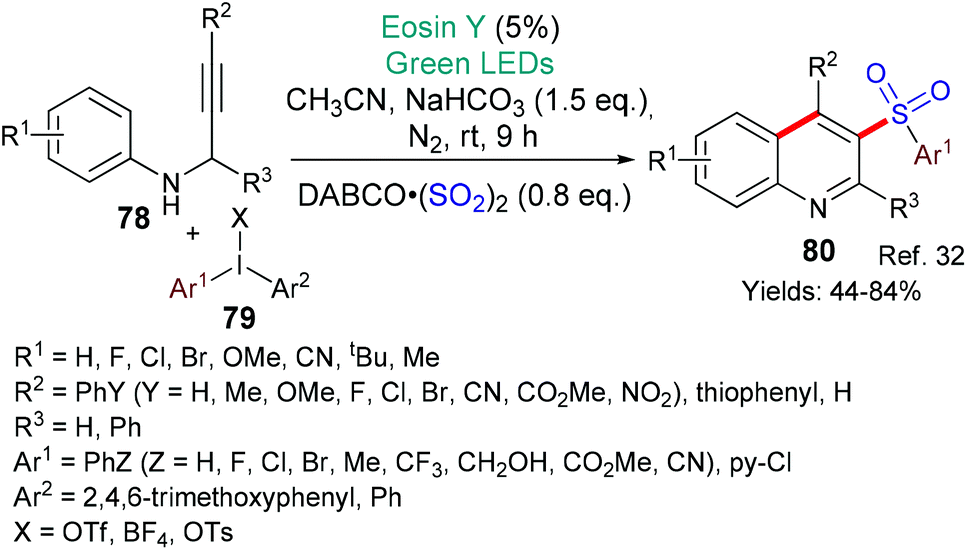 | ||
| Scheme 19 Synthesis of quinolines 80 using eosin-mediated cyclization. | ||
In 2017, Qian et al. reported a new synthesis of sulfonated quinazolines 83 mediated by eosin (Scheme 20).33a Quinazolines can be found in many bioactive natural products, and, additionally, sulfonyl modifications are becoming common in drugs; thus, these sulfonated quinazolines 83 are attractive targets. It is proposed that excited state eosin helps to generate a butoxide radical (tBuO˙) from tertiary-butylhydroperoxide (TBHP) by donation of an electron (*EY → EY˙+). This radical then abstracts a hydrogen atom from the arylsulfinc acid 82 (ArSOOH → ArS˙OO) to form a new radical which adds to the terminal double bond of substrate 81. 5-Exo cyclization onto the cyano moiety is followed by a second cyclization onto the aromatic ring yielding the final compounds 83. The combination of eosin with an arylsulfinic acid (c.f.82) and a peroxide, to generate an ArS˙OO radical, was also used to make certain sulfonylated benzofurans.33b In a curious twist, the same radical (ArS˙OO) was also generated from the sodium salt of the corresponding arylsulfinic acid (ArSO2Na) upon donation of an electron to excited state eosin (in this case, *EY → EY˙−) and then used to make a set of sulfonylated isoquinolinediones.33c
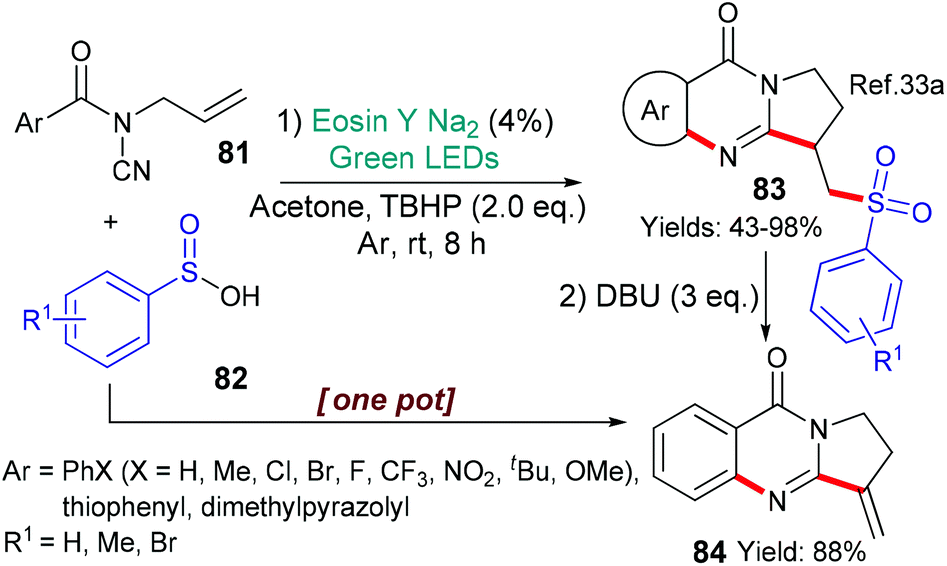 | ||
| Scheme 20 Synthesis of quinazolines 83 and 84 using eosin-mediated cyclization. | ||
The importance of pyrimidines (such as; 88, Scheme 21) in nature and their wide occurrence in drugs (for example, nucleotides & nucleotide analogues) make them highly attractive targets for synthesis. The groups of Shen and Loh chose to investigate including fluorinated side chains in a new regioselective multicomponent synthesis of pyrimidines (using eosin) in order to be able to modify the lipophilicity, solubility and metabolic stability profiles of pyrimidine drug candidates.34 In this case, it is proposed that excited state eosin donates an electron to the fluorinated iodide 87 to generate the alkyl fluoride radical (˙Rf) which adds to the silyl enol ether 86. A single electron oxidation followed by elimination of hydrogen fluoride yields the corresponding fluorinated α,β-unsaturated ketone which then condenses with the amidine 85 to furnish the target pyrimidine 88. Yang and Tang used the same method for generating alkyl fluoride radicals (˙Rf) in their synthesis of perfluoroalkylated oxindoles from N-arylacrylamides.35
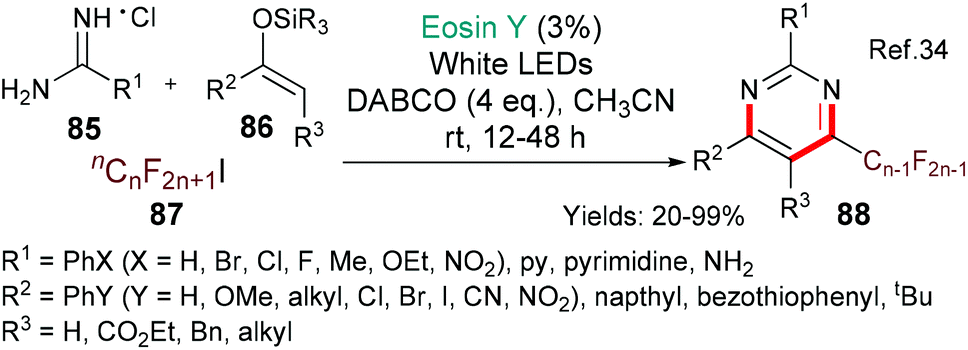 | ||
| Scheme 21 Synthesis of pyrimidines 88 using eosin-mediated cyclization. | ||
In 2018 the group of Guo36 and in 2020 the groups of Sarkar and Murarka,37 published new methods that used phthalimide derivatives 90 as a source of alkyl radicals (Scheme 22). In both cases, it is proposed that excited state eosin donates an electron (*EY → EY˙+) to the phthalamide 90 which subsequently fragments to generate alkyl radicals (˙R3). Again in both cases this alkyl radical adds to the double bond in the substrate (either 89 or 92). After this addition, the two methods diverge mechanistically. Where the intermediate contains an azide (from 89), an iminyl radical is formed as nitrogen is released and this iminyl radical cyclizes onto the biaryl core to yield the products 91.36 In the other example, the radical formed after the addition of ˙R3 to the double bond of 92, cyclizes onto the aldehyde residue affording the products of type 93.37 The phenanthridine 91 and chroman-4-one 93 products were targeted due to their potential for having biological activities of interest.
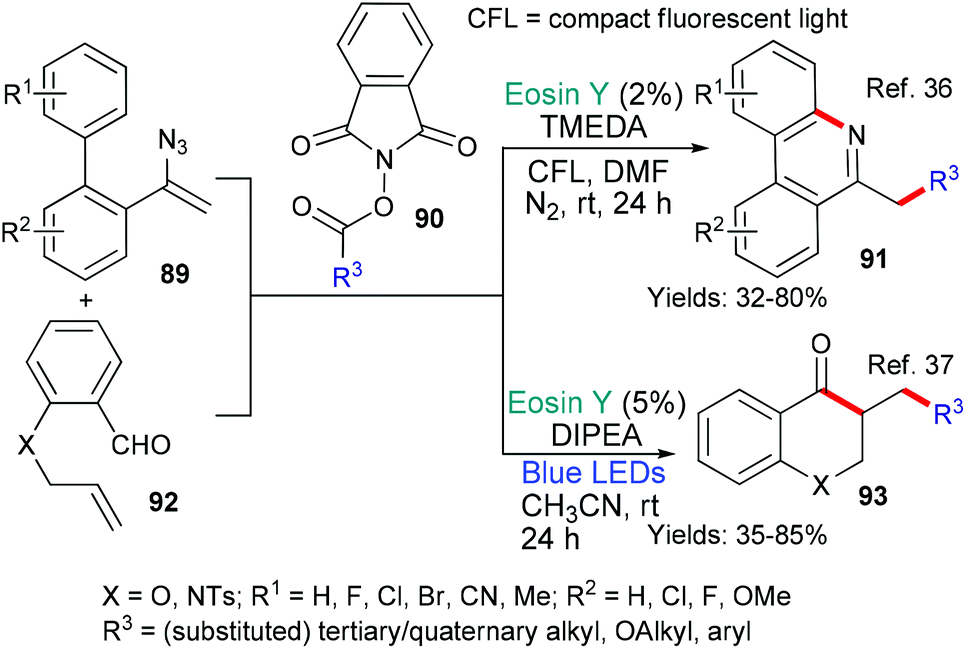 | ||
| Scheme 22 Synthesis of products 91 and 93 using eosin-mediated cyclizations. | ||
In 2017 and 2020, the groups of Jin/Cheng38 and Mkrtchyan/Iaroshenko,39 respectively, published new methods that used aryldiazonium tetrafluoroborates 95 as a source of aryl radicals (Scheme 23). In both cases, it is proposed that excited state eosin donates an electron (*EY → EY˙+) to the aryldiazonium tetrafluoroborate 95 which subsequently fragments to generate the aryl radical (˙Ar). In the first instance, it is proposed that this aryl radical adds to the triple bond in substrate 94 and the resulting vinyl radical cyclizes onto the azide residue with loss of nitrogen to give the indole product 96.38 In the other example, it is proposed that the aryl radical adds to the α,β-unsaturated ketone's double bond to form an intermediate radical adjacent to the amine group. This radical is oxidized to give the imminium cation which is attacked by the proximal phenol to afford the cyclized product. Finally, elimination of Me2NH yields the product 98.39 In both cases, the products were targeted due to their ubiquity; firstly, it is well-known that indoles are extremely common in bioactive natural products and synthetic compounds. Chromones are also privileged heterocycles abundant not only in nature, but in synthetic compounds with a range of applications (from use in the life sciences to uses in the food industry).
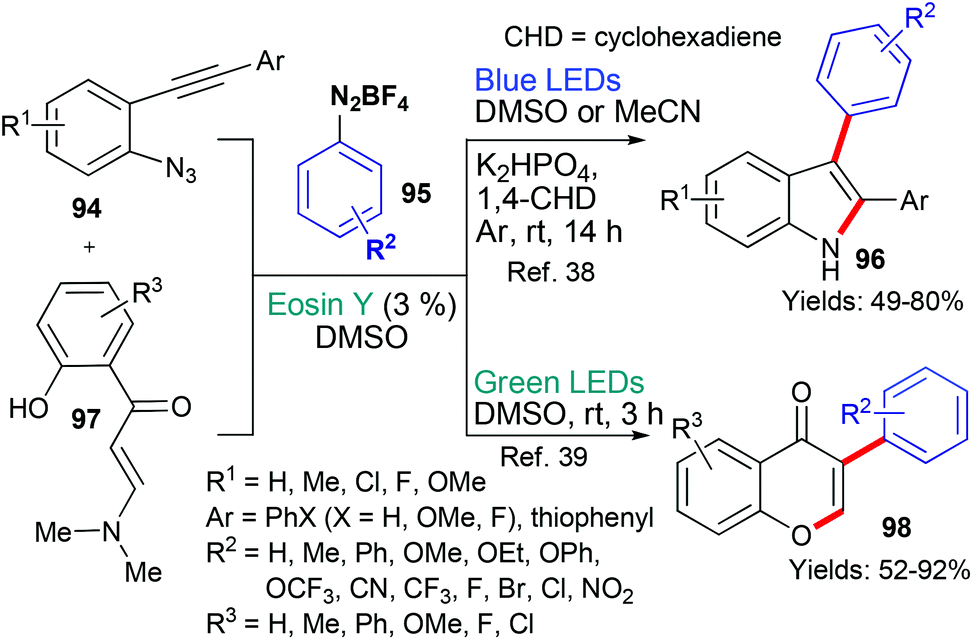 | ||
| Scheme 23 Synthesis of products 96 and 98 using eosin-mediated cyclizations. | ||
Recently (in 2020), the groups of Zhao and Xia published a new methodology for synthesizing isoquinolones 101 from N-substituted pyridinium salts 99 using eosin catalysis (Scheme 24).40 The authors targeted isoquinolones not only for their potential biological activities, but also because they may be used in organic light emitting diodes (LEDs) and as organocatalysts. It is proposed that excited state eosin donates an electron to the pyridinium salt 99 which fragments to afford the corresponding amidyl radical. This radical adds regioselectively to the alkyne 100, with the resulting vinyl radical cyclizing onto the aromatic amide core to furnish the isoquinolone product 101.
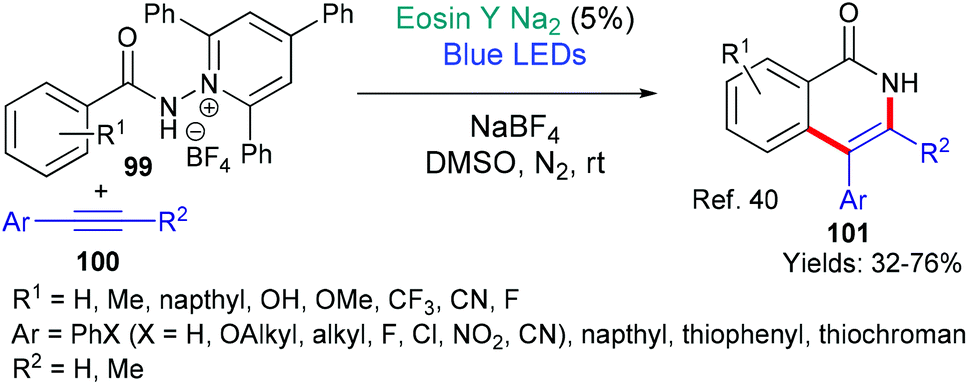 | ||
| Scheme 24 Synthesis of isoquinolones 101 using eosin-mediated cyclization. | ||
Also belonging to this section are reports of an eosin-mediated head-to-head dimerisation of Erlenmeyer azlactones to yield highly functionalized cyclobutanes 102 and 103;41 an eosin-mediated synthesis of 2-mercaptobenzothiazoles 104 from 2-azidoarenediazonium salts and carbon disulfide42 and an eosin-mediated synthesis of phenanthradine derivatives 105 from N-arylacrylamides and alkyl carbazates (Scheme 25).43
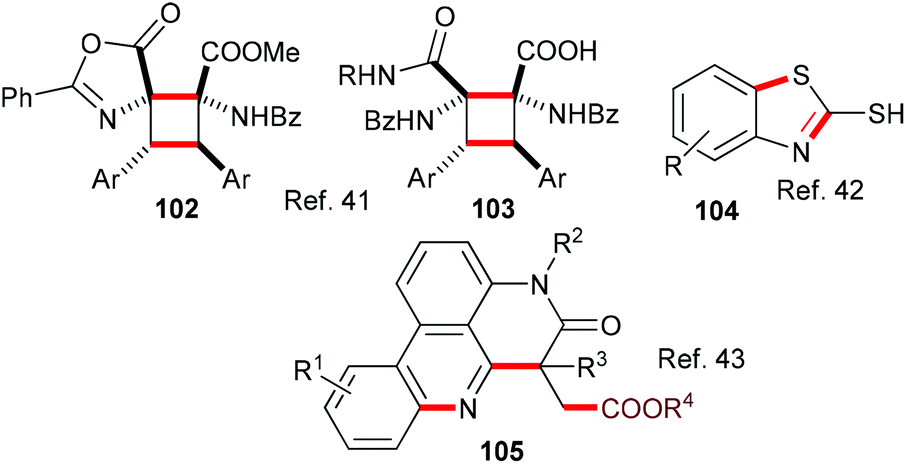 | ||
| Scheme 25 Synthesis of products 102–105 using eosin-mediated cyclizations. | ||
In 2019, Xia et al. published a novel methodology for the synthesis of quinazolinones of type 108 (Scheme 26).44 Quinazolinones are common in both natural products and drugs, indeed, two drugs are synthesized in the paper. In a process with broad scope, it is proposed that eosin initiates the reaction sequence by abstracting one of the hydrogen atoms (*EY → ˙EY–H) from adjacent to the alcohol functionality in 106. Oxidation of the resulting radical to give the corresponding aldehyde (using O2 and eosin) is followed by acid-catalyzed condensation with the o-aminobenzamide 107 to furnish the final products 108.
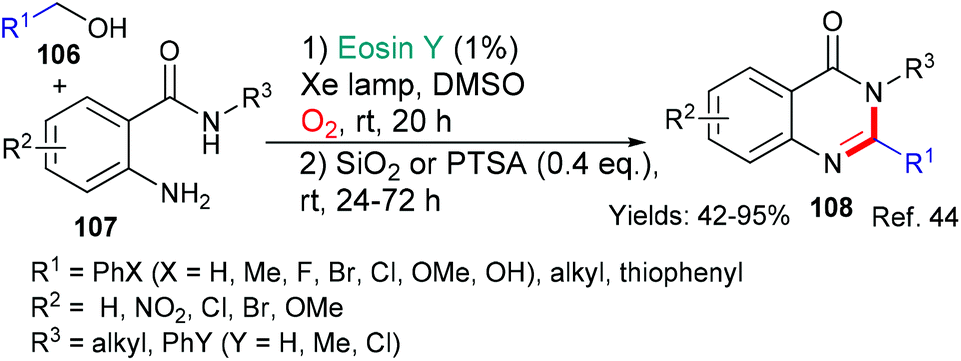 | ||
| Scheme 26 Synthesis of quinazolinones 108 using eosin-mediated cyclization. | ||
Spirooxindoles are complex structures that have been found in natural products and are being included in biologically active synthetic compounds. Recently, the group of Mo and Zhang presented a simple synthesis of a specific group of spirooxindoles of type 112 (Scheme 27).45 In this methodology, it is proposed that excited state eosin abstracts a hydrogen atom from the 1,3-dicarbonyl compound 111. The resulting radical adds to a previously formed adduct of malononitrile 110 and isatin 109. A final cyclization (via a two electron mechanism) unites the ketone and a cyano group to form the pyran ring in the product 112.
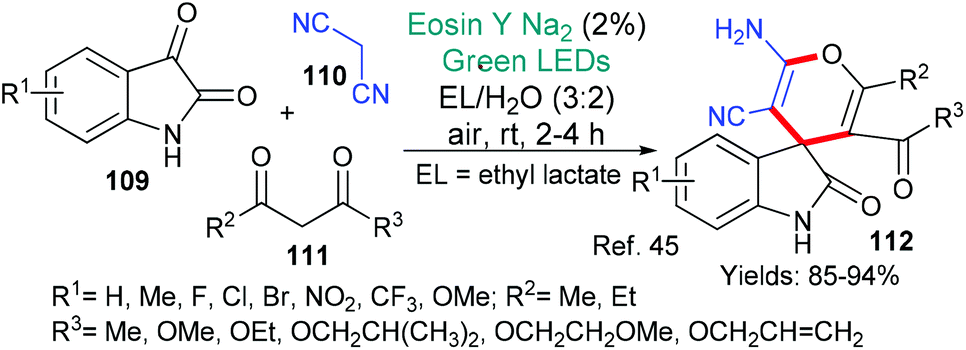 | ||
| Scheme 27 Synthesis of spirooxindoles 112 using eosin-mediated cyclization. | ||
2.3 Eosin-catalyzed C–X bond forming reactions (no ring formed)
Eosin's versatility continues to be seen in this section where we discuss the formation of carbon-heteroatom bonds; excluding, cases where it is part of a cyclization step which were discussed in the previous section (above). The mechanistic roles of eosin are not separated in the section, instead, the reactions are categorized using the hetereoatom's identity. Particularly in this area, this review is not exhaustive as very many minor investigations exist.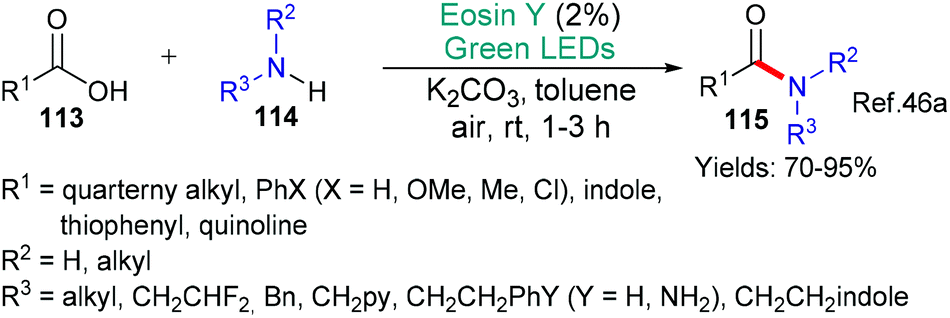 | ||
| Scheme 28 Synthesis of amides 115 using eosin. | ||
Our second example in this section also involves a new methodology for making a fundamentally important motif, in this case it is 1,3-amino alcohols 118 (Scheme 29).47 The method may be said to be particularly useful because it starts from a common substrate class, namely, α,β-unsaturated ketones 116. The authors propose that excited state eosin donates an electron (*EY → EY˙+) to the α,β-unsaturated ketone of substrates 116 to yield the corresponding radical anion. It is proposed that the resulting eosin radical cation assists in the fragmentation of the aminating agent 117 to afford an aminyl radical which combines with the radical anion formed from the α,β-unsaturated ketone yielding, after an additional reduction (enol to alcohol), the product 118.
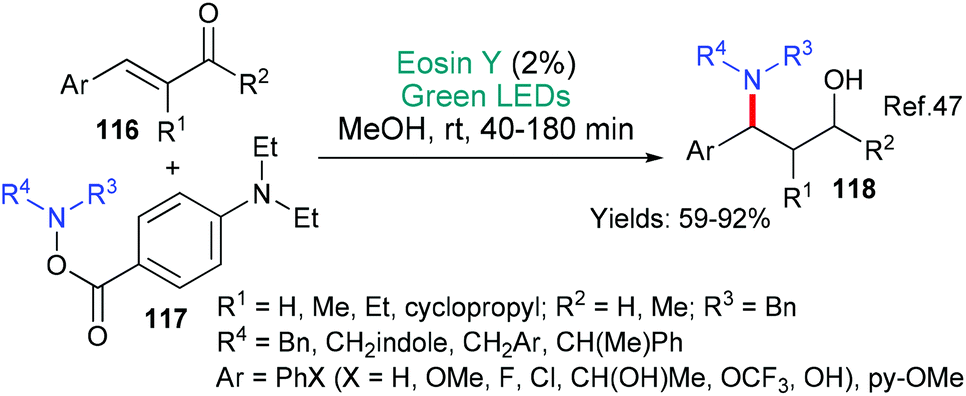 | ||
| Scheme 29 Synthesis of 1,3 amino alcohols 118 using eosin. | ||
In 2018, the groups of Wei and Zhao reported a new metal-free way of aminating quinoxalinones (119 → 121, Scheme 30).48 The authors propose that this simple procedure occurs when the primary or secondary amine 120 donates an electron to excited state eosin (*EY → EY˙−) and the resulting radical cation loses a proton to generate the aminyl radical which adds to the quinoxalinone substrate 119. Following an oxidation (N˙ → N+) facilitated by oxygen, subsequent elimination of an adjacent proton yields the aminated quinoxolinone 121 product.
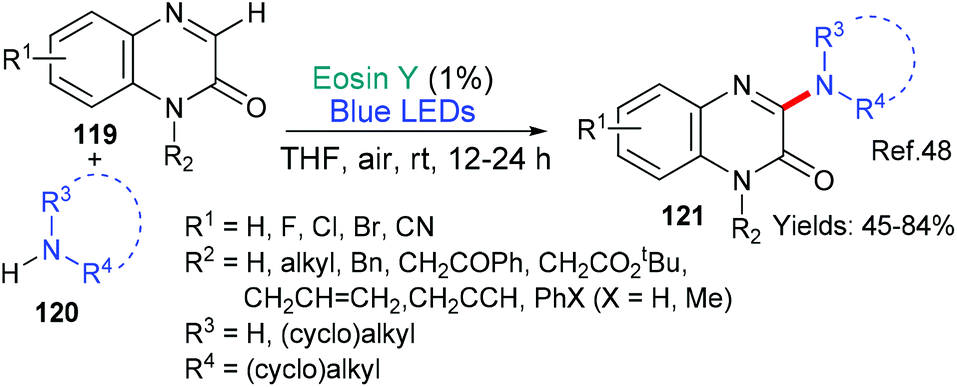 | ||
| Scheme 30 Amination of quinoxalinones 119 using eosin. | ||
The first example from the groups of Noël and Madder is a biocompatible cysteine modification reaction that can be conducted in batch or in microflow; with the latter accelerating the reaction and leading to consistently higher yields (Scheme 31).49 Aryldiazonium salts are formed in situ from readily available anilines 123. The aryldiazonium salts then fragment to form the corresponding aryl radicals (Ar˙) in an eosin-catalzyed process (via*EY → EY˙+ as we have already seen in many examples in this review, see; Schemes 9B, C and 23) and go on to arylate the cysteine residues in small peptide units (122 → 124).
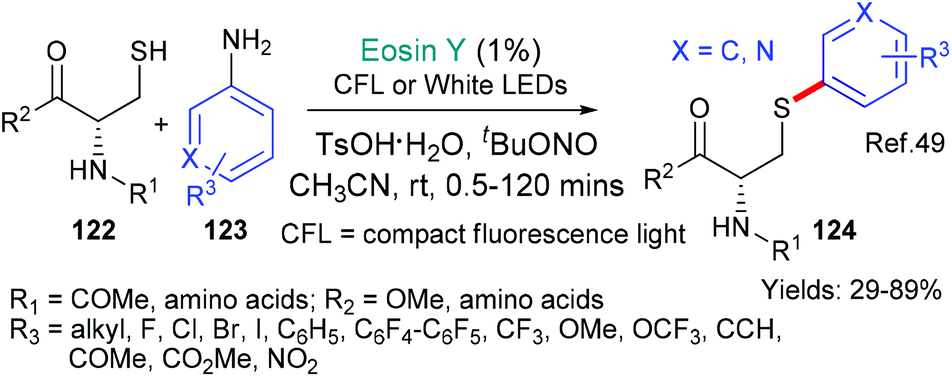 | ||
| Scheme 31 Biocompatible conditions for the arylation of cysteines 122 → 124 in either batch or flow. | ||
In 2019, the group of Singh published a new methodology for the synthesis of aromatic thioethers 127 from hydrazones 125 (Scheme 32).50 The authors propose that the transformation begins when excited state eosin accepts an electron (*EY → EY˙−) from the thiol substrate 126. Following loss of a proton, the RS˙ radical is formed. Two alternative pathways are proposed for the reaction of this radical with the hydrazones 125 with evolution of nitrogen gas to ultimately yield the thioether product 127. Another example of thioether formation was published recently in the form of a protocol for the oxathiacetalization of aldehydes & ketones.51
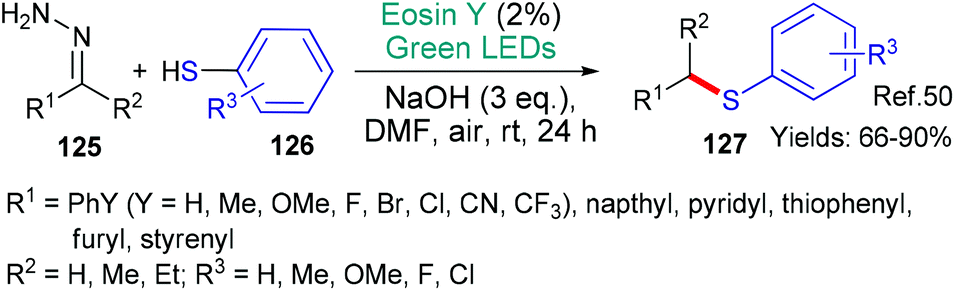 | ||
| Scheme 32 Synthesis of aromatic thioethers 127 using eosin. | ||
Earlier, in 2017, the group of Fraile and Alemán published a synthesis of sulfoxides 130 through an eosin-mediated addition of RS˙ radicals to alkenes 129 (Scheme 33).52 In this case, the mechanism proposed by the authors has eosin fulfilling two distinct roles; firstly, excited state eosin accepts an electron from the thiol 128 which, after loss of a proton, generates the RS˙ radical. This radical adds regioselectively to the alkene substrate 129. In a final step, the sulfur atom is oxidized by singlet oxygen generated through eosin photosensitization.
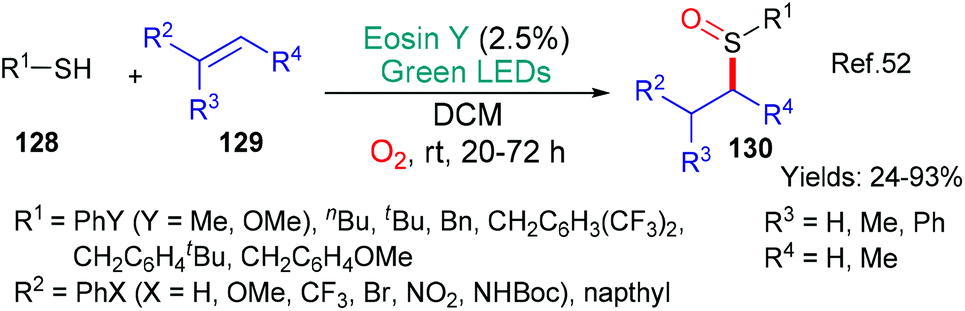 | ||
| Scheme 33 Synthesis of sulfoxides 130 using eosin. | ||
Sulfones were targeted by Chawla and Yadav in 2019 because they represent a common motif within drug candidates (Scheme 34).53 Diazonium salts 131 were used to generate aryl radicals (Ar˙ via*EY → EY˙+ as we have already seen in many examples in this review, see; Schemes 9B, C, 23 and 31) that added to the sodium sulfinate substrates 132 to afford the desired sulfone products 133. The authors showed that the diazonium salts could be formed in situ directly from anilines as had been previously achieved in other protocols (c.f.Scheme 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49).
49).
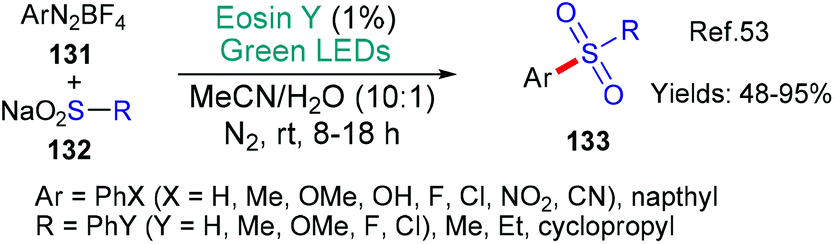 | ||
| Scheme 34 Synthesis of unsymmetrical sulfones 133 using eosin. | ||
Sulfones had also been made earlier, in 2016, in a methodology using eosin that was published by König et al. (Scheme 35).54a In this example, excited state eosin is used to generate a sulfinyl radical by accepting an electron (*EY + R2SO2Na → EY˙− + R2SO2˙). This radical then adds to the substrate 134, ultimately, yielding the products 136. Sulfinyl radicals generated through a variety of eosin-mediated mechanisms54b–d have also been added to double54c,d or triple bonds.54b Yet another interesting example of sulfinyl radical formation was published very recently in a new procedure for the synthesis of enaminosulfones55 which builds on earlier protocols such as the one shown in Scheme 22.36
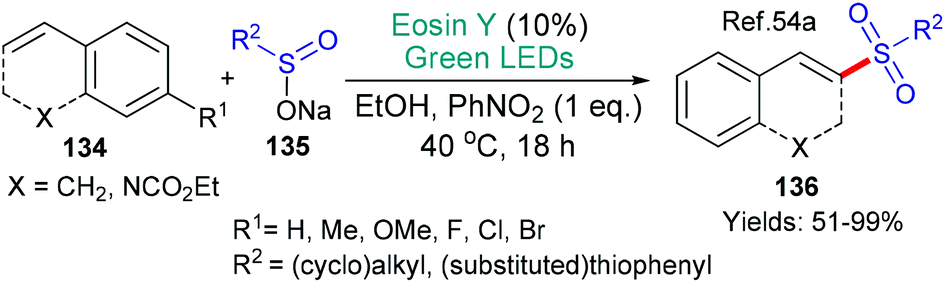 | ||
| Scheme 35 Synthesis of sulfones 136 using eosin. | ||
A mechanistically interesting example of the addition phosphinyl radicals (˙P(O)Ar1Ar2) to alkynes 137 was published by the group of Lei in 2018 (Scheme 36).56a The addition is overwhelmingly Z-selective with substrates where R1 is an aryl group showing the highest selectivity; this enhancement is attributed to π–π stabilization between R1 and Ar2. The authors propose that the reaction proceeds through a proton coupled electron transfer (PCET) mechanism. Thus, the base additive removes a proton from the diarylphosphine oxide 138 at the same time as excited state eosin removes an electron (*EY → EY˙−) generating the phosphinoyl radical that subsequently adds to the radical acceptor alkyne 137. Phosphinyl radicals generated using eosin have also been added to coumarins56b and benzothiazoles.56c
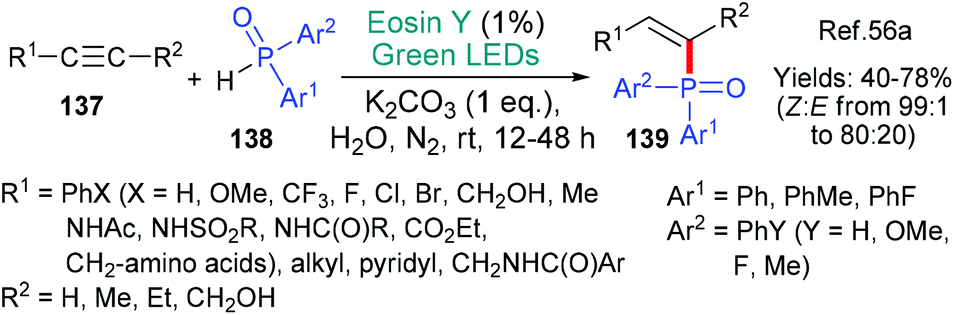 | ||
| Scheme 36 Synthesis of Z-alkenylphosphine oxides 139 using eosin. | ||
2.4 Cooperative catalysis: eosin plus a transition metal-based catalyst
Eosin's versatility is very well showcased in this section about cooperative catalysis being used for the synthesis of privileged scaffolds. In these examples that are summarized in Scheme 37,57–63 eosin is acting in consort with a transition metal to complete the requisite catalytic cycles. Eosin facilitates the use of mild conditions and can widen functional group tolerance. Here, excited state eosin most frequently acts as an oxidant (*EY → EY˙−, Scheme 37A–E),57–61 but there is also an example where it is acting as a reductant (*EY → EY˙+, Scheme 37F)62 and one where it acts as an energy transfer agent to generate singlet oxygen (Scheme 37G).63 An important application of this concept within the critical fields of green chemistry and clean energy chemistry (hydrogen production) was recently reported by the group of Kim.64 In this investigation, a bio- inspired nickel catalyst with eosin as co-oxidant was used to fix carbon dioxide to produce formate ions. The same catalyst system was examined for hydrogen production. | ||
| Scheme 37 Examples of cooperative catalysis between transition metals and eosin for the synthesis of various privileged scaffolds. | ||
2.5 Oxidation and dehydrogenation protocols
Building on precedents, the benzylic oxidation of various benzylamine-containing scaffolds has been shown to be facile using eosin (Scheme 38).65,66 Mechanistically, the reactions are said to proceed via excited state eosin acting as an oxidant (*EY → EY˙−) with the resulting benzylic radical being trapped by a reactive oxygen species (ROS) to afford, ultimately, the oxidized products, dihydroisoquinolones 16065 or picolinamides 162.66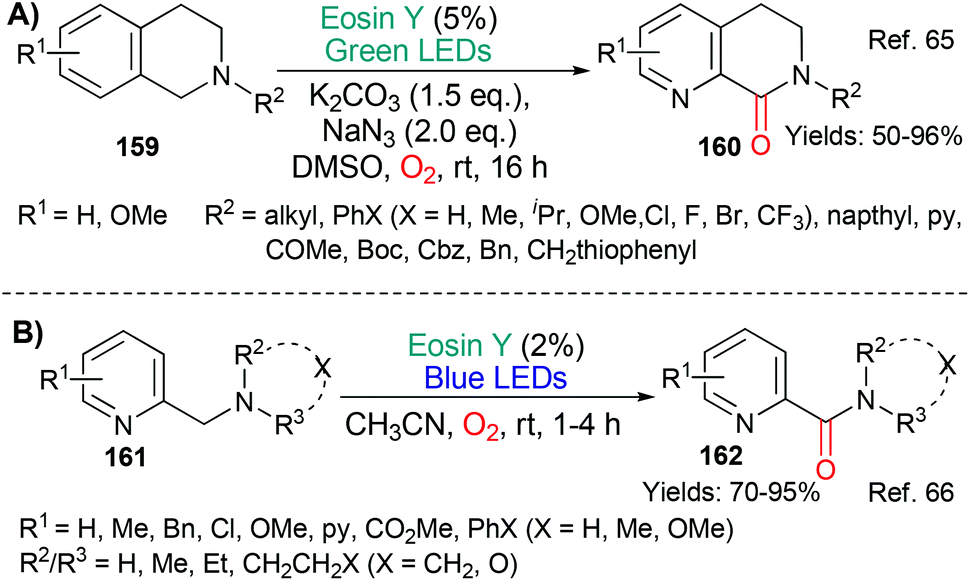 | ||
| Scheme 38 Synthesis of dihydroisoquinolones 160 and picolinamides 162 using eosin. | ||
In 2018, the group of Das published an investigation into the dehydrogenation of a wide range of activated amines (substrates 163–166 afford products 167–170) using excited state eosin (Scheme 39).67 The protocol uses the bases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and requires a carbon dioxide atmosphere. The authors propose a mechanism which includes formation of a DBN-CO2 adduct and excited state eosin acting as an oxidant (*EY → EY˙−).
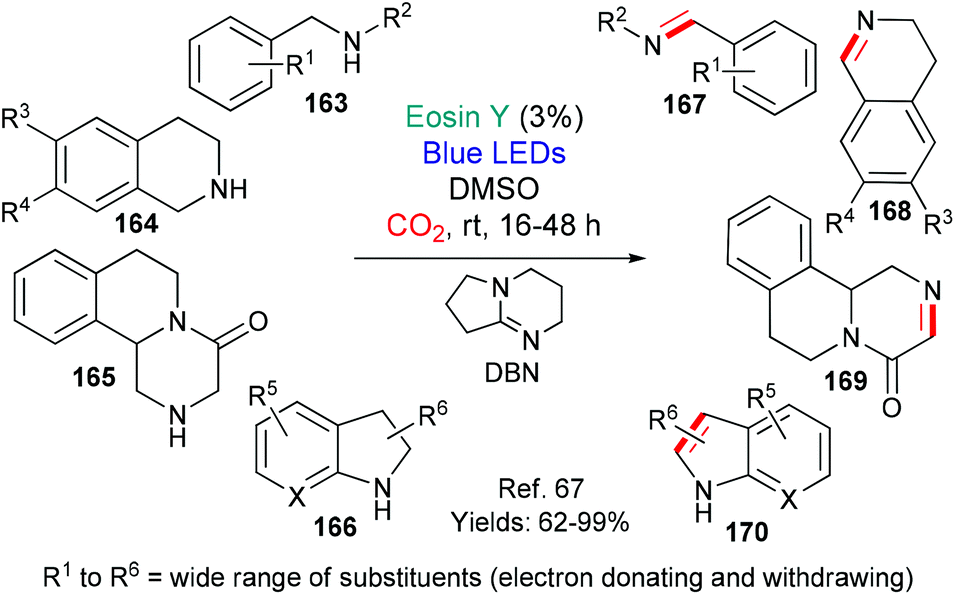 | ||
| Scheme 39 Dehydrogenation of activated amines 163–166 to give privileged structures 167–170 using eosin. | ||
2.6 Unusual uses of eosin
In 2018, the group of Liu published a new method for the synthesis of 4-pyrrolin-2-ones 173 which uses a catalyst combination of eosin and copper acetate in the dark (Scheme 40).69 The authors propose that eosin acts as an electron donor (a reductant) for the transformation of Cu(II) to Cu(I) (EY → EY˙+) and also as an oxidant by accepting an electron from the ketene acetal 172 (EY˙+→ EY) to return to its neutral state. The 4-pyrrolin-2-one products 173 are interesting compounds with many uses.70
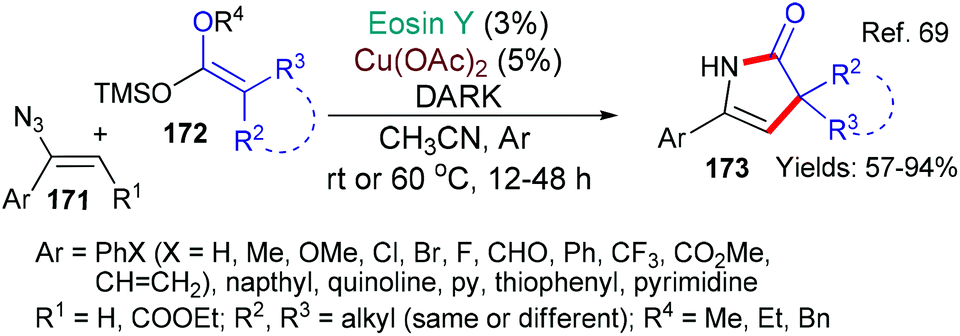 | ||
| Scheme 40 Synthesis of 4-pyrrolin-2-ones 173 using eosin in the dark. | ||
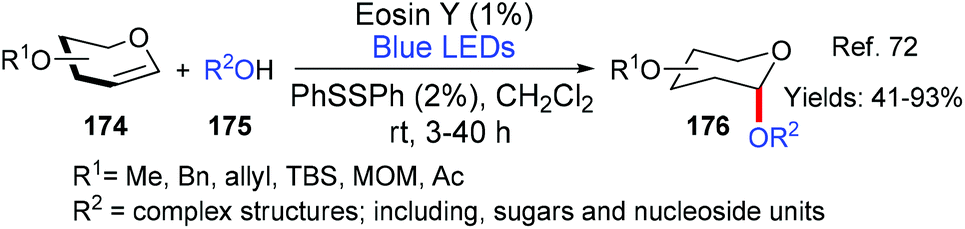 | ||
| Scheme 41 Synthesis of 2-deoxyglycosides 176 using eosin as a photoacid. | ||
3 Conclusion
Here ends our survey of the recent uses of eosin in synthesis. It is not possible for the review to be an exhaustive compilation of works published, not least because with every week that passes new papers appear that add to the body of literature building up around eosin. The main message we would like to convey is the extraordinary versatility of eosin. Eosin has moved far beyond its traditional role as a photosensitizer (energy transfer agent) to encompass so much more. In this review, we have seen eosin act as an oxidant (electron acceptor), as a reductant (electron donor), as an H-atom abstractor, as a photoacid, in cooperative catalysis, in the light and in the dark. We have seen it catalyze the formation of single bonds, multiple bonds, carbon–carbon bonds and many different types of carbon-heteroatom bonds. It is hard to think of another reagent that can mediate so many different types of reaction and fulfill so many different mechanistic roles while at the same time offering excellent functional group tolerance and mild reaction conditions. For these reasons and many more, eosin certainly offers a promising alternative to precious metal catalysis and should be considered as a possible catalyst for new transformations. We expect to see an expansion in the groups that can be oxidized by eosin and in the bonds that can be reduced by eosin to initiate reaction sequences. We also anticipate that the number of radical cascades initiated by eosin will increase significantly in the near future. Thus, eosin's story will continue to develop at a significant pace.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the Greek General Secretariat of Research and Technology in the form of matching (reward) funds (KA: 4143 and KA: 4154).Notes and references
- A. S. Travis, in Tech and Culture, John Hopkins University Press, 1998, vol. 39, issue 1, pp. 105–115 Search PubMed.
- M. Geldof, A. N. P. Gaibor, F. Ligterink, E. Hendriks and E. Kirchner, Heritage Sci., 2018, 6, 17 CrossRef.
- K. C. Nicoloau and T. Montagnon in Molecules that Changed the World, Weinheim, Germany, 2008, pp. 49–56 Search PubMed.
- For a classic example, see: (a) E. J. Corey, D. N. Crouse and J. E. Anderson, J. Org. Chem., 1975, 40, 2140–2141 CrossRef CAS PubMed; For a review containing many examples, see: (b) T. Montagnon, D. Kalaitzakis, M. Triantafyllakis, M. Stratakis and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 15480–15498 RSC.
- (a) C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 2018 CrossRef; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (b) V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC; (c) F. Herbrik, P. C. González, M. Krstic, A. Puglisi, M. Benaglia, M. A. Sanz and S. Rossi, Appl. Sci., 2020, 10, 5596 CrossRef CAS; For important mechanistic discussion, see: (d) M. Majek, F. Filace and A. Jacobi von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed ; For a useful review on metal free C–X bond formation containing many eosin-mediated reactions, see:; (e) L. Ren, M. Ran, J. He, Y. Qian and Q. Yao, Chin. J. Org. Chem., 2019, 39, 1583–1595 CrossRef CAS.
- Y.-T. He, D. Kang, I. Kim and S. Hong, Green Chem., 2018, 20, 5209–5214 RSC.
- J. Jeon, Y.-T. He, S. Shin and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 281–285 CrossRef CAS PubMed.
- A. K. Yadav, A. K. Sharma and K. N. Singh, Org. Chem. Front., 2019, 6, 989–993 RSC.
- (a) P. Dai, X. Yu, P. Teng, W. H. Zhang and C. Deng, Org. Lett., 2018, 20, 6901–6905 CrossRef CAS PubMed ; For a further example where Langois’ reagent is used, see:; (b) J. Fang, W.-G. Shen, G.-Z. Ao and F. Liu, Org. Chem. Front., 2017, 4, 2049–2053 RSC.
- Z. Wei, S. Qi, Y. Xu, H. Liu, J. Wu, H. Li, C. Xia and G. Duan, Adv. Synth. Catal., 2019, 361, 5490–5498 CrossRef CAS.
- W.-K. Tang, Y.-S. Feng, Z.-W. Xu, Z.-F. Cheng, J. Xu, J.-J. Dai and H.-J. Xu, Org. Lett., 2017, 19, 5501–5504 CrossRef CAS PubMed.
- T. Yajima and M. Ikegami, Eur. J. Org. Chem., 2017, 2126–2129 CrossRef CAS.
- M. M. Nebe, D. Loeper, F. Fürmeyer and T. Opatz, Eur. J. Org. Chem., 2018, 2471–2476 CrossRef CAS.
- M.-D. Zhou, Z. Peng, L. Li and H. Wang, Tetrahedron Lett., 2019, 60, 151124 CrossRef CAS.
- S. P. Midya, J. Rana, T. Abraham, B. Aswin and E. Balaraman, Chem. Commun., 2017, 53, 6760–6763 RSC.
- (a) H. Chen, L. Guo and S. Yu, Org. Lett., 2018, 20, 6255–6259 CrossRef CAS PubMed; For a related N-(acyloxy)phthalimide with eosin as an oxidant example, see: (b) Y.-L. Zhang, L. Yang, J. Wu, C. Zhu and P. Wang, Org. Lett., 2020, 22, 7768–7772 CrossRef CAS PubMed.
- Q.-L. Wang, Z. Chen, C.-S. Zhou, B.-Q. Xiong, P.-L. Zhang, C.-A. Yang, Y. Liu and Q. Zhou, Tetrahedron Lett., 2018, 59, 4551–4556 CrossRef CAS.
- K. Wu, L. Wang, S. Colón-Rodríguez, G.-U. Flechsig and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 1774–1778 CrossRef CAS PubMed.
- M. K. Sahoo, S. P. Midya, V. G. Landge and E. Balaraman, Green Chem., 2017, 19, 2111–2117 RSC.
- (a) R. Kapoor, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2019, 60, 805–809 CrossRef CAS; For other examples using diazonium salts and eosin, see: (b) T. Adak, C. Hu, M. Ruldoph, J. Li and A. S. K. Hashmi, Org. Lett., 2020, 22, 5640–5644 CrossRef CAS PubMed; (c) T. Meyer, J.-X. Xu, J. Rabeah, A. Brückner and X.-F. Wu, ChemPhotoChem, 2020, 4, 713–720 CAS.
- C. Tian, L.-M. Yang, H.-T. Tian, G.-H. An and G.-M. Li, J. Fluor. Chem., 2019, 219, 23–28 CrossRef CAS.
- X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 1333–1336 CrossRef CAS.
- X. Liu, Z. Qing, P. Cheng, X. Zheng, J. Zeng and H. Xie, Molecules, 2016, 21, 1690 CrossRef PubMed.
- P. J. Borpatra, M. L. Deb and P. K. Baruah, Tetrahedron Lett., 2017, 58, 4006–4010 CrossRef CAS.
- X. Zhang, Y. He, J. Li, R. Wang, L. Gu and G. Li, J. Org. Chem., 2019, 84, 8225–8231 CrossRef CAS PubMed.
- (a) S. D. Tambe, R. S. Rohokale and U. A. Kshirsagar, Eur. J. Org. Chem., 2018, 2117–2121 CrossRef CAS; (b) l. Gu, C. Jin, W. Wang, Y. He, G. Yang and G. Li, Chem. Commun., 2017, 53, 4203–4206 RSC.
- D.-M. Yan, Q.-Q. Zhao, L. Rao, J.-R. Chen and W.-J. Xiao, Chem. – Eur. J., 2018, 24, 16895–16901 CrossRef CAS PubMed.
- S. Zou, S. Geng, L. Chen, H. Wang and F. Huang, Org. Biomol. Chem., 2019, 17, 380–387 RSC.
- J. Davies, T. D. Svejstrup, D. F. Reina, N. S. Sheikh and D. Leonori, J. Am. Chem. Soc., 2016, 138, 8092–8095 CrossRef CAS PubMed.
- D. Sun, K. Yin and R. Zhang, Chem. Commun., 2018, 54, 1335–1338 RSC.
- (a) P. Qian, Y. Deng, H. Mei, J. Han, J. Zhou and Y. Pan, Org. Lett., 2017, 19, 4798–4801 CrossRef CAS PubMed; (b) L. Wan, M. Zhang, Y. Zhang, Q. Liu, X. Zhao, J.-S. Li, Z. Luo and W. Wei, Chin. Chem. Lett., 2020, 31, 67–70 CrossRef; (c) K.-L. Zuo, Y.-H. He and Z. Guan, Eur. J. Org. Chem., 2019, 939–948 CrossRef CAS.
- X.-Q. Chu, T. Xie, L. Li, D. Ge, Z.-L. Shen and T.-P. Loh, Org. Lett., 2018, 20, 2749–2752 CrossRef CAS PubMed.
- Z. Yang and A. Tang, Synlett, 2019, 30, 1061–1066 CrossRef CAS.
- J.-C. Yang, J.-Y. Zhang, J.-J. Zhang, X.-H. Duan and L.-N. Guo, J. Org. Chem., 2018, 83, 1598–1605 CrossRef CAS PubMed.
- S. Das, S. K. Parida, T. Mandal, L. Sing, S. De Sarkar and S. Murarka, Chem. – Asian J., 2020, 15, 568–572 CrossRef CAS PubMed.
- C. Jin, L. Su, D. Ma and M. Cheng, New J. Chem., 2017, 41, 14053–14056 RSC.
- S. Mkrtchyan and V. O. Iaroshenko, Chem. Commun., 2020, 56, 2606–2609 RSC.
- Y. Zhao, C. Shi, X. Su and W. Xia, Chem. Commun., 2020, 56, 5259–5262 RSC.
- I. F. S. Marra, A. M. de Almeida, L. P. Silva, P. P. de Castro, C. C. Corrêa and G. W. Amarante, J. Org. Chem., 2018, 83, 15144–15154 CrossRef CAS PubMed.
- P. Natarajan, N. Kumar and Priya, ChemistrySelect, 2020, 5, 4862–4865 CrossRef CAS.
- X. Li, X. Fang, S. Zhuang, P. Liu and P. Sun, Org. Lett., 2017, 19, 3580–3583 CrossRef CAS PubMed.
- Q. Xia, Z. Shi, J. Yuan, Q. Bian, Y. Xu, B. Liu, Y. Huang, X. Yang and H. Xu, Asian J. Org. Chem., 2019, 8, 1933–1941 CrossRef CAS.
- M.-N. Chen, J.-Q. Di, J.-M. Li, L.-P. Mo and Z.-H. Zhang, Tetrahedron, 2020, 76, 131059 CrossRef CAS.
- (a) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 40–43 CrossRef CAS; (b) Y. Xu, X. Xu, B. Wu, C. Gan, X. Lin, J. Wang and F. Ke, Asian J. Org. Chem., 2020, 9, 1032–1035 CrossRef CAS.
- V. Srivastava, P. K. Singh, S. Kanaujia and P. P. Singh, New J. Chem., 2018, 42, 688–691 RSC.
- W. Wei, L. Wang, P. Bao, Y. Shao, H. Yue, D. Yang, X. Yang, X. Zhao and H. Wang, Org. Lett., 2018, 20, 7125–7130 CrossRef CAS PubMed.
- C. Bottecchia, M. Rubens, S. B. Gunnoo, V. Hessel, A. Madder and T. Noël, Angew. Chem., Int. Ed., 2017, 56, 12702–12707 CrossRef CAS PubMed.
- S. Chand, A. K. Pandey, R. Singh, S. Kumar and K. N. Singh, Chem. – Asian J., 2019, 14, 4712–4716 CrossRef CAS PubMed.
- Y.-C. Liu, D. M. Reddy, X.-A. Chen, Y.-C. Shieh and C.-F. Lee, Eur. J. Org. Chem., 2020, 2542–2552 CrossRef CAS.
- A. Guerrero-Corella, A. M. Martinez-Gualda, F. Ahmadi, E. Ming, A. Fraile and J. Alemán, Chem. Commun., 2017, 53, 10463–10466 RSC.
- R. Chawla and L. D. S. Yadav, Org. Biomol. Chem., 2019, 17, 4761–4766 RSC.
- (a) A. U. Meyer, K. Straková, T. Slanina and B. König, Chem. – Eur. J., 2016, 22, 8694–8699 CrossRef CAS PubMed; (b) D. Yang, B. Huang, W. Wei, J. Li, G. Lin, Y. Liu, J. Ding, P. Sun and H. Wang, Green Chem., 2016, 18, 5630–5634 RSC; (c) H. Wang, Q. Lu, C.-W. Chiang, Y. Luo, J. Zhou, G. Wang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 595–599 CrossRef CAS PubMed; (d) S. Cai, Y. Xu, D. Chen, L. Li, Q. Chen, M. Huang and W. Weng, Org. Lett., 2016, 18, 2990–2993 CrossRef CAS.
- O. M. Mulina, A. I. Ilovaisky, T. Opatz and A. O. Terent'ev, Tetrahedron Lett., 2021, 64, 152737 CrossRef CAS.
- (a) H. Wang, Y. Li, Z. Tang, S. Wang, H. Zhang, H. Cong and A. Lei, ACS Catal., 2018, 8, 10599–10605 CrossRef CAS; (b) Q. Li, X. Zhao, Y. Li, M. Huang, J. K. Kim and Y. Wu, Org. Biomol. Chem., 2017, 15, 9775–9778 RSC; (c) P. Peng, L. Peng, G. Wang, F. Wang, Y. Luo and A. Lei, Org. Chem. Front., 2016, 3, 749–752 RSC.
- X. Su, F. Yang, Y. Wu and Y. Wu, Org. Biomol. Chem., 2018, 16, 2753–2756 RSC.
- G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C.-H. Tung, L.-Z. Wu and A. Lei, Chem. Commun., 2016, 52, 10407–10410 RSC.
- R. Liu, J. Liu, Y. Wei and M. Shi, Org. Lett., 2019, 21, 4077–4081 CrossRef CAS PubMed.
- D. Kalsi, S. Dutta, N. Barsu, M. Rueping and B. Sundararajua, ACS Catal., 2018, 8, 8115–8120 CrossRef CAS.
- D. Kalsi, N. Barsu, S. Chakrabarti, P. Dahiya, M. Rueping and B. Sundararajua, Chem. Commun., 2019, 55, 11626–11629 RSC.
- X.-J. Yang, Y.-W. Zheng, L.-Q. Zheng, L.-Z. Wu, C.-H. Tung and B. Chen, Green Chem., 2019, 21, 1401–1405 RSC.
- S. P. Sancheti, M. O. Akram, R. Roy, V. Bedi, S. Kundu and N. T. Patil, Chem. – Asian J., 2019, 14, 4601–4606 CrossRef CAS PubMed.
- S. E. Lee, A. Nasirian, Y. E. Kim, P. T. Fard, Y. Kim, B. Jeong, S.-J. Kim, J.-O. Baeg and J. Kim, J. Am. Chem. Soc., 2020, 142, 19142–19149 CrossRef CAS PubMed.
- K. C. C. Aganda, B. Hong and A. Lee, Adv. Synth. Catal., 2019, 361, 1124–1129 CAS.
- V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 151041 CrossRef CAS.
- D. Riemer, W. Schilling, A. Goetz, Y. Zhang, S. Gehrke, I. Tkach, O. Hollóczki and S. Das, ACS Catal., 2018, 8, 11679–11687 CrossRef CAS.
- (a) D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2015, 54, 6283–6287 CrossRef CAS PubMed; (b) D. Kalaitzakis, D. Noutsias and G. Vassilikogiannakis, Org. Lett., 2015, 17, 3596–3599 CrossRef CAS PubMed; (c) D. Kalaitzakis, M. Triantafyllakis, M. Sofiadis, D. Noutsias and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2016, 55, 4605–4609 CrossRef CAS PubMed; (d) M. Sofiadis, J. Sarris, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Eur. J. Org. Chem., 2018, 4523–4526 CrossRef CAS; (e) D. Kalaitzakis, K. Daskalakis, M. Triantafyllakis, M. Sofiadis and G. Vassilikogiannakis, Org. Lett., 2019, 21, 5467–5470 CrossRef CAS PubMed.
- W.-L. Lei, K.-W. Feng, T. Wang, L.-Z. Wu and Q. Liu, Org. Lett., 2018, 20, 7220–7224 CrossRef CAS PubMed.
- T. Montagnon, D. Kalaitzakis, M. Sofiadis and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 180–190 RSC.
- P. D. Dharpure, A. Bhowmick, P. K. Warghude and R. G. Bhat, Tetrahedron Lett., 2020, 61, 151407 CrossRef CAS.
- G. Zhao and T. Wang, Angew. Chem., Int. Ed., 2018, 57, 6120–6124 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |