
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct monitoring of biocatalytic deacetylation of amino acid substrates by 1H NMR reveals fine details of substrate specificity†
Silvia
De Cesare
a,
Catherine A.
McKenna
a,
Nicholas
Mulholland
b,
Lorna
Murray
a,
Juraj
Bella
a and
Dominic J.
Campopiano
 *a
*a
aSchool of Chemistry, University of Edinburgh, David Brewster Road, King's Buildings, Edinburgh, EH9 3FJ, UK. E-mail: Dominic.Campopiano@ed.ac.uk
bSyngenta, Jealott's Hill, Warfield, Bracknell RG42 6EY, UK
First published on 3rd May 2021
Abstract
Amino acids are key synthetic building blocks that can be prepared in an enantiopure form by biocatalytic methods. We show that the L-selective ornithine deacetylase ArgE catalyses hydrolysis of a wide-range of N-acyl-amino acid substrates. This activity was revealed by 1H NMR spectroscopy that monitored the appearance of the well resolved signal of the acetate product. Furthermore, the assay was used to probe the subtle structural selectivity of the biocatalyst using a substrate that could adopt different rotameric conformations.
Introduction
Reaction monitoring remains a pillar of biocatalysis. It has been employed in the study of the activity, selectivity and substrate scope of wild type enzymes and it is a fundamental platform upon which directed evolution can be applied to engineer novel, more efficient biocatalysts.1–4 Therefore, the development of robust and reproducible high-throughput (HTP) assays is necessary to achieve fast and reliable measurement of enzymatic activity, kinetic parameters and reaction conversions.5Hydrolases form a well-characterised class of stereoselective biocatalysts, usually employed to develop kinetic resolution (KR), dynamic kinetic resolution (DKR) or de-symmetrisation processes for the isolation of small, enantiopure molecules.6 Aminoacylases or acylases (EC 3.5.1.14) are important members of this family of enzymes; they specifically catalyse the hydrolysis of the amide bonds of canonical N-acetylated amino acids (N-Ac-AAs) or derivatives, releasing the free amino acid along with acetate into solution (Scheme 1). Given the high stereoselectivity of these class of enzymes, they are usually employed in the resolution of optically active products from racemic starting materials7 and to synthesise a range of amides including the penicillin-derived antibiotics.8 They can be readily combined with N-acetyl amino acid racemases (NAAARs or NSARs) to achieve a DKR that can convert racemic N-acetylated amino acids into enantiopure products.9,10 Moreover, some acylases have also been shown to act “in reverse” to catalyse the formation of amides from amino sugars, amino acids and various carboxylic acids.11,12
 | ||
| Scheme 1 Synthesis of enantiopure D- or L-amino acids via hydrolysis of N-acetylated substrates using a stereoselective D- or L-acylase biocatalyst. | ||
A classic approach to calculate the conversions of these biotransformations is via chiral HPLC analysis.13,14 However, this approach can only be employed when UV-active substrates (e.g. aromatic, heterocyclic) are used. Alternative HPLC assays monitor the kinetics of the hydrolytic reaction by observing the disappearance of the N-acylated starting material.15–17 However, given the weak absorbance of the amide bond in the UV region (214 nm, ε = 103 M−1 cm−1),15 the sensitivity of this analysis is quite poor. To overcome this limitation, various colorimetric and fluorometric assays use two main strategies to monitor biocatalysed deacetylation reactions.
The first strategy consists of exploiting a HTP continuous enzymatic assay.5 For example, three enzymes, an acetate kinase (AK), a pyruvate kinase (PK) and a D-lactate dehydrogenase (LDH) are coupled together in the presence of ATP and phosphoenolpyruvate (PEP) to measure the free acetate formed by linking it directly to the oxidation of NADH to NAD+ (Scheme S1A†).18 Similarly, the formation of L-glutamate (L-Glu) can be easily monitored using an NAD+-dependant L-Glu dehydrogenase (GDH) that catalyses the oxidative deamination of L-Glu to α-ketoglutarate.19 Sánchez Carrón et al.,20 recently reported a HTP colorimetric assay that detects free amino acids by coupling their production with an FAD-dependant L-amino acid oxidase (L-AAO) and a horseradish peroxidase (HRP) (Scheme S1B†). The resulting H2O2 can be linked to the oxidation of o-dianisidine at 436 nm (ε = 5700 M−1 cm−1) or a similar colorimetric reporter. Although these HTP continuous assays are useful when measuring biocatalyst activity and kinetics, they are not suitable for quantitative analysis as the formation of the product is monitored indirectly via a cascade of reactions with a limited linear range.
A second popular strategy often employed for the analysis of free amino acids is the use of derivatization reagents to produce a product with either a strong absorbance or fluorescence in the UV-Vis region. These derivatives are easily detected via HPLC or LC-MS analysis. Such reagents include: ninhydrin,21o-phthalaldehyde (OPA),22,23 Marfey's reagent (MR)24 and 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD).25 Use of MR also allows for the determination of the enantiomeric excess (% ee) of the desired reaction product, since it forms two easily separated diastereoisomers (Scheme S1C†). Amino acids can also be derivatised using various agents, such as alcohols or chloroformates to obtain a volatile product that is readily analysed by GC.26–29 Unfortunately, all derivatization reactions suffer from a lack of reproducibility, given the possible formation of side-products in the assay mixture. Recently, various LC-MS protocols for the quantitation of free amino acids and MR derivatives in solution have also been developed.30–32
Alongside these methods, an ideal candidate for biocatalyst reaction monitoring is 1H NMR spectroscopy. This technique allows the easy calculation of reaction conversions using a relative quantitation method, or the determination of the absolute concentration of starting material and product present in the sample when an appropriate internal standard is employed in a quantitative NMR (qNMR) experiment.33 Given the various advantages of 1H NMR spectroscopy, such as the easy analysis set-up, short measurement time, direct structural information, high accuracy and its non-destructive nature, this technique had already been applied for the quantitative analysis of active ingredients (AI), natural products and complex biological mixtures.33,34 Furthermore, 1H NMR spectroscopy has been used to measure biocatalysed reactions; examples include lipase-catalysed acylations35 and monophosphates ring opening reactions,36 determination of the % ee of hydrolytic KRs,37,38 and to characterise complex carbohydrate derivatives.39,40 Here we report the development, optimization and application of a novel 1H NMR assay to monitor amide hydrolysis catalysed by a highly active deacetylase (Scheme S1D†). This was achieved by direct observation (both time dependent and end-point) of the sharp, well-resolved singlet peak associated with the methyl group of the acetate product. Furthermore, we show the wide utility and applicability of this assay, by applying an optimised 1H NMR protocol to determine the % conversion for a series of N-acetylated, non-canonical amino acids (NCAAs). Finally, we provide insight into the subtle biocatalyst substrate preference of N-acyl amide rotamer conformations.
Results and discussion
1H NMR assay proof of concept
Our target biocatalyst for proof-of-concept studies is the Escherichia coli N-acetyl ornithine (N-Ac-Orn) deacetylase (ArgE, Uniprot code: P23908, Fig. 1A) which is a di-metal (e.g. Zn2+) dependant L-selective acylase with a broad substrate scope.15,161H NMR is ideal for monitoring deacetylations as the 1H NMR spectrum of an N-acetylated amino acid has at least two very distinctive signals, which undergo a shift during hydrolysis (Fig. 1B and C). Catalytic cleavage of the amide bond results in the C-α-proton shifting from 4–5 ppm to 3–4 ppm, while the CH3 group singlet shifts from the amide substrate at 2.1–1.9 ppm to the acetate product at 1.9–1.8 ppm. By comparing the ratios integrals of these signals, it is possible to calculate the relative conversions of the biocatalysed reaction.33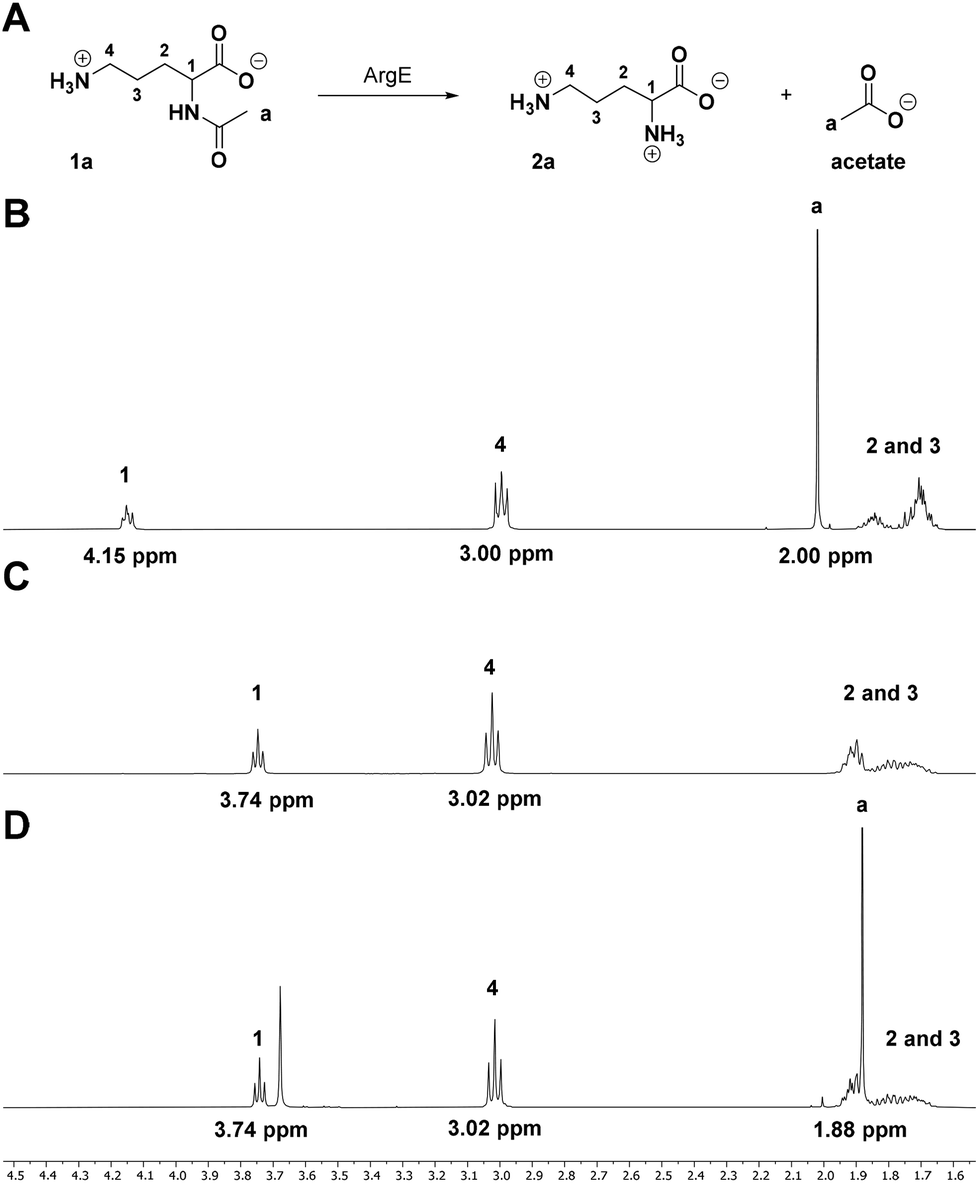 | ||
| Fig. 1 1H NMR assay proof of concept. (A) Scheme for the ArgE-catalysed deacetylation of N-Ac-L-Orn (1a) with 1H NMR spectra with annotated signals. (B) N-Ac-L-Orn (1a) substrate standard. (C) L-Orn (2a) product standard. (D) ArgE biotransformation reaction shows complete conversion of 1a to 2a during 24 h at pH 8.0 (the signal at 3.68 ppm is due to the TRIS buffer used during E. coli ArgE purification). | ||
A biotransformation (Fig. 1A) was set up using the natural substrate of the ArgE deacetylase, N-Ac-L-Orn (1a). Recombinant E. coli ArgE was expressed, purified and characterised as reported in ESI (see ESI, Fig. S1 and S2†). The substrate N-Ac-L-Orn, 1a (20 mM) was incubated with ArgE and CoCl2 at 40 °C for 24 h in a final volume of 500 μL. CoCl2 was added to the reaction mixture, since it has been reported that Co(II) increases the activity of ArgE approximately 2-fold.16 Once L-ornithine (2a) formation was confirmed by LC-TOF MS analysis, the biotransformation was quenched by addition of HClconc to precipitate the enzyme. 100 μL of D2O was mixed in an NMR tube with 400 μL of the crude reaction mixture and the 1H NMR spectrum of the sample was recorded on a Bruker AVA500 (500 MHz) NMR. To achieve a higher signal to noise ratio (S/N), a solvent signal suppression experiment was employed. For reference, an NMR spectrum of a standard samples of substrate 1a and product 2a were also recorded. By comparing the spectra of standards 1a, 2a (Fig. 1B and C) with the reaction mixture (Fig. 1D) it was possible to conclude that the reaction went to completion, following 24 h incubation at 40 °C. This was confirmed by the disappearance of the signals from the C-α proton (multiplet at 4.15 ppm) and the CH3 from the amide (2.00 ppm) (Fig. 1D). This was also accompanied by the clear CH3 signal observed for the acetate product at 1.88 ppm (Fig. 1D). It is worth noting that depending on the nature of the substrate side chain, the C-α proton signal of N-acetylated amino acids (4–5 ppm region) could fall close to or overlap with the residual peak of water (4.79 ppm). This superimposition can alter the intensity of this signal during the solvent suppression experiment, rendering peak integration inaccurate. However, the acetate singlets of the amide substrate fall in a low-field region of the spectrum (2.1–1.7 ppm), far away from the solvent residual peak, thus, it can be integrated with high accuracy to give a good estimate of the reaction conversion. Furthermore, this signal is generated by three equivalent protons, which should deliver a better S/N ratio and higher assay sensitivity; indeed, sodium acetate is a common internal standard used in qNMR experiments for the determination of the concentrations of the species present in the sample.32
Encouraged by the success of our first analysis, we set up a screen of biotransformations following the same reaction protocol, to prove the wide applicability of this assay in the study of enzymatic deacetylations. A panel of nineteen N-acetylated L-proteinogenic amino acids (with the exception of N-acetyl serine, available only as a racemate) was tested as ArgE substrates (Scheme S2†). All reaction conversions were calculated by comparing the intensity of the methyl signal of the N-bound amide substrate and free acetate product (apart from the N-Ac-Gln substrate where we used the signals from the C-α-proton, see ESI†). Amino acid product formation was also confirmed by detection via LC-MS analysis to back up the 1H NMR data. The screening results, reported in Table S1,† are in broad agreement with the data reported in literature which used the less sensitive UV-Vis amide bond assay (λmax = 214 nm) to determine the kinetic parameters of ArgE.14 We found that ArgE displayed a preference for aliphatic and positively charged substrates, whereas acidic (glutamic and aspartic acid) and aromatic (phenylalanine (<2.0% conversion), tyrosine and tryptophan) N-acylated amino acids are not hydrolysed by this L-acylase. In addition, our assay was able to identify some novel substrates, such as N-Ac-L-valine, arginine, histidine and proline. This data shows how 1H NMR spectroscopy can be exploited to obtain reliable assays for these amide substrates with higher sensitivity compared to classic spectrophotometric analysis.
Reaction monitoring
Thus far, we have simply employed 1H NMR spectroscopy to calculate the reaction conversion in an end-point type assay, after prolonged incubation (24 h). However, NMR has been successfully employed to run kinetic experiments to directly monitor various reactions over time.41–43 Similarly, we employed this technique to follow the progress of the ArgE-catalysed deacetylation reaction. To run these real-time, reaction monitoring experiments N-Ac-L-Val (Fig. 2A) was selected as the substrate since this N-acetylated amino acid has two signals that fall at low field in the 1H NMR spectrum. These can be employed to follow the biotransformation progress. The 1H NMR spectrum was recorded for both the N-acyl-L-Val substrate and L-Val reaction product to use as reference (Fig. S3†). We observed both the N-acyl singlet (N-Ac at 2.16 ppm) and the signals generated from the two –CH3 groups of the isopropyl side-chain (iPr, βR at 0.98 pm) for N-Ac-L-Val. | ||
| Fig. 2 Direct time-dependant monitoring of the biocatalytic N-Ac-L-Val deacetylation by 1H NMR spectroscopy. (A) Reaction scheme of the ArgE-catalysed N-Ac-L-Val hydrolysis. The iPr groups of the substrate (βR) and product (βP) are labelled. (B) Superimposition of 1H NMR spectra of the N-Ac-L-Val hydrolysis at 40 °C, pH 8.0 during incubation from 15 (dark red) to 125 min (dark blue). The signals for the substrate (βR at 0.98 ppm and N-Ac at 2.16 ppm) reduce and new signals for the L-Val product (βP 1.12 ppm and Ac, 2.02 ppm) appear. | ||
Two identical reactions were set up in NMR tubes and incubated at 25 °C and 40 °C respectively. A 1H NMR spectrum was recorded on a Bruker AVA 400 (400 MHz) NMR every 10 min for 2.5 h (Fig. 2B); the first time-point measurement was taken 15 min after the addition of ArgE to the reaction mixture. This lag phase was necessary to set up the experiment and to give enough time for the NMR probe to reach the desired temperature. In this initial NMR spectrum, effectively 15 min into the reaction, we observed the appearance of a new signals due to the formation of the acetate (Ac, 2.02 ppm) and the L-Val product (βP at 1.12 pm). Integration of these revealed the reaction progress to be 34%, for the reaction incubated at 25 °C. We then monitored these signals over time (every 10 min) for 3 h. By superimposing the recorded spectra, it was possible to observe the progress of the reaction, with the characteristic signals of the N-Ac-L-Val decreasing over time and reached completion at 125 min (Fig. 2B, coloured dark red at 15 min moving to dark blue). At the same time, we observed an increase in the relative abundance of the L-Val product signals (normalised to the acetate CH3 signal at 2.02 ppm, Fig. 2B). By measuring the integrals of the substrate and product signals for every 1H NMR spectrum recorded, we were able to calculate the percentage conversion of the reaction for each time point at both 25 and 40 °C (Fig. S4A†). We found that 40 °C was the best incubation temperature for ArgE activity, with full conversion (>99%) reached after 2 h incubation, compared with a conversion of 81% for the reaction incubated at room temperature. We also confirmed that the conversion of the substrate follows a linear initial rate after ArgE addition and determined similar specific activities for the reaction monitored by both the 1H NMR and the coupled L-AAO assays (Fig. S4A and S4B†).
N-Acetylated NCAAs library screening
To further emphasise the broad applicability of this 1H NMR assay we prepared and screened a small library of N-acetylated, non-canonical L-amino acids (NCAAs). The substrates used in the screen were prepared synthetically from the free amino acid via acetylation with an excess of acetic anhydride (see ESI†). The screen of the N-Ac-NCAAs substrates was carried out using the standard protocol (see ESI†) and the final conversions, following incubation with ArgE for 24 h, are reported in Table 1.| Substrate | R (side-chain) | Conv (%) | m/z [M + H]+ |
|---|---|---|---|
| 1a | –CH2CH2CH2NH2 | >99.0 | 133 |
| 1b | –CH2CH2SOCH3 | 94.3 | 166 |
| 1c | –CH2CH2SO2CH3 | 76.4 | 182 |
| 1d | –CH2SCH3 | 50.7 | 136 |
| 1e | –CH2CH2CH2NHCNHNHNO2 | >99.0 | 220 |
| 1f | –Cyclopropyl | 60.9 | 116 |
| 1g | –C(CH3)3 | N.D. | N.D. |
| 1h | –CH2C(CH3)3 | N.D. | N.D. |
| 1i | –CH2Cl | >99.0 | 124 |
| 1j | –CHCHCH(CH3)2 | 28.3 | 144 |
No activity was detected towards N-Ac-tert Leu (1g) and N-Ac-tert Bu-Ala (1h), probably caused by the large steric bulk of the tert-butyl group preventing it being a good ArgE substrate. In contrast, good-to-excellent activity was observed with the eight other substrates tested which displayed a broad range of NCAAs side-chain functionalities. The best substrates were N-Ac-Orn (1a), N-Ac-ω-nitro-Arg (1e), N-Ac-β-chloro-Ala (1i) and also included N-Ac-cyclopropyl-Gly (1f). The substrate preference defined by the 1H NMR assay can now be compared with that measured using the coupled L-AAO method (Scheme S1B, Table S2†). This revealed a similar substrate profile however the 1H NMR assay has the advantage that it reports directly on ArgE specificity and removes the impact of the substrate preference of the L-AAO coupling enzyme.20
Conformational analysis of substrate specificity
Finally, we wanted to further explore the selectivity of ArgE using a substrate that displayed the ability to adopt different conformations in solution. We selected trans N-acetyl 2-amino-5-methylhex-3-enoic acid (1j) (Fig. 3A, see ESI†) and observed a series of signals in the double bond region of the 1H NMR spectrum of the starting material (Fig. 3B). N-Acyl amides are known to form two different conformations (rotamers), due to the restricted rotation around the amide bond. The ratio of minor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :major rotamers is dictated by the thermodynamic stability of each conformation.44,45 We assigned the two doublet of doublets at 5.81 and 5.68 ppm (J = 15.6, 6.5 Hz) as Hd and Hb of the minor and major rotamers respectively, with a ratio of 0.7:1 of Hd to Hb (Fig. 3B). These assignments were confirmed by 2D NMR spectroscopy (see ESI† for NMR characterization). In addition, the multiplet at 5.48–5.41 pm is caused by the overlapping signals of Ha and Hc of the major and minor rotamers respectively (Fig. 3B). Upon incubation with ArgE we observed the formation of a new signal at 5.90 ppm, Hf, which we assign to the vinyl 1H of 2j, as well as the reduction of the Hd signal at 5.81 ppm (Fig. 3C). The other vinyl 1H of the product He, appears in the multiplet at 5.48–5.41 ppm (Fig. 3C). Our analysis suggests that ArgE hydrolyses only one of the rotamers (the minor conformation). However, at present we cannot define the absolute conformation of the preferred rotamer (attempts to further define these using variable temperature NMR did not resolve the conformations). This conformational selectivity (atropisomerism) has been observed during the design of N-acyl amide inhibitors and it is now incorporated into the design of pharmaceuticals.46,47 Such detailed substrate analysis is not possible with the HPLC, MS and HTP colorimetric methods.
:major rotamers is dictated by the thermodynamic stability of each conformation.44,45 We assigned the two doublet of doublets at 5.81 and 5.68 ppm (J = 15.6, 6.5 Hz) as Hd and Hb of the minor and major rotamers respectively, with a ratio of 0.7:1 of Hd to Hb (Fig. 3B). These assignments were confirmed by 2D NMR spectroscopy (see ESI† for NMR characterization). In addition, the multiplet at 5.48–5.41 pm is caused by the overlapping signals of Ha and Hc of the major and minor rotamers respectively (Fig. 3B). Upon incubation with ArgE we observed the formation of a new signal at 5.90 ppm, Hf, which we assign to the vinyl 1H of 2j, as well as the reduction of the Hd signal at 5.81 ppm (Fig. 3C). The other vinyl 1H of the product He, appears in the multiplet at 5.48–5.41 ppm (Fig. 3C). Our analysis suggests that ArgE hydrolyses only one of the rotamers (the minor conformation). However, at present we cannot define the absolute conformation of the preferred rotamer (attempts to further define these using variable temperature NMR did not resolve the conformations). This conformational selectivity (atropisomerism) has been observed during the design of N-acyl amide inhibitors and it is now incorporated into the design of pharmaceuticals.46,47 Such detailed substrate analysis is not possible with the HPLC, MS and HTP colorimetric methods.
 | ||
| Fig. 3 Determination of the conformational selectivity of E. coli ArgE. (A) Reaction scheme for the hydrolysis of the substrate trans-2-acetoamido-5-methylhex-3-enoic acid 1j, present in solution as a mixture of rotamers indicated by the rotation arrow. (B) The double bond region (5–6 ppm) of the 1H NMR spectrum of the two trans protons are annotated as follows: Ha and Hb for the major rotamer, Hc and Hd for the minor rotamer. (C) The same chemical shift region as (B) after incubation of 1j with ArgE (0.25 mg mL−1) and CoCl2 (100 μM) at 40 °C for 24 h. The 1H NMR integrals for the major rotamer do not change whereas the integrals for the minor rotamer decrease. The appearance of new signals that correspond to the product 2j are annotated as He and Hf. | ||
Conclusions
In conclusion, by employing 1H NMR spectroscopy we were able to develop a quick and efficient assay to monitor biocatalytic deacetylation of various amides. For proof of concept we have used E. coli N-Ac-L-ornithine deacetylase (ArgE) and a wide range of canonical N-Ac-L-AAs. The comparison of the integrals of the clear signals for the amide starting material and the free acetate product allows for a fast and accurate determination of substrate specificity. The 1H NMR assay can then be employed to run time-dependant experiments to monitor the progress of the hydrolysis over time and determine the % conversion to product. This assay should have broad applicability since hydrolytic enzymes (e.g. acylases), are widely used to resolve racemic mixtures and prepare optically active products.6,7Another strength of the 1H NMR assay is its wide applicability for screening substrates, in contrast to other assays which have a restricted scope due to limitations in product detection. We screened ArgE activity towards a large library of substrates (29 N-Ac-AAs and N-Ac-NCAAs). Furthermore, the 1H NMR assay enabled the conformational selectivity (amide rotamer preference) of the ArgE biocatalyst to be investigated which is beyond the scope of other published methods. Given the simple experimental set-up, fast analysis time and the easy instrument accessibility, this study further supports the potential of 1H NMR based assays in biocatalysis.
The real time monitoring of enzymatic deacetylation reactions could be further improved to achieve accurate time point measurements on a second/minute scale as evidenced by the recent work carried out by Rother and colleagues.48 Moreover, advances in FlowNMR technology and analysis will accelerate application of these methods.49 With the optimised 1H NMR assay at hand we can further expand its potential via monitoring of acylase-coupled processes, such as biocatalytic DKRs.13 A combination of improved tools for substrate screening and bioinformatics-driven discovery, coupled with improved methods for directed evolution, will lead to the increased application of biocatalysts for synthesis.50
Conflicts of interest
The authors declare no conflict on interests.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC), Syngenta and CRITICAT Centre for Doctoral Training for financial support [Ph.D. studentships to S. D. C. and C. A. McK.; Grant Code: EP/L016419/1]. The NMR facility is supported by EPSRC grant EP/R030065/1. We also thank Prof. Guy Lloyd-Jones and Dr Andrew Hall (University of Edinburgh) for their help and useful comments, as well as Dr Dimitrios Papasotiriou and Dr Peter Howe (Syngenta, Bracknell) for their help in the initial optimization of the 1H NMR and LC-MS experimental protocols. We appreciate the insight provided by Dr Manuela Tosin (University of Warwick) and Dr Amanda Jarvis (University of Edinburgh).References
- M. T. Reetz, Angew. Chem., Int. Ed., 2002, 41, 1335–1338 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- U. Markel, K. D. Essani, V. Besirlioglu, J. Schiffels, W. R. Streit, U. Schwaneberg and U. Markel, Chem. Soc. Rev., 2020, 49, 233–262 RSC.
- H. Bisswanger, Perspect. Sci., 2014, 1, 41–55 CrossRef.
- K. Faber, Biotransformations in Organic Chemistry, Springer International Publishing, 2011 Search PubMed.
- B. Wang, Y. Liu, D. Zhang, Y. Feng and J. Li, Tetrahedron: Asymmetry, 2012, 23, 1338–1342 CrossRef CAS.
- K. Buchholz, Appl. Microbiol. Biotechnol., 2016, 100, 3825–3839 CrossRef CAS PubMed.
- S. Martínez-Rodríguez, P. Soriano-Maldonado and J. Antonio, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140377 CrossRef PubMed.
- S. De Cesare and D. J. Campopiano, Curr. Opin. Biotechnol., 2021, 69, 212–220 CrossRef CAS PubMed.
- M. C. Bourkaib, S. Delaunay, X. Framboisier, C. Humeau, J. Guilbot, C. Bize, E. Illous, I. Chevalot and Y. Guiavarc'h, Process Biochem., 2020, 99, 307–315 CrossRef CAS.
- P. Laborda, Y.-M. Lyu, F. Parmeggiani, A.-M. Lu, W.-J. Wang, Y.-Y. Huang, K. Huang, J. Guo, L. Liu, S. L. Flitsch and J. Voglmeir, Angew. Chem., Int. Ed., 2020, 59, 5308–5311 CrossRef CAS PubMed.
- S. Baxter, S. Royer, G. Grogan, F. Brown, K. E. Holt-Tiffin, I. N. Taylor, I. G. Fotheringham and D. J. Campopiano, J. Am. Chem. Soc., 2012, 134, 19310–19313 CrossRef CAS PubMed.
- S. L. Lovelock, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 4652–4656 CrossRef CAS PubMed.
- F. Javid-Majd and J. S. Blanchard, Biochemistry, 2000, 39, 1285–1293 CrossRef CAS PubMed.
- W. C. McGregor, S. I. Swierczek, B. Bennett and R. C. Holz, J. Am. Chem. Soc., 2005, 127, 14100–14107 CrossRef CAS PubMed.
- M. C. Bourkaib, S. Delaunay, X. Framboisier, L. Hôtel, B. Aigle, C. Humeau, Y. Guiavarc'h and I. Chevalot, Enzyme Microb. Technol., 2020, 137, 109536–109548 CrossRef CAS PubMed.
- S. Mukhopadhyay, M. S. Hasson and D. A. Sanders, Bioorg. Chem., 2009, 36, 65–69 CrossRef PubMed.
- E. Schmidt, in Methods of Enzymatic Analysis (Second Edition), ed. H. U. Bergmeyer, Academic Press, 2nd edn., 1974, pp. 650–656 Search PubMed.
- G. Sánchez-Carrón, T. Fleming, K. E. Holt-Tiffin and D. J. Campopiano, Anal. Chem., 2015, 87, 3923–3928 CrossRef PubMed.
- S. Yokoyama and J.-I. Hiramatsu, J. Biosci. Bioeng., 2003, 95, 204–205 CrossRef CAS PubMed.
- R. Rowlett and J. O. E. Murphy, Anal. Biochem., 1981, 112, 163–169 CrossRef CAS PubMed.
- S. L. Zandy, J. M. Doherty, N. D. Wibisono and R. A. Gonzales, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1055–1056, 1–7 CrossRef CAS PubMed.
- R. Bhushan and H. Bru, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed.
- C. Yang, X. Jiang, L. Guo, H. Zhang and M. Liu, J. Sep. Sci., 2007, 30, 3154–3163 CrossRef CAS PubMed.
- M. T. Reetz, K. M. Kühling, S. Wilensek, H. Husmann, U. W. Häusig and M. Hermes, Catal. Today, 2001, 67, 389–396 CrossRef CAS.
- H. Zahradníčková, P. Hušek and P. Šimek, J. Sep. Sci., 2009, 32, 3919–3924 CrossRef PubMed.
- M. Bertrand, A. Chabin, A. Brack and F. Westall, J. Chromatogr. A, 2008, 1180, 131–137 CrossRef CAS PubMed.
- F. Menestrina, J. O. Grisales and C. B. Castells, Microchem. J., 2016, 128, 267–273 CrossRef CAS.
- Y. Nakano, Y. Konya, M. Taniguchi and E. Fukusaki, J. Biosci. Bioeng., 2017, 123, 134–138 CrossRef CAS PubMed.
- A. R. Klein, K. A. B. Hanson and L. Aristilde, Environ. Chem. Lett., 2020, 18, 229–235 CrossRef CAS.
- N. J. Ayon, A. D. Sharma and W. G. Gutheil, J. Am. Soc. Mass Spectrom., 2019, 30, 448–458 CrossRef CAS PubMed.
- S. K. Bharti and R. Roy, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- A. M. Tomlins, P. J. D. Foxall, M. J. Lynch, J. Parkinson, J. R. Everett and J. K. Nicholson, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 367–380 CrossRef CAS.
- R. Legner, A. Wirtz and M. Jaeger, J. Spectrosc., 2018, 2018, 11 Search PubMed.
- M. Akimoto, T. Yu, K. Moleschi, K. Van, G. S. Anand and G. Melacini, Chem. Commun., 2020, 56, 8091–8094 RSC.
- M. T. Reetz, A. Eipper, P. Tielmann and R. Mynott, Adv. Synth. Catal., 2002, 344, 1008–1016 CrossRef CAS.
- K. A. Farley, U. Reilly, D. P. Anderson, B. P. Boscoe, M. W. Bundesmann, D. A. Foley, M. S. Lall, C. Li, M. R. Reese and J. Yan, Magn. Reson. Chem., 2017, 55, 348–354 CrossRef CAS PubMed.
- S. C. Cosgrove, A. P. Mattey, M. Riese, M. R. Chapman, W. R. Birmingham, A. J. Blacker, N. Kapur, N. J. Turner and S. L. Flitsch, ACS Catal., 2019, 9, 11658–11662 CrossRef CAS.
- R. Cairns, A. Gomm, J. Ryan, T. Clarke, E. Kulcinskaja, K. Butler and E. O. Reilly, ACS Catal., 2019, 9, 1220–1223 CrossRef CAS.
- S. Meier, Catal. Commun., 2020, 135, 105894 CrossRef CAS.
- C. M. L. Lau, G. Jahanmir and Y. Chau, Acta Biomater., 2020, 101, 219–226 CrossRef CAS PubMed.
- M. J. Smith, C. B. Marshall, F.-X. Theillet, A. Binolfi, P. Selenko and M. Ikura, Curr. Opin. Struct. Biol., 2015, 32, 39–47 CrossRef CAS PubMed.
- T. Lanyon-Hogg, M. Ritzefeld, N. Masumoto, A. I. Magee, H. S. Rzepa and E. W. Tate, J. Org. Chem., 2015, 80, 4370–4377 CrossRef CAS PubMed.
- J. S. Laursen, J. Engel-Andreasen, P. Fristrup, P. Harris and C. A. Olsen, J. Am. Chem. Soc., 2013, 135, 2835–2844 CrossRef CAS PubMed.
- S. R. LaPlante, P. Forgione, C. Boucher, R. Coulombe, J. Gillard, O. Hucke, A. Jakalian, M. Joly, G. Kukolj, C. Lemke, R. McCollum, S. Titolo, P. L. Beaulieu and T. Stammers, J. Med. Chem., 2014, 57, 1944–1951 CrossRef CAS PubMed.
- J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed.
- C. Claaßen, K. Mack and D. Rother, ChemCatChem, 2020, 12, 1190–1199 CrossRef PubMed.
- A. M. R. Hall, J. C. Chouler, A. Codina, P. T. Gierth, J. P. Lowe and U. Hintermair, Catal. Sci. Technol., 2016, 6, 8379–8576 RSC.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00122a |
| This journal is © The Royal Society of Chemistry 2021 |