
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-templated synthesis of rigid and conformationally restricted cyclic bisporphyrins: specific retention times on a cyanopropyl-modified silica gel column†
Masaya
Hashimoto
a,
Yusuke
Kuramochi
 ab,
Sayumi
Ito
b,
Yuho
Kinbara
a and
Akiharu
Satake
*ab
ab,
Sayumi
Ito
b,
Yuho
Kinbara
a and
Akiharu
Satake
*ab
aDepartment of Chemistry, Graduate School of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: asatake@rs.tus.ac.jp
bDepartment of Chemistry, Faculty of Science Division II, Tokyo University of Science, Japan
First published on 15th March 2021
Abstract
A series of rigid and conformationally restricted cyclic bis(zinc porphyrin)s connected via 2,2′-bipyridine and phthalamide, isophthalamide, or terephthalamide moieties were prepared by metal-templated synthesis. The yields were significantly improved when compared with those obtained under metal-free conditions. In particular, phthalamide and terephthalamide derivatives were obtained only by metal-templated synthesis. Structural analyses and dynamics of the exchange between the conformers in each cyclic porphyrin were examined by NMR spectroscopy. Although the distances between the two zinc porphyrins were extended in the order of phthalamide, isophthalamide, and terephthalamide derivatives, the order of the specific retention of the cyclic porphyrins on cyanopropyl-modified silica gel (CN-MS) chromatography columns varied. Thus, this order was reversed in the isophthalamide and terephthalamide derivatives. Based on the rigid structure of the terephthalamide derivative, the origin of the specific retention on the CN-MS chromatography column was attributed to both the distance and rigidity of the cyclic porphyrins.
Introduction
Cofacial bis(metalloporphyrin) derivatives are known as molecule-based functional materials. The functionalities are spread over various areas, such as chiral sensing,1,2 molecular recognition,3 supramolecular polymers,4 oxygen activation,5,6 and catalysts.6–8 Because their functions are related to the distances and dihedral angles between the two porphyrins as well as conformational freedom, the preparation of various cofacial bis(metalloporphyrin)s has been beneficial.9–11 Previously, we reported the synthesis of bis(zinc porphyrin)s connected through 2,2′-bipyridyl at 4,4′-positions. Their conformations were controlled to the cofacial orientation by complexation with metal ions on the 2,2′-bipyridyl moieties.12,13Herein, we report the synthesis of a series of bis(zinc porphyrin)s connected through 2,2′-bipyridyl at 6,6′-positions, followed by covalent linkage of the opposite meso-positions of the bis(zinc porphyrin)s. Thus, the two porphyrins are connected to different positions of a 2,2′-bipyridyl moiety by comparing them with the previous ones, and subsequently fixing them. These compounds are designed for different purposes from the previous one. The conceptual molecular structure of this project is shown in Fig. 1A. Here, a 2,2′-bipyridyl moiety is connected to the 6 and 6′-positions at the meso-position of two metalloporphyrins, which are linked via an appropriate linker at the opposite meso-positions. The introduction of appropriate linkers regulates the distances and dihedral angles between the two metalloporphyrins, as well as their conformational freedom. In the linked bismetalloporphyrin, the association constants of metal ions on the 2,2′-bipyridyl moiety increase because the conformational freedom of the bipyridyl moiety is significantly decreased by the linkage. This is advantageous for assessing various metal ions with the 2,2′-bipyridyl moiety. In addition, an unusual orientation of metal complexes or organometallic complexes is expected on such a spatially restricted ligand.14 In porphyrin moieties, various metal ions can also be introduced to systematically afford various functional multinuclear complexes. For instance, Mn, Fe, Co, Ru, and Rh porphyrins act as photoredox catalysts for olefin epoxidation,15,16 CO2 reduction,17–19 oxidation of hydrocarbons,20,21 water oxidation,22 syntheses of N-acyl sulfimides and sulfoximines,23 and carbon–carbon σ-bond oxidation.24 Zinc porphyrin acts as a photosensitizer in photochemical reactions, for example, photoinduced α-functionalization of aldehydes,25 photoinduced living polymerization,26 light-driven O2 reduction,27 and photocatalytic CO2 reduction.28,29 Thus, because the bismetalloporphyrin, as shown in Fig. 1A, has a coordination site by the 2,2′-bipyridyl part inside the restricted space, a systematic introduction of metal ions on the 2,2′-bipyridyl part will be beneficial for improving their catalytic activities or inducing successive catalytic reactions.
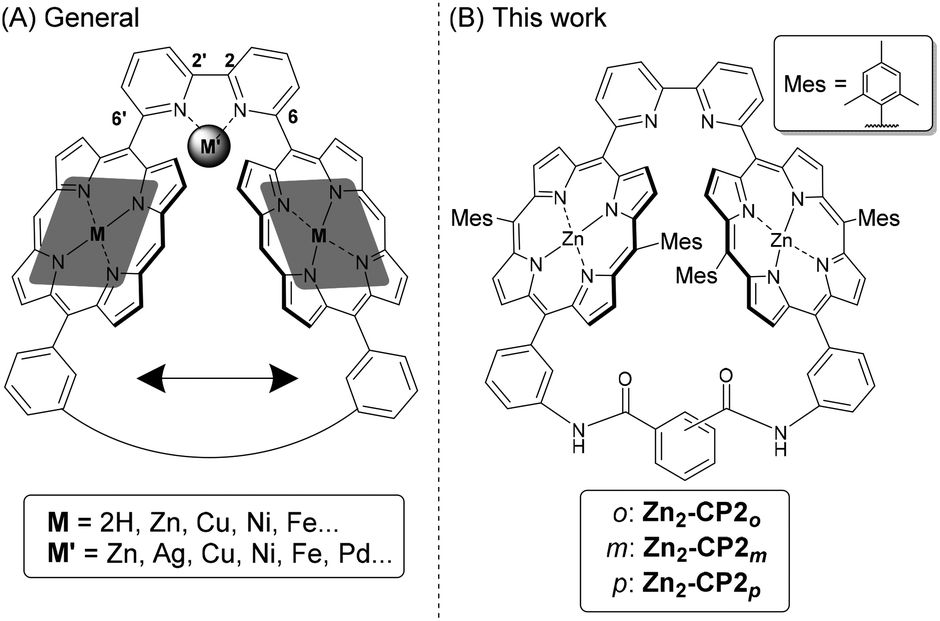 | ||
| Fig. 1 (A) A conceptual molecular structure of cofacial cyclic bisporphyrin. (B) Structures of cofacial cyclic bisporphyrins (Zn2-CP2o,m,p). | ||
The particular molecular structures described in this paper are shown in Fig. 1B. We planned to synthesize a series of bis(zinc porphyrin) derivatives, Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p, in which the two porphyrins are connected via phthalamide, isophthalamide, and terephthalamide, respectively. Among the bisporphyrin derivatives, the distances and dihedral angles between the two porphyrin moieties varied systematically. Previously, we reported the specific and efficient chromatographic isolation of cofacial cyclic trisporphyins from mixtures containing similar linear porphyrin derivatives on functionalized silica gel.32 Indeed, this chromatography was useful for isolating cyclic bisporphyrin derivatives, even though they exist as minor products in crude mixtures. Another purpose of this work was to understand the principle of unique chromatographic behavior. The preparation of a series of similar bisporphyrins is useful for discussing the relationships between structures and retention behaviors on functionalized silica gels.
In this paper, we report the preparation of cofacial bisporphyrin derivatives Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p by metal-templated macrocyclic formation followed by their efficient separation on cyanopropyl-modified silica gel (CN-MS). In the subsequent sections, the structural analyses and dynamics of the conformational changes of the bisporphyrin derivatives are described. Finally, a new insight into the origin of the specific retentions is discussed based on the relationships between retention behaviors and structural factors of a series of cyclic bisporphyrin derivatives.
Results and discussion
Two synthetic routes were considered for the synthesis of Zn2-CP2o,m,p, as shown in Scheme 1. In the first route (A→B→D), nickel-mediated homocoupling is applied to produce B, followed by cross-linking with phthalic, isophthalic, or terephthalic dichloride to produce Zn2-CP2o,m,p (D), respectively. In the other route (A→C→D), the order of nickel-mediated homocoupling and cross-linking with acid dichloride is reversed.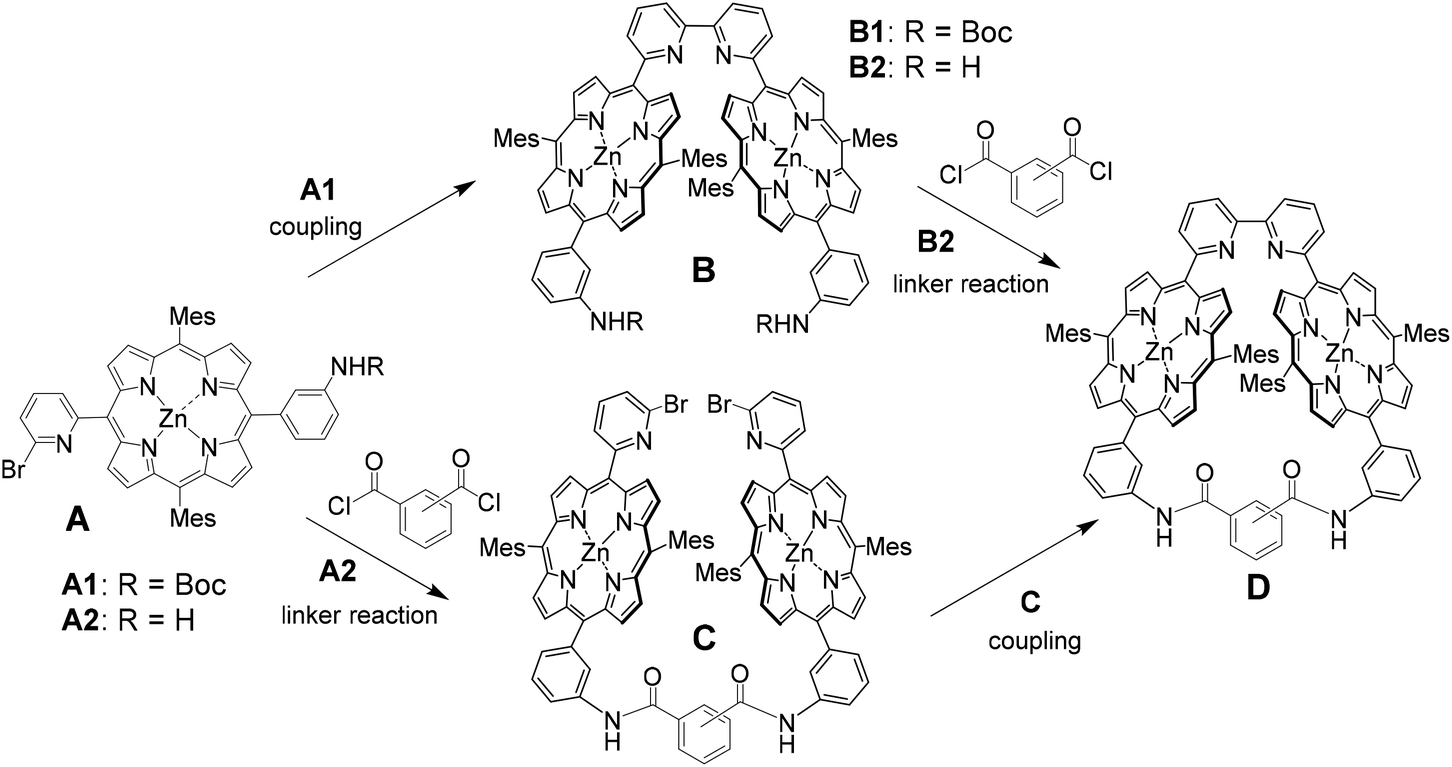 | ||
| Scheme 1 Two synthetic plans to construct cofacial cyclic bisporphyrins (Zn2-CP2o,m,p). | ||
Monoporphyrin 4Fb was synthesized by the condensation of aldehydes 1 and 2 and dipyrromethane 3, followed by oxidation with p-chloranil. After treatment with Zn(OAc)2, 4Zn was isolated in 25% yield in two steps. The homocoupling reaction of 4Zn was performed to result in the bisporphyrin 5Zn2. Because the solubility of 5Zn2 was very low in various solvents, the purification of 5Zn2 was difficult at this stage. Therefore, the zinc ions were removed in the presence of trifluoroacetic acid (TFA) to afford free-base bisporphyrin 5Fb2, 5Fb2 was isolated by preparative gel permeation chromatography (GPC). The Boc-protecting groups in 5Fb2 were subsequently removed with neat TFA to afford 6Fb2. Finally, zinc ions were re-introduced into the two porphyrin moieties of 6Fb2 to afford 6Zn2 in 69% yield in four steps (Scheme 2).
 | ||
| Scheme 2 Synthesis of acyclic bisporphyrin 6Zn2; bpy = 2,2′-bipyridine and cod = 1,5-cyclooctadiene. | ||
Macrocyclization of 6Zn2 with isophthaloyl dichloride was examined under various conditions to produce the diamide Zn2-CP2m. To a diluted diamine solution of 6Zn2 in dichloromethane (0.5 or 0.2 mM), 1.5 equivalents of isophtaloyl dichloride solution (0.9 mM) were slowly added with a syringe pump for 12 h in the presence of pyridine (4 equiv.) as a base. The reaction mixtures were analyzed by analytical high-performance liquid chromatography (HPLC) with a CN-MS column using a pyridine-based eluent, followed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. An HPLC chart and a MALDI-TOF mass spectrum are shown in Fig. 2 and Fig. S9 and S10, respectively, in the ESI.†
 | ||
| Fig. 2 HPLC chart of a crude mixture after reaction of 6Zn2 (0.5 mM) with isophthaloyl dichloride. Column: Cosmosil 5CN-MS (7.5 mm I.D. × 5 cm). Eluent: toluene/pyridine (15/85). Flow rate: 1.0 mL min−1. Monitored at 563 nm. Cyclic diamide Zn2-CP2m was eluted at a RT of 2.9 min (HPLC yield: 15%). The most other components were eluted up to a RT of 1.5 min without retention. | ||
In the HPLC chart, only the target macrocyclic compound Zn2-CP2m was retained on the column (retention time (RT) 2.9 min), and the linear compounds and the larger macrocyclic compound Zn4-CP4m were eluted in the initial RT region up to 1.5 min. In the MALDI-TOF mass spectrum, the relative intensities did not reflect their yields. In general, cyclic compounds such as Zn2-CP2m and Zn4-CP4m have a significant appearance as compared to linear porphyrin derivatives.
Therefore, the mass spectral data were used only for the characterization of the fractions by HPLC analysis. The HPLC-based yields of Zn2-CP2m were estimated to be 15 and 28%, respectively, for 0.5 and 0.2 mM solutions without metal-template conditions. Although the dilution condition of 0.2 mM was better than that of 0.5 mM, further dilution of the system was not realistically applicable to scale up the system. Therefore, metal-templated synthesis was tested at a concentration of 0.2 mM in the presence of one equivalent of zinc, silver, and copper ions to improve the yield of the target macrocycle Zn2-CP2m. The yields of Zn2-CP2m determined from HPLC (Fig. S11 in the ESI†) are presented in Table 1. Addition of 1 equiv. of cationic Zn(OTf)2 and Ag(OTf) improved the yield to approximately 50%. When 1 equiv. of zinc chloride, zinc acetate, and excess amounts of Zn(OTf)2 were used, the yields decreased compared with those in the absence of metal ions. Cu(OTf)2 was ineffective in yielding Zn2-CP2m. In the cases of Cu(OTf)2 and 5 equiv. of Zn(OTf)2, some precipitates (probably insoluble complexes composed of metal ions and porphyrin derivatives) were produced, which were separated before the HPLC analysis. One of the reasons for the low yields was the formation of precipitates.
To obtain insight into the effects of metal ions on macrocyclization, UV–vis titration experiments for 5Zn2 (7.5 × 10−6 M) with Ag(OTf), Zn(OTf)2, and Cu(OTf)2 were conducted in CHCl3 (Fig. 3a, and Fig. S12 and S13†). In the case of Ag(OTf) and Zn(OTf)2, complexation of 5Zn2 with metal ions converged to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 even in the presence of 1 equiv. of metal ions under diluted conditions (the insets of Fig. 3a and S12†), whereas further complexation or transformation occurred after the formation of a 1:1 complex in the presence of Cu(OTf)2 (inset of Fig. S13†). These results suggest that one equivalent of Ag(OTf) and Zn(OTf)2 is sufficient and appropriate to form 1:1 complexes.
:1 even in the presence of 1 equiv. of metal ions under diluted conditions (the insets of Fig. 3a and S12†), whereas further complexation or transformation occurred after the formation of a 1:1 complex in the presence of Cu(OTf)2 (inset of Fig. S13†). These results suggest that one equivalent of Ag(OTf) and Zn(OTf)2 is sufficient and appropriate to form 1:1 complexes.
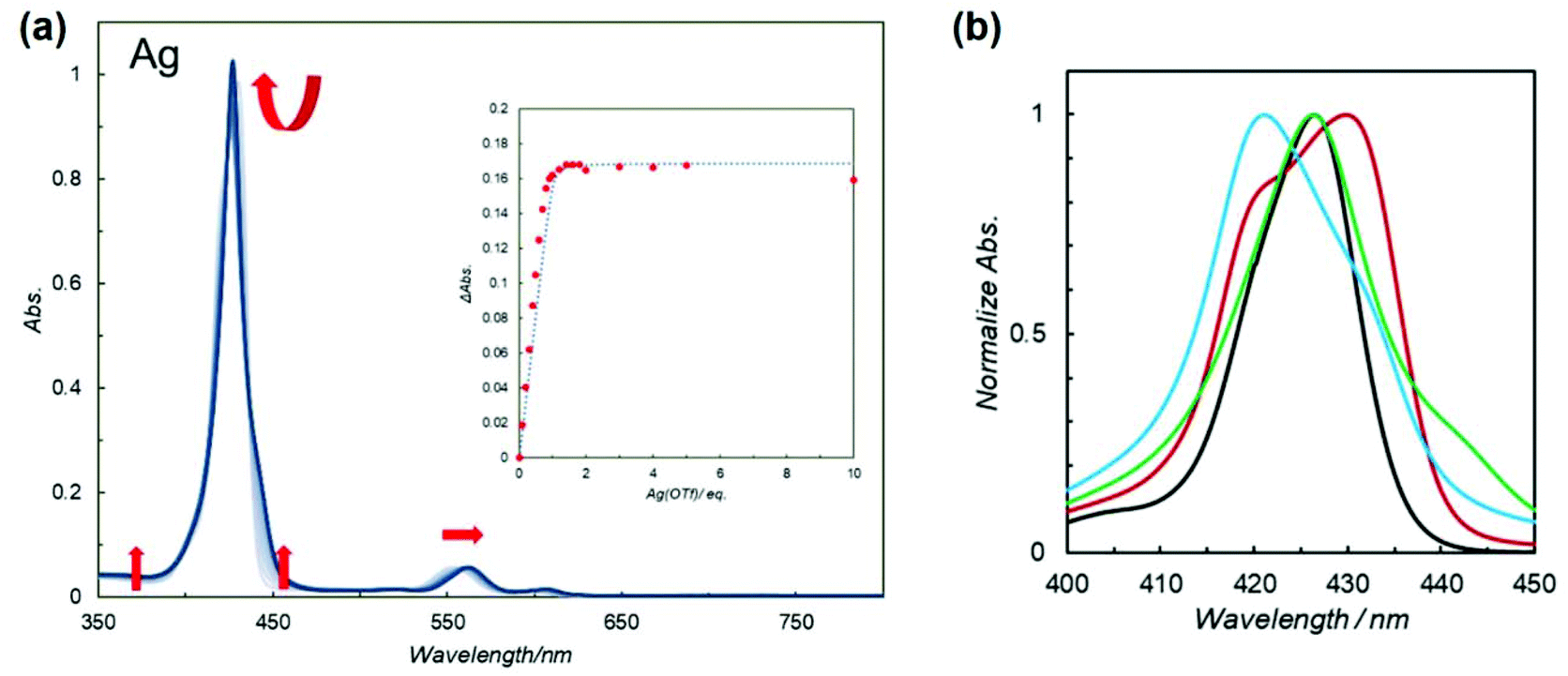 | ||
| Fig. 3 (a) UV–vis spectral change of 5Zn2 (7.5 × 10−6 M) during titration with Ag(OTf) in CHCl3. (Inset) The relationship between the change of absorption at 450 nm and equivalents of added Ag(OTf). A curve fitting analysis is shown as a dotted line. (b) Normalized overlaid UV–vis spectra (in CHCl3) of 4Zn (black), 5Zn2 (red), 5Zn2 with 1 equiv. of Zn(OTf)2 (cyan), 5Zn2 with 1 equiv. of Ag(OTf) (green) between 400 and 450 nm. The whole spectra (350–650 nm) are presented in Fig. S14† as (A)–(D). | ||
The UV–vis spectra of 4Zn and 5Zn2 in the absence and presence of 1 equiv. of Zn(OTf)2 or Ag(OTf) recorded in CHCl3 are shown in Fig. S14.† The spectral shapes and peak maxima of the Soret bands varied slightly. To compare the Soret bands, the overlaid spectra are shown in Fig. 3b. In bis(porphyrin) systems, the exciton coupling model10,30,31 is useful for qualitatively discussing the relative orientation between the two porphyrins. As compared to the UV–vis spectrum of zinc porphyrin monomer 4Zn (427 nm), in which there is no exciton coupling, metal-free bipyridine 5Zn2 gave split Soret bands at 420 and 430 nm, whose peaks were both blue and red-shifted. On the one hand, the red-shifted band is ascribed to the head-to-tail type of excitonic coupling between the two transition dipoles of X in the anti-conformation of 5Zn2, as shown in Fig. 4. On the other hand, the blue-shifted band is because of excitonic couplings of the parallel orientation between the two transition dipoles of Y and the oblique orientation of X in the syn conformation (Fig. 4). Therefore, the metal-free bipyridine 5Zn2 adopts both anti- and syn-conformations by freely rotating along the C–C bond in the bipyridyl moiety. In the presence of 1 equiv. of Zn(OTf)2, the red-shifted Soret band significantly decreased, whereas the blue-shifted band increased. These results indicate that the syn conformation was predominantly formed by the coordination of zinc ions on the bipyridyl part to fix the rotation along the C–C bond. In the case of Ag(OTf), since the red-shifted band decreased, the decrease in the anti-conformation was confirmed by the coordination of silver ions on the bipyridyl. However, the blue-shifted Soret band was not remarkable. This result probably because of the differences in ion radii of Zn(II) (0.60 Å) and Ag(I) (0.67 Å). Exciton coupling is highly sensitive to the relative distance between the transition dipoles. Both 1 equiv. of Zn(OTf)2 and Ag(OTf) acted as preferable metal templates to fix the syn conformation of 5Zn2. Notably, real macrocyclization was performed in 6Zn2, in which two amino groups exist on the terminal parts. Therefore, selective interactions between Zn(OTf)2 (or Ag(OTf)) and the 2,2′-bipyridyl moiety in 6Zn2 are required, because the cationic metal ions may also interact with the terminal amino groups. If more than 1 equiv. of the metal template is added, the undesired interactions with the amino groups result in precipitates, and the reactivity of the amines interacting with acyl chlorides may decrease. Therefore, excess amounts of Zn(OTf)2 and Ag(OTf) should not be added to the metal-templated macrocyclization system.
 | ||
| Fig. 4 Interaction of transition dipole moments of the bisporphyrin. | ||
Another route (A→C→D in Scheme 1) to obtain Zn2-CP2m was also examined, as shown in Scheme 3. Removal of Boc groups on 4Zn was performed by treatment with TFA to obtain 7Fb. Condensation of 7Fb (2 equiv.) with isophthaloyl dichloride in dichloromethane gave bisamide 8Fb2, which was reacted with zinc acetate to afford 8Zn2 in 38% yield in three steps. As a key step, nickel-mediated macrocyclization was applied to 8Zn2 under several conditions. However, the target macrocyclic compound, Zn2-CP2m, was obtained in a low yield (3% from HPLC in Fig. S15†). Therefore, the first synthetic route, A→B→D, shown in Scheme 1, was developed.
 | ||
| Scheme 3 Alternative synthetic route to cyclic bisporphyrin Zn2-CP2m. | ||
To obtain Zn2-CP2m, metal-templated synthesis of 6Zn2 with isophthaloyl dichloride in the presence of 1 equiv. of Zn(OTf)2 or Ag(OTf) was the most appropriate. Therefore, the same conditions were applied to obtain Zn2-CP2p and Zn2-CP2o using terephthaloyl and phthaloyl dichlorides, instead of isophthaloyl dichloride. The yields of Zn2-CP2p and Zn2-CP2o were determined by HPLC using a CN-MS column. The chromatograms of the crude reaction mixtures are shown in Fig. 5, and the reaction yields are summarized in Table 2. On the CN-MS column using pyridine as the eluent, only the target cyclic compounds were significantly retained. The RTs were 9.2 and 15.6 min for Zn2-CP2p and Zn2-CP2o, respectively, with a flow rate of 0.5 mL min−1. The RTs of Zn2-CP2p and Zn2-CP2o were significantly longer than those of Zn2-CP2m (4.0 min) recorded under the same conditions (Fig. 5d). It is noted that all other compounds, except for the cyclic dimers, were eluted at RT 2 min without retention.
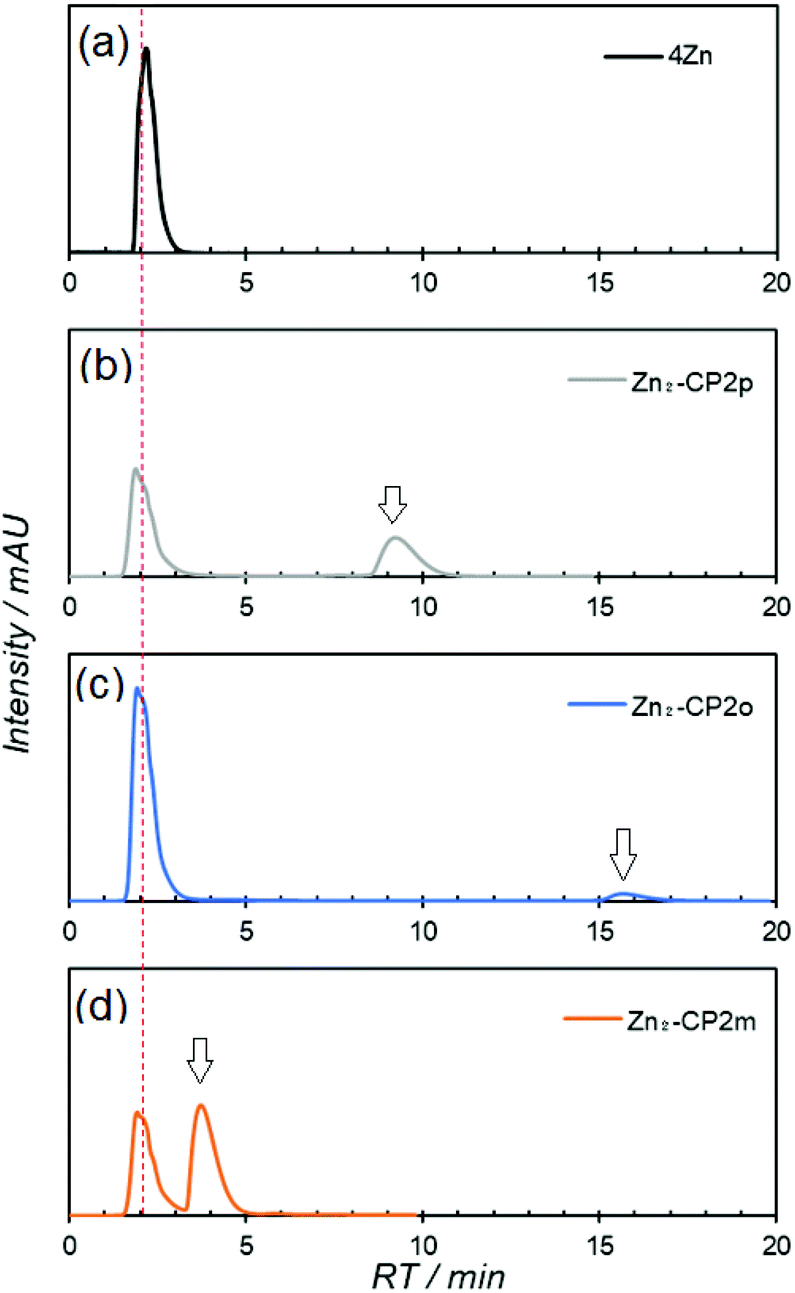 | ||
| Fig. 5 HPLC charts of (a) 4Zn2, crude mixtures after the reaction of 6Zn2 (0.5 mM) in the presence of 1 equiv. of Zn(OTf)2 with (b) terephthaloyl dichloride, (c) phthaloyl dichloride, and (d) isophthaloyl dichloride. Column: Cosmosil 5CN-MS (7.5 mm I.D. × 5 cm), eluent: pyridine 100%, flow late: 0.5 mL min−1, monitored at 563 nm. Arrows indicate the peaks corresponding to (b) Zn2-CP2p, (c) Zn2-CP2o, and (d) Zn2-CP2m, respectively. | ||
| Entry | Linker (1.5 eq.) | Metal (1.0 eq.) | Zn2-CP2p or Zn2-CP2o HPLC yield (%) |
|---|---|---|---|
| 1 | Terephthaloyl dichloride (p) | — | 0 |
| 2 | Terephthaloyl dichloride (p) | Zn(OTf)2 | 19 |
| 3 | Terephthaloyl dichloride (p) | Ag(OTf) | 24 |
| 4 | Phthaloyl dichloride (o) | — | 0 |
| 5 | Phthaloyl dichloride (o) | Zn(OTf)2 | 5 |
| 6 | Phthaloyl dichloride (o) | Ag(OTf) | 3 |
In both cases using terephthaloyl and phthaloyl dichlorides, no macrocyclic compounds were obtained in the absence of Zn(OTf)2 or Ag(OTf), whereas macrocyclic compounds, Zn2-CP2p and Zn2-CP2o, were observed in the mixtures prepared under metal-templated conditions. The yield of Zn2-CP2p was approximately 20%, whereas that of Zn2-CP2o was less by several percent. The reason for the low yields of Zn2-CP2o is that phthalimide derivatives, such as 9 in Fig. S16,† were formed significantly, which were detected by MALDI-TOF mass spectrometry (Fig. S17†). While the yield of Zn2-CP2o was very low, it was never obtained in the absence of a metal template. Therefore, the metal-template method is especially effective for the preparation of the kinetically disadvantageous phthaldiamide Zn2-CP2o, compared with phthalimide derivatives, such as 9, which are immediately formed from an amine part and phthaloyl dichloride. The yield of Zn2-CP2p was slightly increased using Ag(OTf) instead of Zn(OTf)2, suggesting that the larger Ag(I) ion is beneficial for obtaining bis(porphyrin) Zn2-CP2p, in which the two amide parts are located more apart than those in Zn2-CP2m.
Efficient isolation
After the metal-templated macrocyclization, Zn2-CP2m, Zn2-CP2o, and Zn2-CP2p were obtained as mixtures, including other linear oligomers. Although the yields of Zn2-CP2m, Zn2-CP2o and Zn2-CP2p were moderate or significantly low compared with other byproducts, isolation of the cyclic compounds was accomplished efficiently using a preparative CN-MS column using pyridine as an eluent. Under these conditions, all the compounds, except for the cyclic compounds, were eluted first without any retention, and then, almost only the cyclic compounds were eluted independently. As a result, tens of milligrams of pure Zn2-CP2m and Zn2-CP2p were isolated, and a few milligrams of Zn2-CP2o were obtained. It is noted that HPLC yields determined from the crude mixtures and the corresponding isolated yields were almost the same, indicating that the decrease in the isolated yields during the purification process was suppressed.Structural analyses of macrocycles on 1H NMR
To estimate the conformational structures of the macrocyclic porphyrins Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p, their 1H NMR spectra in pyridine-d5 are compared in Fig. 6. In pyridine-d5, two pyridine molecules coordinate to the two zinc parts to form five-coordinated complexes. Because the exchange rate of the coordinating pyridine molecules to peripheral non-coordinating pyridine is high at room temperature (ca. 23–25 °C), it is difficult to detect the coordinating pyridine in NMR spectroscopy.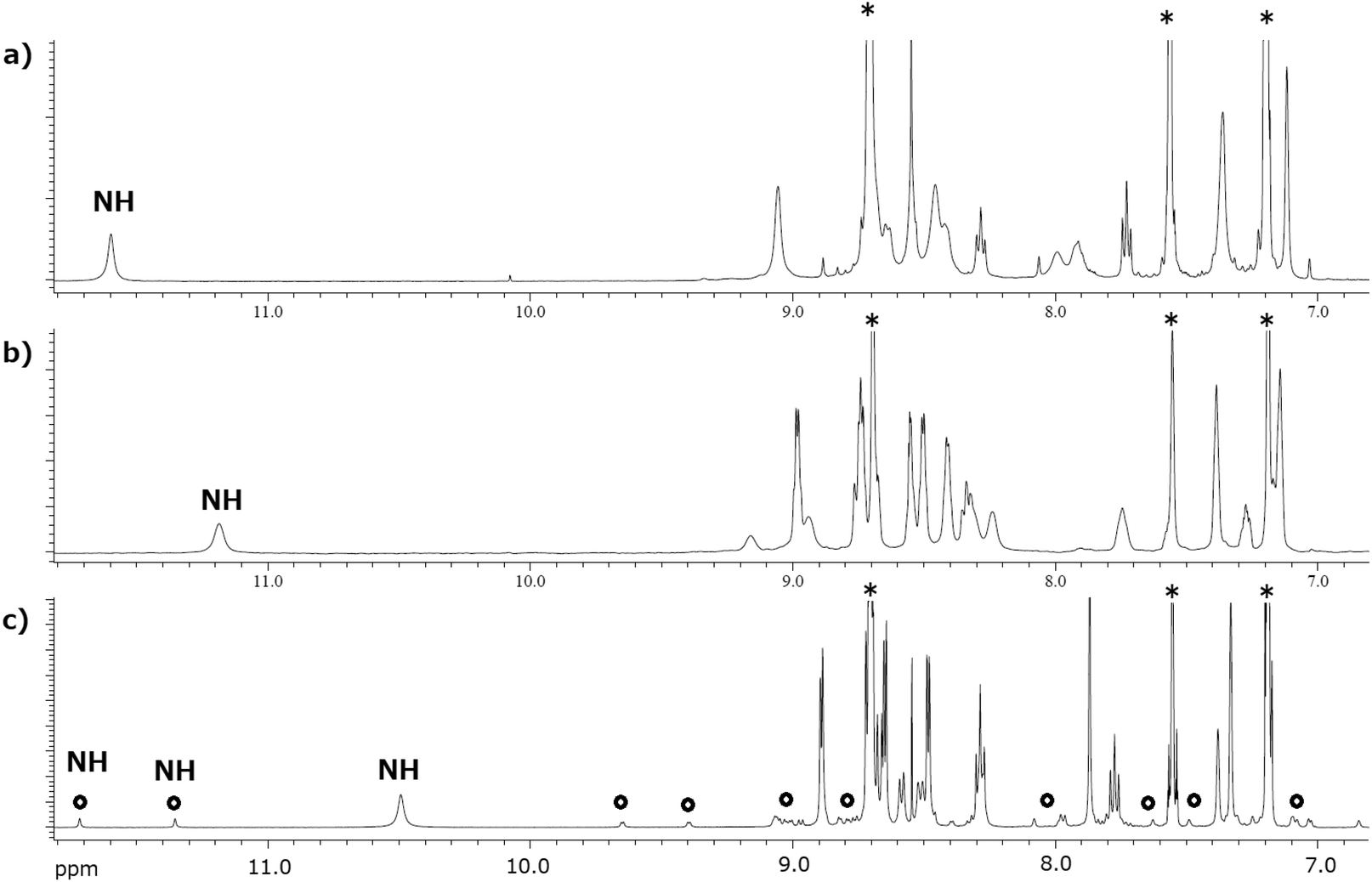 | ||
| Fig. 6 1H NMR (500 MHz) spectra of (a) Zn2-CP2o, (b) Zn2-CP2m, and (c) Zn2-CP2p in pyridine-d5 (12–7 ppm). Asterisks indicate the signals of pyridines. Open circles in (c) indicate the signals of the minor isomer. | ||
Consequently, the labile coordination behavior of pyridine eliminates the anisotropy of coordination. Therefore, only the conformational changes in the macrocyclic frames can be considered in the NMR spectra. The 1H NMR spectra of Zn2-CP2o (Fig. 6a) and Zn2-CP2m (Fig. 6b) show a pseudo C2v symmetric structure at room temperature, respectively, indicating that the two porphyrin planes are cofacial on average. In contrast, two conformational isomers were observed for Zn2-CP2p (Fig. 6c). The major isomer is a pseudo C2v symmetric structure, whereas the minor isomer (indicated by open circles in Fig. 6c) is not symmetric. The chemical shifts of amide-NH groups in Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p (for the major isomer) are observed at 11.58, 11.17, and 10.48 ppm, respectively. These differences occur when the two cofacial porphyrins are close to each other, and as a result, the amide protons are affected by the ring current produced by the nearby porphyrin moieties. In particular, the amide protons of Zn2-CP2o are deshielded by existing just outside the ring currents. In the case of the amide protons in Zn2-CP2m and Zn2-CP2p, the degree of deshielding effects was smaller. Therefore, the order of the lower chemical shifts of the NH groups in Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p (for the major isomer) suggests that the average distances of the two porphyrins in the cofacial porphyrins increase in the same order (Zn2-CP2o < Zn2-CP2m < Zn2-CP2p).
The assignments of all the 1H and 13C NMR (Fig. S19†) signals of Zn2-CP2m were obtained using various two-dimensional NMR techniques, namely HH-correlation spectroscopy (HH-COSY) (15 mM) (Fig. S20†), heteronuclear single quantum correlation (HSQC) (15 mM) (Fig. S21†), heteronuclear multiple bond coherence (HMBC) (15 mM) (Fig. S22†), and nuclear Overhauser effect spectroscopy (NOESY) (6 mM) (Fig. S23†). The assignments and observed NOE correlations are shown in Fig. S24.† The chemical shifts of aromatic parts, Ar-15, Ar(m)-18, 19, and 20 varied slightly in the different NMR samples (15 and 6 mM) prepared from the same Zn2-CP2m material. The chemical shifts are presented in Table S1 in the ESI.† These results suggest that the proton signals of the aromatic parts are significantly affected by the circumstances and concentrations.
Because stronger NOE correlation signals were observed between NH-17 and Ar-16, and NH-17 and Ar(m)-20, the major conformer was determined as syn-1 in Fig. 7, in which the amide groups exist on the near side of Ar-16 and Ar(m)-20. Meanwhile, relatively weaker correlation signals were also observed between NH-17 and Ar-15 and between NH-17 and Ar(m)-18. These NOE correlations indicate the existence of syn-2 and anti isomers as minor components. Distinct conformers could not be observed at room temperature. Rapid interconversion (conformational changes) between syn-1, anti, and syn-2 was observed at room temperature.
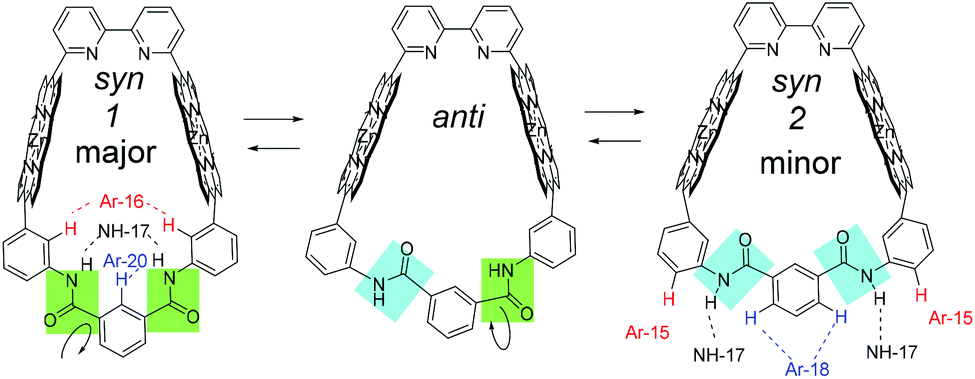 | ||
| Fig. 7 Exchange among conformers, syn1 (major), anti, and syn2 of Zn2-CP2m. | ||
In the case of Zn2-CP2p, two conformational isomers were clearly separated in the 1H NMR spectra of pyridine-d5 at a ratio of 85:15 (Fig. 6c and Fig. S25 and S26†). The signal pattern of the major conformer is simple, indicating a symmetric structure, whereas the pattern of the minor isomer is complicated (Fig. S26†). For assignments of the 1H and 13C NMR (Fig. S27†) signals of the major isomer of Zn2-CP2p, various two-dimensional NMR techniques, namely HH-COSY (Fig. S28†), HSQC (Fig. S29†), and NOESY (Fig. S30, S31, and S32†), were performed. The assignments and observed NOE correlations in the major conformer are shown in Fig. S33.† As the intense NOE correlation between 10.44 and 7.33 was observed in the major conformer (Fig. S30†), the NH groups in the amide moieties are directed to the porphyrin groups, as illustrated in Fig. 8 (syn).
 | ||
| Fig. 8 Schematics of two conformers of Zn2-CP2p. (left) Major symmetric syn form and (right) minor unsymmetric anti form. | ||
Parts of correlation signals between the major and minor conformers were observed in the NOESY (Fig. S30, S31, and S32†). The correlations were because of the chemical exchange between the two conformational isomers. The correlated signals among the amide groups, the β-protons, and the methyl groups on the mesityl moieties are summarized in Table S2.† Other minor signals could not be assigned because of overlapping with the major signals. In the minor conformer, the two NH groups are separately observed at 11.67 and 11.30 ppm. The inner methyl group in mesityl moieties in the major conformer at 1.47 ppm was correlated with four methyl groups in the minor conformer at 2.56, 1.98, 0.61, and −1.04 ppm. They are widely distributed. Two of the four methyl groups in the minor conformer are upper-field shifted (0.61 and −1.04 ppm), whereas the other two methyl groups in the minor conformer are lower-field shifted (2.56 and 1.98 ppm). These results indicate that the two porphyrin moieties are twisted in the minor conformer, and the former two methyl groups are shielded to a significant extent by the opposite facial porphyrin, whereas the latter two are shielded to a lesser extent. A schematic of the two conformational isomers is shown in Fig. 8 (anti). In the major symmetric form (syn), the NH groups were directed to the inside of the ring. In the minor non-symmetric form (anti), the two porphyrin groups are twisted, resulting in four methyl groups that are not identical. In the case of Zn2-CP2p, the exchange rate between the two conformational isomers was slower than that in Zn2-CP2m. The observations are used in the discussion in the last section.
Structural analyses of macrocycles on UV–vis spectra
In contrast to the NMR spectra, UV–vis spectra reflect the populations of conformational isomers, because the timescale of the measurements is considerably faster than the exchange rate among the conformational isomers. The UV–vis spectra in pyridine are shown in Fig. 9E to compare the Soret bands (410–450 nm) for monomer 4Zn, and Zn2-CP2o,m,p. The highest absorption peak in each spectrum was normalized in the spectra. All three cyclic dimers (Zn2-CP2o,m,p) have blue-shifted Soret bands compared with the monomer 4Zn, indicating that the two zinc porphyrins in Zn2-CP2o,m,p face each other, as shown in Fig. 10. The excitonic couplings between the two parallel transition dipole components of Y and the oblique transition dipole component X contribute to the blue shift. The degrees of the blue shifts and full width at half maximum (FWHM) are different among the three compounds, suggesting that the orientation and distance between the two porphyrins in each cyclic dimer are different. In the three blue-shifted spectra, the spectrum of Zn2-CP2m was blue-shifted to the highest extent, and the FWHM was narrow. These features indicate that the relative positions of the two porphyrins in Zn2-CP2m are constant and parallel. Although the existence of syn-2 and anti forms of Zn2-CP2m was suggested by the 1H NMR spectrum (Fig. 7), the population of the minor anti isomer was small. In the case of Zn2-CP2p, the absorption in the Soret band region was broadened. This is ascribed to the overlapping of the UV–vis spectra of conformers, syn and anti, on the amide groups (Fig. 8), as observed in the 1H NMR spectrum (Fig. 6). The degree of the blue shift of Zn2-CP2o was lowest in the three cyclic porphyrins, although the distance between the two porphyrin groups was expected to be the closest among the three conformers. The UV–vis spectrum of Zn2-CP2o indicates that the two porphyrins adopt a twisted conformation, in which the transition dipoles of Y adopt an oblique orientation.30 In Fig. 11, the possible conformers of Zn2-CP2o are presented. The anti-conformers are expected to be twisted. Since the 1H NMR spectrum of Zn2-CP2o showed a relatively simple pattern corresponding to a pseudo-symmetric structure (Fig. 6a), the exchange between the two anti conformers must occur in the NMR timescale. | ||
| Fig. 9 UV–vis spectra in pyridine of (A) 4Zn, (B) Zn2-CP2m, (C) Zn2-CP2o, and (D) Zn2-CP2p (E) Overlaid spectra of (A)–(D) in the Soret band region. | ||
 | ||
| Fig. 10 Interaction of transition dipole moments of the cyclic bisporphyrin. | ||
 | ||
| Fig. 11 Schematics of exchange among conformers of Zn2-CP2o. (left and right) anti forms and (center) syn form. | ||
Calculated models of Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p
In Fig. 12, S35, and S36,† the energy-minimized molecular models of conformers of Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p obtained by density-functional theory (DFT) calculations (B3LYP/6-31G(d)) are shown. In the case of Zn2-CP2o, anti form is more stable than the syn form by 4.3 kcal mol−1 (18.0 kJ mol−1) in a vacuum. | ||
| Fig. 12 Molecular structures of syn- and anti-conformers of Zn2-CP2o optimized with DFT calculations (B3LYP/6-31G(d) in vacuo), and the energy difference. | ||
Intramolecular hydrogen bonding is observed in the anti form, and the two porphyrin moieties are twisted (Fig. 12), as discussed in the previous section.
Two syn forms are compared in Zn2-CP2m (Fig. S35†), and syn-1 is more stable by 6.3 kcal mol−1 (26.4 kJ mol−1) in a vacuum. This result is consistent with the NOESY spectrum of Zn2-CP2m, in which a stronger correlation signal was observed between Ar-16 and NH-17, and Ar-20 and NH-17 (Fig. S23†).
In the case of Zn2-CP2p, syn and anti forms are compared in Fig. S36,† and syn form is more stable by 5.8 kcal mol−1 (24.3 kJ mol−1) in a vacuum. In pyridine-d5 at 298 K, the ratio of syn to anti is 85:15, corresponding to ΔG° = 4.3 kJ mol−1. The difference in the ΔG° values (ΔΔG° 20.0 kJ mol−1) corresponds to the solvation energy of pyridine. It is noted that the signals of syn and anti conformers in the 1H NMR spectrum of Zn2-CP2p are ascribed to the difference in the activation energy between the syn- and anti-conformers of Zn2-CP2p from those of Zn2-CP2m. The former must be larger than that of Zn2-CP2m.
The distances between the two zinc ions in Zn2-CP2o (anti form), Zn2-CP2m (syn form) and Zn2-CP2p (syn form) are estimated to be 7.87, 8.30 A, and 9.27 A, respectively.‡ The order of the distances between the two porphyrins is consistent with the order of the lower chemical shifts of the NH groups in Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p (for the major isomer) (Fig. 13). The NH protons are located at the edges of the porphyrin moieties. Thus, they are sensitive to the ring current of the nearby porphyrins. In the molecular model of Zn2-CP2o, the contribution of the deshielding effect to the NH groups seems large.
 | ||
| Fig. 13 Molecular structures and the distances between two zinc ions of (a) anti form of Zn2-CP2o, (b) syn-1 form of Zn2-CP2m, and (c) syn form of Zn2-CP2p optimized with DFT calculations (B3LYP/6-31G(d) in vacuo). The calculated distances between two zinc ions are presented. | ||
Consideration of retention times on the CN-MS column
Specific retention phenomena were observed for Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p by CN-MS chromatography. Our group previously observed the specific retention of cyclic zinc porphyrin trimers32 and cyclic nickel porphyrin dimers.14 Therefore, this report is the third example of a specific retention phenomenon. For specific retention, the structural requirements become obvious. Thus, the compounds are cyclic and have two porphyrin moieties connected via bipyridine. However, these two structural requirements are not sufficient conditions. At present, the mechanism of specific retention remains unclear. To discuss this mechanism, we believe that the collection of more examples of relationships between structures and retention behaviors is necessary. In this report, we have prepared three new cyclic bisporphyrins, Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. Retention behavior is now discussed.As shown in Fig. 5, the retention times were extended in the following order: Zn2-CP2m, Zn2-CP2p, and Zn2-CP2o. Unexpectedly, the order does not follow the distances between the two zinc porphyrins in Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. From the comparison of cyclic zinc porphyrin trimers32 and cyclic nickel porphyrin dimers,14 we expected that a shorter distance between the two zinc porphyrins would be advantageous for longer retention. However, the order of Zn2-CP2m and Zn2-CP2p was inverted, and the retention time of Zn2-CP2p was significantly longer. This result cannot be explained by differences in the static structures prepared by molecular models (Fig. 13). In the 1H NMR analyses, conformational isomers (syn and anti) were present in both Zn2-CP2m and Zn2-CP2p. The exchange rate between the syn and anti isomers was slower for Zn2-CP2p. The slower exchange rate was ascribed to the restriction of conformational freedom in Zn2-CP2p. Here, we assume that cyanopropyl moieties on the modified silica gel interact with the major syn forms of both Zn2-CP2m and Zn2-CP2p, and the retention is determined by the period of the interaction. The period of the interaction depends on the rigidity of the cyclic porphyrins. Thus, Zn2-CP2p is advantageous for longer retention than Zn2-CP2m. In the case of Zn2-CP2o, the close distance between the two porphyrin moieties probably contributes to the significantly longer retention observed in Fig. 5.
This new insight into the mechanism of the unique chromatographic behavior of the CN-MS column was proposed by preparing a series of cyclic porphyrin dimers, Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. The discussion that the retention depends on the rigidity of the cyclic porphyrins requires further studies.
Conclusions
Metal-templated bisamide linking of bis(zinc porphyrin) with two aniline groups connected through 2,2′-bipyridyl at 6,6′-positions with phthaloyl dichloride, isophthaloyl dichloride, and terephthaloyl dichloride was accomplished to afford Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. The yields were significantly improved compared to those obtained under metal-free conditions. In particular, phthalimide and terephthalamide derivatives were obtained only by metal-templated synthesis.Although the target macrocyclic compounds Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p were obtained as mixtures containing significant amounts of linear derivatives, the target compounds were isolated using a preparative CN-MS column using pyridine as an eluent, without any loss during the purification process. This interesting result is because of the specific retention of the cyclic porphyrin compounds on the CN-MS column.
Structural analyses of Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p were performed by NMR and UV–vis spectroscopies as well as DFT calculations. In the UV–vis method, populations of conformational isomers and the relative distances and orientations between the two zinc porphyrins in each cyclic porphyrin dimer could be estimated. It was seen that the two porphyrin moieties were twisted in Zn2-CP2o, whereas they were parallel in Zn2-CP2m. The distances between the two zinc porphyrins were extended in the following order: Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. This was confirmed by 1H NMR and DFT calculations.
In the NMR methods, exchanges between the conformers in each cyclic porphyrin were observed. The relative exchange rate was slow in Zn2-CP2p, as compared with that of Zn2-CP2m. As a result, syn and anti conformers were observed separately in the 1H NMR spectrum of Zn2-CP2p.
The order of specific retention of the cyclic porphyrins on a CN-MS chromatography column was not followed by the distances between the two zinc porphyrins in Zn2-CP2o, Zn2-CP2m, and Zn2-CP2p. Based on the rigid structure of the terephthalamide derivative, Zn2-CP2p, the origin of the specific retention on a CN-MS chromatography column was attributed to both the distance and rigidity of the cyclic porphyrins.
Experimental
General procedure
All chemicals and solvents were of commercial reagent quality and were used without further purification unless otherwise stated. Aldehyde 1, (3-formyl-phenyl)-carbamic acid tert-butyl ester, was prepared according to the literature.33N,N-Dimethylformamide (DMF) was dried over molecular sieves of size 4 Å and distilled under reduced pressure. The reactions were performed on silica gel 60F254 TLC plates (Merck). The silica gel utilized for column chromatography was purchased from Kanto Chemical Co. Inc.: Silica gel (Spherical, Neutral) 40–100 μm and (Flash) 40–50 μm. Cyanopropyl-modified silica gel (CN-MS, Cyanogel) (dp: 40 μm, pore size: 60 Å) was purchased from Yamazen, and a mixture of toluene and pyridine (85/15 v/v) and 100% pyridine were used as eluents. 1H and 13C NMR, distortionless enhancement by polarization transfer 135 (DEPT 135), COSY, HSQC, and NOESY spectra were recorded using a JEOL JNM-ECS-500. Chemical shifts were recorded in parts per million (ppm) relative to tetramethylsilane. MALDI–TOF mass spectra were recorded on a JEOL JMS S-3000 system with dithranol as a matrix with sodium iodide (NaI). UV-vis absorption spectra were collected using a square cell (path length = 1.0 cm) on a JASCO V-650 spectrometer. Steady-state emission spectra were collected on a Hitachi F-4500 spectrometer and corrected for the response of the detector system. The fluorescence intensities were normalized at the absorbance of the excitation wavelength.High-performance liquid chromatographies (HPLCs)
HPLCs were carried out by using the following three systems: [System 1] JASCO PU-2080plus and MD-2018plus systems equipped with two TSK G2500HHR (Tosoh, 7.8 mm × 30 cm, exclusion limit: 20000 Da) and one TSK G2000HHR (Tosoh, 7.8 mm × 30 cm, exclusion limit: 10000 Da) columns using pyridine as an eluent. [System 2] JASCO PU-2089 and MD-44010 systems equipped with a Cosmosil 5CN-MS column (nacalai tesque, 7.5 mm I.D. × 5 cm) using toluene/pyridine (15/85) or 100% pyridine as an eluent. [System 3: Preparative HPLC] LC-908 (JAI) attached to one TSK G2500HHR column and one G2000HHR column eluted with pyridine as an eluent.
Computational methods
DFT calculations were carried out using the Gaussian 09 package of programs.34 Each structure was fully optimized using the B3LYP functional with the 6-31G(d) basis set. The stationary points were verified using vibrational analysis.Synthesis of 4Zn
A 500 mL three-necked round-bottom flask was charged with aldehyde 1 (0.312 g, 1.42 mmol), 6-bromo-2-pyridinecarboxaldehyde 2 (0.266 g, 1.42 mmol), dipyrromethane 3 (0.751 g, 2.84 mmol), and Ar-saturated CHCl3 (285 mL). Then, 1 mL of CHCl3 solution containing BF3·OEt2 (100 µL, 0.796 mmol) was slowly added to it, and the mixture was stirred at rt under an Ar atmosphere in the dark. After 0.5 h, 1 mL of CHCl3 solution containing Et3N (200 µL, 1.59 mmol) and p-chloranil (1.05 g, 4.26 mmol) were added to it, and the mixture was stirred at room temperature for 19 h. The solvent was evaporated to dryness. The black-purple residue was purified twice with flush silica gel columns (eluent: CHCl3), giving a purple solid containing a symmetric byproduct porphyrin and 4Fb (0.616 g). The solid was placed in a 100 mL flask and dissolved in CHCl3 (20 mL). A small amount (∼1 mL) of methanol containing Zn(OAc)2 (0.632 g, 3.44 mmol) was added to it, and the mixture was stirred for 42 h. The resulting solution was washed with water (×2) and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated to dryness. The crude purple solid (0.625 g) was purified with a flush silica gel column (eluent: toluene:ethyl acetate = 70:1→30:1→10:1), giving pure target porphyrin 4Zn (0.286 g, 0.299 mmol) with 21% yield (based on aldehyde 1). 1H NMR (400 MHz, CDCl3) δ/ppm = 8.87 (d, J = 4.4 Hz, 2H), 8.82 (d, J = 4.4 Hz, 2H), 8.80 (d, J = 4.4 Hz, 2H), 8.75 (d, J = 4.4 Hz, 2H), 8.25 (d, J = 6.8 Hz, 1H), 7.94–7.86 (m, 4H), 7.60 (s, 1H), 7.28 (s, 4H), 2.63 (s, 6H), 1.81 (s, 12H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ/ppm = 162.70, 150.35, 149.99, 149.87, 149.60, 143.48, 140.71, 139.36, 139.04, 137.97, 137.58, 136.81, 132.66, 131.61, 131.51, 130.86, 129.64, 129.13, 128.79, 128.32, 127.75, 127.11, 126.76, 125.39, 119.69, 117.74, 116.24, 80.91, 29.81, 28.25, 21.57. MALDI-TOF MS of 4Znm/z 854.2242 (M)+, calcd for C54H47BrN6O2Zn 854.2232.
Synthesis of 5Zn2
A 200 mL Schlenk tube was charged with 4Zn (108 mg, 0.112 mmol), 2,2′-bipyridine (22.0 mg, 0.141 mmol). After the Schlenk tube was evacuated and filled with Ar gas, dry DMF degassed with freeze–thaw cycles (100 mL) and 1,5-cyclooctadiene (42 μL, 0.342 mmol) were added to the tube. After the mixture was stirred for 10 min at room temperature, Ni(cod)2 (960 mg, 3.49 mmol) was added. The reaction mixture was sonicated and stirred at rt in the dark. After overnight stirring, the resulting mixture was added to a small amount of CHCl3 (∼20 mL) in a perfluoroalkoxy alkane (PFA)-coated flask. The Schlenk tube was washed with CHCl3 (∼20 mL). The combined CHCl3 solution was washed with 0.3 M citric acid solution (×4), 25% ammonium solution (×3), and dried over anhydrous Na2SO4. The solution was passed through a membrane filter (Omnipore, 0.1 μm) to remove the precipitates. The filtrate was concentrated to dryness, giving a crude purple solid containing 5Zn2 (110 mg). Retention times of HPLC (columns: TOSOH G2500HHR × 2 + G2000HHR × 1, eluent: pyridine, flow rate: 1.0 mL min−1): 21.62 min for 5Zn2 and 23.68 min for monomers containing 4Zn. UV-vis absorption of a pure sample obtained by Zn insertion to a pure 5Fb2: λmax (CHCl3)/nm = 430 (ε = 6.7 × 105 M−1 cm−1) and 553 (ε = 3.4 × 104 M−1 cm−1). NMR spectroscopic analysis could not be performed due to its poor solubility in any solvent. MALDI-TOF MS of 5Zn2 (C108H94N12O4Zn2, exact MS 1750.61) gave only fragment signals corresponding to de-Boc derivatives: m/z 1551.49 (5Zn2 − (Boc)2 + (H)3)+, calcd for C98H78N12Zn2 1551.51.Synthesis of 5Fb2
A 20 mL flask was charged with a crude purple solid containing 5Zn2 (110 mg) and CHCl3 (20 mL). Trifluoroacetic acid (TFA) (0.1 mL, 1.31 mmol) was slowly added to it and the mixture was stirred at rt for 1.5 h. The resulting solution was slowly poured into a saturated NaHCO3 aqueous solution (50 mL) in an ice-bath. The organic layer was transferred into a PFA-coated funnel and the aqueous layer was extracted with CHCl3 (10 mL × 3). The combined organic layer was washed with water and dried over anhydrous Na2SO4. The solution was passed through a membrane filter (Omnipore, 0.1 μm) to remove the precipitates, and the filtrate was concentrated to dryness. The resulting residue was purified with a preparative HPLC using pyridine as an eluent, giving a purple solid, 5Fb2 (57.8 mg, 35.5 μmol) in 69% yield (2 steps from 4Zn). 1H NMR (400 MHz, CDCl3) δ/ppm = 9.02–9.00 (m, 6H), 8.86 (d, J = 4.0 Hz, 4H), 8.79 (d, J = 4.0 Hz, 4H), 8.72 (d, J = 4.0 Hz, 4H), 8.26 (d, J = 4.0 Hz, 2H), 8.11 (t, J = 6.8 Hz, 4H), 7.96 (s, 4H), 7.67 (t, J = 6.8 Hz, 2H), 7.30 (s, 8H), 6.76 (s, 2H), 2.64 (s, 12H), 1.88 (s, 24H), 1.53 (s, 18H), −2.53 (s, 4H); 13C NMR (75 MHz, CDCl3) δ/ppm = 160.19, 155.37, 153.13, 142.87, 139.55, 138.57, 137.89, 137.00, 135.90, 130.63, 129.74, 127.92, 127.44, 124.86, 123.89, 120.46, 119.48, 118.62, 118.03, 117.74, 80.86, 28.52, 21.82, 21.62. MALDI-TOF MS of 5Fb2 (C108H98N12O4; C108H94N12O4Zn2, exact MS 1626.78) gave only fragment signals corresponding to [5Fb2 − (Boc) − (t-butoxy) + (H)2]+ (m/z 1454.70) and [5Fb2 − (Boc)2 + (H)2]+ (m/z 1426.68).Synthesis of 6Fb2
TFA (2.0 mL) was slowly added to 5Fb2 (26.5 mg, 16.3 μmol) in a 25 mL flask, and the mixture was stirred for 1 h. The resulting solution was slowly poured into a saturated NaHCO3 aqueous solution (50 mL) in an ice-bath. The organic layer was transferred to a PFA-coated funnel and the aqueous layer was extracted with CHCl3 (10 mL × 3). The combined organic layer was washed with water and dried over anhydrous Na2SO4. The solvent was evaporated to dryness, giving a purple solid 6Fb2 (22.0 mg). TLC (toluene:EtOAc = 30:1) Rf = 0.50; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.99–9.03 (m, 6H), 8.93 (d, J = 4.4 Hz, 4H), 8.79 (d, J = 4.4 Hz, 4H), 8.72 (d, J = 4.4 Hz, 4H), 8.25 (d, J = 7.8 Hz, 2H), 8.12 (t, J = 7.8 Hz, 2H), 7.75–7.40 (m (br), 6H), 7.31 (s, 8H), 7.10 (d, J = 7.8 Hz, 2H), 2.65 (s, 12H), 1.89 (s, 24H), −2.53 (s, 4H); UV-vis absorption: λmax (CHCl3)/nm = 425, 516, 551, 589; fluorescence (λex = 515 nm, CHCl3): λmax/nm = 648, 713. MALDI-TOF MS of 6Fb2 (C98H82N12; M.W. 1427.8): found m/z 1428.0 (M)+.
Synthesis of 6Zn2
A 20 mL flask was charged with 6Fb2 (22.0 mg) and CHCl3 (5 mL). Zn(OAc)2 (56.8 mg, 310 μmol) in a small amount of methanol (∼1 mL) was added to it and the mixture was stirred at rt for 19 h. The resulting solution was transferred to a PFA-coated funnel and washed with water (×2) and brine. The organic layer was passed through Phase Separator paper (Whatman). The solvent was evaporated to dryness, giving a purple solid 6Zn2 (28.3 mg, 18.2 μmol) quantitatively from 5Fb2. TLC (toluene:EtOAc = 30:1) Rf = 0.13; UV-vis absorption: λmax (CHCl3)/nm = 423, 559, 600; fluorescence (λex = 515 nm, CHCl3) λmax/nm = 607, 651. NMR spectroscopic analysis could not be performed due to the poor solubility in any solvent. MALDI-TOF MS of 6Zn2m/z 1551.50 (M + H)+, calcd for C98H79N12Zn2 1551.51.
Synthesis of Zn2-CP2o,m,p (metal templated synthesis)
A 10 mL flask was charged with 6Zn2 (5.0 mg, 3.2 μmol) and dry CH2Cl2 (5 mL). A CH3CN (∼1 mL) solution containing the metal ion (3.2 μmol) was slowly added to it, and the resulting solution was transferred to a 300 mL three-necked round bottom flask and diluted with dry CH2Cl2 to be 0.2 mM of 6Zn2. A dry CH2Cl2 solution (5 mL) containing dichloride (4.8 μmol) and pyridine (1 μL, 12 μmol) was slowly added to the 6Zn2 solution for 12 h with a microsyringe pump (KD Scientific, KDS 100). The solution was poured into a saturated NaHCO3 aqueous solution (50 mL) in a PFA-coated flask. The organic layer was washed with water (×3), and passed through Phase Separator paper (Whatman). The solvent was evaporated to dryness, giving a purple solid. The solids obtained by the reactions with three kinds of dichlorides were analyzed with HPLCs to estimate the reaction yields.Purification of Zn2-CP2o,m,p
The crude mixtures containing the target cyclic dimers were purified on cyanopropyl modified silica gel columns (YFLC Gel, CN, Yamazene Co. pore size: 40 μm) with a mixed solvent of toluene and pyridine (15/85 v/v) and 100% pyridine as an eluent. The solvent was evaporated to dryness, giving pure target products.Zn2-CP2o 1H NMR (500 MHz, pyridine-d5) δ/ppm = 11.59 (s, 2H), 9.05 (s, 2H), 8.73 (s, 4H), 8.64 (s, 4H), 8.45–8.42 (8H), 8.28 (t, J = 8.0 Hz, 2H), 8.06–7.91 (m, 4H), 7.72 (t, J = 7.8 Hz, 2H), 7.36 (s, 8H), 7.11 (s, 4H), 2.53 (s, 12H), 1.79 (s, 12H), 1.50 (s, 12H); MALDI-TOF-MS (dithranol) m/z 1680.4970 (M)+, calcd for [C106H81N12O2Zn2]+ 1680.5110.
Zn2-CP2m 1H and 13C NMR assignments are shown in Fig. S23.† MALDI-TOF-MS (dithranol) m/z 1681.5197 (M + H)+, calcd for [C106H81N12O2Zn2]+ 1681.5183.
Zn2-CP2p 1H and 13C NMR assignments are shown in Fig. S31.† MALDI-TOF-MS (dithranol) m/z 1703.4968 (M + Na)+, calcd for [C106H80N12O2Zn2Na]+ 1703.5003.
Synthesis of 7Fb
TFA (4.0 mL) was slowly added to 4Zn (50.4 mg, 52.7 μmol) in a 25 mL flask, and the mixture was stirred at rt for 1 h. The resulting solution was poured into a saturated NaHCO3 aqueous solution (50 mL) in an ice-bath. The organic layer was transferred to a PFA-coated funnel and the aqueous layer was extracted with CHCl3 (10 mL × 3). The combined organic layer was washed with water and dried over anhydrous Na2SO4. The solvent was evaporated to dryness, giving a purple solid 7Fb (37.7 mg, 47.5 μmol) in 91% yield. 1H NMR (400 MHz, CDCl3) δ/ppm = 8.89 (d, J = 4.8 Hz, 2H), 8.78 (d, J = 4.8 Hz, 2H), 8.72 (d, J = 4.8 Hz, 2H), 8.67 (d, J = 4.8 Hz, 2H), 8.25 (dd, J = 6.4, 2.0 Hz, 1H), 7.91–7.86 (m, 2H), 7.61 (d, J = 7.8 Hz, 2H), 7.54 (s, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.27 (s, 4H), 7.05 (m, 1H), 3.92 (s (br), 2H), 2.62 (s, 6H), 1.82 (s, 12H), −2.65 (s, 2H). MALDI-TOF MS of 7Fbm/z 792.24 (M)+, calcd for C49H41BrN6 792.26.Synthesis of 8Fb2
A 50 mL two-necked flask was charged with 7Fb (17.1 mg, 21.5 μmol), 4-dimethyl-aminopyridine (DMAP, 2.8 mg, 22.9 μmol) and dry pyridine (2 mL). A dry CH2Cl2 solution (4 mL) containing isophthaloyl chloride (3.2 mg, 15.8 µmol) was slowly added to the 7Fb solution with a microsyringe pump (kd Scientific, KDS 100). After 15.5 h, the solution was poured into a saturated NaHCO3 aqueous solution in a PFA-coated flask. The organic layer was washed with water (×3), and passed through Phase Separator paper (Whatman). The solvent was evaporated to dryness, giving a purple solid (16.6 mg). The crude solid was purified with a flush silica gel column (eluent: CHCl3), affording a pure target compound 8Fb2 (7.8 mg, 4.5 μmol) in 42% yield. TLC (CHCl3:MeOH = 50:1) Rf = 0.63; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.81 (d, J = 4.8 Hz, 4H), 8.78 (d, J = 4.8 Hz, 4H), 8.71 (d, J = 4.8 Hz, 4H), 8.66 (d, J = 4.8 Hz, 4H), 8.49 (s, 1H), 8.34 (s, 2H), 8.21 (s, 1H), 8.15 (dd, J = 8.0, 1.2 Hz, 2H), 8.01–7.87 (m, 8H), 7.71–7.67 (m, 2H), 7.51–7.41 (m, 2H), 7.25 (s, 4H), 7.21 (s, 4H), 2.59 (s, 12H), 1.81 (s, 12H), 1.76 (s, 12H), −2.68 (s, 4H); MALDI-TOF-MS (dithranol) m/z 1715.5282 (M + H)+, calcd for [C106H85Br2N12O2]+ 1715.5285; UVvis absorption: λmax (CHCl3)/nm = 420, 517.
Synthesis of 8Zn2
A 10 mL flask was charged with 8Fb2 (7.8 mg, 4.5 μmol) and CHCl3 (3 mL). Zn(OAc)2 (8.2 mg, 45 μmol) in a small amount of methanol was added to it and the mixture was stirred at room temperature for 5 h. The resulting solution was transferred to a PFA-coated funnel and washed with water (×3). The organic layer was passed through Phase Separator paper (Whatman). The solvent was evaporated to dryness, giving a purple solid 8Zn2 (8.0 mg, 4.3 μmol) in 96% yield. TLC (CHCl3:MeOH = 50:1) Rf = 0.13; UV-vis absorption: λmax (CHCl3)/nm = 426, 554. MALDI-TOF MS of 8Zn2m/z 1838.34 (M)+, calcd for C106H80Br2N12O2Zn2 1838.35.
Synthesis of Zn2-CP2m (coupling)
A 20 mL Schlenk tube was charged with 8Zn2 (3.0 mg, 1.6 μmol) and 2,2′-bipyridine (0.3 mg, 1.9 μmol). After the Schlenk tube was evacuated and filled with Ar gas, dry DMF degassed with freeze–thaw cycles (10 mL) and 1,5-cyclooctadiene (0.6 μL, 1.9 μmol) were added to the tube. After the mixture was stirred for 10 min at rt, Ni(cod)2 (32.7 mg, 119 μmol) was added. The reaction mixture was sonicated and stirred at 50 °C in the dark. After overnight stirring, the resulting mixture was added to a small amount of CHCl3 in a perfluoroalkoxy alkane (PFA)-coated flask. The Schlenk tube was washed with CHCl3. The combined CHCl3 solution was washed with 0.3 M citric acid solution (×4) and 25% ammonium solution (×3), and dried over anhydrous Na2SO4. The solution was passed through a membrane filter (Omnipore, 0.1 μm) to remove the precipitates. The filtrate was concentrated to dryness, giving a purple solid (3.5 mg). The solid was analyzed by HPLC to estimate the reaction yield.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ENEOS Hydrogen Trust Fund. We would like to thank Editage for English language editing and the Tokyo University of Science for financial support.Notes and references
- V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Am. Chem. Soc., 2001, 123, 2979–2989 CrossRef CAS PubMed.
- T. Kurtán, N. Nesnas, Y.-Q. Li, X. Huang, K. Nakanishi and N. Berova, J. Am. Chem. Soc., 2001, 123, 5962–5973 CrossRef PubMed.
- V. Valderrey, G. Aragay and P. Ballester, Coord. Chem. Rev., 2014, 258–259, 137–156 CrossRef CAS.
- K. Kinjo, T. Hirao, S.-I. Kihara, Y. Katsumoto and T. Haino, Angew. Chem., Int. Ed., 2015, 54, 14830–14834 CrossRef CAS PubMed.
- Z. N. Zahran, E. A. Mohamed, A. A. Haleem and Y. Naruta, Chem. – Eur. J., 2018, 24, 10606–10611 CrossRef CAS PubMed.
- J. Rosenthal, B. J. Pistorio, L. L. Chng and D. G. Nocera, J. Org. Chem., 2005, 70, 1885–1888 CrossRef CAS PubMed.
- R. K. Totten, P. Ryan, B. Kang, S. J. Lee, L. J. Broadbelt, R. Q. Snurr, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2012, 48, 4178–4180 RSC.
- P. Mondal, S. Sarkar and S. P. Rath, Chem. – Eur. J., 2017, 23, 7093–7103 CrossRef CAS PubMed.
- P. D. Harvey, C. Stern, C. P. Gros and R. Guilard, Coord. Chem. Rev., 2007, 251, 401–428 CrossRef CAS.
- A. Satake and Y. Kobuke, Org. Biomol. Chem., 2007, 5, 1679–1691 RSC.
- S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
- Y. Tomohiro, A. Satake and Y. Kobuke, J. Org. Chem., 2001, 66, 8442–8446 CrossRef CAS PubMed.
- S. Fukuda, A. Satake and Y. Kobuke, Thin Solid Films, 2006, 499, 263–268 CrossRef CAS.
- A. Satake, Y. Katagami, Y. Odaka, Y. Kuramochi, S. Harada, T. Kouchi, H. Kamebuchi and M. Tadokoro, Inorg. Chem., 2020, 59, 8013–8024 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kishi, H. Kotani, Y.-M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed.
- S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734–5740 CrossRef CAS PubMed.
- (a) H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS; (b) J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- (a) A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 4867–4874 CrossRef CAS; (b) D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 5870–2877 CrossRef.
- (a) E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Mater., 2017, 29, 7140–7150 CrossRef CAS; (b) E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef CAS PubMed.
- E. Vanover, Y. Huang, L. Xu, M. Newcomb and R. Zhang, Org. Lett., 2010, 12, 2246–2249 CrossRef CAS PubMed.
- (a) T. Nakazono, A. R. Parent and K. Sakai, Chem. – Eur. J., 2015, 21, 6723–6726 CrossRef CAS PubMed; (b) T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- V. Bizet, L. Buglioni and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 5639–5642 CrossRef CAS PubMed.
- (a) S. Y. Lee, H. S. Fung, S. Feng and K. S. Chan, Organometallics, 2016, 35, 2480–2487 CrossRef CAS; (b) S. Y. Lee and K. S. Chan, Organometallics, 2013, 32, 5391–5401 CrossRef CAS.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef PubMed.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- T. Lazarides, L. V. Sazanovich, A. J. Simaan, M. C. Kafentzi, M. Delor, Y. Mekmouche, B. Faure, M. Reglier, J. A. Weinstein, A. G. Coutsolelos and T. Tron, J. Am. Chem. Soc., 2013, 135, 3095–3103 CrossRef CAS PubMed.
- (a) P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Chem. – Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed; (b) C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Chem. Sci., 2015, 6, 6847–6864 RSC; (c) K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667–11673 CrossRef CAS.
- (a) Y. Kuramochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed; (b) Y. Kuramochi and A. Satake, Chem. – Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- A. Osuka and K. Maruyama, J. Am. Chem. Soc., 1988, 110, 4454–4456 CrossRef CAS.
- Y. Ohkoda, A. Asaishi, T. Namiki, T. Hashimoto, M. Yamada, K. Shirai, Y. Katagami, T. Sugaya, M. Tadokoro and A. Satake, Chem. – Eur. J., 2015, 21, 11745–11756 CrossRef CAS PubMed.
- (a) J. M. Hooker, A. Datta, M. Botta, K. N. Raymond and M. B. Francis, Nano Lett., 2007, 7, 2207–2210 CrossRef CAS PubMed; (b) T. Schadendorf, C. Hoppmann and K. Rück-Braun, Tetrahedron Lett., 2007, 48, 9044–9047 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of 4Zn, 5Fb2, 6Fb2, 7Fb, 8Fb2, Zn2-CP2m, and Zn-CP2p. See DOI: 10.1039/d1ob00088h |
| ‡ The B3LYP/6-31G(d) level of theory was used and the contributions from dispersion interactions were not considered for the calculations. Please refer to Fig. S37 in the ESI,† which presents models constructed using the B3LYP-D3/6-31G(d) level of theory (in vacuo). Contributions from dispersion interactions were considered for the calculations |
| This journal is © The Royal Society of Chemistry 2021 |