
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative azidations of phenols and ketones using iodine azide after release from an ion exchange resin†
Teresa
Kösel
,
Gerald
Dräger
 and
Andreas
Kirschning
*
and
Andreas
Kirschning
*
Institute of Organic Chemistry, Leibniz University Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: andreas.kirschning@oci.uni-hannover.de
First published on 9th March 2021
Abstract
The oxidative oligoazidation of phenols and ketones using iodine azide (IN3) provided by its release from an ion exchange resin is reported. Preliminary mechanistic studies indicate a previously unknown reactivity of iodine azide toward phenols and ketones.
Introduction
The iodination of alkenes and aromatics is complicated by the reversibility of this process, which is caused by the formation of HI in the presence of the iodine used. Therefore, ways must be found to remove hydrogen iodide as soon as it has formed in order to achieve the best results. For this purpose HgO, HNO3, HIO3 and H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 have been used to oxidise hydrogen iodide. Other ways to achieve efficient iodination of phenols are mixtures consisting of sodium iodide and either tert-butyl hypochlorite or chloramine T.2 Okamoto and collaborators reported a dichloroiodate(I) agent 1 formed from ICl and benzyltrimethylammonium chloride that allows ortho-iodination of phenols under mild conditions.3 It proved superior to ICl. It has also been reported that the presence of methanol is beneficial and postulated that the active reagent is methyl hypoiodite 2.
1 have been used to oxidise hydrogen iodide. Other ways to achieve efficient iodination of phenols are mixtures consisting of sodium iodide and either tert-butyl hypochlorite or chloramine T.2 Okamoto and collaborators reported a dichloroiodate(I) agent 1 formed from ICl and benzyltrimethylammonium chloride that allows ortho-iodination of phenols under mild conditions.3 It proved superior to ICl. It has also been reported that the presence of methanol is beneficial and postulated that the active reagent is methyl hypoiodite 2.
We reported on the synthesis of electrophilic halonium reagents 3 that are structurally related to 1. These are obtained by iodine(III)-mediated oxidation of organic ammonium bromides or iodides.4 The resulting acylated haloate(I) complexes 35 can be further diversified by ligand exchange using silylated nucleophiles yielding ate(I) anions 4. They have been used in a variety of reactions6 including activation of thioglycosides7 and dithioacetals8 and as cooxidants for TEMPO-mediated oxidations.9 Chemically, these haloate(I) anions 3 [Br(OAc)2−, I(OAc)2−, I(O2CCF3)2−, I(N3)2−] behave like Br-OAc, I-OAc, I-OTf and I-N3.10 A practical advantage is that the starting ammonium halide can be an ion exchange resin (e.g. Amberlyst A-26), so that polymer-bound versions of haloate(I) anions are available. If the nucleophile (Nu) is azide and the halogen atom is iodine, the orange polymer is a stable and safe form of iodine azide. This polymer is in fact non-explosive and can be stored in the dark under an argon atmosphere at −15 °C for several months without loss of activity. We recently showed that this form of iodine azide can also serve as a precursor for azide radicals.11 The polymeric by-product (ion exchange resin-azide form), which is formed during transformations with 4a, is removed by simple filtration (Scheme 1). The polymer-bound version of reagents 4 can be regenerated by ion exchange with iodide, where PhI(OAc)2 promotes oxidation to 3 (R2 = CH3) and ligand exchange with TMSN3 yields the regenerated polymer.12
 | ||
| Scheme 1 Iodination with benzyltrimethylammonium dichloroiodate(I) in methanol according to Okamoto et al.3 and haloate(I) reagents producible by iodine(III) oxidation of halide anions. | ||
The close relationship between dichloroiodate(I) 1 and bisazidoiodate(I) 4a led us to investigate the reactivity of 4a with phenols.
Results and discussion
Exposing phenol 5 to the functionalised polymer 4a resulted in 2,4,6-triiodophenol (6) in 90% yield (Scheme 2, eqn (1)). This result is consistent with the iodination protocol reported by Okamoto (Scheme 1). However, when extending the protocol to other phenols, we found that this first example is rather an exception. In a second series of experiments, ethinylestradiol (7) was selected as the phenolic substrate, which led to an unexpected result, the formation of three estradiol derivatives 8–10 (eqn (2)). While the formation of diiodide 8 was expected, the formation of bis- and trisazido adducts 9 and 10 revealed a new chemistry of iodine azide. By increasing the equivalents of 4a, it could be determined that first the iodination product 8 is formed, followed by phenol 9. Finally dienone 10 with three azido groups becomes the only product, if a sufficient amount of reagent is used. | ||
| Scheme 2 Iodination of phenol 5 and ethinylestradiol 7 with functionalised polymer 4a (the configuration at C-10 in 10 was related to that found in 15 by X-ray crystallographic analysis). aRatios determined by 1H NMR spectroscopic analysis, bequivalents refer to theoretical presence of ion exchange sites reported by the vendor, cisolated yields are given; the presence of blue LED light did in principal not lead to altered results. | ||
Next, this unprecedented reaction was extended to other phenols to afford azides 11–20. The structure including the configuration of the newly formed stereogenic center in 15 (formed from estrone) was unequivocally confirmed by X-ray crystallographic analysis. In view of the structural and stereochemical similarity of the starting steroids (estradiol, estrone and estriol), it can be assumed that the four products 16–19 all have the β-oriented azide substituent at C-10 as determined for 15. The chemical shift δ for H1 at 6.53–6.57 ppm was used as a diagnostic feature for this purpose.
The conversions of several phenols to azides 10–15 consistently proceeded with excellent yields (Scheme 3). A brief investigation of the influence of the solvent showed that methanol can be used in addition to acetonitrile, an observation that fits the results of Okamoto et al.,3 so it can be assumed that hypoiodite 2 is also formed as a reactive intermediate in this case. A similar yield was found for azidodienone 15 with extended reaction time. Interestingly, the use of an arene with three methoxy groups gave the para-substituted mono-azidodienone 12 in a remarkable yield of 96%. The steroidal hormone equilenin (21), first isolated from the urine of pregnant mares,13 shows diverse biological activities, including antiseborrheic, lipid metabolism regulating or neurological disorders treating properties. The conversion to azide 20 is synthetically useful as it reveals a new protocol for derivatizations of this type of steroids. Remarkably, we found that the aromatic B ring remained intact in the presence of iodine azide.
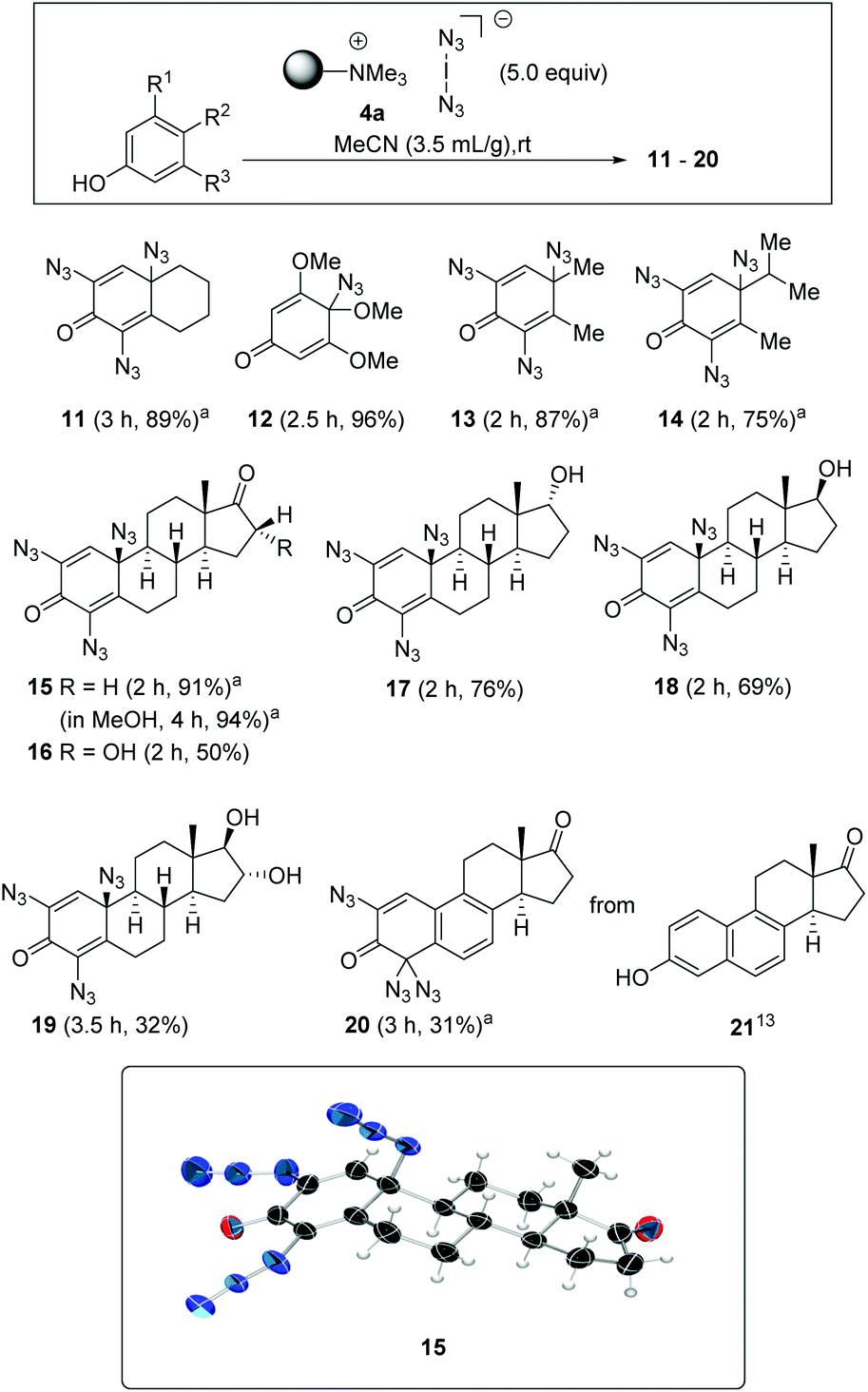 | ||
| Scheme 3 Azidation of phenols and formation of 11–20 (isolated yields are given; in some cases, the lower yield could be due to the phenol being bound to the cation exchange resin; the configuration at C-10 in 16–19 are proposed based on X-ray crystallographic analysis of 15: C = black, N = blue, O = red). aThe characterization of the new azides was accompanied by IR spectroscopy. However, we encountered difficulties in the preparation of mass spectra for a few oligoazides. | ||
To gain a mechanistic insight into the stepwise formation of azidodienone 10, the reaction was monitored in an NMR tube. For this purpose we performed the conversion of ethinylestradiol (7) with 5.0 equiv. of 4a in deuterated acetonitrile and collected a sample directly after setting up the reaction. After only five seconds the formation of diiodide 8 (δ = 7.67 ppm) and the formation of bis- (δ = 6.89 ppm) and trisazides (δ = 6.71 ppm) 9 and 10 could be detected by 1H-NMR spectroscopy (Scheme 4). However, because the reaction proceeded very rapidly, this experiment did not provide clear evidence on the time course of the formation of individual intermediates and on the question of whether mechanistically parallel pathways to the iodides and azides might exist. We were able to isolate and characterize all products 8–10.
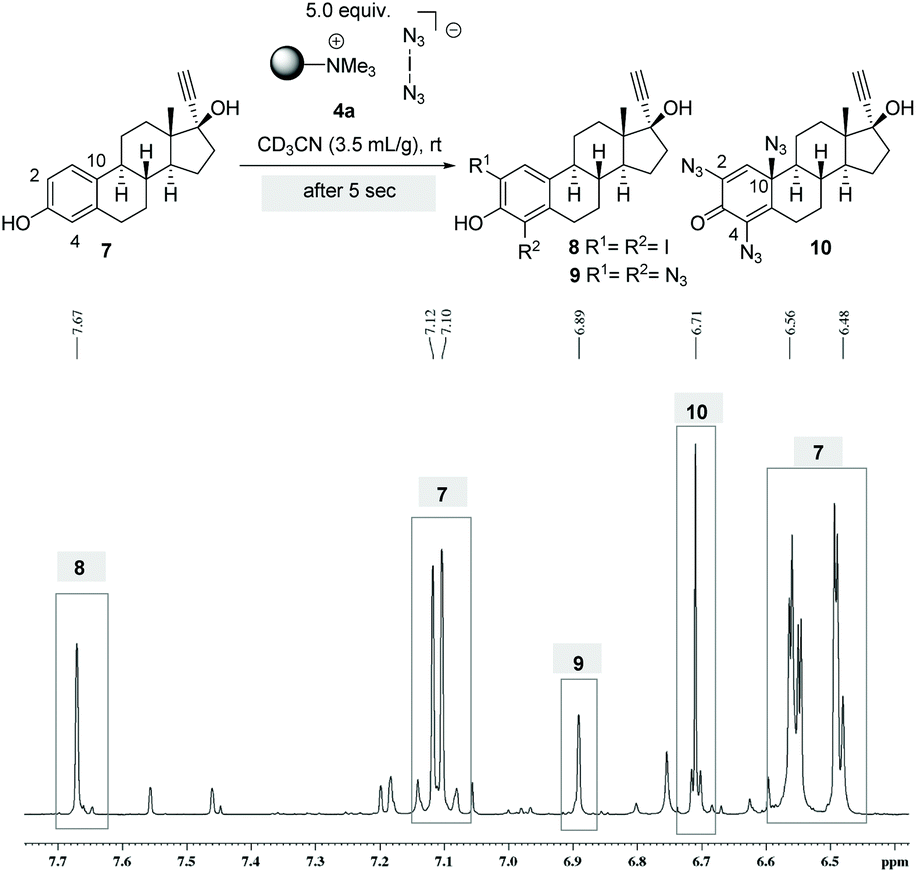 | ||
| Scheme 4 Reaction tracking of ethinylestradiol (7) with 4a by 1H NMR spectroscopic analysis. | ||
To collect further information on azidodienone formation and possible precursor iodides, we repeated the reaction tracking for the reaction of estrone (22), whose structure was clearly secured by X-ray analysis, with only 1.0 equiv. of 4a in deuterated acetonitrile at 0 °C and collected samples over a period of 1 h (Scheme 5). This experiment proceeded analogous to that for phenol 7, but it allowed the observation of the time course of the formation of intermediates.
 | ||
| Scheme 5 Reaction monitoring of estrone (22) with 4a by 1H-NMR spectroscopic analysis; samples were taken according to given times (bisazide 24 could not be isolated, but the signal at δ = 6.7 ppm was assigned to bisazide 24 by comparison with the corresponding signal at δ = 6.89 ppm for bisazide 9 in Scheme 4). | ||
To collect evidence that azidodienone formation occurs via the initial electrophilic iodination of ortho positions, we independently prepared diiodide 22 (see ESI†) which quantitatively yielded the expected trisazido adduct 15 under our standard conditions (Scheme 6).
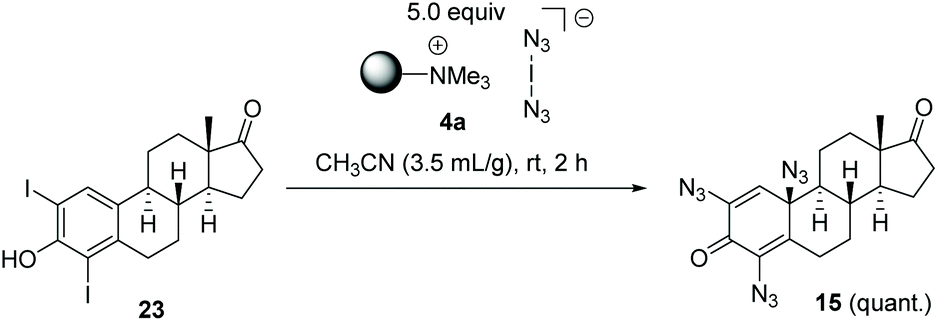 | ||
| Scheme 6 Synthesis of azidodienone 15 starting from diiodide 23. | ||
After only five minutes, the appearance of a strong signal at δ = 7.66 ppm was detected, which is characteristic of diiodide 23 (Scheme 5). Next, after 30 minutes, a signal at δ = 6.89 ppm is clearly visible, which is characteristic of bisazido adduct 24. From these findings, it can be concluded that diiodide 23 (δ = 7.66 ppm) must most likely be a precursor for bisazido product 24 and the latter for azidodienone 15.
These analytical studies allow us to propose a possible mechanism for the formation of azidodienone 15 from estrone (22, Scheme 7). After electrophilic iodination of both ortho positions, which provides diiodide 23, we propose an activation of the phenolic position by iodine azide, which gives hypoiodite 26. Next, the nucleophilic addition of azide leads to the formation of dienone 27, which after iodonium migration can form a new hypoiodite 28. Aromatization could be the driving force for this step. After this sequence, azide is introduced two more times, the last addition is facilitated by the strong electrophilic character of dienone 25.
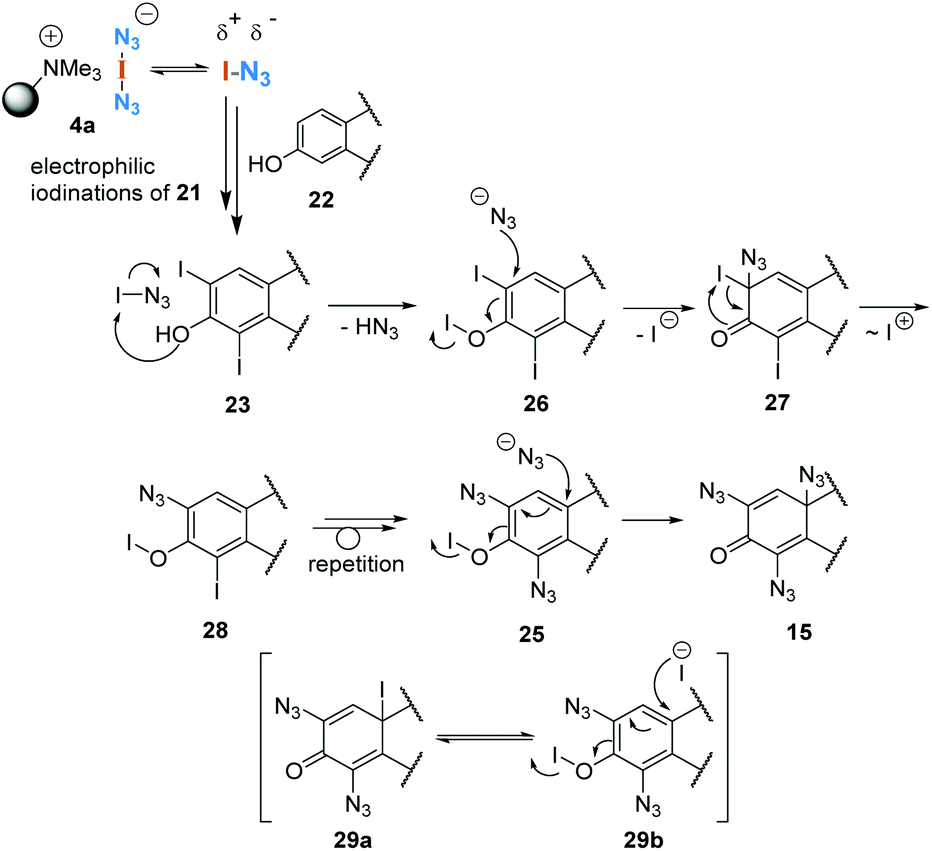 | ||
| Scheme 7 Postulated mechanism for oligoazide formation. | ||
With this mechanistic proposal in hand, we concluded that enols might behave similarly to phenols in the presence of iodoazide. In a first proof of concept study we chose acylarenes as substrates and treated these with an excess of polymer-bound iodine azide 4a in acetonitrile at 83 °C (Scheme 8). As expected, mono- and diazidations occurred in the α-positions within 15 h to 3 d after treatment and azides 30a–d were isolated in yields ranging from 57% to 95%. Geminal bisazides can be directly converted into nitrogen-containing heterocycles such as tetrazoles or triazoles, which play an important role in pharmaceutical research.14 For this protocol, we used a thiosulfate ion exchange resin for reductive workup to remove by-products such as iodine and IN3, obviating the need for hydrolytic workup. Mechanistically, this oxidation could be initiated by hypoiodite formation, similar to that proposed in Scheme 7 for phenol oxidation.
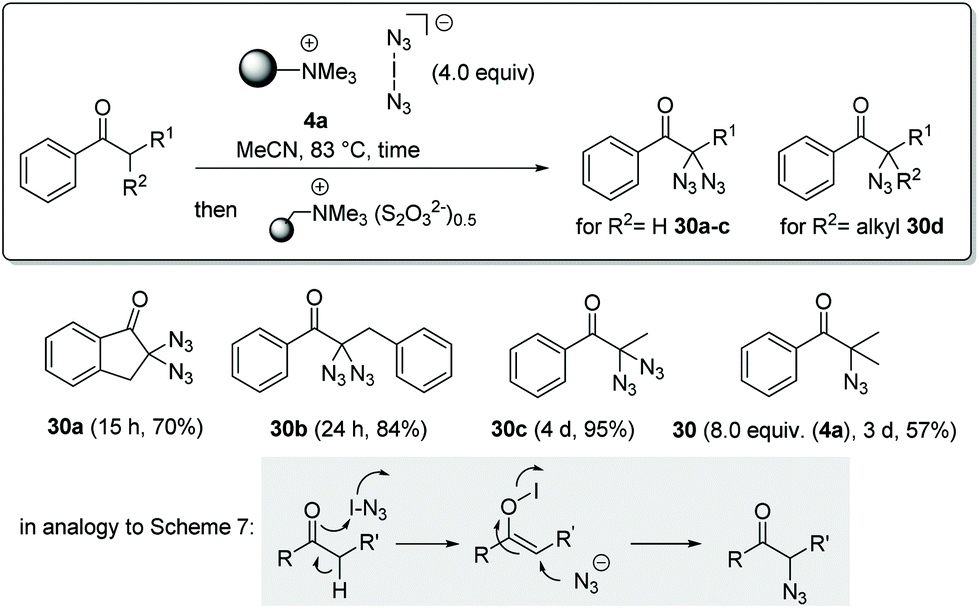 | ||
| Scheme 8 Azidation of acylarenes (isolated yields are given). | ||
Experimental section
General procedure for the oxidative azidation of phenols
A mixture of the phenol (0.5 mmol, 1.0 equiv.) and polymer-bound iodine azide (4a, 1.19 g, 2.50 mmol, 5.0 equiv. according to theoretical functionalisation) was stirred in absolute MeCN (4.16 mL, 3.5 mL g−1 polymer) at ambient temperature under an argon atmosphere. After complete consumption of the reactant as judged by TLC, the reaction was terminated by filtration and the resin was washed with EtOAc and the combined filtrates were concentrated under reduced pressure. E.g., this protocol provided 4-azido-3,4,5-trimethoxycyclohexa-2,5-dien-1-one (12) (107.8 mg, 479 μmol; 96% yield) starting from 3,4,5-trimethoxyphenol. 1H-NMR (CDCl3, 400 MHz): δ [ppm] 5.51 (s, 2H, 2 × CH), 3.77 (s, 6H, 3 × C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) COCH3), 3.18 (s, 3H, N3-COCH3); 13C-NMR (CDCl3, 100 MHz): δ [ppm] 185.5 (q, CO), 165.3 (q, 2 × C=COCH3), 103.2 (t, 2 × CH), 85.2 (q, N3-COCH3), 56.5 (p, 2 × CCOCH3), 53.0 (p, N3-CCH3); IRνmax [cm−1] 2116 ν(N3); ESI-MS (ESI+) m/z calculated for C9H11N3O4Na+ [M + Na]+ 248.0647; found 248.0644
COCH3), 3.18 (s, 3H, N3-COCH3); 13C-NMR (CDCl3, 100 MHz): δ [ppm] 185.5 (q, CO), 165.3 (q, 2 × C=COCH3), 103.2 (t, 2 × CH), 85.2 (q, N3-COCH3), 56.5 (p, 2 × CCOCH3), 53.0 (p, N3-CCH3); IRνmax [cm−1] 2116 ν(N3); ESI-MS (ESI+) m/z calculated for C9H11N3O4Na+ [M + Na]+ 248.0647; found 248.0644
Conclusions
In summary, we have uncovered a new reactivity of iodine azide toward phenols and ketones, which is initiated by the release of this highly reactive agent from an ion-exchange resin into the organic solution. Mechanistically, we propose the formation of hypoiodite intermediates as a key step in achieving oxidative azidation of phenols and ketones.15 We believe that the extension of the synthetic potential of iodine azide under safe conditions will open new amination pathways for pharmaceutically relevant phenols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Bundesministerium für Bildung und Forschung (BMBF, project SILVIR: 16GW0202).Notes and references
- R. D. C. Gallo, K. S. Gebara, R. M. Muzzi and C. Raminelli, J. Braz. Chem. Soc., 2010, 21(no. 4), 770–774 CrossRef CAS.
- T. Kometani, D. Watt, T. Ji and T. Fitz, J. Org. Chem., 1985, 50, 5384–5387 CrossRef CAS.
- S. Kajigaeshi, T. Kakinami, H. Yamasaki, S. Fujisaki, M. Kondo and T. Okamoto, Chem. Lett., 1987, 16, 2109–2112 CrossRef.
- A. Kirschning, C. Plumeier and L. Rose, Chem. Commun., 1998, 33–34 RSC.
- The structure of bisacyliodate(I) salts was recently confirmed by X-ray analysis: K. Muñiz, B. García, C. Martínez and A. Piccinelli, Chem. – Eur. J., 2017, 23, 1539–1545 CrossRef PubMed.
- (a) Md. A. Hashem, A. Jung, M. Ries and A. Kirschning, Synlett, 1998, 195–197 CrossRef; (b) A. Kirschning, Md. A. Hashem, H. Monenschein, L. Rose and K.-U. Schöning, J. Org. Chem., 1999, 64, 6522–6526 CrossRef CAS; (c) H. Monenschein, G. Sourkouni-Argirusi, K. M. Schubothe, T. O'Hare and A. Kirschning, Org. Lett., 1999, 1, 2101–2105 CrossRef CAS; (d) S. Domann, G. Sourkouni-Argirusi, N. Merayo, A. Schönberger and A. Kirschning, Molecules, 2001, 6, 61–66 CrossRef CAS; (e) A. Kirschning, E. Kunst, M. Ries, L. Rose, A. Schönberger and R. Wartchow, ARKIVOC, 2003, 145–162 Search PubMed.
- S. Luiken and A. Kirschning, J. Org. Chem., 2008, 73, 2018–2020 CrossRef CAS PubMed.
- (a) A. Kirschning, A. Schönberger and M. Jesberger, Org. Lett., 2001, 3, 3623–3626 CrossRef CAS PubMed; (b) J. Jaunzems, G. Sourkouni-Argirusi, M. Jesberger and A. Kirschning, Tetrahedron Lett., 2003, 44, 637–639 CrossRef CAS.
- (a) A. Kirschning, G. Sourkouni-Argirusi and M. Brünjes, Adv. Synth. Catal., 2003, 345, 635–642 CrossRef; (b) K. Kloth, M. Brünjes, E. Kunst, F. Gallier, A. Adibekian and A. Kirschning, Adv. Synth. Catal., 2005, 347, 1423–1434 CrossRef CAS.
- (a) A. Kirschning, H. Monenschein and C. Schmeck, Angew. Chem., Int. Ed., 1999, 38, 2594–2596 CrossRef CAS; (b) A. Kirschning and H. Monenschein, Polymer-bound ammonium bisazidoiodate(I), e-EROS Encycl. Reagents Org. Synth., 2002 DOI:10.1002/047084289X.rn00019.
- T. Kösel, G. Schulz, G. Dräger and A. Kirschning, Angew. Chem., 2020, 132, 12475–12479 ( Angew. Chem. , 2020 , 59 , 12376–12380 ) CrossRef.
- S. Cludius-Brandt, L. Kupracz and A. Kirschning, Beilstein J. Org. Chem., 2013, 9, 1745–1750 CrossRef PubMed.
- (a) A. Girard, G. Sandulesco, A. Fridenson and J. J. Rutgers, C. R. Acad. Sci., 1932, 195, 981 CAS; (b) V. M. Dembitsky, N. Savidov1, V. V. Poroikov, T. A. Gloriozova and A. B. Imbs, Appl. Microbiol. Biotechnol., 2018, 102, 4663–4674 CrossRef CAS PubMed.
- I. E. Celik and S. F. Kirsch, Eur. J. Org. Chem., 2021, 53–63 CrossRef CAS.
- Recently, a mixture of chlorimidazolinium chloride and sodium azide was shown to be capable of oxidatively azidate phenols: M. Kitamura, K. Murakami, T. Koga, T. Eto, A. Ishikawa, H. Shimooka and T. Okauchi, Eur. J. Org. Chem., 2019, 5824–5827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and spectroscopic data including copies of NMR spectra of synthetic intermediates. CCDC 2049506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob00083g |
| This journal is © The Royal Society of Chemistry 2021 |