
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a 1,2-difluorofucoside activity-based probe for profiling GH29 fucosidases†
Yvette M. C. A.
Luijkx
ab,
Seino
Jongkees
a,
Karin
Strijbis
b and
Tom
Wennekes
 *a
*a
aDepartment Chemical Biology and Drug Discovery, Utrecht Institute for Pharmaceutical Sciences and Bijvoet Center for Biomolecular Research, Utrecht University, Utrecht, The Netherlands. E-mail: t.wennekes@uu.nl
bDepartment Biomolecular Health Sciences, Division Infectious diseases and Immunology, Faculty of Veterinary Medicine, Utrecht University, Utrecht, The Netherlands
First published on 15th March 2021
Abstract
GH29 α-L-fucosidases catalyze hydrolysis of terminal α-L-fucosyl linkages with varying specificity and are expressed by prominent members of the human gut microbiota. Both homeostasis and dysbiosis at the human intestinal microbiota interface have been correlated with altered fucosidase activity. Herein we describe the development of a 2-deoxy-2-fluoro fucosyl fluoride derivative with an azide mini-tag as an activity-based probe (ABP) for selective in vitro labelling of GH29 α-L-fucosidases. Only catalytically active fucosidases are inactivated by this ABP, allowing their functionalization with a biotin reporter group via the CuAAC reaction and subsequent in-gel detection at nanogram levels. The ABP we present here is shown to be active against a GH29 α-L-fucosidase from Bacteroides fragilis and capable of labeling two other GH29 α-L-fucosidases with different linkage specificity, illustrating its broader utility. This novel ABP is a valuable addition to the toolbox of fucosidase probes by allowing identification and functional studies of the wide variety of GH29 fucosidases, including those in the gut microbiota.
Introduction
Retaining α-L-fucosidases (GH29 fucosidases) are exo-glycosidases that catalyze the hydrolysis of terminal α-L-fucopyranosides from cellular glycans.1 This glycosidase family is widespread in bacteria, fungi and mammals, and implicated in several critical biological processes such as immune response, signal transduction and adhesion of pathogens.1–3 GH29 fucosidases have a diverse substrate specificity. They commonly hydrolyze α-(1,2) linkages from fucose to galactose or α-(1,3), α-(1,4), and α-(1,6) linkages to N-acetylglycosamine. Based on their substrate specificity they can be divided in two main subfamilies (A and B).4 Subfamily A fucosidases have relatively relaxed substrate specificity. For example the human GH29 fucosidase is assigned to subfamily A as it is active on α-1,2/3/4/6-linked fucosyl substrates.5 Similar to this the GH29 fucosidase Bt2970 from the intestinal bacterium Bacteroides thetaiotaomicron shows a broad substrate specificity.6 In contrast, subfamily B is more specific towards α-(1,3) or α-(1,4) linkages. Several other GH29 fucosidases, among which BT2192 and BT3798 (from B. thetaiotaomicron) show a more stringent specificity, placing them in subfamily B.4,6,7 We are interested in elucidating the role of this diverse set of bacterial fucosidases specifically in the human gut where they have been implicated in host-microbe interplay, microbiota cross feeding and colonization resistance.8–10 For example, it has been established that infection efficiency of the common foodborne pathogens, Salmonella typhimurium and Campylobacter jejuni, relies on fucosidases secreted by other residing bacteria.2,11,12 To study the exact function of fucosidases produced by pathogens and commensals we first have to identify them and determine their spatial and temporal distribution.13 Investigations into the functions of different bacterial fucosidases will benefit from the availability of chemical probes that enable the screening, isolation and study of catalytically-active fucosidases.GH29 fucosidases hydrolyze their substrate with overall retention of configuration at the anomeric center through a classic Koshland double displacement mechanism (Scheme 1A).14 The reaction mechanism involves two carboxylates from aspartic acids in the active site, one acting as the catalytic nucleophile and the other acting as the catalytic acid–base. The mechanism proceeds through a covalent fucosyl-enzyme intermediate accessed via a pair of oxocarbenium ion-like transition states. Quinone methide based probes for glycosidases have been reported to generate a reactive electrophile upon hydrolysis of their glycosidic bond, of which one example covalently labeled a human fucosidase (Scheme 1B).15–18 Although these probes have shown their merit in several studies, their major limitation is that the affinity for the enzyme is lost by cleavage of the glycosidic bond resulting in possible non-specific labeling of non-associated proteins. 2-Deoxy-2-fluoro glycosides and cyclophellitol epoxides or azirdines are the two main classes of covalent inhibitors that, opposed to the quinone methide based probes, can inactivate and label a glycosidase by trapping the covalent enzyme intermediate.19,20 By altering the stereochemistry, substituents and warhead of the cyclophellitol scaffold, Overkleeft and co-workers have developed ABPs for a wide range of different retaining glycosidases.21–24 A study by Davies and co-workers provided the first structural basis for the inactivation of a β-glycosidase by cyclophellitol, employing an electrophilic trap mechanism to label the target enzyme (Scheme 1C).25
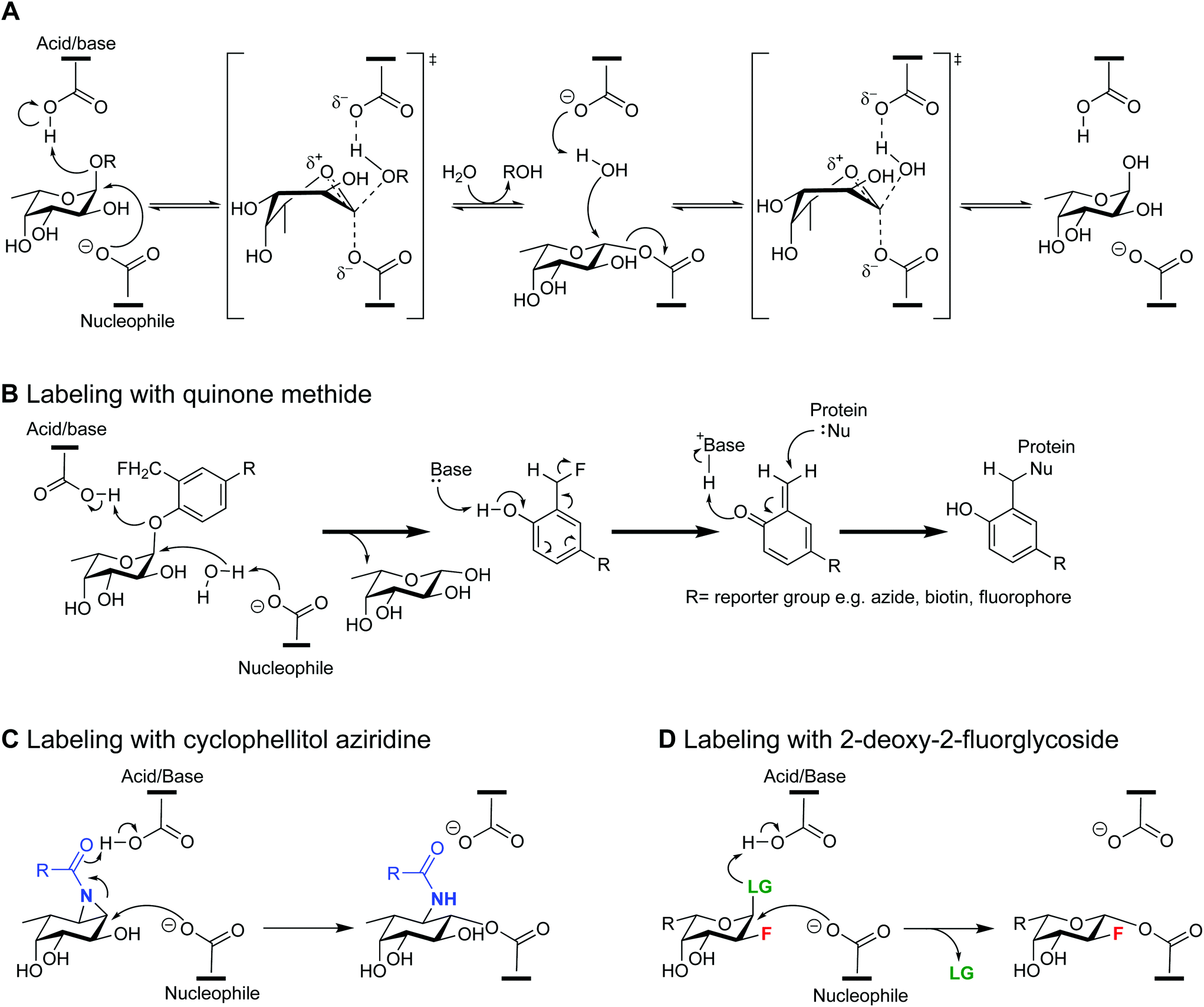 | ||
| Scheme 1 (A) Classic Koshland double displacement mechanism for GH29 fucosidases. Labeling mechanism of a fucosidase for a quinone methide based probe (B), a cyclophellitol aziridine based probe (C) and a 2-deoxy-2-fluoro fucoside-based probe (D). | ||
2-Deoxy-2-fluoro glycosides were originally introduced by Withers and co-workers.26,27 The substitution of a highly electronegative fluorine atom for the C2 hydroxyl group destabilizes the oxocarbenium ion-like states, resulting in a decreased rate for both the formation of the glycosyl–enzyme intermediate and its subsequent hydrolysis. By employing a suitably reactive leaving group at the anomeric site the formation of the covalent intermediate will still proceed. Essentially, these act as a very slow substrate and result in accumulation of the glycosyl–enzyme intermediate (Scheme 1D). Activated 2-deoxy-2-fluoro glycosides have been used to identify and label catalytic nucleophiles of a variety of retaining glycosidases and this class of inhibitors has been extensively reviewed over the past decades.20,21,28–30 The inhibition of specific glycosidases by 2-deoxy-2-fluoro glycoside-based inhibitors has been demonstrated in live animals, and showed promise in future imaging applications by applying radiolabelled inactivators.31,32 Furthermore, activated 2-deoxy-2-fluoro glycosides were the basis for activity-based protein profiling (ABPP) probes to target β-D-glucosaminidases, β-galactosidases, and β-glycanases, when conjugated to a reporter group such as biotin or a fluorophore.20,33–36 The application in this technique allowed for the first successful selective labelling of β-galactosidases in a complex proteome.34
In 2003 Withers et al. set a precedent for mechanism-based inhibitors of retaining fucosidase as they reported 2-deoxy-2-fluoro-fucosyl fluoride as a selective retaining fucosidase inhibitor and identified the catalytic nucleophile of the GH29 fucosidase of the thermophilic bacterium Thermogata maritima.37 This was followed in 2015 by Overkleeft et al. who developed fucopyranose-configured cyclophellitol aziridine ABPs for selective labelling of GH29 fucosidases in bacteria, mice and man.22 Although the reported cyclophellitol based ABPs have shown to be more potent inhibitors compared to their 2-deoxy-2-fluoro-glycoside analogues, their multi-step synthesis remains a major hurdle.
One of the long-term aims within our research program is to identify the role of bacterial fucosidases in both homeostasis and dysbiosis in the human gut. We have previously reported that Campylobacter jejuni fuc+ strains are dependent on exogenous microbial fucosidases for increased growth and invasion.12 This highlights the importance of fucosidases in the development of infections and shows the merit for the development of different ABPs to further study these processes and identify the responsible enzymes and organisms. In this study our aim was therefore to investigate if we could transform the synthetically more readily accessible 2-deoxy-2-fluor-fucosides into an ABP that can detect microbial α-L-fucosidases in a time- and concentration-dependent manner.
We describe here the successful synthesis of a 2-deoxy-2-fluoro-fucosyl fluoride-based ABP (Scheme 2, YL209) compatible with click chemistry via an azide mini-tag and demonstrate the versatility of this probe by applying it to GH29 fucosidases with varying linkage specificity.
 | ||
Scheme 2 Synthesis of YL209 and the structure of 2FFucF. Reagents and conditions: (a) ZnCl2, cat. H2SO4, acetone, 85%; (b) TsCl, DMAP, pyridine, 0 °C to rt, 86%; (c) (i) TFA/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); (ii) Ac2O, pyridine, 92%; (d) (i) HBr (37% in AcOH), DCM 0 °C to rt; (ii) sat. aq. NaH2PO4, Zn, Acetone, 77%; (e) NaN3, DMF, 90 °C, 95%; (f) XeF2, BF3-OEt2, benzene, diethyl ether, −10 °C to rt, 57%; (g) NaOMe, MeOH, 80%. :1); (ii) Ac2O, pyridine, 92%; (d) (i) HBr (37% in AcOH), DCM 0 °C to rt; (ii) sat. aq. NaH2PO4, Zn, Acetone, 77%; (e) NaN3, DMF, 90 °C, 95%; (f) XeF2, BF3-OEt2, benzene, diethyl ether, −10 °C to rt, 57%; (g) NaOMe, MeOH, 80%. | ||
Results and discussion
Our first aim was to convert the 2-deoxy-2-fluoro-fucosyl fluoride inhibitor into an ABP by developing a synthetic route that allows for the introduction of a functional group that can be used to form a covalent linkage to a reporter group such as a biotin or fluorophore. There is a precedent for fucosides with substituents at the 6-position to serve as a substrate for different fucosidases,38 and various other glycosidases also seem to tolerate different substitutions at the 6-position.39–41 Therefore we decided to introduce an azide mini-tag on the C6 position of the fucoside. This position additionally retains free rotation around the C6 methylene group that may permit the azide to adopt a favorable position in the fucosidase active site, which is necessary in downstream clicking to the reporter group.We aimed to synthesize the ABP via the fluorination of a glycal intermediate (Scheme 2) starting from L-galactose. First, all secondary hydroxy groups were protected as di-isopropylidene acetals to afford 1 in 85% yield. The free C6 hydroxyl in intermediate 1 was converted into C6 tosylate 2. Next, the di-isopropylidene acetals were hydrolyzed and the liberated hydroxyls protected as acetyl esters to provide compound 3 in 79% yield over the three steps. Our initial approach to obtain the 6-azido-galactal 5 was to perform an azide substitution on the C6 tosylate of 3 followed by anomeric bromination and zinc mediated reductive elimination to the 6-azido glycal. This however did not lead to the desired product, but instead resulted in substitution of the C6 azide by a bromide. We therefore decided to perform the bromination and elimination prior to introducing the azide. Bromination of 3 with HBr gave the anomeric bromide intermediate that was immediately treated with zinc dust following a classical Fisher–Zach reductive elimination to give intermediate glycal 4 in 77% yield. Substitution of the tosylate of 4 with sodium azide at elevated temperatures gave compound 5 in an excellent 95% yield. Treatment of the protected L-galactal derivative 5 with XeF2 in the presence of a Lewis acid resulted in difluorination of the alkene from the top face to provide an equatorial C2 fluorine and as the main product the favorable α anomeric fluorine. Deacetylation of 6 yielded the desired probe YL209. To investigate the effect of the azide at C6 on inactivation of fucosidases we also synthesized the known fucosidase inhibitor 2FFucF (structure in Scheme 2), with a methyl group as C6, according to a literature procedure.37,42
The leaving group at C1 of the fluorinated glycosides has previously been shown to play an important role in the effectiveness of these probes as ABPs. Overkleeft et al. reported that the installation of an anomeric N-phenyl trifluoroacetimidate on 2-fluoro-β-glycopyranose-based ABPs drastically improved their potency.43 We therefore also attempted to synthesize the N-phenyl trifluoroacetimidate on our 2-fluoro-6-azido α-L-fucosyl based ABP. Although we could obtain the penultimate C2, C3 levulinoyl ester protected intermediate, in our hands we were unable to obtain the final molecule due to decomposition during deprotection (ESI Fig. 1†). This result can perhaps be explained by the relatively high instability/reactivity of galactosides and fucopyranosides due to electron withdrawing properties of their axial 4-OH.44
With the putative mechanism-based inhibitor YL209 in hand, we sought to evaluate its ability to inactivate a microbial fucosidase. We selected a fucosidase expressed by the well-known gut commensal, Bacteroides fragilis, as the initial target enzyme. The selectivity of BfFucH has already been reported and shows a broad substrate recognition. Incubation of BfFucH with various concentrations of YL209 revealed a time and concentration-dependent inactivation of the enzyme that could be fit to a single exponential decay equation (Fig. 1A). However, incubation of BfFucH with various concentrations YL209 never resulted in complete inactivation. When incubated with 5, 10, 20, 40, and 80 mM YL209 respectively 43%, 48%, 60%, 70%, and 76% inactivation could be achieved. This prompted us to investigate whether YL209 itself was stable under the conditions used. TLC analysis of the probe incubated in PBS over time however showed no degradation of the probe (ESI Fig. 2†). We next investigated whether the YL209 covalent intermediate with the enzyme was hydrolyzing at an appreciable rate, thereby reactivating the enzyme. This would result in activity reaching an equilibrium based on competing rates of inactivation by YL209 and enzyme reactivation, and thus explaining the incomplete inactivation. BfFucH was incubated with excess YL209 and initial hydrolysis rates were determined using the 4-nitrophenyl-α-L-fucopyranoside after excess inhibitor was removed (Fig. 1B). Time-dependent reactivation of a glycosidase that had been inactivated with a similar difluoro glycoside based probe was described before.45 Similar to what was described by Kim and co-workers we observed an increase of the initial hydrolysis rate over time, according to a hyperbolic function, suggesting turnover of the enzyme-intermediate playing a role in the earlier observed equilibrium.
 | ||
Fig. 1 Time-dependent inactivation of BfFucH. (A) Experimental data represents the BfFucH activity with YL209: 0 ( ), 5 ( ), 5 ( ), 10 ( ), 10 ( ), 20 ( ), 20 ( ), 40 ( ), 40 ( ), 80 ( ), 80 ( ) mM. Curves are the nonlinear fits of data to a single-exponential decay equation. (B) Reactivation kinetics for covalently inhibited BfFucH, control samples (absence of inhibitor) ( ) mM. Curves are the nonlinear fits of data to a single-exponential decay equation. (B) Reactivation kinetics for covalently inhibited BfFucH, control samples (absence of inhibitor) ( ), pre-incubated with 50 mM YL209 ( ), pre-incubated with 50 mM YL209 ( ). (C) Plot of the inactivation rate constant (kobs) as function of YL209 for BfFucH. (D) Plot of the inactivation rate constant (kobs) as function of 2FFucF for BfFucH. ). (C) Plot of the inactivation rate constant (kobs) as function of YL209 for BfFucH. (D) Plot of the inactivation rate constant (kobs) as function of 2FFucF for BfFucH. | ||
We next investigated the inactivation kinetics at several concentrations of YL209 and the kinetic parameters (kinact and Ki) were determined by plotting kobsversus [I] (Fig. 1C). Inactivation of BfFucH by YL209 exhibited a good fit to all data by non-linear regression using the equation kobs = kinact/(1 + Ki/[I]), from which a Ki value of 9.6 ± 2.7 mM and a kinact value of 0.029 ± 0.002 min−1 could be deduced. We next determined the Ki and kinact for the related known inactivator, 2FFucF, to be respectively 26.4 ± 10.1 mM and 0.009 ± 0.001 min−1. The inhibition constant Ki for YL209 shows it to be a more potent inhibitor compared to 2FFucF, as the concentration needed to produce half maximum inhibition is lower. This is supported by the kinact value indicating a higher rate of inactivation for YL209 compared to 2FFucF. The second-order rate constant for ABP YL209 (kinact/Ki = 3.0 ± 0.003 M−1 min−1) that governs inactivation compared well to 2FFucF (kinact/Ki = 0.3 ± 0.010 M−1 min−1) (Fig. 1D). The azide mini-tag at C-6 thus appears to have a beneficial effect on the inactivation process. These experiments show that YL209 inactivates BfFucH on a time scale appropriate for practical applications and suggests that this compound can be used to detect GH29 fucosidases in more complex samples.
To explore the practical possibilities of the probe to detect fucosidases, we incubated the recombinant BfFucH with YL209 overnight at 37 °C followed by SDS PAGE gel and electrophoretic transfer to a nitrocellulose blot. For the visualization of the ABP-BfFucH signal we conducted the functionalization with an alkyne-biotin on the surface of the nitrocellulose blot. This method has previously been shown to have similar sensitivity compared to regular imaging methods and it eliminates the need for labor intensive separation steps to remove excess of probe before attachment of the visualization handle.46 Incubation of YL209 with BfFucH followed by SDS-PAGE and Western Blot analysis revealed no signal when normal electrophoresis protocols were followed. It has previously been discussed in literature that this result can occur due to hydrolysis of the ester linkage of the glycosyl–enzyme intermediate under the basic conditions (pH 8.8) of the buffered gel.33 A short electrophoresis protocol (90 V for 20 minutes and 200 V for 8 minutes) to circumvent this was developed by Stubbs et al. However, this protocol was not reproducible in our hands and we therefore selected a different gel system with a more favorable running pH. In contrast to the conventional Tris-glycine SDS-PAGE gels, the BIS-Tris gels have a running pH of 7.2 and this near neutral pH diminishes the reactivity of amino and sulfhydryl groups.47
A BIS-TRIS MOPS gel was used in conjunction with the Western Blot analysis above and on blot click reaction showed that these adjusted conditions proved successful for labelling of BfFucH (Fig. 2A). To confirm that the alkyne-biotin was reacting specifically with the azide mini-tag of the BfFucH inactivated with YL209 and not aspecifically with the protein itself, various controls were performed (Fig. 2A and B). The results from these controls show that the inactivation and subsequent biotin CuAAc are highly specific and neither reagent on its own results in labelling of the enzyme. Incubating BfFucH with YL209 revealed the lower limit of detection of BfFuc to be approximately 300 ng (Fig. 2C). This is in a similar range as was previously reported for another 2-deoxy-2-fluoro-glycoside ABP.34
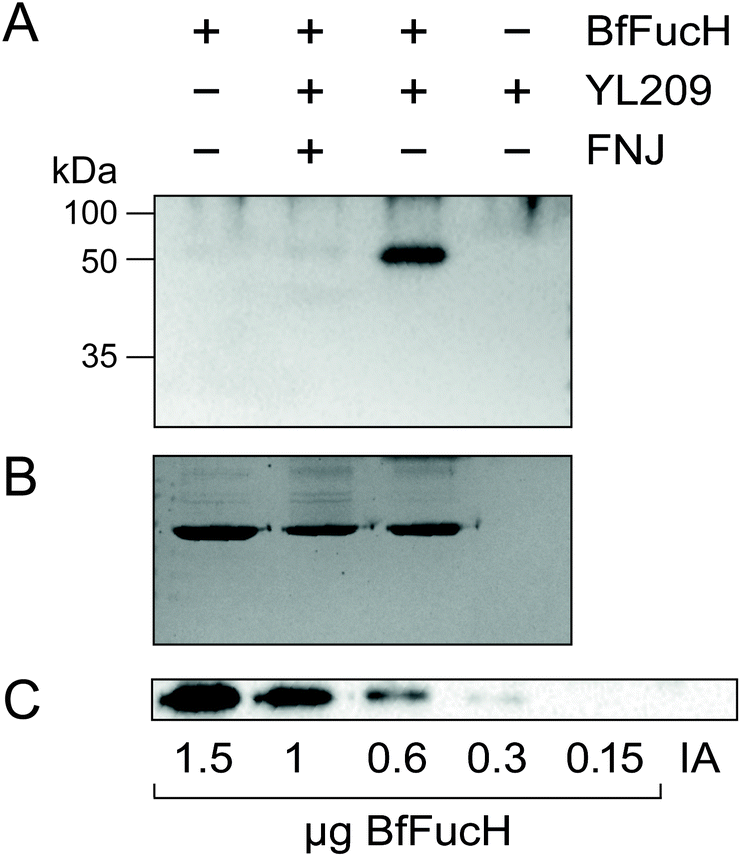 | ||
| Fig. 2 Visualization of YL209 labelled BfFucH. After inactivation, the samples were run on BIS-TRIS, blotted on nitrocellulose, labelled by CuAAC on blot with 100 μM alkyne-biotin and analyzed using anti-biotin-HRP. (A) TRIS-BIS-western blot with the appropriate controls. (B) PageBlue stained TRIS-BIS gels, showing equal loading of lanes. (C) Detection limit of YL209 for specified amount of BfFucH; IA: 1.5 μg BfFucH pre incubated with 100 μM FNJ. | ||
To demonstrate the versatility of our approach we applied YL209 to two commercially available recombinant GH29 fucosidases. One GH29 fucosidase from a bacterial source, Thermotoga maritima (E-FUCTM) with specificity for α-(1,2)-L-fucoside residues and one from a homo sapiens source (E-FUCHS) with a broad specificity for α-(1-2,3,4,6)-L-fucoside residues. In both cases we observed specific labeling of the enzymes when incubated with YL209 (Fig. 3). Noticeable, for E-FUCTM we also see specific labelling of a higher running spot that might correspond to the naturally occurring hexamer conformation of this enzyme.48 These results shows that our probe is capable of labeling GH29 fucosidases independent of linkage specificity.
 | ||
| Fig. 3 Labelling of three GH29 fucosidases by using YL209 in conjunction with in-blot CuAAC. (A) Western blot analysis of samples of recombinant BfFucH (Lane 1), Homo Sapiens fucosidase (Lane 2), Thermotoga maritima fucosidase (Lane 3). (B) TRIS-BIS PageBlue (PB) analysis of the samples shown in panel A. | ||
Having established the activity of YL209, we looked further into future applications of this probe such as in fucosidase identification in microbiota members. To expand the potential applicability of YL209 we validated the in-solution labeling of the probe–enzyme complex with a reporter, as opposed to on-blot labeling that we used up to this point. We incubated purified BfFucH with YL209 overnight at 37 °C. The sample was dialyzed to remove excess probe and the protein was denatured under mild reducing conditions at pH 3.5 so that both the azide moiety and the ester linkage of the covalent intermediate remained intact. Treatment of the mixture with alkyne-biotin under CuI catalysis followed by western blot analysis revealed a clear selective labelling of the BfFucH enzyme (Fig. 4). In a control we observed that labeling could be lowered by the presence of the active site competitor iminosugar FNJ during incubation with the probe.
 | ||
| Fig. 4 Western Blot analysis of BfFucH inactivated with YL209 in conjunction with in-solution CuAAC. (A) TRIS-BIS-western blot with the appropriate controls. (B) TRIS-BIS PageBlue analysis of the samples shown in (A). | ||
Conclusions
We have successfully synthesized an α-L-fucosidase targeting ABP and developed a strategy to label active retaining fucosidases by using an azide on C6 as a small robust chemical mini-tag. We have demonstrated that the approach can be used to detect several glycosidases with different substrate specificities and with on-blot as well as in-solution labelling. Furthermore, we believe that the readily accessible synthesis described here for YL209 will pave the way for further investigations into the effect of different C1 or C6 modifications on labeling sensitivity and specificity for α-L-fucosidases. In particular, the leaving group on C1 of the 2-deoxy-2-fluorinated glycosides has previously shown to play an essential role in the effectiveness of these molecules as ABPs, although our attempted installation of an anomeric N-phenyltrifluoroacetimidate proved not possible in this case. Together, these properties make this ABP a suitable candidate to further derivatise, identify and study bacterial fucosidases of pathogenic and commensal bacteria in future studies.Experimental
General methods
All chemicals were obtained from commercial suppliers and used as received unless stated otherwise. Organic solvents for reactions were dried for at least 2 days over molecular sieves (3 or 4 Å). 1H and 13C NMR spectra were recorded on either an Agilent 400 instrument (400 and 101 MHz) or a Bruker Avance Neo 600 spectrometer (600 and 125 MHz). Chemical shifts are reported in δ values relative to TMS and J coupling constants are reported in Hertz (Hz). Silica column chromatography was performed using silica gel SiliaFlash P60 (SiliCycle, Canada, 40–63 μm, 239–400 mesh). TLC analysis was conducted on SiliaPlate TLC Aluminium Backed TLC F254 (SiliCycle) with examination under UV light (254 nm) where applicable, and with 5% sulfuric acid in ethanol or with ceric ammonium molybdate, followed by heating. 2-Deoxy-2-fluoro-α-L-fucosyl fluoride (2FFucF) was synthesized according to known methodology.421,2,3,4-Di-O-isoproylidene-α-L-galactopyranose
To a solution of ZnCl2 (800 mg, 5.92 mmol, 2.1 equiv.) in acetone (15 mL) was added catalytic H2SO4 (27 μL, 0.5 mmol) and L-galactose (500 mg, 2.80 mmol, 1 equiv.) at room temperature. The mixture was stirred at room temperature for 5 h, and then quenched with sat. aq. NaHCO3. The resulting suspension was filtrated over a pad of Celite that was washed twice with acetone. The filtrate was concentrated under reduced pressure and extracted with diethylether. The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by silica flash chromatography (7/3 = PE/EtOAc) to afford 1 (620 mg, 2.38 mmol) as a colorless oil in a 85% yield.R f = (PE/EtOAc: 8/2 = 0.14); 1H NMR (400 MHz, CDCl3) δ 5.51 (d, J = 5.0 Hz, 1H), 4.56 (dd, J = 7.9, 2.4 Hz, 1H), 4.28 (dd, J = 5.0, 2.4 Hz, 1H), 4.22 (dd, J = 8.0, 1.8 Hz, 1H), 3.90–3.54 (m, 3H), 1.48 (s, 3H), 1.40 (s, 3H), 1.28 (d, J = 1.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 109.83, 109.03, 96.65, 71.97, 71.12, 70.93, 68.42, 62.71, 26.37, 26.28, 25.28, 24.65.
Identical with reported data.49
1,2,3,4-Di-O-isoproylidene-6-O-(p-toluenesulfonyl)-α-L-galactopyranose
A solution of p-toluenesulfonyl chloride (650 mg, 3.41 mmol, 1.8 equiv.) in DCM (1 mL) was added to a cooled (−5 °C) solution of 1 (500 mg, 1.92 mmol, 1 equiv.) and 4-(dimethylamino)pyridine (40 mg, 0.34 mmol, 0.18 equiv.) in anhydrous pyridine (5 mL). The solution was warmed to room temperature and stirred overnight. The solvent was removed under reduced pressure and the residue dissolved in water and extracted with EtOAc (three times). The organic layer was washed with 0.1 M HCl, saturated bicarbonate, and brine, and dried over sodium sulfate, filtered and evaporated. The residual syrup was purified by silica flash chromatography (8/2 = PE/EtOAc) to afford 2 (685 mg, 1.65 mmol) as a yellow oil in a 86% yield.R f = (PE/EtOAc: 8/2 = 0.25); 1H NMR (400 MHz, CDCl3) δ 7.84–7.30 (m, 4H), 5.45 (d, J = 4.9 Hz, 1H), 4.59 (dd, J = 7.9, 2.6 Hz, 1H), 4.29 (dd, J = 5.0, 2.5 Hz, 1H), 4.23–4.19 (m, 2H), 4.10–4.01 (m, 2H), 2.44 (s, 3H), 1.50 (s, 3H), 1.34 (s, 3H), 1.31 (s, 3H), 1.28 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 145.08, 133.20, 130.08, 128.47, 109.92, 109.29, 96.47, 70.87, 70.75, 70.72, 68.52, 66.21, 26.32, 26.15, 25.26, 24.69, 21.97. HR MS (m/z) calcd for C19H26O8S [M + Na]+, 437.1192; found, 437.1243.
Identical with reported data.50
1,2,3,4-Tetra-O-acetyl-6-O-(p-toluenesulfonyl)-α-L-galactopyranose
Compound 2 (800 mg, 1.93 mmol, 1 equiv.) was dissolved in DCM (2 mL) and added dropwise to a vigorously stirred mixture of H2O and TFA (v/v, 10 mL). The reaction mixture was stirred for 1 h at room temperature, after which the solvents were evaporated under reduced pressure and co evaporation with toluene. Dry pyridine (5 mL) and dry acetic anhydride (5 mL) were added to the residual syrup and this reaction mixture was stirred at room temperature for 16h. The solvents were removed under reduced pressure and the resulting residue was dissolved in CH2Cl2. The organic layer was washed with water and brine, dried over sodium sulfate, filtered and concentrated. The residual syrup was purified by silica flash chromatography (PE/EtOAc = 7/3) to afford 3 (892 mg, 1.78 mmol) as a yellow oil in 92% yield over 2 steps.R f = (PE/EtOAc:7/3 = 0.19); 1H NMR (400 MHz, CDCl3) δ 7.82–7.70 (m, 2H), 7.37–7.31 (m, 2H), 5.65 (dd, J = 8.3, 1.2 Hz, 1H), 5.40 (dd, J = 2.9, 1.7 Hz, 1H), 5.32–5.21 (m, 1H), 5.03 (ddd, J = 10.4, 3.4, 1.2 Hz, 1H), 4.14–4.07 (m, 1H), 4.06–3.97 (m, 2H), 2.45 (s, 3H), 2.10 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.16, 170.11, 169.67, 169.11, 145.64, 132.56, 130.32, 130.28, 128.45, 92.38, 71.63, 70.91, 67.94, 66.75, 65.63, 22.02, 21.08, 20.96, 20.84, 20.82.
3,4-Di-O-acetyl-6-O-(p-toluenesulfonyl)-L-galactal
HBr (37% in AcOH, 2 mL) was added dropwise to a cooled (−5 °C) solution of 3 (850 mg, 1.69 mmol, 1 equiv.) in DCM (2 mL). The reaction was stirred for 1 h at room temperature after which it was quenched by pouring into ice water. This mixture was extracted with EtOAc (three times). And the combined organic layers were washed with sat. aq. NaHCO3, brine, dried over sodium sulfate, filtered and concentrated. The residual crude syrup was dissolved in acetone (5 mL). Sat. aq. NaH2PO4 (7 mL) and Zn (dust, <10 μm, ≥98%) (1.6 gr, 24.47 mmol, 24 equiv.) were added to the mixture and stirred for 3 h at room temperature. The suspension was filtrated over a pad of Celite, the Celite was washed twice with acetone. The filtrate was concentrated under reduced pressure to remove excess acetone and dissolved in EtOAc. The combined organic layers were washed with NaHCO3, brine, dried over sodium sulfate and concentrated to obtain 4 in 77% yield over 2 steps. The product 4 was used without further purification.R f = (PE/EtOAc:8/2 = 0.16); 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.39–7.30 (m, 2H), 6.33 (dd, J = 6.2, 1.7 Hz, 1H), 5.46 (ddd, J = 4.5, 2.2, 1.1 Hz, 1H), 5.35 (dd, J = 3.6, 1.9 Hz, 1H), 4.70 (ddd, J = 6.3, 3.1, 1.2 Hz, 1H), 4.35–4.04 (m, 3H), 2.44 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 145.14, 129.89, 128.02, 98.85, 72.43, 67.02, 66.65, 63.69, 63.33, 21.64, 20.51, 20.51. HR MS (m/z) calcd for C17H20O8S [M + Na]+, 407.0792; found, 407.0764.
3,4-Di-O-acetyl-6-azido-α-L-galactal
NaN3 (845 mg, 13 mmol, 10 equiv.) was added to a solution of 4 (500 mg, 1.30 mmol, 1 equiv.) in dry DMF (5 mL) and stirred at 90 °C, with a condenser attached to the reaction vessel, until TLC showed complete conversion. H2O and EtOAc were added. The water layer was washed three times more with EtOAc. The organic layers were concentrated under reduced pressure and purified by silica flash chromatography (PE/EtOAc = 9/1) to afford 5 (315 mg, 1.24 mmol) as an colorless oil in a 95% yield.R f = (PE/EtOAc:8/2 = 0.5); 1H NMR (400 MHz, CDCl3) δ 6.48 (d, J = 6.2 Hz, 1H), 5.55 (dd, J = 2.9, 1.5 Hz, 1H), 5.43–5.33 (m, 1H), 4.77–4.73 (m, 1H), 4.25–4.15 (m, 1H), 3.69–3.27 (m, 2H), 2.14 (s, 3H), 2.03 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.17, 170.07, 145.35, 98.93, 74.08, 64.29, 63.95, 50.53, 20.76, 20.62. HR MS (m/z) calcd for C10H13N3O5 [M + Na]+, 278.0792; found, 278.0763.
3,4-Di-O-acetyl-6-azido-2-fluoro-α-L-galactopyranosyl fluoride
A solution of 5 (560 mg, 2.19 mmol, 1 equiv.) in dry benzene (20 mL) was added dropwise (over 5 minutes) to a stirred and cooled (−5/−10 °C) solution of XeF2 (320 mg, 1.89 mmol, 0.9 equiv.) and BF3-etherate in dry ether (270 μL, 2.19 mmol, 1 equiv.). The solution was allowed to warm up to room temperature and stirred for another 2 h. The solution was washed with sat. aq. NaHCO3. After which the aqueous layers were extracted with ether. The combined organic layers were washed with H2O, dried over sodium sulfate and concentrated. The concentrate was subsequently purified by silica flash chromatography (DCM/ether = 98/2) to afford 6 (366 mg, 1.25 mmol) as a white solid in 57% yield.R f = (DCM/Ether:98/2 = 0.45); 1H NMR (400 MHz, CDCl3) δ 5.86 (dd, J = 53.0, 2.9 Hz, 1H), 5.51 (d, J = 3.5 Hz, 1H), 5.44–5.36 (m, 1H), 4.94–4.60 (m, 1H), 4.31 (dddd, J = 7.5, 5.3, 1.5, 0.8 Hz, 1H), 3.53–3.17 (m, 2H), 2.16 (s, 3H), 2.05 (s, 3H).13C NMR (101 MHz, CDCl3) δ 105.02, 103.48, 85.63, 84.19, 70.49, 68.70, 67.71, 50.37, 20.77, 20.70. 19F NMR (400 MHz, CDCl3) δ −152.03, −211.06. HR MS (m/z) calcd for C10H13F2N3O5 [M + Na]+, 316.0715; found, 316.0714.
6-Azido-2,6-dideoxy-2-fluoro-α-L-galactopyranosyl fluoride
To a cooled solution of 6 (366 mg, 1.25 mmol, 1 equiv.) in MeOH (10 mL) NaOMe was added dropwise. The reaction was stirred for 2 h at room temperature. Amberlyte H + resins were added until pH = 7. The solution was filtrated and solvents were evaporated under reduced pressure. After which it was purified by silica flash chromatography (DCM/ether = 9/1) to afford YL209 as a colorless oil. Lyophilization was performed to obtain YL209 (209 mg, mmol) as a white solid in 80% yield.R f = (DCM/ether:9/1 = 0.31); 1H NMR (600 MHz, MeOD) δ 5.81 (dd, J = 54.3, 2.9 Hz, 1H), 4.66 (dddd, J = 48.9, 24.5, 9.9, 3.0 Hz, 1H), 4.12 (ddd, J = 8.2, 4.8, 1.3 Hz, 1H), 4.02 (ddd, J = 12.7, 9.8, 3.4 Hz, 1H), 3.96 (td, J = 3.8, 1.3 Hz, 1H), 3.64–3.36 (m, 2H). 13C NMR (151 MHz, MeOD) δ 105.54, 88.77, 72.31, 69.78, 67.40, 50.69. 19F NMR (400 MHz, MeOD) δ −153.33, −214.63. HR MS (m/z) calcd for C6H9F2N3O3 [M + Na]+, 232.0504; found, 232.0595.
Inactivation kinetics
The time-dependent inactivation of BfFucH by YL209 and 2FFucF was monitored measuring the residual enzyme activity over time. This was accomplished by incubation of the enzyme (0.031 mg mL−1 BfFucH) in 100 μL of PBS buffer (+0.1% BSA) containing either inactivator YL209 or 2FFucF. Five inactivation concentrations (0, 5, 10, 20, 40, 80 mM) were used. The control mixtures contained the same amount of enzyme but no inactivator. Both inactivation and control samples were incubated at room temperature and at several time intervals an aliquot of each inactivation mixture was added to a solution of the substrate, 4-nitrophenyl α-L-fucopyranoside in PBS, so that the final assay contained BfFucH at a concentration of 3.1 μg mL−1 and 2.2 mM substrate (5 times Km) in PBS;51 the total volume was 100 μL. The initial rates were measured under steady-state conditions by spectrophotometric monitoring of the release of 4-nitrophenolate at 400 nm. Pseudo first order rate constants (kobs) were determined by fitting the decay curve to a single exponential decay equation using Graphpad PRISM 8. The first and second order rate constants (kinact and kinact/Ki) were determined by fitting the pseudo first order rate constants versus the inactivator concentrations data to the equation: kobs = kinact/(1 + Ki/[I]).Reactivation kinetics
The time-dependent reactivation of BfFucH inactivated by YLl209 was monitored measuring the residual enzyme activity over time. This was accomplished by incubation of the enzyme (0.031 mg mL−1, 620 nM BfFucH) in 100 μL of PBS buffer (+0.1% BSA) containing inactivator YL209 (50 mM final concentration) at 37 °C for 3 h so that 75% inactivation had occurred. The control mixtures contained the same amount of enzyme but no inactivator. Excess inactivator was then removed by extensive dialysis at 4 °C against large volumes of PBS using a microconcentrator (Pierce, microconcentrator 10.000 MWCO). Reactivation was monitored by addition of an aliquot, at specific time intervals, to a solution of the substrate, 4-nitrophenyl α-L-fucopyranoside in PBS, so that the final assay contained BfFucH at a concentration of 3.1 μg mL−1 and 2.2 mM substrate in PBS with a total volume of 100 μL. The initial rates were measured under steady-state conditions by spectrophotometric monitoring of the release of 4-nitrophenolate at 400 nm. The curve was fitted to a single exponential association equation using Graphpad PRISM 8.Chemoselective labelling on nitrocellulose blot using alkyne-biotin
An aliquot of the enzyme (final [BfFucH] = 100 μg mL−1, 2 μM) was treated with a solution of YL209 in PBS (final [YL209] = 50 mM) and incubated overnight at 37 °C. The commercially available fucosidases Thermotoga maritima (E-FUCTM, Megazyme) and homo sapiens (E-FUCHS, Megazyme) were analyzed in the same way as the recombinant BfFucH. The samples were mixed with 3× loading buffer and the samples were loaded without heating onto a BIS-TRIS polyacrylamide gel. After a short electrophoresis of 20 minutes at 200V the samples were blotted to a nitrocellulose membrane (TransBlot Turbo Transfer System, Biorad). Allowing full protein transfer within 7 minutes to limit exposure to the basic transfer buffer. The membrane was washed for 2 minutes in PBS after which it was incubated for 2h at room temperature in the presence of 100 μM alkyne-biotin, 2.5 mM L-ascorbate and 0.5 mM Cu2SO4 in PBS. The click reaction was followed by 2 times 5 minutes wash with PBS containing 0.1% Tween-20 (PBS-T). The membrane was blocked using 5% skimmed milk in PBS-T with rocking for 1 h at room temperature. The blocking solution was decanted and a solution 1% skimmed milk in PBS-T containing αbiotin-HRP (1:10.000, Jackson ImmunoResearch) was added and incubated for 1h at room temperature. Membranes were washed for 3 × 10 minutes with PBS-T. Detection of membrane-bound αbiotin-HRP conjugates was accomplished by chemiluminescent detection using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and imaged in a Gel-Doc system (Bio-Rad).
Chemoselective labeling in solution using alkyne-biotin
An aliquot of the enzyme (final [BfFucH] = 100 μg mL−1, 2 μM) was treated with a solution of YL209 in PBS (final [YL209] = 50 mM) and incubated overnight at 37 °C. The inactivated samples were then dialysed overnight (Pierce Slide-A-lyzer Mini dialysis unit, 10.000 MWCO) at 4 °C. After dialysis solutions of 1 M pH 2.0 sodium phosphate and saturated urea (one third of the volume) were added to yield a final pH of 3.5. One volume of alkyne-biotin in water (100 μM final concentration) with 2.5 mM L-ascorbate and 0.5 mM Cu2SO4 was added to the mixture and incubated overnight at RT. A microconcentrator (Pierce, microconcentrator 10.000 MWCO) was pre-washed with 0.1% PBS-T and centrifuged 15 minutes 15000g. This was continued by the concentration of the sample in said microconcentrator to a final volume of 50 μL. The mixture was mixed with 3× laemlli buffer before loading 20 μL on a TRIS-BIS polyacrylamide gel. After electrophoresis, the gel was blotted to a nitrocellulose membrane (TransBlot Turbo Transfer System, Biorad). The membrane was blocked using 5% skimmed milk in PBS-T with rocking for 1 h at room temperature. The blocking solution was decanted and a solution 1% skimmed milk in PBS-T containing αbiotin-HRP (1:10000, Jackson ImmunoResearch) was added and incubated for 1 h at room temperature. Membranes were washed for 3 × 10 minutes with PBS-T. Detection of membrane bound αbiotin-HRP conjugates was accomplished by chemiluminescent detection using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and imaged in a Gel-Doc system (Bio-Rad).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from The Netherlands Organization for Scientific Research (NWO, VIDI grant 723.014.005, to T. W.). K. Strijbis has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (ERC-2019-STG 852452). We would like to thank Gerlof Bosman for the expression of the recombinant protein BfFucH.References
- J. Intra, M. E. Perotti, G. Pavesi and D. Horner, Gene, 2007, 392, 34–46 CrossRef PubMed.
- K. M. Ng, J. A. Ferreyra, S. K. Higginbottom, J. B. Lynch, P. C. Kashyap, S. Gopinath, N. Naidu, B. Choudhury, B. C. Weimer, D. M. Monack and J. L. Sonnenburg, Nature, 2013, 502, 96–99 CrossRef PubMed.
- J. M. Garber, H. Nothaft, B. Pluvinage, M. Stahl, X. Bian, S. Porfirio, A. Enriquez, J. Butcher, H. Huang, J. Glushka, E. Line, J. A. Gerlt, P. Azadi, A. Stintzi, A. B. Boraston and C. M. Szymanski, Commun. Biol., 2020, 3, 1–11 CrossRef PubMed.
- F. A. Shaikh, A. Lammerts van Bueren, G. J. Davies and S. G. Withers, Biochemistry, 2013, 52, 5857–5864 CrossRef PubMed.
- R. A. DiCioccio, J. J. Barlow and K. L. Matta, J. Biol. Chem., 1982, 257, 714–718 CrossRef.
- H. Sakurama, E. Tsutsumi, H. Ashida, T. Katayama, K. Yamamoto and H. Kumagai, Biosci. Biotechnol. Biochem., 2012, 76, 1022–1024 CrossRef PubMed.
- D. A. Sela, D. Garrido, L. Lerno, S. Wu, K. Tan, H. J. Eom, A. Joachimiak, C. B. Lebrilla and D. A. Mills, Appl. Environ. Microbiol., 2012, 78, 795–803 CrossRef PubMed.
- J. M. Pickard and A. V. Chervonsky, J. Immunol., 2015, 194, 5588–5593 CrossRef PubMed.
- H. Wu, O. Rebello, E. H. Crost, C. D. Owen, S. Walpole, C. Bennati-Granier, D. Ndeh, S. Monaco, T. Hicks, A. Colvile, P. A. Urbanowicz, M. A. Walsh, J. Angulo, D. I. R. Spencer and N. Juge, Cell. Mol. Life Sci., 2020, 1, 3 Search PubMed.
- M. T. Sorbara and E. G. Pamer, Mucosal Immunol., 2019, 12, 1 CrossRef CAS.
- M. Stahl, L. M. Friis, H. Nothaft, X. Liu, J. Li, C. M. Szymanski and A. Stintzi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7194–7199 CrossRef CAS.
- Y. M. C. A. Luijkx, N. M. C. Bleumink, J. Jiang, H. S. Overkleeft, M. M. S. M. Wösten, K. Strijbis and T. Wennekes, Cell. Microbiol., 2020, 22, e13252 CrossRef CAS.
- F. C. Pereira and D. Berry, Environ. Microbiol., 2017, 19, 1366–1378 CrossRef.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS.
- H. Hinou, M. Kurogochi, H. Shimizu and S. I. Nishimura, Biochemistry, 2005, 44, 11669–11675 CrossRef CAS.
- M. Kurogochi, S. I. Nishimura and Y. C. Lee, J. Biol. Chem., 2004, 279, 44704–44712 CrossRef CAS.
- M. Nandakumar, Y. L. Hsu, J. C. Y. Lin, C. Lo, L. C. Lo and C. H. Lin, ChemBioChem, 2015, 16, 1555–1559 CrossRef CAS.
- Y. L. Hsu, M. Nandakumar, H. Y. Lai, T. C. Chou, C. Y. Chu, C. H. Lin and L. C. Lo, J. Org. Chem., 2015, 80, 8458–8463 CrossRef CAS.
- T. J. Sminia, H. Zuilhof and T. Wennekes, Carbohydr. Res., 2016, 435, 121–141 CrossRef CAS PubMed.
- B. P. Rempel and S. G. Withers, Glycobiology, 2008, 18, 570–586 CrossRef CAS.
- L. I. Willems, J. Jiang, K. Y. Li, M. D. Witte, W. W. Kallemeijn, T. J. N. Beenakker, S. P. Schröder, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chem. – Eur. J., 2014, 20, 10864–10872 CrossRef CAS.
- J. Jiang, W. W. Kallemeijn, D. W. Wright, A. M. C. H. C. H. van den Nieuwendijk, V. Coco Rohde, E. Colomina Folch, H. van den Elst, B. I. Florea, S. Scheij, W. E. Donker-Koopman, M. Verhoek, N. Li, M. Schürmann, D. Mink, R. G. Boot, J. D. C. C. Codée, G. A. van der Marel, G. J. Davies, J. M. F. G. Aerts and H. S. Overkleeft, Chem. Sci., 2015, 6, 2782–2789 RSC.
- J. Jiang, C. L. Kuo, L. Wu, C. Franke, W. W. Kallemeijn, B. I. Florea, E. van Meel, G. A. van der Marel, J. D. C. Codée, R. G. Boot, G. J. Davies, H. S. Overkleeft and J. M. F. G. Aerts, ACS Cent. Sci., 2016, 2, 351–358 CrossRef CAS.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C. L. Kuo, M. Artola, W. Dai, C. van Elk, M. van Eijk, G. A. van der Marel, J. D. C. Codée, B. I. Florea, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 CrossRef CAS.
- T. M. Gloster, R. Madsen and G. J. Davies, Org. Biomol. Chem., 2007, 5, 444–446 RSC.
- S. G. Withers, K. Rupitz and I. P. Street, J. Biol. Chem., 1988, 263, 7929–7932 CrossRef CAS.
- S. J. Williams and S. G. Withers, Carbohydr. Res., 2000, 327, 27–46 CrossRef CAS.
- M. T. C. Walvoort, G. A. van der Marel, H. S. Overkleeft and J. D. C. Codée, Chem. Sci., 2013, 4, 897–906 RSC.
- N. Xu, Y. Lin, S. A. Hofstadler, D. Matson, C. J. Call, B. Ren, E. M. Mosi and S. G. Withers, 64 Enzyme Kinetics and Mechanism [4] [4] Trappingof β-GlycosidaseIntermediates, 1998, vol. 70 Search PubMed.
- S. S. Macdonald, A. Patel, V. L. C. Larmour, C. Morgan-Lang, S. J. Hallam, B. L. Mark and S. G. Withers, J. Biol. Chem., 2018, 293, 3451–3467 CrossRef CAS.
- J. D. McCarter, M. J. Adam, N. G. Hartman and S. G. Withers, Biochem. J., 1994, 301, 343–348 CrossRef CAS.
- J. D. McCarter, M. J. Adam and S. G. Withers, J. Labelled Compd. Radiopharm., 1992, 31, 1005–1009 CrossRef CAS.
- K. A. Stubbs, A. Scaffidi, A. W. Debowski, B. L. Mark, R. V. Stick and D. J. Vocadlo, J. Am. Chem. Soc., 2008, 130, 327–335 CrossRef CAS.
- D. J. Vocadlo and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 5338–5342 CrossRef CAS PubMed.
- O. Hekmat, Y. W. Kim, S. J. Williams, S. He and S. G. Withers, J. Biol. Chem., 2005, 280, 35126–35135 CrossRef CAS.
- S. J. Williams, O. Hekmat and S. G. Withers, ChemBioChem, 2006, 7, 116–124 CrossRef CAS.
- C. A. Tarling, S. He, G. Sulzenbacher, C. Bignon, Y. Bourne, B. Henrissat and S. G. Withers, J. Biol. Chem., 2003, 278, 47394–47399 CrossRef CAS.
- T. W. Liu, C. W. Ho, H. H. Huang, S. M. Chang, S. D. Popat, Y. T. Wang, M. S. Wu, Y. J. Chen and C. H. Lin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14581–14586 CrossRef CAS PubMed.
- J. D. McCarter, M. J. Adam and S. G. Withers, Biochem. J., 1992, 286, 721–727 CrossRef CAS PubMed.
- U. Grabowska, D. A. MacManus, K. Biggadike, M. I. Bird, S. Davies, T. Gallagher, L. D. Hall and E. N. Vulfson, Carbohydr. Res., 1997, 305, 351–361 CrossRef CAS.
- D. A. MacManus, U. Grabowska, K. Biggadike, M. I. Bird, S. Davies, E. N. Vulfson and T. Gallagher, J. Chem. Soc., Perkin Trans. 1, 1999, 295–305 RSC.
- W. Korytnyk, S. Valentekovic-Horvath and C. R. Petrie, Tetrahedron, 1982, 38, 2547–2550 CrossRef CAS.
- C. D. Jeroen, C. Codée, H. S. Overkleeft, M. T. C. Walvoort, W. W. Kallemeijn, L. I. Willems, M. D. Witte, J. M. F. G. Aerts, G. A. van der Marel and J. D. C. Codeé, Chem. Commun., 2012, 4848841, 10357–10456 Search PubMed.
- H. H. Jensen and M. Bols, Org. Lett., 2003, 5, 3419–3421 CrossRef CAS.
- J. H. Kim, R. Resende, T. Wennekes, H. M. Chen, N. Bance, S. Buchini, A. G. Watts, P. Pilling, V. A. Streltsov, M. Petric, R. Liggins, S. Barrett, J. L. McKimm-Breschkin, M. Niikura and S. G. Withers, Science, 2013, 340, 71–75 CrossRef CAS.
- J. Ohata, F. Vohidov and Z. T. Ball, Mol. Biosyst., 2015, 11, 2846–2849 RSC.
- J. P. Hachmann and J. W. Amshey, Anal. Biochem., 2005, 342, 237–245 CrossRef CAS.
- G. Sulzenbacher, C. Bignon, T. Nishimura, C. A. Tarling, S. G. Withers, B. Henrissat and Y. Bourne, J. Biol. Chem., 2004, 279, 13119–13128 CrossRef CAS PubMed.
- B. Doboszewski and P. Herdewijn, Tetrahedron Lett., 2012, 53, 2253–2256 CrossRef CAS.
- N. Lunau, K. Seelhorst, S. Kahl, K. Tscherch, C. Stacke, S. Rohn, J. Thiem, U. Hahn and C. Meier, Chem. – Eur. J., 2013, 19, 17379–17390 CrossRef CAS.
- T. I. Tsai, S. T. Li, C. P. Liu, K. Y. Chen, S. S. Shivatare, C. W. Lin, S. F. Liao, C. W. Lin, T. L. Hsu, Y. T. Wu, M. H. Tsai, M. Y. Lai, N. H. Lin, C. Y. Wu and C. H. Wong, ACS Chem. Biol., 2017, 12, 63–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00054c |
| This journal is © The Royal Society of Chemistry 2021 |