
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-fused dimethoxybenzimidazole-benzimidazolequinone (DMBBQ): tunable halogenation and quinone formation using NaX/Oxone†
Darren
Conboy
 a,
Patrick
Kielty
b,
Joseph C.
Bear
c,
Jeremy K.
Cockcroft
d,
Pau
Farràs
b,
Patrick
McArdle
b,
Richard J.
Singer
c,
Dennis A.
Smith
b and
Fawaz
Aldabbagh‡
*ab
a,
Patrick
Kielty
b,
Joseph C.
Bear
c,
Jeremy K.
Cockcroft
d,
Pau
Farràs
b,
Patrick
McArdle
b,
Richard J.
Singer
c,
Dennis A.
Smith
b and
Fawaz
Aldabbagh‡
*ab
aDepartment of Pharmacy, School of Life Sciences, Pharmacy and Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames, KT1 2EE, UK. E-mail: F.Aldabbagh@kingston.ac.uk
bSchool of Chemistry, National University of Ireland Galway, University Road, Galway, H91 CF50, Ireland
cDepartment of Chemical and Pharmaceutical Sciences, School of Life Sciences, Pharmacy and Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames, KT1 2EE, UK
dDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 26th February 2021
Abstract
Ring-fused benzimidazolequinones are well-known anti-tumour agents, but dimeric ring-fused adducts are new. The alicyclic [1,2-a] ring-fused dimethoxybenzimidazole-benzimidazolequinone (DMBBQ) intermediate allows late-stage functionalization of bis-p-benzimidazolequinones. DMBBQs are chlorinated and brominated at the p-dimethoxybenzene site using nontoxic sodium halide and Oxone in HFIP/water. X-ray crystallography is used to rationalize site preference in terms of the discontinuity in conjugation in the DMBBQ system. Quinone formation occurs by increasing in situ halogen generation and water. Conversely, radical trifluoromethylation occurs at the quinone of the DMBBQ.
Introduction
Alicyclic ring-fused benzimidazolequinones are potent anti-tumour agents (Scheme 1a), studied as synthetic alternatives to mitomycin C.1–4 Cerium(IV) ammonium nitrate oxidizes p-dimethoxybenzenes to p-benzoquinones,5 and to dimeric adducts.6 Bis-chloromethylbenzimidazolequinone 1 was reported in 38% yield (Scheme 1b) from the oxidation of 2-(chloromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole using CAN (2.9 equiv.).7 Bis-p-quinone 1 exhibited greater cytotoxicity against solid tumour cell lines than monomeric benzimidazolequinones. Recently, the CAN oxidation of 4,7-dimethoxybenzimidazole alkoxyamine 2 was used to give the bis-benzimidazolequinone 3 (Scheme 1c).8 Dimethoxybenzimidazole-benzimidazolequinone (DMBBQ) 4a was obtained in high yield (86%) when using less CAN, and in contrast to bis-p-quinone 3, DMBBQ 4a was insensitive to light. Radical trifluoromethylation of 4a using Langlois’ reagent (NaSO2CF3) occurred at the quinone to give 4b in 54% yield.8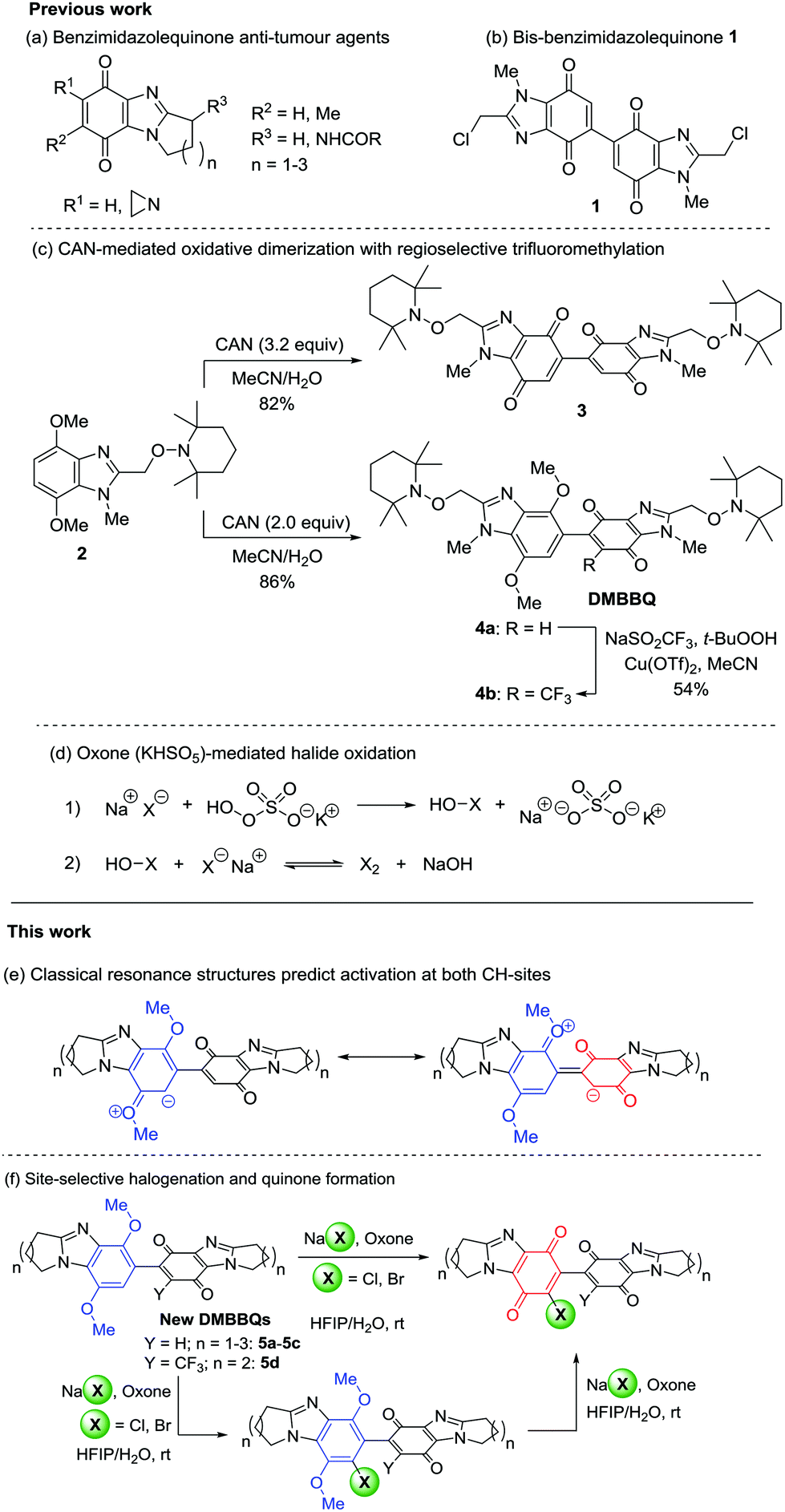 | ||
| Scheme 1 Development of DMBBQs as precursors to difunctionalized bis-benzimidazolequinones. | ||
Elemental chlorine and bromine are corrosive, toxic, and ordinarily inconvenient for synthesis. The useful halogen is however generated in situ from the oxidation of a benign and inexpensive alkali metal halide.9 Oxone (2KHSO5·KHSO4·K2SO4) is a stable, safe and cheap external oxidant.10 The active ingredient of Oxone is potassium peroxymonosulfate (KHSO5) and when accompanied by stoichiometric equivalents of halide (NaX) can generate molecular halogen (X2) for electrophilic chlorination and bromination (Scheme 1d). The halide with Oxone gives oxidative transformations with an accompanying halogenation of naphthols and phenols,11 and with and without halogenation for indoles.12,13 Other metal-free oxidations, such as N-bromosuccinimde (NBS) with a small amount of H2SO4,14,15 and PhI(OCOCF3)2 (PIFA),8,16,17 are reported to induce conversion of p-dimethoxybenzenes to p-quinones without halogenation. The disadvantage of using NBS and PIFA is the generation of organic waste products. Moreover, the combination of H2O2 with HX gives chlorinated and brominated alicyclic [1,2-a] ring-fused benzimidazolequinones from p-dimethoxybenzene precursors.18 The accompanying aromatic halogenation is considered advantageous, since it offers further functionalization opportunities. However, unlike monomeric benzimidazoles, milder room-temperature conditions are required for Cl2 and Br2 generation when using DMBBQs as the substrates (see below), which led us to use NaX/Oxone. The use of Oxone is unprecedented in oxidative demethylations to give p-quinones. Herein, we disclose selective chlorination and bromination of novel CAN-derived alicyclic ring-fused DMBBQs at the aromatic site, despite possible activation at the quinone (Scheme 1e). Electrophilic aromatic halogenation of DMBBQs 5a–5d (Scheme 1f) is accompanied by one-pot quinone formation.
Results and discussion
Five to seven-membered alicyclic ring-fused dimethoxybenzimidazoles 6a–6c are readily available,17 and used to prepare new DMBBQs 5a–5c. DMBBQ 5b was prepared in 73% yield by optimizing the conditions for CAN-oxidation of 6b in MeCN/H2O (Scheme 2).5–8 The latter solvent system also gave pyrido[1,2-a]benzimidazolequinone 7 in 15% yield. Replacing the MeCN with hexafluoroisopropanol (HFIP) improved the yield of the required DMBBQ 5b to 87% with no monomeric benzimidazolequinone 7 detected. Similar high yields (88 and 84%) using HFIP/H2O were obtained for the formation of pyrrolo- and azepino ring-fused DMBBQs 5a and 5c from 6a and 6c, respectively. The formation of 7 requires the intervention of a water molecule at the CAN generated radical cation to prevent dimerization.6,18 The persistence of radical cation intermediates in HFIP,19 and their shielding from water due to the formation of heterogeneous environments,20 may account for the exclusive CAN-mediated dimerization.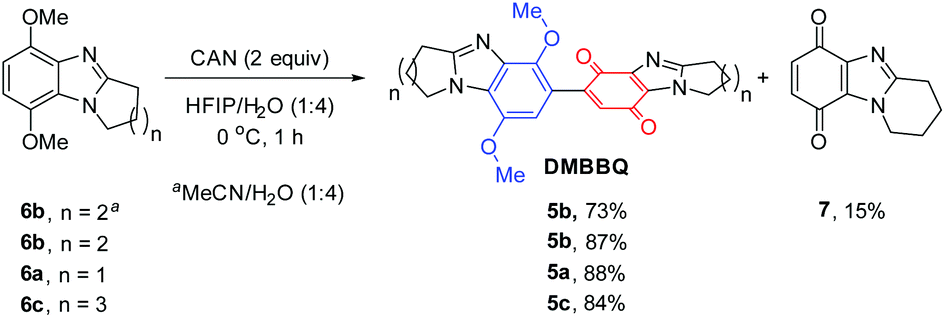 | ||
| Scheme 2 Synthesis of alicyclic ring-fused DMBBQs. | ||
DMBBQs 5a–5c were insufficiently crystalline for X-ray crystallography. However, the premise for the regioselective halogenation was supported by the preparation of bis(chloromethyl)-substituted DMBBQ 8 (76% yield), in an analogous fashion to 1 (see above),7,8 but using less CAN (2.0 equiv.). The crystal structure of dimethylchloride 8 showed that the aromatic and quinone rings adopt an out-of-plane conformation with respect to each other (Scheme 3 and Fig. S1†). The torsion angle (θ) of 67.6° describes the angle between the planes of the benzimidazole and benzimidazolequinone, which prevents effective orbital overlap over both rings, ruling out resonance activation of the 6-quinone position. Cyclic voltammetry and TD-DFT on DMBBQ 4a, provides further support for the site-selectivity for the electrophilic substitution with localized HOMO and LUMO at the respective dimethoxybenzimidazole and benzimidazolequinone motifs.8
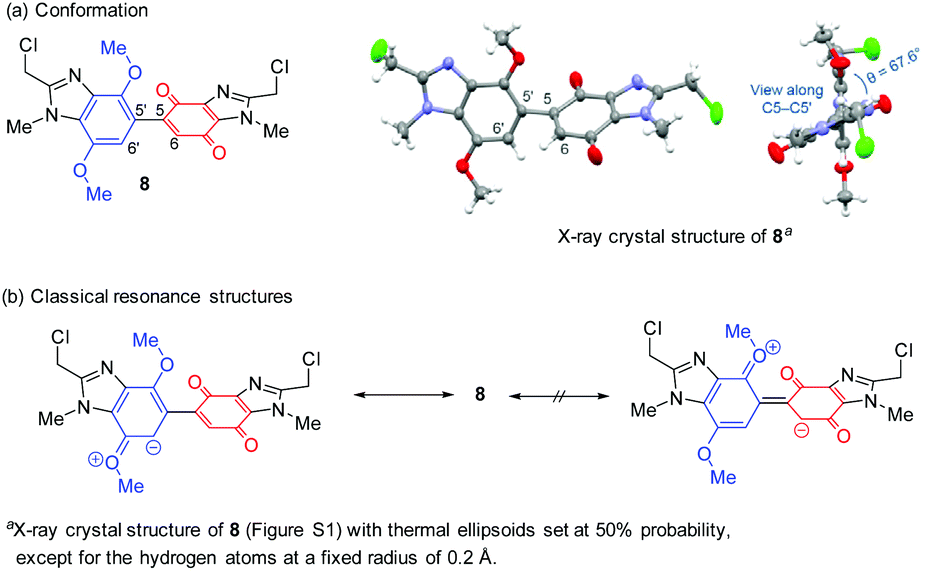 | ||
| Scheme 3 Evidence for MeO-activation at only the aromatic-CH. | ||
Chlorination of pyrrolo-fused dimer 5a was initially attempted using H2O2 with HCl under conditions which mediated the regioselective chlorination of anilines (Table 1).17 Reaction monitoring by TLC indicated the complete consumption of 5a after 1 h of reflux, but a low yield of 7′-chloride 9a (21%) was attributed to the formation of intractable baseline decomposition products. The requirement for milder conditions, led us to investigate the room temperature Oxone/NaCl reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | NaCl (mmol) | KHSO5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (mmol) b (mmol) |
Solvent | Yieldc (%) of 9a | Yieldc (%) of 10a |
| a Conditions: DMBBQ 5a (0.20 mmol), MeCN or HFIP (2 mL), H2O (0.1 mL). b Used as Oxone. c Isolated yields. Values in parentheses recovered 5a. d H2O2 (4 mmol), HCl (0.20 mmol), MeCN (2 mL), reflux, 1 h. e Intractable mixture. f 16 h. g HFIP (2 mL), H2O (0.2 mL), 9 h. | |||||
| 1d | — | — | MeCN | 21 | 0e |
| 2 | 0.20 | 0.24 | MeCN/H2O | 39 (52) | 0 |
| 3 | 0.20 | 0.24 | HFIP/H2O | 51 (43) | 0 |
| 4 | 0.40 | 0.24 | HFIP/H2O | 79 | 12 |
| 5f | 0.80 | 0.80 | HFIP/H2O | 15 | 71 |
| 6g | 0.80 | 0.80 | HFIP/H2O | 0 | 89 |
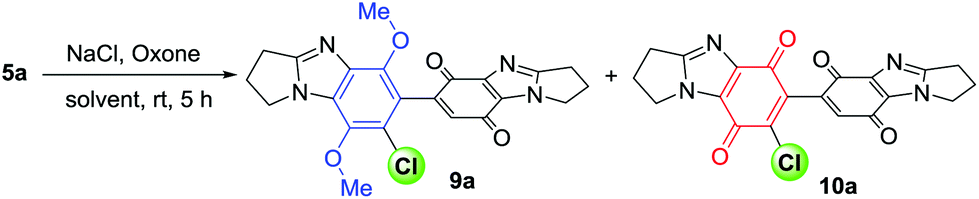
Addition of an aqueous solution of NaCl (1 equiv.) and Oxone (KHSO5, 1.2 equiv.) to 5a in MeCN gave chloride 9a in 39% yield, with column chromatography providing 52% recovery of 5a (Table 1). Polyfluorinated alcohols improve yields for NaCl/Oxone-mediated aliphatic and aromatic chlorinations,21 and are known to accelerate aromatic chlorination in systems that generate in situ Cl2.22 The reaction solvent was thus changed from MeCN to HFIP, with a greater consumption of substrate 5a observed, and 9a isolated in an improved yield of 51%. The highest yield of 9a (79%) after 5 h was obtained using a greater amount of NaCl (2 equiv.) with chlorination also accompanied by oxidation to give the bis-p-quinone 10a in 12% yield. Cl2 and water are responsible for conversion of the p-dimethoxybenzimidazole to benzimidazolequinone.18 Thus, optimized conditions for one-pot chlorination and quinone formation were achieved by increasing the amount of KHSO5 (4 equiv.) and NaCl (4 equiv.), so increasing the likelihood of NaCl reaction with HOCl, over HOCl reaction with the substrate 5a (Scheme 1d). These conditions favoured Cl2 formation and gave bis-benzimidazolequinone 10a in 71% yield, with an increase in water from 5 to 10% relative to HFIP giving exclusively 10a in 89% yield after 9 h.
The conditions which favored selective chlorination over quinone formation (entry 4, Table 1) were applied to pyrido and azepino-ring fused DMBBQs 5b and 5c to respectively give 9b (75%) and 9c (72%) (Method A, Scheme 4). In these cases, column chromatography removed co-eluting mixtures of substrates 5b and 5c with respective bis-p-quinones 10b and 10c. The optimized conditions for quinone formation (entry 6, Table 1) were applied to 5b and 5c to respectively give 10b and 10c in isolated yields of 88 and 74%, with only the latter giving some dimethoxybenzene intermediate, 9c in 12% yield (Method B, Scheme 4).
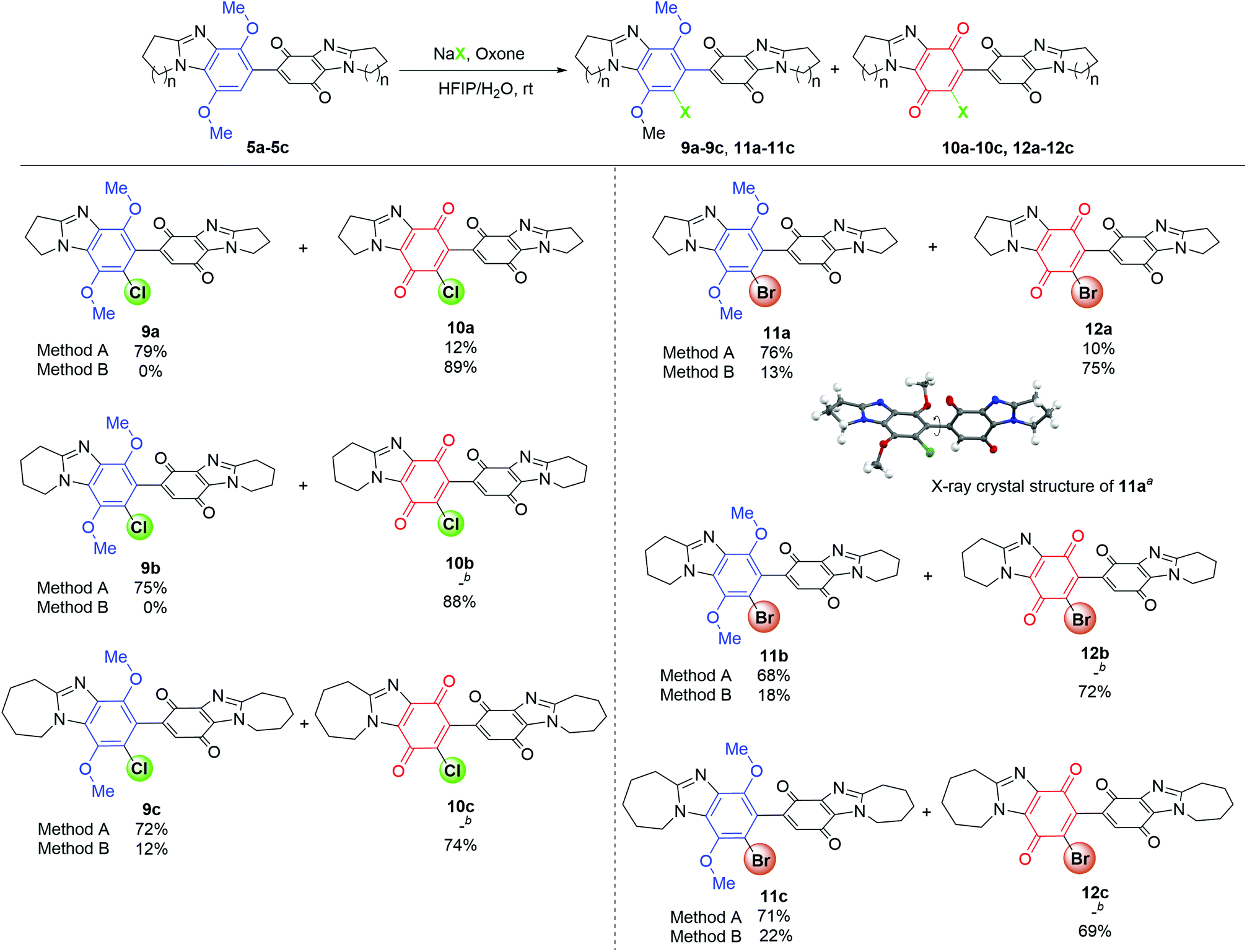 | ||
| Scheme 4 Halogenation and quinone formation. Conditions: Method A is the same as Table 1, entry 4. Method B is the same as Table 1, entry 6. Isolated yields. aX-ray crystal structure (Fig. S2†) with thermal ellipsoids set at 50% probability, except for the hydrogen atoms at a fixed radius of 0.2 Å. The torsional angle highlighted is 79.2°. bInseparable with 5b or 5c. | ||
NaBr replaced NaCl to facilitate regioselective bromination of 5a–5c (using entry 4, Table 1 conditions). There was a marginal decrease in the tendency for bromination compared to chlorination. Complete consumption of 5a occurred with the difference in Rf values allowing separation of 7′-bromo-DMBBQ 11a from bis-p-quinone 12a by column chromatography in respective yields of 76% and 10% (Method A, Scheme 4). X-ray crystallographic structure determination for 11a supported the site for bromination (Scheme 4 and Fig. S2†).
Under the same conditions, the pyrido- and azepino[1,2-a]-fused dimers 5b and 5c gave selectively brominated DMBBQs 11b and 11c in respective yields of 68% and 71%. In a similar manner to the chlorinations, ring-expanded DMBBQs 5b and 5c were less reactive towards bromination than the pyrrolo-fused DMBBQ 5a with unreacted material separated by column chromatography as inseparable mixtures with respective bis-p-quinones 12b and 12c. One-pot bromination and quinone formation (entry 6, Table 1 conditions) gave mostly the brominated dimeric quinones 12a–12c (69–75% yield), however column chromatography separated some dimethoxybenzimidazole intermediates 11a–11c (13–22% yield). The lower yields (with Method B) of brominated quinones 12a–12c are presumably due to the inferior oxidizing ability of Br2 relative to Cl2.23 The presence of a red-brown vapour in the NaBr/Oxone-mediated oxidative brominations is evidence for the in situ formation of Br2 (Fig. S3†).
Functionalization at both available alicyclic ring-fused DMBBQ CH-positions was established. Trifluoromethylation at the quinone of 5b gave substrate 5d in a similar moderate yield (51%, Scheme 5) to that reported for DMBBQ 4b.8 Using the Langlois’ reagent, intractable baseline products were observed and Baran et al. have attributed peroxide reaction with sulfinate as a significant wastage reaction.24 The ˙CF3 radical is considered weakly nucleophilic,25 and reported CH-alkylations occur at the ‘electron-poor’ quinone.26,27 Chlorination with quinone formation to give the fully functionalized bis-p-quinone 14 can be carried out in one or two steps with 5d depending on the desire to isolate more of the intermediate chloride 13. Using the optimized conditions for chlorination only (Table 1, entry 4 conditions) gave 13 in 73% yield, with the regioselectivity confirmed by the X-ray crystal structure (Scheme 5 and Fig. S4†). The torsional angle is almost identical to that of 8, indicating the substituents of 13 have no influence on the adopted solid-state geometry. Oxidation of isolated DMBBQ 13 using NaCl and Oxone gave bis-p-quinone 14 in 85% yield. Increasing the amount of NaCl and Oxone to favour one pot oxidative chlorination (Table 1, entry 6 conditions), gave bis-p-quinone 14 from 5d in 68% yield, with some intermediate DMBBQ 13 (19%) separated by column chromatography. The smaller yields for chlorides 13 and 14 reflect the deactivation of the trifluoromethylated DMBBQs 5d and 13 towards the electrophilic oxidizing mixture.
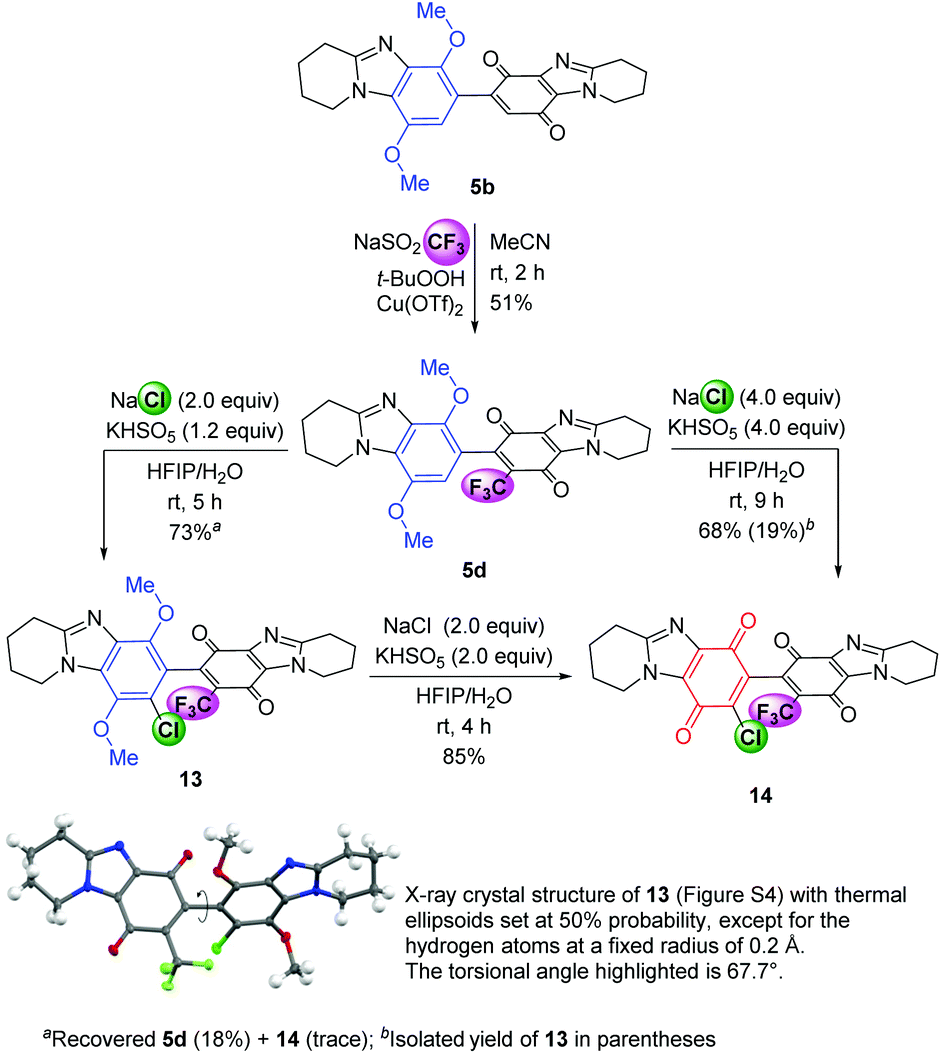 | ||
| Scheme 5 Radical and electrophilic functionalization. | ||
NaX/Oxone system is clearly an efficient system for ring-fused DMBBQs, allowing one-pot halogenation and quinone formation. The reality is transformations developed for our specific heterocyclic system (ring-fused DMBBQs) are not readily transferable to other similar systems. According to the literature,28 halogenation using NaX/Oxone would not be site-selective where there are other available heterocyclic CH-positions, e.g. the non-functionalised imidazole-2-position. Nevertheless, future work will aim to take advantage of the difference in reactivity between the two DMBBQ-CHs to increase scope to other site-selective functionalizations.
Conclusions
New alicyclic [1,2-a] ring-fused DMBBQs undergo one-pot electrophilic aromatic substitution and oxidative demethylation to give halogenated bis-p-quinones using the harmless and inexpensive NaX/Oxone system. The tuneable system gives mainly the site-selective chlorination and bromination intermediate at lower halogen concentrations. The torsion angle in DMBBQs supports the chemoselectivity by lack of MeO-activation at the quinone-CH. HFIP enhances the oxidative aromatic halogenation and for the first time is used in CAN-mediated oxidative aromatic couplings, which formed DMBBQs. Overall, this article details the synthesis in good to excellent yields of new ring-fused bis-benzimidazolequinone heterocyclic systems, and provides reliable late-stage methods for incorporating two different substituents into bis-p-quinones.Experimental
Materials
All chemicals obtained from commercial sources used as received. 5,8-Dimethoxy-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazole (6a), 6,9-dimethoxy-1,2,3,4-tetrahydropyrido[1,2-a]benzimidazole (6b), and 1,4-dimethoxy-7,8,9,10-tetrahydro-6H-azepino[1,2-a]benzimidazole (6c) were prepared in 89%, 72% and 91% yield respectively, according to our reported oxidative ring-closure of the appropriate 3,6-dimethoxy-2-(cycloamino)aniline.17 1,2,3,4-Tetrahydropyrido[1,2-a]benzimidazole-6,9-dione (7) has melting point, chromatographic and spectroscopic data consistent with that previously reported.29 2-(Chloromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole was prepared in 85% yield by N-methylation and chlorination of (4,7-dimethoxy-1H-benzimidazol-2-yl)methanol.8 Thin layer chromatography (TLC) was performed on TLC silica gel 60 F254 plates. Flash column chromatography was carried out on silica gel (Apollo Scientific 60/40–63 μm) and dry column vacuum chromatography used Apollo Scientific silica gel (ZEOprep 60 and 15–35 µm particle size).30Measurements
All melting points were measured on a Stuart Scientific melting point apparatus SMP1. IR spectra were recorded using a PerkinElmer Spec 1 with ATR attached.All NMR spectra were recorded using a Bruker Avance III 400 MHz spectrometer equipped with a 5 mm BBFO+, broadband autotune probe and controlled with TopSpin 3.5.7 acquisition software and IconNMR 5.0.7 automation software Copyright © 2017 Bruker BioSpin GmbH. The exception was 2,2′-bis(chloromethyl)-4′,7′-dimethoxy-1,1′-dimethyl-1H,1′H-[5,5′-bibenzimidazole]-4,7-dione (8) recorded on a JEOL ECX 400 MHz NMR spectrometer equipped with a DEC AXP 300 computer workstation. The chemical shifts are in ppm relative to TMS. 13C NMR spectra at 100 MHz are with complete proton decoupling. NMR assignments are supported by DEPT-135. 19F NMR spectra were obtained at 376 MHz using automatic digital lock correction in the absence of an internal standard.
HRMS spectra were obtained at the National Mass Spectrometry Facility at Swansea University using a Waters Xevo G2-S or Thermo Scientific LTQ Orbitrap XL 1 mass spectrometer with an Atmospheric Solids Analysis Probe (ASAP), except for DMBBQ 8 obtained at NUI Galway using ESI time-of-flight mass spectrometer (TOFMS) in positive mode using a Waters LCT Mass Spectrometer. The precision of all accurate mass measurements was better than 5 ppm.
A single crystal of 8 was grown by slow evaporation from 10% MeOH/CH2Cl2 at room temperature (Fig. S1†). Single crystal data for 8 was collected at 299 K using an Oxford Diffraction Xcalibur (equipped with a Mo Kα X-ray source and a Sapphire detector) using the CrysAlisPro software at NUI Galway.
Single crystals of 7′-bromo-5′,8′-dimethoxy-2,2′,3,3′-tetrahydro-1H,1′H-[6,6′-bipyrrolo[1,2-a]benzimidazole]-5,8-dione (11a) and 8′-chloro-6′,9′-dimethoxy-8-(trifluoromethyl)-1,1′,2,2′,3,3′,4,4′-octahydro[7,7′-bipyrido[1,2-a]benzimidazole]-6,9-dione (13) were grown at room temperature by slow evaporation from 1,2-dichloroethane and vapour diffusion of hexane into 1,2-dichloroethane, respectively (Fig. S2 and S4†). X-ray diffraction data on single crystals of compounds 11a and 13 were collected using an Agilent Oxford Diffraction SuperNova (equipped with a microfocus Cu Kα X-ray source, a Cryojet5®, and an Atlas CCD detector) using the CrysAlisPro software at University College London.
The crystal structures of 8, 11a, and 13 were solved using SHELXT31 and refined using SHELXL,32 both of which were operated from within either the Oscail33 or the OLEX234 software packages. For compound 11a, a split atom model for the disorder of the methoxy group containing atom O(3) was used to model the disorder, which refined to 72.5% for the methyl group of C(13A) compared to 27.5% for the methyl group of C(13B).
Crystallographic data for compounds 8, 11a and 13 were deposited with the Cambridge Data Centre with deposition numbers CCDC 1948449, 2022191, and 2022190,† respectively.
Synthesis of alicyclic ring-fused DMBBQs (5a–5c)
CAN (8.793 g, 16.04 mmol) in H2O (80 mL) was added over 30 min to ring-fused benzimidazoles 6a–6c (8.00 mmol) in HFIP (20 mL) at 0 °C. The solution was stirred for 30 min and evaporated to dryness. H2O (200 mL) was added, and the mixture extracted with CH2Cl2 (4 × 150 mL). The organic extracts were dried (MgSO4), evaporated, and purified by flash column chromatography using gradient elution of CH2Cl2/MeOH.:1 CH2Cl2/MeOH); mp 189–191 °C; νmax (neat, cm−1) 1674 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1650 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.64 (s, 1H, 7-H), 6.42 (s, 1H, 7′-H), 4.33–4.25 (m, 4H), 4.16 (s, 3H, Me), 3.88 (s, 3H, Me), 3.07–2.97 (m, 4H), 2.80–2.65 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.0, 177.9 (both CO), 160.7, 160.3, 147.6, 146.4, 143.5, 141.7, 141.6 (all C), 133.0 (7-CH), 129.5, 125.7, 117.2 (all C), 104.3 (7′-CH), 61.2, 55.9 (both Me), 45.1, 45.0, 26.54, 26.51, 23.4, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C22H21N4O4 405.1563; found 405.1557; m/z 407 (4%), 406 (27), 405 (C22H21N4O4, 100).
:1 CH2Cl2/MeOH); mp 176–177 °C; νmax (neat, cm−1) 1675 (CO), 1645 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.64 (s, 1H, 8-H), 6.41 (s, 1H, 8′-H), 4.44 (t, J = 5.3 Hz, 2H), 4.39–4.32 (m, 2H), 4.13 (s, 3H, Me), 3.86 (s, 3H, Me), 3.10–2.99 (m, 4H), 2.11–1.92 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.3, 178.4 (both CO), 151.7, 150.9, 146.7, 143.3, 142.7, 141.7, 136.5 (all C), 133.9 (8-CH), 130.0, 127.3, 117.3 (all C), 104.5 (8′-CH), 61.3, 55.9 (both Me), 45.6, 45.4, 25.7, 24.9, 23.1, 22.3, 20.5, 19.8 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H25N4O4 433.1876; found 433.1871; m/z 435 (5%), 434 (27), 433 (C24H25N4O4, 100).
:1 CH2Cl2/MeOH); mp 169–171 °C; νmax (neat, cm−1) 1674 (CO), 1650 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.63 (s, 1H, 2-H), 6.45 (s, 1H, 2′-H), 4.70–4.55 (m, 4H), 4.16 (s, 3H, Me), 3.88 (s, 3H, Me), 3.12–3.01 (m, 4H), 1.98–1.72 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.4, 179.0 (both CO), 158.1, 157.4, 146.3, 143.7, 142.0, 141.1, 136.2 (all C), 134.2 (2-CH), 130.4, 127.8, 116.6 (all C), 105.1 (2′-CH), 61.4, 55.9 (both Me), 46.1, 45.8, 30.9, 30.8, 29.8, 29.3, 28.9, 28.3, 25.6, 25.0 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C26H29N4O4 461.2189; found 461.2187; m/z 463 (5%), 462 (29), 461 (C26H29N4O4), 431 (5).
:1 EtOAc:hexanes); νmax (neat, cm−1) 1655 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.70 (s, 1H), 6.47 (s, 1H), 4.79 (s, 2H), 4.76 (s, 2H), 4.15 (s, 3H, OMe), 4.09 (s, 3H, NMe), 4.07 (s, 3H, NMe), 3.89 (s, 3H, OMe); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.7, 178.7 (both CO), 149.6, 148.7, 147.0, 144.2, 142.7, 141.1, 136.1 (all C), 134.5 (CH), 131.8, 128.8, 117.1 (all C), 105.9 (CH), 61.6, 56.1 (both OMe), 36.8, 35.6 (both CH2), 33.0, 32.6 (both NMe); HRMS (ESI) m/z: [M + H]+ calcd for C20H19N4O435Cl2 449.0783; found 449.0778.
O), 1650 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.64 (s, 1H, 7-H), 6.42 (s, 1H, 7′-H), 4.33–4.25 (m, 4H), 4.16 (s, 3H, Me), 3.88 (s, 3H, Me), 3.07–2.97 (m, 4H), 2.80–2.65 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.0, 177.9 (both CO), 160.7, 160.3, 147.6, 146.4, 143.5, 141.7, 141.6 (all C), 133.0 (7-CH), 129.5, 125.7, 117.2 (all C), 104.3 (7′-CH), 61.2, 55.9 (both Me), 45.1, 45.0, 26.54, 26.51, 23.4, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C22H21N4O4 405.1563; found 405.1557; m/z 407 (4%), 406 (27), 405 (C22H21N4O4, 100).
:1 CH2Cl2/MeOH); mp 176–177 °C; νmax (neat, cm−1) 1675 (CO), 1645 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.64 (s, 1H, 8-H), 6.41 (s, 1H, 8′-H), 4.44 (t, J = 5.3 Hz, 2H), 4.39–4.32 (m, 2H), 4.13 (s, 3H, Me), 3.86 (s, 3H, Me), 3.10–2.99 (m, 4H), 2.11–1.92 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.3, 178.4 (both CO), 151.7, 150.9, 146.7, 143.3, 142.7, 141.7, 136.5 (all C), 133.9 (8-CH), 130.0, 127.3, 117.3 (all C), 104.5 (8′-CH), 61.3, 55.9 (both Me), 45.6, 45.4, 25.7, 24.9, 23.1, 22.3, 20.5, 19.8 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H25N4O4 433.1876; found 433.1871; m/z 435 (5%), 434 (27), 433 (C24H25N4O4, 100).
:1 CH2Cl2/MeOH); mp 169–171 °C; νmax (neat, cm−1) 1674 (CO), 1650 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.63 (s, 1H, 2-H), 6.45 (s, 1H, 2′-H), 4.70–4.55 (m, 4H), 4.16 (s, 3H, Me), 3.88 (s, 3H, Me), 3.12–3.01 (m, 4H), 1.98–1.72 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 180.4, 179.0 (both CO), 158.1, 157.4, 146.3, 143.7, 142.0, 141.1, 136.2 (all C), 134.2 (2-CH), 130.4, 127.8, 116.6 (all C), 105.1 (2′-CH), 61.4, 55.9 (both Me), 46.1, 45.8, 30.9, 30.8, 29.8, 29.3, 28.9, 28.3, 25.6, 25.0 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C26H29N4O4 461.2189; found 461.2187; m/z 463 (5%), 462 (29), 461 (C26H29N4O4), 431 (5).
:1 EtOAc:hexanes); νmax (neat, cm−1) 1655 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.70 (s, 1H), 6.47 (s, 1H), 4.79 (s, 2H), 4.76 (s, 2H), 4.15 (s, 3H, OMe), 4.09 (s, 3H, NMe), 4.07 (s, 3H, NMe), 3.89 (s, 3H, OMe); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.7, 178.7 (both CO), 149.6, 148.7, 147.0, 144.2, 142.7, 141.1, 136.1 (all C), 134.5 (CH), 131.8, 128.8, 117.1 (all C), 105.9 (CH), 61.6, 56.1 (both OMe), 36.8, 35.6 (both CH2), 33.0, 32.6 (both NMe); HRMS (ESI) m/z: [M + H]+ calcd for C20H19N4O435Cl2 449.0783; found 449.0778.
H2O2/HCl-mediated chlorination
H2O2 (50% w/v in water, 0.227 mL, 4.0 mmol) and HCl (37% w/v in water, 17 μL, 0.20 mmol) were sequentially added dropwise to a stirred solution of DMBBQ 5a (81 mg, 0.20 mmol) in MeCN (2 mL), and heated at reflux for 1 h. The solution was cooled and EtOAc (2 mL) added, and washed with Na2CO3 (satd., 2 mL). The organic extract was dried (MgSO4), evaporated, and the residue purified by flash column chromatography using gradient elution of CH2Cl2/MeOH to give 7′-chloro-5′,8′-dimethoxy-2,2′,3,3′-tetrahydro-1H,1′H-[6,6′-bipyrrolo[1,2-a]benzimidazole]-5,8-dione (9a) (18 mg, 21%); orange crystals; Rf 0.41 (19:1 CH2Cl2/MeOH); mp (decomp >206 °C); νmax (neat, cm−1) 1674 (CO), 1653 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.58 (s, 1H, 7-H), 4.30 (t, J = 7.2 Hz, 4H), 4.17 (s, 3H, Me), 3.94 (s, 3H, Me), 3.09–2.97 (m, 4H), 2.80–2.70 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.4, 177.7 (both CO), 160.9, 160.8, 146.3, 145.7, 144.5, 140.1, 137.2 (all C), 135.5 (7-CH), 129.7, 128.8, 119.2, 117.3 (all C), 62.6, 61.3 (both Me), 45.1, 44.5, 26.6, 26.5, 23.3, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C22H20N4O435Cl 439.1173; found 439.1171; m/z 442 (9%), 441 (C22H20N4O437Cl, 36), 440 (25), 439 (C22H20N4O435Cl, 100).
NaX/Oxone-mediated halogenation (method A)
Oxone (74 mg, 0.12 mmol, containing KHSO5, 0.24 mmol) and NaCl (23 mg, 0.40 mmol) or NaBr (41 mg, 0.40 mmol) in H2O (0.1 mL) were added over the course of 5 min to a stirred solution of DMBBQ 5a–5d (0.20 mmol) in HFIP (2 mL) at room temperature, and stirred for 5 h.NaX/Oxone-mediated halogenation and quinone formation (method B)
Oxone (0.246 g, 0.40 mmol, containing KHSO5, 0.80 mmol) and NaCl (47 mg, 0.80 mmol) or NaBr (82 mg, 0.80 mmol) in H2O (0.2 mL) were added over the course of 5 min to a stirred solution of DMBBQ 5a–5d (0.20 mmol) in HFIP (2 mL) at room temperature, and stirred for 9 h.Reaction work-up for methods A and B
H2O (5 mL) was added, and the mixture extracted with CH2Cl2 (3 × 5 mL). The organic extracts were dried (MgSO4), evaporated to dryness, and purified by flash column chromatography using gradient elution of CH2Cl2/MeOH.:1 CH2Cl2/MeOH); mp (decomp >207 °C); νmax (neat, cm−1) 1659 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.55 (s, 1H, 7′-H), 4.31 (q, J = 7.6 Hz, 4H), 3.06–2.98 (m, 4H), 2.83–2.73 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.2, 176.3, 176.2, 168.9 (all CO), 162.2, 161.3, 146.4, 145.8, 141.6, 140.6, 138.1 (all C), 135.3 (7′-CH), 129.6, 128.7 (both C), 45.5, 45.2, 26.5, 26.4, 23.1, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C20H14N4O435Cl 409.0703; found 409.0703; m/z 412 (8%), 411 (C20H14N4O437Cl, 35), 410 (23), 409 (C20H14N4O435Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp >197 °C); νmax (neat, cm−1) 1676 (CO), 1653 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.56 (s, 1H, 8-H), 4.46–4.32 (m, 4H), 4.13 (s, 3H, Me), 3.94 (s, 3H, Me), 3.11–3.01 (m, 4H), 2.16–1.95 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.6, 178.2 (both CO), 151.7, 151.5, 145.5, 143.8, 141.6, 138.1 (all C), 136.3 (8-CH), 135.1, 130.24, 130.19, 119.6, 117.7 (all C), 62.5, 61.4 (both Me), 45.4, 44.8, 25.7, 25.0, 23.0, 22.3, 20.4, 19.8 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H24N4O435Cl 467.1486; found 467.1482; m/z 471 (2%), 470 (9), 469 (C24H24N4O437Cl, 35), 468 (27), 467 (C24H24N4O435Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp >212 °C); νmax (neat, cm−1) 1655 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.54 (s, 1H, 8′-H), 4.37 (q, J = 5.9 Hz, 4H), 3.04 (t, J = 6.3 Hz, 4H), 2.13–1.96 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.4, 176.8, 176.4, 169.1 (all CO), 153.3, 152.3, 142.4, 141.7, 141.2, 139.9, 137.4 (all C), 136.2 (8′-CH), 130.2, 129.3 (both C), 45.8, 45.5, 25.0, 24.9, 22.2, 22.1, 19.7, 19.5 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C22H18N4O435Cl 437.1017; found 437.1016; m/z 440 (8%), 439 (C22H18N4O437Cl, 35), 438 (25), 437 (C22H18N4O435Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp >234 °C); νmax (neat, cm−1) 1675 (CO), 1651 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.56 (s, 1H, 2-H), 4.73–4.46 (m, 4H), 4.15 (s, 3H, Me), 3.90 (s, 3H, Me), 3.15–3.01 (m, 4H), 1.99–1.74 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.7, 178.7 (both CO), 158.2, 157.9, 145.7, 143.4, 140.9, 137.5 (all C), 136.7 (2-CH), 134.7, 130.5, 130.3, 120.1, 117.1 (all C), 61.59, 61.56 (both Me), 45.9, 45.3, 30.9, 30.8, 29.7, 29.3, 28.9, 28.1, 25.5, 24.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C26H28N4O435Cl 495.1799; found 495.1801; m/z 499 (2%), 498 (10), 497 (C26H28N4O437Cl, 36), 496 (29), 495 (C26H28N4O435Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp >237 °C); νmax (neat, cm−1) 1659 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.50 (s, 1H, 2′-H), 4.75–4.61 (m, 2H), 4.61–4.51 (m, 2H), 3.16–2.99 (m, 4H), 2.03–1.69 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.4, 177.3, 176.3, 169.7 (all CO), 159.7, 158.8, 142.6, 141.1, 140.5, 139.5, 137.0 (all C), 136.5 (2′-CH), 130.5, 129.7 (both C), 46.3, 46.0, 30.73, 30.65 (all CH2), 29.3 (2 × CH2), 28.1, 28.0, 24.8, 24.7 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H22N4O435Cl 465.1330; found 465.1326; m/z 468 (9%), 467 (C24H22N4O437Cl, 36), 466 (27), 465 (C24H22N4O435Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp >209 °C); νmax (neat, cm−1) 1674 (CO), 1652 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.55 (s, 1H, 7-H), 4.31 (t, J = 7.2 Hz, 4H), 4.16 (s, 3H, Me), 3.93 (s, 3H, Me), 3.09–2.99 (m, 4H), 2.81–2.71 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.3, 177.8 (both CO), 160.81, 160.79, 146.3, 146.1, 145.9, 140.8, 138.2 (all C), 135.4 (7-CH), 129.7, 128.9, 118.9, 109.6 (all C), 62.8, 61.4 (both Me), 45.1, 44.5, 26.6, 26.5, 23.3, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C22H20N4O479Br 483.0668; found 483.0670; m/z 487 (5%), 486 (24), 485 (C22H20N4O481Br, 100), 484 (24), 483 (C22H20N4O479Br, 97).
:1 CH2Cl2/MeOH); mp (decomp >209 °C); νmax (neat, cm−1) 1662 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.53 (s, 1H, 7′-H), 4.31 (q, J = 6.6 Hz, 4H), 3.07–2.97 (m, 4H), 2.83–2.71 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.1, 176.4, 175.7, 169.0 (all CO), 162.0, 161.3, 146.2, 145.8, 142.5, 141.9, 136.8 (all C), 134.8 (7′-CH), 129.7, 128.4 (both C), 45.4, 45.2, 26.5, 26.4, 23.1, 22.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C20H14O4N479Br 453.0193; found 453.0180; m/z 456 (22%), 455 (C20H14O4N481Br, 97), 454 (22), 453 (C20H14O4N479Br, 100).
:1 CH2Cl2/MeOH); mp (decomp >186 °C); νmax (neat, cm−1) 1675 (CO), 1652 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.54 (s, 1H, 8-H), 4.47–4.34 (m, 4H), 4.11 (s, 3H, Me), 3.93 (s, 3H, Me), 3.11–3.02 (m, 4H), 2.16–1.96 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.6, 178.2 (both CO), 151.8, 151.5, 145.6, 145.4, 141.6, 139.1 (all C), 136.2 (8-CH), 135.8, 130.4, 130.2, 119.4, 110.1 (all C), 62.7, 61.5 (both Me), 45.4, 44.8, 25.7, 25.0, 23.1, 22.3, 20.4, 19.8 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H24N4O479Br 511.0981; found 511.0984; m/z 515 (6%), 514 (26), 513 (C24H24N4O481Br, 100), 512 (26), 511 (C24H24N4O479Br, 97).
:1 CH2Cl2/MeOH); mp (decomp >220 °C); νmax (neat, cm−1) 1655 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.51 (s, 1H, 8′-H), 4.37 (t, J = 5.6 Hz, 4H), 3.05 (t, J = 6.3 Hz, 4H), 2.14–1.96 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.3, 176.9, 175.9, 169.3 (all CO), 153.2, 152.3, 141.7, 141.6, 141.19, 141.17, 137.9 (all C), 135.6 (8′-CH), 130.2, 129.0 (both C), 45.7, 45.5, 25.0, 24.9, 22.2, 22.1, 19.7, 19.5 (all CH2); HRMS (ASAP) m/z: calcd for [M + H]+ C22H18N4O479Br 481.0511; found 481.0512; m/z 485 (3%), 484 (24), 483 (C22H18N4O481Br, 97), 482 (26), 481 (C22H18N4O479Br, 100).
:1 CH2Cl2/MeOH); mp (decomp >243 °C); νmax (neat, cm−1) 1675 (CO), 1651 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.53 (s, 1H, 2-H), 4.73–4.45 (m, 4H), 4.14 (s, 3H, Me), 3.89 (s, 3H, Me), 3.15–3.00 (m, 4H), 1.99–1.69 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 179.7, 178.8 (both CO), 158.3, 157.9, 145.9, 145.0, 140.9, 138.4 (all C), 136.6 (2-CH), 135.5, 130.5, 130.4, 118.8, 110.8 (all C), 61.7, 61.6 (both Me), 45.9, 45.3, 30.89, 30.85, 29.7, 29.4, 28.9, 28.2, 25.5, 24.9 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C26H28N4O479Br 539.1294; found 539.1297; m/z 543 (5%), 542 (27), 541 (C26H28N4O481Br, 100), 540 (28), 539 (C26H28N4O479Br, 97).
:1 CH2Cl2/MeOH); mp (decomp >232 °C); νmax (neat, cm−1) 1659 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.49 (s, 1H, 2′-H), 4.78–4.64 (m, 2H), 4.62–4.48 (m, 2H), 3.16–2.99 (m, 4H), 2.05–1.68 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.4, 177.3, 175.9, 169.8 (all CO), 159.6, 158.7, 141.3, 141.0, 140.8, 140.5, 138.2 (all C), 135.9 (2′-CH), 130.5, 129.3 (both C), 46.2, 46.0, 30.74, 30.66 (all CH2), 29.3 (2 × CH2), 28.1, 28.0, 24.8, 24.7 (all CH2); HRMS (ASAP) m/z: [M + H]+ calcd for C24H22N4O479Br 509.0824; found 509.0826; m/z 513 (4%), 512 (26), 511 (C24H22N4O481Br, 100), 510 (26), 509 (C24H22N4O479Br, 97).
:1 CH2Cl2/MeOH); mp 177–179 °C; νmax (neat, cm−1) 1682 (CO), 1663 (CO); 1H NMR (400 MHz, CDCl3) δ: 6.24 (s, 1H, 8′-H), 4.52–4.32 (m, 4H), 4.12 (s, 3H, Me), 3.84 (s, 3H, Me), 3.06 (q, J = 6.5 Hz, 4H), 2.13–1.92 (m, 8H); 13C{1H} NMR (100 MHz, CDCl3) δ: 178.5, 174.1 (both CO), 153.1, 150.8, 147.73, 147.70, 142.3, 141.0, 135.6 (all C), 130.2 (q, 2JC–F = 27.2 Hz, 8-C), 129.9, 127.4 (C), 121.8 (q, 1JC–F = 276.6 Hz, CF3), 114.4 (C), 103.6 (d, J = 2.1 Hz, 8′-CH), 61.0, 55.9 (both Me), 45.7, 45.6, 25.7, 25.0, 23.1, 22.2, 20.5, 19.6 (all CH2); 19F NMR (376 MHz, CDCl3) δ: −56.48; HRMS (ASAP) m/z: [M + H]+ calcd for C25H24N4O4F3 501.1750; found 501.1752; m/z 503 (5%), 502 (28), 501 (C25H24N4O4F3, 100).
:1 CH2Cl2/MeOH); mp 163–166 °C; νmax (neat, cm−1) 1683 (CO), 1668 (CO); 1H NMR (400 MHz, CDCl3) δ: 4.48–4.36 (m, 4H), 4.15 (s, 3H, Me), 3.93 (s, 3H, Me), 3.07 (q, J = 6.6 Hz, 4H), 2.15–2.06 (m, 4H), 2.06–1.96 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 177.7, 173.7 (both CO), 153.1, 151.4 (both C), 144.29–144.17 (m, C), 144.1, 141.0, 137.7, 134.3 (all C), 131.7 (q, 2JC–F = 27.5 Hz, 8-C), 130.4, 130.1 (both C), 121.5 (q, 1JC–F = 276.4 Hz, CF3), 118.5, 115.2 (both C), 62.6, 61.1 (both Me), 45.7, 44.8, 25.7, 25.0, 23.0, 22.2, 20.4, 19.6 (all CH2); 19F NMR (376 MHz, CDCl3) δ: −58.95; HRMS (ASAP) m/z: [M + H]+ calcd for C25H23N4O4F335Cl 535.1360; found 535.1357; m/z 539 (2%), 538 (9), 537 (C25H23N4O4F337Cl, 35), 536 (27), 535 (C25H23N4O4F335Cl, 100).
:1 CH2Cl2/MeOH); mp (decomp. >239 °C); νmax (neat, cm−1) 1669 (CO); 1H NMR (400 MHz, CDCl3) δ: 4.39 (t, J = 5.9 Hz, 4H), 3.05 (t, J = 6.2 Hz, 4H), 2.15–2.06 (m, 4H), 2.06–1.96 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ: 175.7, 175.5, 172.1, 168.6 (all CO), 153.7, 153.4, 141.6, 140.7, 140.5, 139.8, 137.1 (all C), 132.0 (q, 2JC–F = 28.3 Hz, 8′-C), 130.2, 129.5 (both C), 121.0 (q, 1JC–F = 277.0 Hz, CF3), 45.9, 45.8, 25.00, 24.99 (all CH2), 22.1 (2 × CH2), 19.51, 19.48 (both CH2); 19F NMR (376 MHz, CDCl3) δ: −59.03; HRMS (ASAP) m/z: [M + H]+ calcd for C23H17N4O4F335Cl 505.0891; found 505.0889; m/z 508 (12%), 507 (C23H17N4O4F337Cl, 34), 506 (25), 505 (C23H17N4O4F335Cl, 100).
Oxidation of DMBBQ 13
Oxone (74 mg, 0.12 mmol, containing KHSO5, 0.24 mmol) and NaCl (14 mg, 0.24 mmol) in H2O (0.12 mL) were added over the course of 5 min to a stirred solution of DMBBQ 13 (64 mg, 0.12 mmol) in HFIP (1.2 mL) at room temperature, and stirred for 4 h. H2O (3 mL) was added, and the mixture extracted with CH2Cl2 (3 × 3 mL). The organic extracts were dried (MgSO4), evaporated to dryness, and purified by flash column chromatography using gradient elution of CH2Cl2/MeOH to give bis-p-quinone 14 (51 mg, 85%); orange crystals; spectral data and melting point were consistent with the above.Author contributions
D. Conboy – investigation, methodology and writing; P. Kielty – investigation and visualization; J. C. Bear – investigation (X-ray); J. K. Cockcroft – investigation (X-ray); P. Farràs – supervision; P. McArdle – investigation (X-ray); R. J. Singer – resources; D. A. Smith – supervision; F. Aldabbagh – conceptualization, funding acquisition, main supervision and writing.Conflicts of interest
There are no conflicts to declare.Note after first publication
This article replaces the version published on 05 March 2021, in which part of Scheme 1 was missing.Acknowledgements
We thank Kingston University for a PhD Scholarship for Darren Conboy and the Irish Research Council for an Enterprise Partnership Postgraduate Scholarship for Patrick Kielty.Notes and references
- A. Ghodousi, X. Huang, Z. Cheng and E. B. Skibo, J. Med. Chem., 2004, 47, 90 CrossRef CAS PubMed.
- M. Lynch, S. Hehir, P. Kavanagh, D. Leech, J. O'Shaughnessy, M. P. Carty and F. Aldabbagh, Chem. – Eur. J., 2007, 13, 3218 CrossRef CAS PubMed.
- E. B. Skibo, A. Jamil, B. Austin, D. Hansen and A. Ghodousi, Org. Biomol. Chem., 2010, 8, 1577 RSC.
- K. Fahey, L. O'Donovan, M. Carr, M. P. Carty and F. Aldabbagh, Eur. J. Med. Chem., 2010, 45, 1873 CrossRef CAS PubMed.
- P. Jacob III, P. S. Callery, A. T. Shulgin and N. Castagnoli Jr., J. Org. Chem., 1976, 41, 3627 CrossRef PubMed.
- B. E. Love and A. L. Simmons, Tetrahedron Lett., 2016, 57, 5712 CrossRef CAS.
- A. Gellis, H. Kovacic, N. Boufatah and P. Vanelle, Eur. J. Med. Chem., 2008, 43, 1858 CrossRef CAS PubMed.
- P. Kielty, P. Farràs, P. McArdle, D. A. Smith and F. Aldabbagh, Chem. Commun., 2019, 55, 14665 RSC.
- R. K. Dieter, L. E. Nice and S. E. Velu, Tetrahedron Lett., 1996, 37, 2377 CrossRef CAS.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329 CrossRef CAS PubMed.
- M. Uyanik, N. Sahara and K. Ishihara, Eur. J. Org. Chem., 2019, 27 CrossRef CAS.
- J. Xu and R. Tong, Green Chem., 2017, 19, 2952 RSC.
- J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754 CrossRef PubMed.
- D. W. Kim, H. Y. Choi, K.-J. Lee and D. Y. Chi, Org. Lett., 2001, 3, 445 CrossRef CAS PubMed.
- L. O'Donovan, M. P. Carty and F. Aldabbagh, Chem. Commun., 2008, 43, 5592 RSC.
- H. Tohma, H. Morioka, Y. Harayama, M. Hashizume and Y. Kita, Tetrahedron Lett., 2001, 42, 6899 CrossRef CAS.
- D. Conboy, S. I. Mirallai, A. Craig, P. McArdle, A. A. Al-Kinani, S. Barton and F. Aldabbagh, J. Org. Chem., 2019, 84, 9811 CrossRef CAS PubMed.
- M. Sweeney, L.-A. J. Keane, M. Gurry, P. McArdle and F. Aldabbagh, Org. Lett., 2018, 20, 6970 CrossRef CAS PubMed.
- L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Chem. Commun., 1996, 2105 RSC.
- O. Hollóczki, A. Berkessel, J. Mars, M. Mezger, A. Wiebe, S. R. Waldvogel and B. Kirchner, ACS Catal., 2017, 7, 1846 CrossRef.
- M. Zhao and W. Lu, Org. Lett., 2017, 19, 4560 CrossRef CAS PubMed.
- R. Ben-Daniel, S. P. de Visser, S. Shaik and R. Neumann, J. Am. Chem. Soc., 2003, 125, 12116 CrossRef CAS PubMed.
- A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed.
- J. Yining, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411 CrossRef PubMed.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251 CrossRef PubMed.
- D. R. Sutherland, M. Veguillas, C. L. Oates and A.-L. Lee, Org. Lett., 2018, 20, 6863 CrossRef CAS PubMed.
- V. M. Lakshmireddy, Y. N. Veera, T. J. Reddy, V. J. Rao and B. C. Raju, Asian J. Org. Chem., 2019, 8, 1380 CrossRef CAS.
- M. Sweeney, M. Gurry, L.-A. J. Keane and F. Aldabbagh, Tetrahedron Lett., 2017, 58, 3565 CrossRef CAS.
- L. M. Harwood, Aldrichimica Acta, 1985, 18, 25 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- P. McArdle, J. Appl. Crystallogr., 2017, 50, 320 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S4 and X-ray crystallography Tables S1–S3 and 1H, 13C and 19F NMR spectra (PDF). CIF files available for compounds 8, 11a and 13. CCDC 1948449 2022191 2022190. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob00032b |
| ‡ Present address: Kingston University. |
| This journal is © The Royal Society of Chemistry 2021 |