
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivorce in the two-component BVMO family: the single oxygenase for enantioselective chemo-enzymatic Baeyer–Villiger oxidations†
Robert
Röllig
ab,
Caroline E.
Paul
 c,
Magalie
Claeys-Bruno
d,
Katia
Duquesne
a,
Selin
Kara
b and
Véronique
Alphand
*a
c,
Magalie
Claeys-Bruno
d,
Katia
Duquesne
a,
Selin
Kara
b and
Véronique
Alphand
*a
aAix Marseille Univ, CNRS, Centrale Marseille, iSm2 UMR 7313, Marseille, France. E-mail: v.alphand@univ-amu.fr
bAarhus University, Denmark
cDelft University of Technology, The Netherlands
dAix Marseille Univ, Univ Avignon, CNRS, IRD, IMBE UMR7263, Marseille, France
First published on 22nd March 2021
Abstract
Two-component flavoprotein monooxygenases consist of a reductase and an oxygenase enzyme. The proof of functionality of the latter without its counterpart as well as the mechanism of flavin transfer remains unanswered beyond doubt. To tackle this question, we utilized a reductase-free reaction system applying purified 2,5-diketocamphane-monooxygenase I (2,5-DKCMO), a FMN-dependent type II Baeyer–Villiger monooxygenase, and synthetic nicotinamide analogues (NCBs) as dihydropyridine derivatives for FMN reduction. This system demonstrated the stand-alone quality of the oxygenase, as well as the mechanism of FMNH2 transport by free diffusion. The efficiency of this reductase-free system strongly relies on the balance of FMN reduction and enzymatic (re)oxidation, since reduced FMN in solution causes undesired side reactions, such as hydrogen peroxide formation. Design of experiments allowed us to (i) investigate the effect of various reaction parameters, underlining the importance to balance the FMN/FMNH2 cycle, (ii) optimize the reaction system for the enzymatic Baeyer–Villiger oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one, rac-camphor, and rac-norcamphor. Finally, this study not only demonstrates the reductase-independence of 2,5-DKCMO, but also revisits the terminology of two-component flavoprotein monooxygenases for this specific case.
Introduction
The Baeyer–Villiger (BV) oxidation converts ketones to esters and lactones by oxidative cleavage of the C–C bond adjacent to the carbonyl group.1,2 The (chemical) usage of peroxide derivatives is, without a doubt, a powerful methodology for this C–C bond-breaking process; however, it also suffers from a lack of enantioselectivity. As often observed with biological processes, the enzyme-catalysed version of BV oxidation overcomes this handicap.3,4Oxidative enzymes are capable of catalysing a large range of reactions, but often require cofactors, which have to be recycled to enable their catalytic usage. Among these enzymes, the class of oxygenases is immensely diverse5–8 and contains inter alia flavoprotein monooxygenases (FPMOs), a family in which are found enzymes that catalyse BV oxidation. These monooxygenases belong to the groups A, B or C of FPMOs9 and are single- or two-component enzymes (Scheme 1), respectively called type I and type II Baeyer–Villiger monooxygenases (BVMOs).9–11
 | ||
| Scheme 1 Flavin-dependent single- and two-component monooxygenase systems. (a) One-component FPMO system: the reductase and oxygenase domains are gathered in a single protein. Flavin is tightly bound as a prosthetic group in the active site of the enzyme. (b) Two-component FPMO system: reductase and oxygenase components are in two separated proteins. The flavin reduction catalysed by the reductase is driven by NAD(P)H oxidation, whereas flavin-H2 oxidation is driven by the oxygenase. The mode of flavin-H2 transport depends on the two-component monooxygenase, achieved either by free diffusion or via reductase-oxygenase mediation.12 | ||
Despite their structural differences, the one- and two-component oxygenase systems share a dependency on the expensive nicotinamide adenine dinucleotide cofactor (NAD(P)H). Moreover, the stability of NAD(P)H can become a hurdle (under basic conditions), as the adenine dinucleotide moiety is susceptible to hydrolysis.13–15
Synthetic nicotinamide analogues (nicotinamide coenzyme biomimetics, NCBs) address both obstacles, as they can be synthesized at lower costs while remaining stable.16 Moreover, they have been established to replace their natural templates for numerous flavin-dependent enzymes17–19 with a flavin shuttled electron mediation.20–22
Most of the studies on enzymatic BV oxidation focused on one-component FPMOs from group B (also known as type I BVMOs). These enzymes are available in high numbers in protein databanks, easily detectable by consensus sequence research23 and subsequently more frequently investigated and applied in many catalytic processes.3,4,24–26 In contrast, two-component systems referred to as type II BVMOs (from group C of FPMOs) are rare. Only two representatives are known so far, 2,5-diketocamphane 1,2-monooxygenase I (2,5-DKCMO; EC 1.14.14.108) and 3,6-diketocamphane 1,6-monooyxgenase (2,6-DKCMO; EC 1.14.14.155),27–32 probably because a two-component system is not a trivial biocatalytic arrangement to identify. The mechanism of hydride transfer and the balance of the reductase and oxygenase reactions are still unknown. Consequently, the question of whether the oxygenase can work independently of the reductase component and nicotinamide cofactor remains to be determined.
In this in vitro study, we showed the promising catalytic capacity of the oxygenase component and established its mode of reaction. We report an innovative and simplified reductase-free enzymatic system for BV oxidation, driven by NCB hydride donation for flavin mononucleotide (FMN) reduction and further electron mediation through the flavin. The purified type II BVMO 2,5-DKCMO from Pseudomonas putida ATCC 17453![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30,31,33–35 was applied in a reductase-free fashion and compared with a reductase-oxygenase two-component system. The regio- and enantioselective BV oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one (rac-1) was used as the model reaction. The reductase-free system was further investigated focusing on potential limits of the approach. Design of Experiments (DoE) was executed to estimate the impact of the seven reaction parameters and optimize the final system. The utilization of NCBs allowed us to address the previous question and demonstrated the compatibility of the monooxygenase with artificial hydride donors without a loss of activity. Finally, the tunability of the system to avoid flavin degradation is discussed and the concept of flavin recycling as the adjusting wheel of two-component systems is highlighted and validated by carrying out a complete reaction.
30,31,33–35 was applied in a reductase-free fashion and compared with a reductase-oxygenase two-component system. The regio- and enantioselective BV oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one (rac-1) was used as the model reaction. The reductase-free system was further investigated focusing on potential limits of the approach. Design of Experiments (DoE) was executed to estimate the impact of the seven reaction parameters and optimize the final system. The utilization of NCBs allowed us to address the previous question and demonstrated the compatibility of the monooxygenase with artificial hydride donors without a loss of activity. Finally, the tunability of the system to avoid flavin degradation is discussed and the concept of flavin recycling as the adjusting wheel of two-component systems is highlighted and validated by carrying out a complete reaction.
Results and discussion
Model reaction system
The great interest on type II BMVOs is due to their simple character, as they are separated reductase and oxygenase components. This feature may allow the transfer of the preformed FMNH2 to the oxygenase component by rapid diffusion without the need for any protein–protein interaction. It has been shown previously that multiple FMN reductases can act as alternative distal donors36 to the monooxygenase component. Consequently, it is reasonable to assume that it can accept FMNH2 from an origin other than enzymatically reduced if no protein–protein interaction with a reductase is required. Herein, the possibilities to tune such enzymatic type II BVMO reactions by the substitution of a reductase-mediated reduction of the FMN are very promising, since simplified reaction systems are of great potential for further applications, for instance, for an external electron supply through a variety of electron donors.The type II BVMO applied in this study is the well-known 2,5-DKCMO from P. putida ATCC 17453 (NCIMB 10007),30,31,33–35 which was produced via heterologous expression as an E. coli-codon-optimized version with an N-terminal His-tag for affinity chromatography purification (see ESI section A for further details†). A fast substrate profiling showed that rac-bicyclo[3.2.0]hept-2-en-6-one 1, an established substrate for the evaluation of enantioselective BV oxidation37 (Fig. 1), displayed the highest conversion rate among the assayed compounds (see ESI, Fig. S4†). The biotransformation of rac-1 by 2,5-DKCMO was already investigated by Lau and co-workers,34 as well as the group of Bornscheuer,35,38,39 but no complete characterization of the reaction had been made. Thus, we selected this compound as the model substrate for our study.
 | ||
| Fig. 1 Enantioselective and regioselective enzymatic BV oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one 1. Each enantiomer of the ketone is the precursor of two regioisomeric lactones 2 and 3. The thickness of the arrows represents the preference of 2,5-DKCMO. | ||
Biocatalyst formulations
Interestingly, the biocatalyst was stored at −20 °C for 21 days in a frozen as well as lyophilized form with minimal loss of activity. Both formulations maintained approx. 70–80% of the conversion rate of the ‘fresh’ biocatalyst, determined directly after purification. In the experiments with ‘fresh’ oxygenase, 84 ± 5% of the model substrate 1 was converted after two hours, whereas in the frozen enzyme system and the lyophilized one, 59 ± 5% and 67 ± 7% of conversion were observed respectively (see ESI, Fig. S6†). This observation indicates that 2,5-DKCMO is particularly resistant against lyophilization and freezing conditions compared to most BVMOs for which low stability is often the greatest hurdle.25,40 From a practical point of view, this quality was highly desirable since it allowed the easy storage of the biocatalyst, and consequently facilitated the comparison of various reaction conditions, avoiding frequent enzyme production and/or tedious normalization of the activity.Screening of oxygenase-catalysed reactions with NCBs
We investigated the potential of two synthetic nicotinamide analogues41 for FMN reduction. The direct FMN reduction requires stoichiometric amounts of hydride donors, which will be oxidized. The inexpensive cost of synthesising NCBs makes them attractive for this use. Moreover, in terms of electron transfer efficiency and atom economy, the NCBs are superior to the natural system.20 NCB-driven approaches were documented for other flavoenzymes, such as very recently for a regioselective aromatic hydroxylation,41 and other two-component FMPOs.20–22 These systems outperformed the natural enzymatic regeneration system proceeding at four times faster initial reaction rates21 and NCBs showed a very high Km compared with the natural nicotinamide cofactor.41 However, they have never applied to type II BVMO.The two partially water-soluble and stable NCBs, 1-benzyl-1,4-dihydronicotinamide (BNAH), and 1-(2-carbamoylmethyl)-1,4-dihydronicotinamide (AmNAH, caricotamide), were selected. To ensure a homogeneous reaction system, both NCBs were supplied from a stock solution in DMSO. The first experiments, with both NCBs and without reductase, showed a slow enantioselective conversion of ketone 1 with an increase of the enantiomeric excess in favour of the (+)-enantiomer and a preferential formation of (+)-(1R,5S)-2-oxabicyclo[3.3.0]oct-6-en-3-one 2, also called (+)-normal lactone. The reaction system is shown in Scheme 2. Both NCBs provided the hydride donation for the reduction of the flavin cofactor FMN before FMNH2 diffusion into the active site of the oxygenase. However, a difference of performance was observed as shown in Fig. 2 and AmNAH appears as the most suitable hydride donor. Both a higher ee of the substrate and a higher conversion were observed at the two tested concentrations (10 and 50 mM).
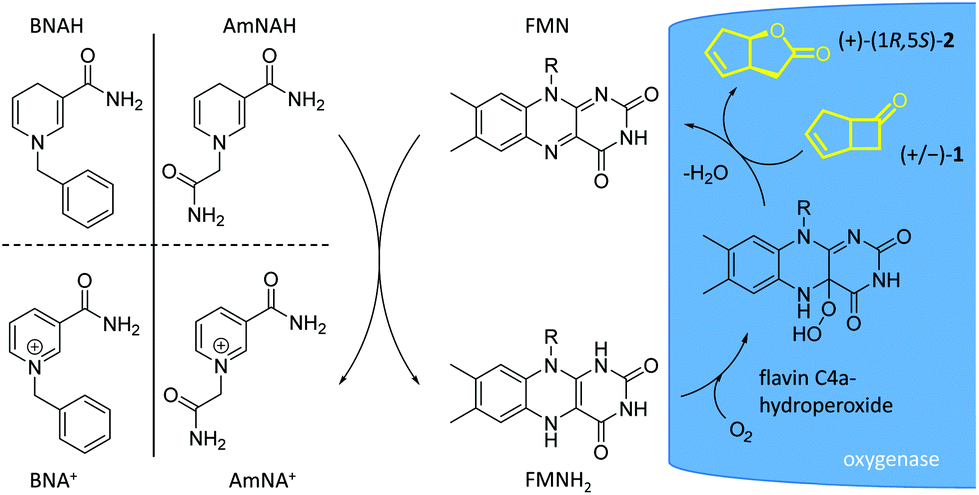 | ||
| Scheme 2 Reductase-free NCB-driven BV oxidation by the oxygenase of a two-component type II BVMO. The nicotinamide analogues BNAH and AmNAH transfer a hydride to FMN to give its fully reduced form in the absence of any reductase. The reduced FMNH2 diffuses into the active site of 2,5-DCKMO, where the flavin C4a-hydroperoxide is formed and the enantioselective BV oxidation takes place. The fully oxidized FMN diffuses into the reaction medium and is accessible for further cycles of chemical reduction by NCB and enzymatic (re)oxidation by monooxygenase. | ||
 | ||
| Fig. 2 Screening of the NCB-driven chemo-enzymatic enantioselective BV oxidation of rac-1 by 2,5-DCKMO. Conversion (%, ▲) and ketone 1 ee (%, red bars) after, respectively, two and four hours as the average of duplicates. Experimental conditions: 5 mM rac-1, 0.55 mg mL−1 purified and lyophilized oxygenase, 0.5 mg mL−1 catalase, and 100 μM FMN in 1 mL of Tris–HCl buffer (50 mM, pH 7.5 at 22 °C). | ||
The preliminary results did not show the full potential of the approach. The presence of reduced and oxidized flavin, as well as oxygen in the aqueous medium, is a potentially delicate composition.42–46 In absence of the catalase, a change of the colour of the reaction from pale yellow to orange was observed with AmNAH as shown in Fig. 3. This indicates the formation of anionic semiquinone flavin radicals and other flavin decomposition products, due to an uncoupling of the reduced flavins into radicals and hydrogen peroxide.47,48 H2O2 is known to be formed with flavoprotein monooxygenases, including two component styrene monooxygenase and halogenases.20,21 On the other hand, it is also known to oxidize substrates as ketone 1 in racemic lactone 2.49 In our case, the addition of catalase improved the biotransformation (Fig. 3) as illustrated by the complete disappearance of the (−)-ketone 1 in six hours along with a better ee of normal lactone 2 (82% and 55% respectively with and without catalase after two hours of reaction). Consequently, it can be concluded that catalase addition alleviates the unselective chemical BV oxidation by H2O2. In this regard, the stability of the enzyme in the presence of H2O2 was also evaluated.‡ After its storage in up to 50 mM H2O2 for four hours, no significant decline of activity was observed (data not shown), confirming the great stability of the oxygenase.
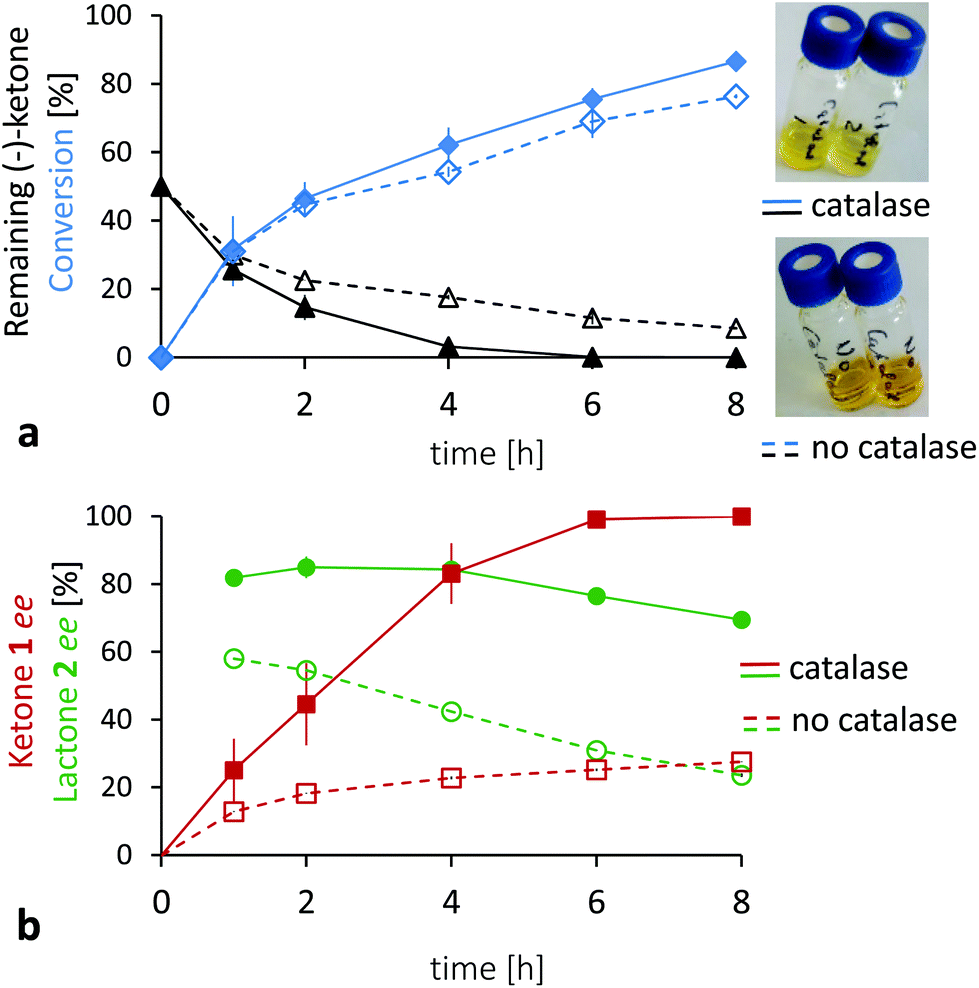 | ||
Fig. 3 Chemo-enzymatic enantioselective BV oxidations of rac-1 with AmNAH in the presence and absence of catalase. Experiments with catalase: plain lines —, full marks; without catalase: dashed lines - - -, empty marks. Graph (a): Conversion:  , ,  ; Remaining (−)-ketone 1: ▲, Δ. Graph (b): Normal lactone 2 ee: ; Remaining (−)-ketone 1: ▲, Δ. Graph (b): Normal lactone 2 ee:  , ,  ; Ketone 1 ee: ; Ketone 1 ee:  , ,  . The error bars indicate the standard deviation of two independent reactions. The pictures on the right show the reaction mixtures after 8 hours. Experimental conditions: 4 mM rac-1, 50 μM FMN, 50 mM AmNAH in 1 mL of tricine buffer (50 mM, pH 8.5) at 15 °C. 0.3 mg mL−1 lyophilized 2,5-DCKMO and, if mentioned, a great excess (2.5 mg mL−1) of catalase, but no flavin reductase added. . The error bars indicate the standard deviation of two independent reactions. The pictures on the right show the reaction mixtures after 8 hours. Experimental conditions: 4 mM rac-1, 50 μM FMN, 50 mM AmNAH in 1 mL of tricine buffer (50 mM, pH 8.5) at 15 °C. 0.3 mg mL−1 lyophilized 2,5-DCKMO and, if mentioned, a great excess (2.5 mg mL−1) of catalase, but no flavin reductase added. | ||
Control experiments for flavin reductase and AmNAH
To transfer electron mediators from one (bio)molecule to another, two basic mechanisms have evolved in nature: (i) either driven in an active fashion e.g. via protein–protein interaction as with cytochrome P450 enzymes (this interaction mediates the electron transfer from the flavin cytochrome P450 reductase to the heme of the oxygenase50) or (ii) passively by simple free diffusion as it has been shown for the two-component FPMO bacterial luciferase.51 In other two-component FPMOs, this transfer has been a pivotal question for the last decades that remains to be clarified,12,52 although the concept of free-diffusion of the reduced flavin to the oxygenase was predominant for most of the described systems.12,53,54 On the other hand, in the case of type I BVMO, it has been shown that NADPH contributes to enzymatic catalysis and interferes positively with enantioselectivity,55,56not making its substitution desirable.To determine the behaviour of our model type II BVMO system in the presence and absence of a reductase, purified flavin reductase (Fre) from E. coli was selected to act as the reductase counterpart. This enzyme was previously described as a suitable supplier of reduced FMN for 2,5-DKCMO.30 The Fre enzyme was applied in our experiments in an equimolar amount (compared to the monooxygenase) using NADH as the hydride donor. To evaluate the potential impact of Fre on the catalytic performance of the reaction system, identical setups without Fre served as control experiments (Fig. 4).
 | ||
Fig. 4 Enantioselective BV oxidation of the model substrate rac-1 by 2,5-DKCMO with various hydrogen donor systems. Graph (a): time course of experiments driven by AmNAH without Fre (plain lines —) and experiments with the Fre reductase and NADH (dashed lines - - -). Conversion of the rac-1: ▲, ketone ee:  . Graph (b): ketone ee versus conversion curve for experiments with AmNAH and without Fre (■ and —) as well as experiments with NADH and with Fre ( . Graph (b): ketone ee versus conversion curve for experiments with AmNAH and without Fre (■ and —) as well as experiments with NADH and with Fre ( ), and without the Fre reductase ( ), and without the Fre reductase ( ). In the green box frame: lactone ee versus the conversion curve, applying the same assignment. Experimental conditions: 4 mM rac-1, 25 mM NADH or AmNAH respectively, 5.0 μM FMN, 1 mL of tricine buffer (50 mM, pH 8.5) at 30 °C. 0.4 mg mL−1 frozen 2,5-DCKMO, 2.5 mg mL−1 catalase, and if required, lyophilized Fre was added in equimolar amounts to the oxygenase. Reactions were performed in duplicate. ). In the green box frame: lactone ee versus the conversion curve, applying the same assignment. Experimental conditions: 4 mM rac-1, 25 mM NADH or AmNAH respectively, 5.0 μM FMN, 1 mL of tricine buffer (50 mM, pH 8.5) at 30 °C. 0.4 mg mL−1 frozen 2,5-DCKMO, 2.5 mg mL−1 catalase, and if required, lyophilized Fre was added in equimolar amounts to the oxygenase. Reactions were performed in duplicate. | ||
Surprisingly, in terms of substrate conversion, the reductase-free system applying AmNAH as the hydride donor outperformed the reductase setup using NADH over time (see Fig. 4a). Similar results were obtained when five times more Fre enzyme was applied§ (data not shown). The NADH-driven Fre-system did not exceed a final conversion of 63 ± 0% (reached in less than two hours) in the equimolar Fre system, or 56 ± 2% (reached after three hours) when Fre was applied in five times excess, respectively. The reaction system without the reductase, driven by the direct FMN reduction via AmNAH, reached nearly full conversion (97 ± 1%) after six hours. The evolution of ee of ketone 1 was also in agreement with this observation.
Additionally, we compared the two systems with the reaction only driven by NADH (without Fre). To succeed in comparing the enantioselectivity of the three complex systems, the effect of different FMN reduction rates has to be eliminated. Thus, Fig. 4b shows the substrate ee as a function of the conversion. The plotted curves of the reductase-free reaction setup with AmNAH, as well as with NADH were perfectly supposable, and in agreement with the experiment associating the reductase and NADH.
With these experiments, we demonstrated the independence of 2,5-DKCMO from any flavin reductase enzyme exemplary by its independence from Fre. The oxygenating component of the chosen type II BVMO system maintained its enzymatic capacity in the absence of the reductase. Moreover, the nicotinamide cofactor was substituted by AmNAH, demonstrating that the hydride donor for in situ FMN reduction can be easily replaced. Therefore, we conclude that the mechanism of reduced flavin transfer in this two-component monooxygenase system does not require a protein–protein interaction but is achieved passively by free diffusion.
Characterization of the reductase-free reaction system
Since the stability of the oxygenase is not questioned, we ascribe the early cessation of the reaction observed in some experiments to an imbalance of the reaction system. Indeed, FMN recycling has a particularly delicate balance, as it depends on fine adjustments of the concentrations of the hydride donor, the enzyme, and the flavin itself. To investigate this hypothesis and to further assess the characteristics of the system, we took advantage of the adaptability of 2,5-DKCMO regarding the reductase and hydride donor for FMN reduction.The impact of different reaction parameters in the reductase-free setup was tested using a DoE (Fig. 5) comprising NCB type and concentration, FMN and oxygenase concentration as well as temperature and buffer type, and pH (for DOE details, see ESI, Table S1†).
 | ||
| Fig. 5 Screening by DoE of parameter effects on reductase-free 2,5-DKCMO-mediated enantioselective BV oxidations of rac-1. The (i) enantiomeric excess of the ketone 1 at three hours and (ii) total conversion after 22 hours were selected to calculate the DoE output. The parameters are shown to the left, the levels are shown to the right: e.g. [FMN] is the parameter, and 25–150 μM are the levels. The length of the bars represents the impact of the parameter levels on ketone ee and conversion. Greater effects are shown by larger bars. Blue numbers show some changes in the values of outputs compared to the reference level (black bars and dashed lines). Further explanations are provided in the ESI.† Experimental conditions: catalase was added in great excess (2.5 mg mL−1), NCBs were supplied from a DMSO stock solution (1 M) to a total volume of 0.5 mL, other parameters were modified as shown in the figure. | ||
The ee of the ketone after three hours and the final conversion were chosen as data for the DoE calculation. They allowed respectively an insight into (i) the reaction rate (activity) of the system in the early stages of the reaction, by monitoring the enantioselectivity of the enzyme, and (ii) the durability of the system by monitoring the capacity of the enzyme to transform the substrate in 22 hours. The latter one is of particular interest because it depends on the process stability of all species present in its environment.
The DoE results are shown in Fig. 5. It is important to note that the length of bars in the figure is not proportional to the quantitative outcomes, but represents the influence of a given parameter on the outcomes. From the DoE results (Fig. 5), we concluded that the buffer (both nature and pH) had a relatively small effect on both outputs, and hence on the whole system. Unsurprisingly, the temperature and oxygenase concentration affected the system during the entire reaction. All other tested parameters affected the response largely but with varying degrees of intensity in the early and later states. The same observation was observed for the FMN concentration, but with a greater effect of lower concentrations at the early stage to a lower impact in the late stage.
The NCBs revealed a clear influence of their nature and concentration on the performance of the system. Interestingly, the impact of the N-substituent type, an amide (AmNAH) or phenyl group (BNAH), was the strongest parameter at the early stage of the reaction (ketone ee at three hours), as shown by the AmNAH bar being considerably longer than its BNAH counterpart. We attribute the early stage-impact to the dissimilar flavin reduction rate (NCB-driven hydride transfer) evoked by different (electro)chemical characteristics e.g. redox potential of the NCBs,57 but also their chemical stability in aqueous solution. Although BNAH is the stronger reducing agent, its half-life time in aqueous media (approx. 1.5 hours) is more than ten times shorter compared to AmNAH.16,41 At the end of the reaction, the impact of both NCBs decreased, as the rate of hydride transfer to FMN became negligible, and therefore insignificant. Hydride donation was not only affected by the type of hydride donor (NCBs) but also by the concentration, as we observed an impact of this parameter at the early stage and the end of the reaction. Interestingly, this shifts over time with an increasing impact of lower concentrations at the later stage of the reaction. Thus, lower NCB concentrations did not result in higher conversion. However, it must be noted that the interpretations gained from the DoE are only valid within its boundaries, e.g. an impact of a pH value below 7.5, or above 8.5 might be considerable.
The analysis of DoE results shows that all parameters with a great impact on the reaction system are associated directly with the FMN/FMNH2 cycle (see Fig. 6). This applies to the reduction of the flavin, e.g. the type and concentration of the NCBs (see Fig. 6 in blue), but also to its enzymatic FMNH2 (re)oxidation for which the oxygenase concentration (see Fig. 6 in red) is crucial.
 | ||
| Fig. 6 Impact on the reaction parameter of the reductase-free NCB driven model reaction. The parameters affecting the (abiotic) reduction of the FMN are the type and concentration of the NCB (in blue). The oxygenase concentration catalyses the (re)oxidation of the fully reduced flavin (in red) and hence is the important counterpart in this cycle. Temperature (not shown) and FMN concentration (in black, in the centre) contribute to both chemical reduction and enzymatic (re)oxidation of the flavin. | ||
The temperature was also identified as a relevant parameter in the system, which is in accordance with our interpretation, as it affects both flavin reduction and (re)oxidation, albeit to a different extent (reduction = chemically versus oxidation = enzymatically). In some experiments, an increased chemical oxidation of ketone but no bioconversion was observed. Therefore, the impact of the parameter on both the chemical and chemoenzymatic performance of the reaction system was also evaluated.
As a consequence of the identification of the key role of the flavin and its function as the adjusting wheel of the reaction, we assessed the FMN reduction/(re)oxidation cycle to be crucial for a (long-time) catalysis. The reduction and oxidation rates of the flavin need to be balanced to ensure the greater stability of the system's components, in particular, to minimize the instability of the reduced flavin in the presence of oxygen.
Improvement of the system
A second DoE was performed with AmNAH, identified to give the highest conversions of our model substrate rac-ketone 1. This DoE aimed to optimise the reaction for maximum conversion and high enantioselectivity. Herein, we also included the other two oxygenase substrates, rac-camphor (rac-4) and rac-norcamphor (rac-5) (see ESI, Fig. S4†). Maintaining the reaction parameters affecting FMN reduction (NCB type and concentration), as well as the flavin (re)oxidation (oxygenase concentration) constant, we investigated the optimum FMN concentration and the temperature for the three substrates, respectively within the intervals of 10–25 μM and 12–30 °C. The outputs, conversion and ketone ee were measured after two hours for all the compounds (Fig. 7). | ||
| Fig. 7 Optimization by DoE of the enzymatic BV oxidation of rac-1 (a and d), rac-camphor 4 (b and e), and rac-norcamphor 5 (c and f) in a reductase-free system. The two parameters, [FMN] and temperature, varied from 12 °C to 30 °C and from 10 μM to 25 μM. The outputs are (i) the total conversion (a–c), and (ii) the enantiomeric excess (d–f) after two hours. Note: different scales are displayed for the same colour code regarding the DoE output. Experimental conditions: 5 mM substrate, 0.19 mg mL−1 purified and frozen oxygenase, 25 mM AmNAH, 2.5 mg mL−1 catalase in 50 mM tricine buffer at pH 8.5. | ||
Among the tested substrates, 2,5-DKCMO showed the highest activity towards the model substrate rac-1 with a maximum conversion of approx. 75% (Fig. 7a), followed by rac-4 with >35% (Fig. 7b) with an exclusive conversion of (1R,4R)-(+)-camphor. rac-5 is a rather poor substrate achieving conversions less than 20% (Fig. 7c) within the first two hours under the given conditions. For all tested substrates we observed greater conversions (Fig. 5a–c) at higher temperatures. For rac-1, low and medium FMN concentration (up to 20 μM) resulted in better performances. Similar results were observed with rac-4 (or more precisely (1R,4R)-(+)-camphor) and rac-5 despite significantly lower rates.
Regarding the ee of the substrates (Fig. 7d–f) we observed (i) an effect of the temperature on the rac-1, resulting in highest ee values at low to moderate temperatures and (ii) an effect of the FMN concentration on rac-4 with a clear trend for higher ee at low FMN concentrations. In this context, we expect the optimal FMN concentration for the reaction to be lower than our tested range. The results of rac-5 however, are difficult to evaluate due to the low conversion in the DOE condition window.
Summarizing the results of the second DoE, we observed the general trend for better enzymatic performance at lower to mediate temperatures and flavin concentrations.
These data support our hypothesis of the flavin/flavin-H2 cycle being the bottleneck and hence the major adjusting wheel in the reductase-free setup. It is also noticeable that the optima for the three compounds are slightly different, which reflects the influence of different enzymatic oxidation rates. Therefore, in a virtually optimized reaction setup, we expect to accomplish experimental conditions, in which flavin reduction and the (enzymatic) (re)oxidation are appropriately balanced.
Enantioselective in vitro biotransformation of rac-1 by 2,5-DKCMO
The biotransformation of rac-1 by 2,5-DKCMO was already tested34,35,38,39 but no complete description had been made. A typical gas chromatogram of the enzymatic bioconversion is shown in Fig. S5 in the ESI.† The knowledge gained from the DoEs allowed us to apply improved conditions for full conversion and high enantioselectivity as shown in Fig. 8 to characterize the model reaction in detail. This, however, is not trivial, as enzyme reaction rates may slightly vary from batch to batch, which requires the adaption of the flavin reduction rate to ensure the stability of the system. A set of adapted conditions using AmNAH allows examining the evolution of all compounds present in the reaction as reported in Fig. 8. As no alcohol formation was observed, reduction of the ketone by AmNAH was excluded. | ||
| Fig. 8 Time-course of the model biotransformation of rac-1 catalysed by 2,5-DKCMO. The curves represent the conversion and the remaining ketone enantiomer percentage. Bars show the lactone yields. The standard deviations were calculated from duplicates. Experimental conditions: 5 mM rac-1, 0.19 mg mL−1 purified and frozen oxygenase, 2.5 mg mL−1 catalase, 25 mM AmNAH, 5.0 μM FMN in 1 mL of tricine buffer (50 mM, pH 8.5) at 14 °C. | ||
The BV oxidation of each enantiomer of 1 offers theoretically two possibilities, as shown in Fig. 1. (−)-Ketone 1 can be transformed into either the so-called (+)-normal lactone 2 or (−)-abnormal lactone 3, while (+)-ketone delivers the opposite enantiomers. The temperature of the biotransformation was lowered to 14 °C for a better vision of the evolution of each enantiomer. Indeed, as suggested in Fig. 7d and in accordance with a behaviour already described for various enzymes,58,59 enantioselectivity was favourably affected by a drop in temperature. Under our reaction conditions, 2,5-DKCMO converted (−)-(1S,5R)-enantiomer 1 was first obtained as shown in Fig. 8. The beginning of the bioconversion was therefore characterized by the formation of (almost exclusively) (+)-normal lactone 2. This demonstrates the high enantio- and regioselectivity of the reaction. Surprisingly, once the (−)-(1S,5R)-1 was (almost) consumed, the oxygenase converted also the (+)-(1R,5S)–1 at similar rates compared to the enantiomeric counterpart. This conversion gave birth to both (−)-normal lactone 2, as well as the (+)-abnormal lactone 3. To the best of our knowledge, such sequential oxidation of enantiomers of the rac-1 has only been described for the one-component phenylacetone monooxygenase (PAMO) with non-natural FAD derivates.60
Experimental details
Note: a detailed description of procedures is available in the ESI section A – Material and Methods.†Chemicals
rac-1, rac-4, and rac-5 as well as the other tested substrates were obtained from chemical suppliers. Authentic lactone samples were synthesized by microbiological biotransformation according to described procedures.61,62 NCBs were synthesized as described in the literature41 and used from a stock solution in DMSO (1 M).Preparation of the oxygenase
Biotransformation and gas chromatography analyses
Sealed 2 mL glass vials were used for all bioconversions. Typically, the total reaction volume was set to 1 mL or 0.5 mL, respectively. Samples were taken from the same batch over time, directly from the reaction medium (buffer), and extracted in ethyl acetate solution containing 0.5 g L−1 of tridecane as the internal standard, in a volumetric ratio of 1 to 2.GC analyses of the three ketones and their corresponding lactones were carried out on a Cyclosil-B column (Agilent J&W GC columns). Product identification was ensured by the comparison of GC retention times with those of authentic samples (see ESI A†).
Design of experiments (DoE)
AZURAD software (http://www.azurad.fr) was applied for the DoEs using a polynomial model with linear, quadratic, and interaction effects. Multilinear regression was applied to calculate the coefficients in the modelling step. The details are shown in ESI A in Tables S1, S2 and Fig. S7.†Conclusions
The high stability of the oxygenase part of the two-component BVMO system of 2,5-DKCMO, as well as its independence from the reductase, offers multiple possibilities to apply this biocatalyst in a modular fashion, as long as the availability of fully reduced flavin is provided. In this regard, it is essential to balance (i) FMN reduction, (ii) diffusion-driven transport, and (iii) (re)oxidation of the oxygenase. FMN diffusion is the most difficult to customize. Adjusting the FMN reduction rate, or the (specific) activity of the oxygenase, respectively, offers a more convenient strategy. Notably, tuning should be adopted to the other parameters to ensure the balanced state of the flavin recycling.The reductase-free setup of the redox system finally demonstrated that the transport mechanism of reduced flavin to the oxygenase in this study proceeded by free diffusion. This reductase-independent enzymatic BV oxidation enables the reduction of the complexity of this two-component enzyme. In addition, it questions the terminology of the system(s), since the reductase might only be required in the natural (in vivo) context, but can be removed in artificial systems, still maintaining the oxygenase functionality. 2,5-DKCMO might be characterized as one component with the ability to utilize (hydroquinone) electron mediators, or flavin hydroquinone-dependent monooxygenase. Therefore, for other representatives of two-component FPMOs, each oxygenase component could advantageously be investigated separately from its reductase counterpart.
We gave herein an additional example of the synthetic potential of two-component flavoprotein monooxygenases. The proof of the independence of 2,5-DCKMO from any reductase enzyme is the foundation of further reductase-free bioconversion concepts for BV oxidations. Although the enzyme in this study has a narrow substrate scope, we are convinced our work will encourage the search for more promiscuous enzymes of this family.
We see the great potential of similar systems, applying alternative FMN reduction strategies directly mediated in the solution, to elaborate simplified and efficient enzymatic redox reaction systems. Such a reductase-free system, associated with potentially cost-efficient hydride donors can serve as a promising platform. Thus, innovative and sustainable flavin-dependent biotransformations driven by in situ flavin reduction, as for example with transition metal-catalysed hydride transfers, can be target applications for further studies.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 764920. NCBs were synthesized41 and kindly provided by MSc. Alice Guarneri (Wageningen University, The Netherlands).References
- G. R. Krow, Org. React., 1993, 43, 251–296 CrossRef
.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef
- H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef PubMed
- M. J. L. J. Fürst, A. Gran-Scheuch, F. S. Aalbers and M. W. Fraaije, ACS Catal., 2019, 9, 11207–11241 CrossRef
-
O. Hayaishi, Y. Ishimura, T. Nakazawa, M. Nozaki and R. W. Estabrook, in Biochemie des Sauerstoffs, Springer-Verlag Berlin, 1968, pp. 196–228 Search PubMed
-
O. Hayaishi, in Encyclopedia of Biological Chemistry, Elsevier, 2nd edn, 2013, pp. 371–374 Search PubMed
- Z. Li, J. B. van Beilen, W. A. Duetz, A. Schmid, A. de Raadt, H. Griengl and B. Witholt, Curr. Opin. Chem. Biol., 2002, 6, 136–144 CrossRef
- V. B. Urlacher and R. D. Schmid, Curr. Opin. Chem. Biol., 2006, 10, 156–161 CrossRef PubMed
- M. M. E. Huijbers, S. Montersino, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Arch. Biochem. Biophys., 2014, 544, 2–17 CrossRef
- A. Willetts, Trends Biotechnol., 1997, 15, 55–62 CrossRef
- W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef
- J. Sucharitakul, R. Tinikul and P. Chaiyen, Arch. Biochem. Biophys., 2014, 555–556, 33–46 CrossRef PubMed
- N. J. Oppenheimer, Mol. Cell. Biochem., 1994, 138, 245–251 CrossRef PubMed
- B. M. Anderson and C. D. Anderson, J. Biol. Chem., 1963, 238, 1475–1478 CrossRef
- C. O. Schmakel, K. S. V. Santhanam and P. J. Elving, J. Am. Chem. Soc., 1975, 97, 5083–5092 CrossRef PubMed
- R. J. Knox, T. C. Jenkins, S. M. Hobbs, S. Chen, R. G. Melton and P. J. Burke, Cancer Res., 2000, 60, 4179–4186 Search PubMed
- C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernández, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180–183 CrossRef
- T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033–1039 CrossRef
- S. A. Löw, I. M. Löw, M. J. Weissenborn and B. Hauer, ChemCatChem, 2016, 8, 911–915 CrossRef
- C. E. Paul, D. Tischler, A. Riedel, T. Heine, N. Itoh and F. Hollmann, ACS Catal., 2015, 5, 2961–2965 CrossRef
- M. Ismail, L. Schroeder, M. Frese, T. Kottke, F. Hollmann, C. E. Paul and N. Sewald, ACS Catal., 2019, 9, 1389–1395 CrossRef CAS
- J. Phonbuppha, R. Tinikul, T. Wongnate, P. Intasian, F. Hollmann, C. E. Paul and P. Chaiyen, ChemBioChem, 2020, 21, 2073–2079 CrossRef CAS PubMed
- A. Riebel, H. M. Dudek, G. de Gonzalo, P. Stepniak, L. Rychlewski and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2012, 1–11 Search PubMed
- S. Ménil, J. Petit, E. Courvoisier-Dezord, A. Debard, V. Pellouin, T. Reignier, M. Sergent, V. Deyris, K. Duquesne, V. Berardinis and V. Alphand, Biotechnol. Bioeng., 2019, 116, 2852–2863 CrossRef
- M. Bučko, P. Gemeiner, A. Schenkmayerová, T. Krajčovič, F. Rudroff and M. D. Mihovilovič, Appl. Microbiol. Biotechnol., 2016, 100, 6585–6599 CrossRef
- T. Reignier, V. de Berardinis, J.-L. Petit, A. Mariage, K. Hamzé, K. Duquesne and V. Alphand, Chem. Commun., 2014, 50, 7793 RSC
- A. Willetts, Microorganisms, 2019, 7, 395 CrossRef CAS
- A. Willetts, Microorganisms, 2018, 7, 1 CrossRef
- A. Willetts, P. Masters and C. Steadman, Microorganisms, 2018, 6, 41 CrossRef
- M. Kadow, K. Balke, A. Willetts, U. T. Bornscheuer and J.-E. Bäckvall, Appl. Microbiol. Biotechnol., 2014, 98, 3975–3986 CrossRef CAS PubMed
- D. G. Taylor and P. W. Trudgill, J. Bacteriol., 1986, 165, 489–497 CrossRef CAS
- E. Conrad, R. DuBus, M. J. Namtvedt and I. C. Gunsalus, J. Biol. Chem., 1965, 240, 495–503 CrossRef
- A. Willetts and D. Kelly, Microorganisms, 2016, 4, 38 CrossRef PubMed
- H. Iwaki, S. Grosse, H. Bergeron, H. Leisch, K. Morley, Y. Hasegawa and P. C. K. Lau, Appl. Environ. Microbiol., 2013, 79, 3282–3293 CrossRef CAS PubMed
- M. Kadow, S. Saß, M. Schmidt and U. T. Bornscheuer, AMB Express, 2011, 1, 1–8 CrossRef
- A. Willetts, Microorganisms, 2019, 7, 1 CrossRef CAS
- A. Watanabe, T. Uchida, R. Irie and T. Katsuki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5737–5742 CrossRef CAS PubMed
-
M. Kadow, Dissertation, Ernst-Moritz-Arndt-Universität Greifswald, 2012
- M. Kadow, K. Loschinski, S. Saß, M. Schmidt and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2012, 1–11 Search PubMed
- K. Balke, A. Beier and U. T. Bornscheuer, Biotechnol. Adv., 2018, 36, 247–263 CrossRef CAS
- A. Guarneri, A. H. Westphal, J. Leertouwer, J. Lunsonga, M. C. R. Franssen, D. J. Opperman, F. Hollmann, W. J. H. Berkel and C. E. Paul, ChemCatChem, 2020, 12, 1368–1375 CrossRef CAS
- Q. Gibson and J. Hastings, Biochem. J., 1962, 83, 368–377 CrossRef CAS
- J. H. Swinehart, J. Am. Chem. Soc., 1966, 88, 1056–1058 CrossRef CAS
- A. Ehrenberg, F. Muller and P. Hemmerich, Eur. J. Biochem., 1967, 2, 286–293 CrossRef CAS
- N. V. Beaudette and N. Langerman, Arch. Biochem. Biophys., 1974, 161, 125–133 CrossRef CAS
- J. Lind and G. Merényi, Photochem. Photobiol., 1990, 51, 21–27 CrossRef CAS
- G. M. Kishore and E. E. Snell, Biochem. Biophys. Res. Commun., 1979, 87, 518–523 CrossRef CAS
- D. Holtmann and F. Hollmann, ChemBioChem, 2016, 17, 1391–1398 CrossRef CAS
- Y. Tsuda, T. Tanno, A. Ukai and K. Isobe, Tetrahedron Lett., 1971, 12, 2009–2012 CrossRef
- T. Iyanagi, C. Xia and J.-J. P. Kim, Arch. Biochem. Biophys., 2012, 528, 72–89 CrossRef CAS
- R. Tinikul, W. Pitsawong, J. Sucharitakul, S. Nijvipakul, D. P. Ballou and P. Chaiyen, Biochemistry, 2013, 52, 6834–6843 CrossRef CAS
- D. Tischler, R. Kermer, J. A. D. Groning, S. R. Kaschabek, W. J. H. van Berkel and M. Schlomann, J. Bacteriol., 2010, 192, 5220–5227 CrossRef CAS
- B. N. Webb, J. W. Ballinger, E. Kim, S. M. Belchik, K.-S. Lam, B. Youn, M. S. Nissen, L. Xun and C. Kang, J. Biol. Chem., 2010, 285, 2014–2027 CrossRef CAS
- Z. T. Campbell and T. O. Baldwin, J. Biol. Chem., 2009, 284, 8322–8328 CrossRef CAS
- G. de Gonzalo, G. Ottolina, G. Carrea and M. W. Fraaije, Chem. Commun., 2005, 3724 RSC
- A. Alfieri, E. Malito, R. Orru, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6572–6577 CrossRef CAS
- C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2014, 4, 788–797 CrossRef CAS
- R. S. Phillips, Enzyme Microb. Technol., 1992, 14, 417–419 CrossRef CAS
- V. Kasche, B. Galunsky, A. Nurk, E. Piotraschke and A. Rieks, Biotechnol. Lett., 1996, 18, 455–460 CrossRef CAS
- C. Martinoli, H. M. Dudek, R. Orru, D. E. Edmondson, M. W. Fraaije and A. Mattevi, ACS Catal., 2013, 3, 3058–3062 CrossRef CAS
- I. Hilker, M. C. Gutiérrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3, 546–554 CrossRef CAS
- L. Butinar, M. Mohorčič, V. Deyris, K. Duquesne, G. Iacazio, M. Claeys-Bruno, J. Friedrich and V. Alphand, Phytochemistry, 2015, 117, 144–153 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00015b |
| ‡ Assay conditions: 50 μL mL−1 oxygenase, 50 μM FMN, 25 mM NADH as the hydride source, 50 mM Tris-HCl buffer at pH 7.5, 20 °C and 160 rpm. |
| § Five times excess compared with the monooxygenase using the same NADH concentration. |
| This journal is © The Royal Society of Chemistry 2021 |