
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the mechanism of the chiral phosphoric acid-catalyzed aza-Cope rearrangement†
Bruno N.
Falcone
 a and
Matthew N.
Grayson
*b
a and
Matthew N.
Grayson
*b
aSSPC, the SFI Research Centre for Pharmaceuticals, School of Chemistry, University College Cork, Cork, T12 K8AF, Ireland
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: M.N.Grayson@bath.ac.uk
First published on 30th March 2021
Abstract
The first catalytic enantioselective aza-Cope rearrangement was reported in 2008 by Rueping et al. The reaction is catalyzed by a 1,1′-bi-2-naphthol-derived (BINOL-derived) phosphoric acid and achieved high yields and enantioselectivities (up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er with 75% yield). This work utilizes Density Functional Theory to understand the mechanism of the reaction and explain the origins of the enantioselectivity. An extensive conformational search was carried out to explore the different activation modes by the catalyst and, the Transition State (TS) leading to the major product was found to be 1.3 kcal mol−1 lower in energy than the TS leading to the minor product. The origin of this stabilization was rationalized with NBO and NCI analysis: it was found that the major TS has a greater number of non-bonding interactions between the substrate and the catalyst, and shows stronger H-bond interactions between H atoms in the substrate and the O atoms in the phosphate group of the catalyst.
:3 er with 75% yield). This work utilizes Density Functional Theory to understand the mechanism of the reaction and explain the origins of the enantioselectivity. An extensive conformational search was carried out to explore the different activation modes by the catalyst and, the Transition State (TS) leading to the major product was found to be 1.3 kcal mol−1 lower in energy than the TS leading to the minor product. The origin of this stabilization was rationalized with NBO and NCI analysis: it was found that the major TS has a greater number of non-bonding interactions between the substrate and the catalyst, and shows stronger H-bond interactions between H atoms in the substrate and the O atoms in the phosphate group of the catalyst.
Introduction
In 2008, Rueping et al.1 reported the first catalytic enantioselective aza-Cope rearrangement2 (Fig. 1), which consists in a sigmatropic rearrangement3–6 involving a nitrogen-containing species of which a diastereoselective version had previously been reported.7 This important reaction provides a valuable route for the synthesis of chiral homoallylic amines, useful intermediates in the synthesis of natural products,8 and other organic compounds such as β-amino acids, aminoalcohols, aminoepoxides, pyrrolidines, and piperidines.9–11 The reaction is catalyzed by a 1,1′-bi-2-naphthol-derived (BINOL-derived) phosphoric acid. This kind of catalyst has been utilized in enantioselective versions of a wide range of reactions,12,13 and has been the focus of studies of several computational works,14–20 including many in our group.21–24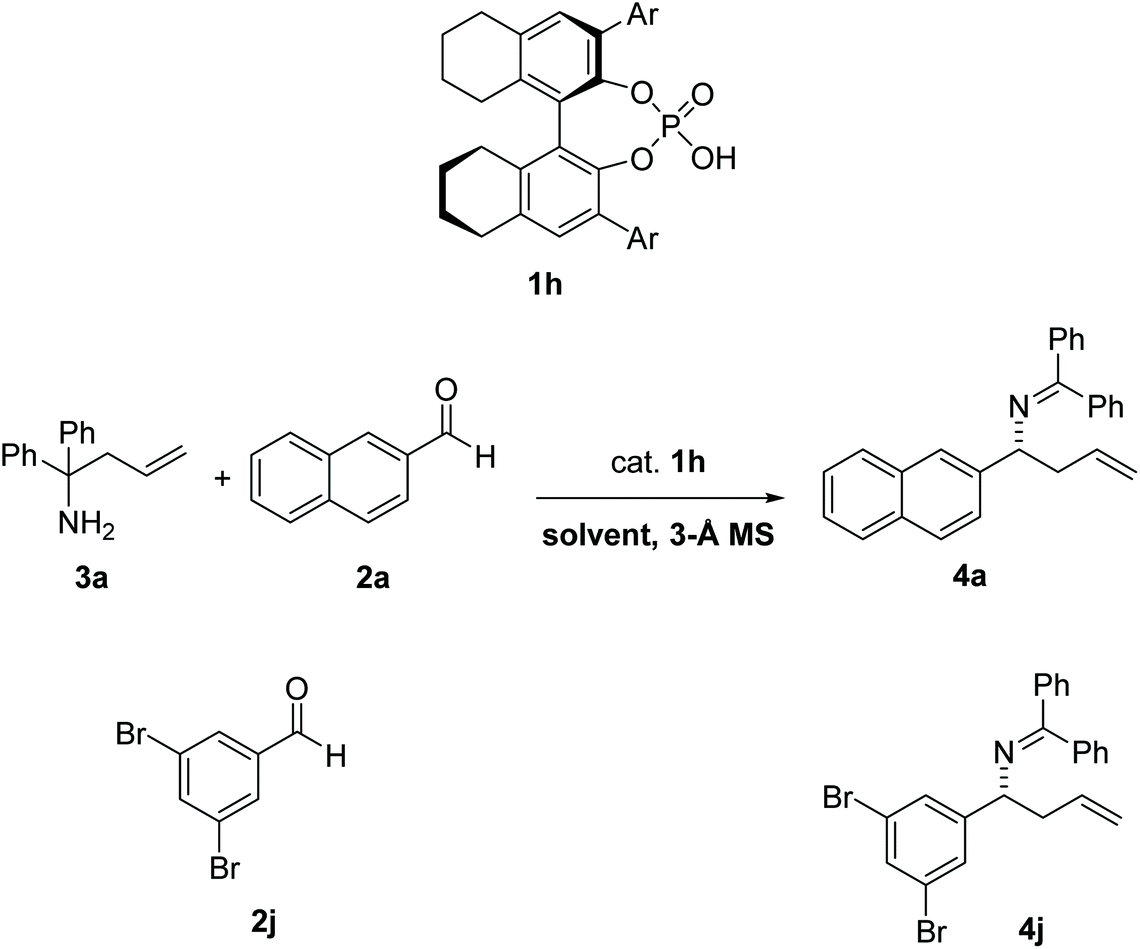 | ||
| Fig. 1 Reaction conditions for the phosphoric-acid catalyzed 2-aza-Cope rearrangement. Ar: 2-Naphthyl. | ||
In this work, we studied the mechanism of the reaction using Density Functional Theory (DFT). An extensive conformational search was carried out to explore the different activation modes by the catalyst and the transition states (TSs) of the C–C forming step were located, different activation modes were analyzed, and the origin of the enantioselectivity was explained. NCI plots showed a greater number of non-bonding interactions and Natural Bond Analysis showed stronger H-bond interactions, between the substrate and catalyst for the TS leading to the major product.
Computational methods
We have performed DFT calculations with Gaussian 0925 using the M06-2X26 functional. An ultrafine integration grid was used for all calculations. Optimizations were carried out with the 6-31G* basis set in the gas phase. Single point energies27 were obtained for the optimized structures using the M06-2X functional with the polarized, triple-zeta valence quality def2-TZVPP basis set28 and the SMD29 solvent model. Dibutylether was used as a model solvent for MTBE, since it was previously found to give valid results.30 Reported energies refer to single point energies for the model system, and to free energies for the full system (i.e. single point energies with thermal corrections arising from translational, rotational, vibrational and electronic motions as implemented in Gaussian 09, calculated at the M06-2X 6-31G* level).Conformational searches were carried out with MacroModel version 12.331 using the OPLS3 force field31 in the gas phase and a mixed torsional Monte Carlo Multiple Minimum (MCMM) and low frequency mode (LMOD) sampling. A sufficient number of steps were chosen so as to obtain each conformation at least five times. In order to minimize the number of conformations of the full catalyst, the cyclohexyl rings were fixed in the lowest energy conformation, as was previously reported.22 The H-bond between the NH in the substrate and the phosphate in the catalyst was fixed at 1.8 Å, and the breaking and forming bonds were fixed at 1.7 Å and 2.0 Å respectively, which correspond to the TS distances consistent with previous work32 and model calculations. The Paton group have investigated sampling ion-pairs with explicitly solvated MD simulations to great effect.33 We opted for constrained conformational sampling to enable us to proceed straight to TS optimization in Gaussian.
Results and discussion
A chiral phosphoric-acid catalyzed 2-aza-Cope rearrangement reaction has recently been reported.1 The reaction has been tested with a variety of aldehydes (11 substrates) and amine 3a. The largest er was observed with aldehyde 2j leading to product 4j, in the presence of 10% catalyst 1h. The reaction was also evaluated in different solvents with substrate 2a, leading to product 4a, and the highest er was obtained in toluene (87.5:12.5 er) and methyl tert-butyl ether (MTBE, 91:9 er) (Table 1).
| Entry | Aldehyde | Product | Solvent | Yield | er |
|---|---|---|---|---|---|
| 1 | 2a | 4a | Toluene | n/a | 87.5:12.5 |
| 2 | 2a | 4a | MTBE | 77% | 91:9 |
| 3 | 2j | 4j | MTBE | 75% | 97:3 |
The proposed mechanism of the 2-aza-Cope rearrangement (Fig. 2) starts with the formation of an imine by reaction between the starting amine and the aldehyde. The Brønsted-acid catalyzed rearrangement proceeds via a six-membered transition state, activated by the formation of an H-bond between the intermediate imine and the catalyst, in which a new C–C bond is formed with the terminal alkene acting as the nucleophile and the imine as the electrophile, generating a new tetrahedral carbon center. The newly formed product then loses the association with the phosphoric acid, regenerating the catalyst.
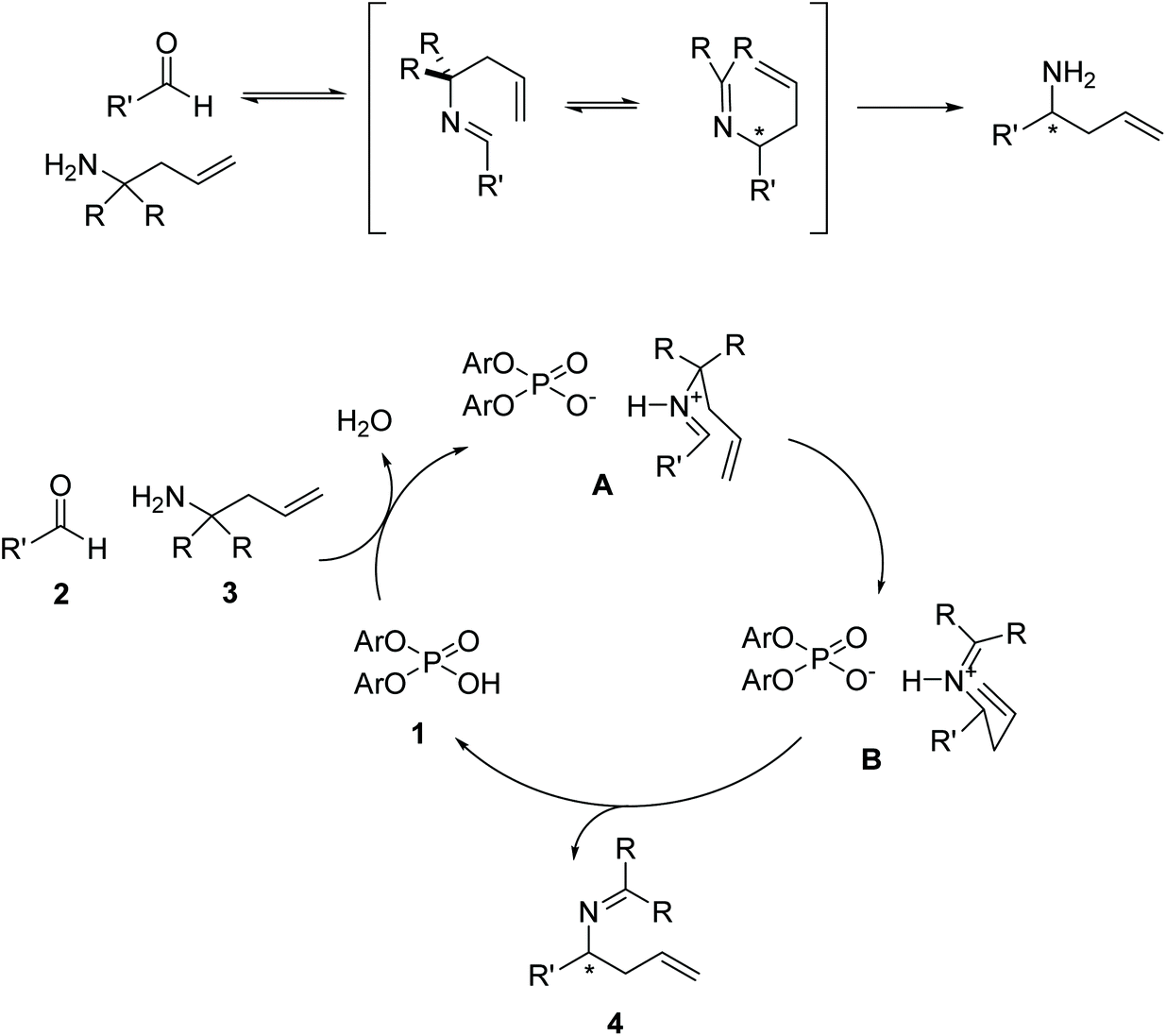 | ||
| Fig. 2 Mechanism of the phosphoric acid catalyzed 2-aza-Cope rearrangement. | ||
In this work, the origin of the enantioselectivity in the A to B rearrangement step was studied using DFT. First, different activation modes of the transition state were located through a comprehensive conformational search followed by optimization, by using butadiene phosphoric acid as a model catalyst and a phenyl substituent as R′ (Fig. 3). Only TS structures with a chair conformation of the six membered ring, and with the bulky R′ group in equatorial position were considered, as both literature and our calculations showed that conformations in boat conformation or with the R′ group in axial position were too high in energy.34 73 such conformations were located, in which different H-bond patterns are observed between the H atoms of the reacting substrate and the O atoms of the catalyst. The numbering of atoms is shown in Fig. 3. H-bonds were considered to be formed if the O–H distance was below 2.6 Å. The simplest activation pattern TS-I is observed by the formation of a single H-bond between the N–H atom and the negatively charged O atom, with a 1.52 Å distance (Fig. 4). However, this conformation was found to be 7.8 kcal mol−1 higher in energy than the lowest energy conformation, therefore a substantial extra stabilization is observed by the formation of additional H-bonds. The lowest energy conformation TS-A shows four H-bonds, one between N–H and O1, two H-bonds between both H2 and H3 and O2, and a further H-bond between H2 and the bridging O3. All other activation modes show either three or four total H-bonds, with the highest energy conformation TS-H being 3.1 kcal mol−1 above TS-A.
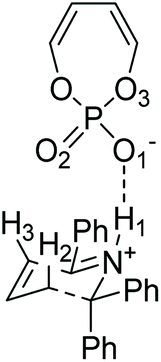 | ||
| Fig. 3 Model catalyst and substrate to study the different activation modes in the TS. | ||
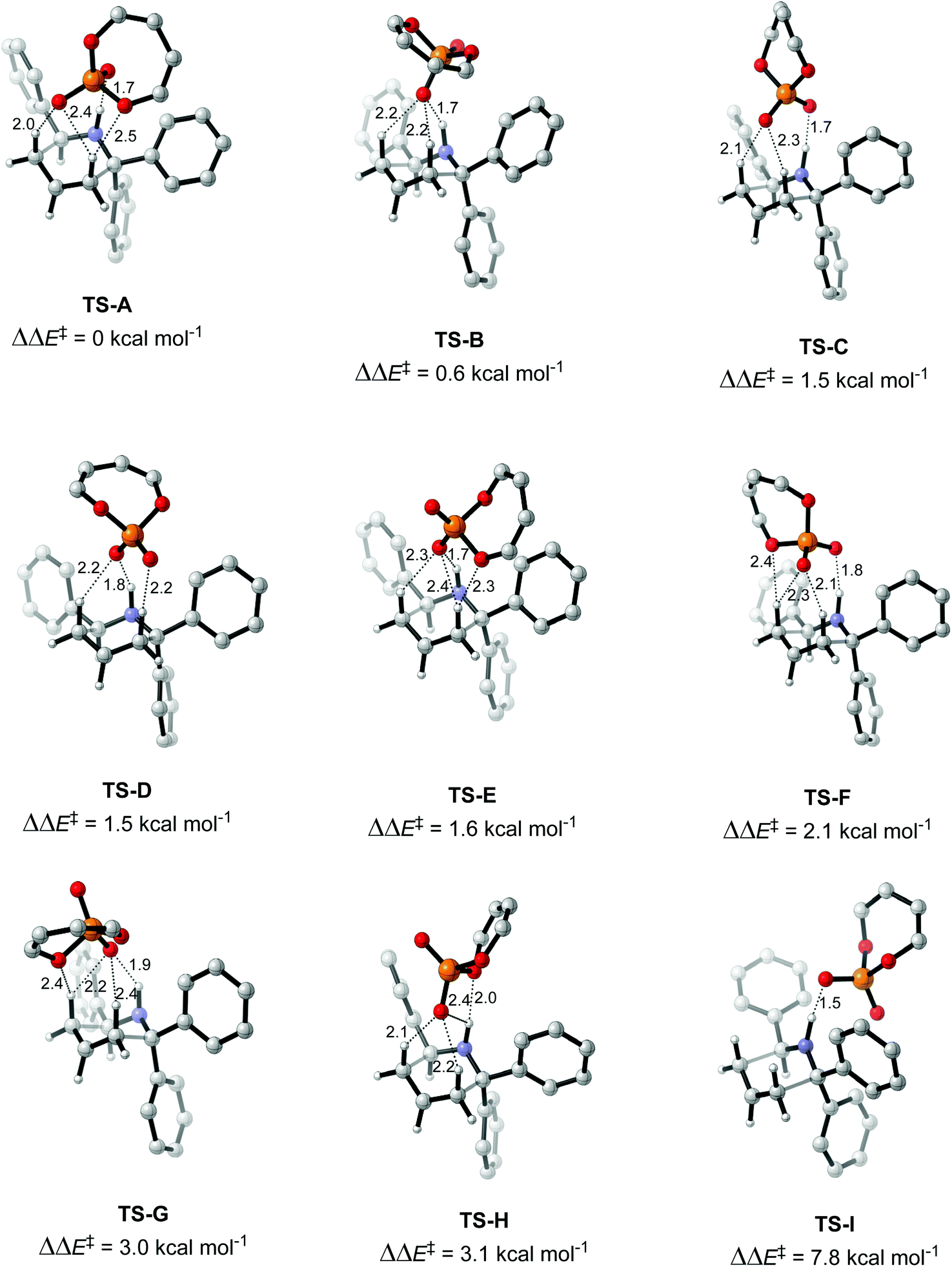 | ||
| Fig. 4 Activation modes obtained for the model system consisting of a butadiene catalyst, diphenyl-substituted amine and benzaldehyde. Optimized with M06-2X 6-31G*, with single point M06-2X def2-TZVPP in toluene (SMD). Hydrogen bonds between the substrate and the catalyst below 2.6 Å are shown as dashed lines and distances in Å. | ||
Once the different activation modes were investigated, a conformational search and optimization was carried out for the full catalyst 1h and the transition state leading to product 4j. 31 structures were located for the TS leading to the major product, and 32 for the TS leading to the minor product. The lowest energy TS structures leading to the major product (TS-1-major) and the minor product (TS-1-minor) are shown in Fig. 5 and 6. TS-1-major was found to be 1.21 kcal mol−1 and 1.26 kcal mol−1 lower in energy in toluene and dibutylether (as a model for MTBE) respectively than TS-1-minor.
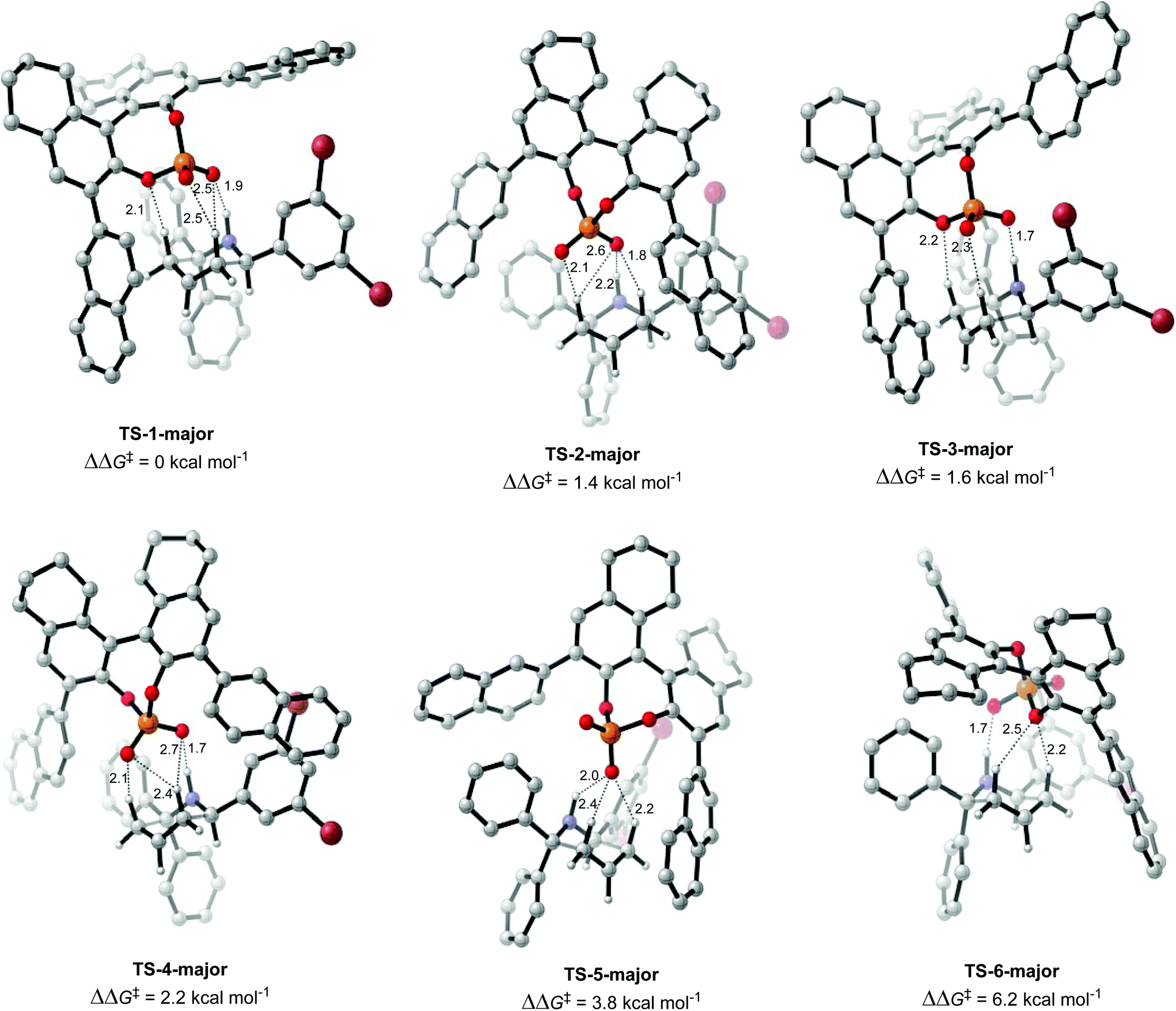 | ||
| Fig. 5 TS structures leading to the major product, optimized with M06-2X 6-31G*, single point with M06-2X def2-TZVPP in dibutylether (SMD), and thermal corrections with M06-2X 6-31G*. Hydrogen bonding interactions are shown as dashed lines and distances in Å. | ||
 | ||
| Fig. 6 TS structures leading to the minor product, optimized with M06-2X 6-31G*, single point with M06-2X def2-TZVPP in dibutylether (SMD), and thermal corrections with M06-2X 6-31G*. Hydrogen bonding interactions are shown as dashed lines and distances in Å. | ||
An analysis of the activation modes observed for the major and minor TSs, in comparison with those observed for the model catalyst, can bring important conclusions. Firstly, all activation modes for the full catalyst show three or four H-bonds between the phosphate and the C–H or N–H atoms. Activation mode TS-I, which showed only one H-bond, was not observed in the full catalyst system. However, as this conformation was found to be 7.8 kcal mol−1 higher in energy, it is not expected to be significant in the full system. Given the higher steric hindrance of the full catalyst, and extra bromine atoms in the substrate, the relative order of the different activation modes changes. For instance, activation mode TS-B in the model catalyst (ΔΔE‡ of 0.6 kcal mol−1), which shows three simultaneous H-bonds between the O atom from the phosphate and the three H atoms from the substrate, was found to be much higher in energy in the full catalyst system: TS-5-major (ΔΔG‡ of 3.79 kcal mol−1) and TS-6-minor (ΔΔG‡ of 2.77 kcal mol−1).
To further investigate the mechanism of the enantiodetermining step, the structures of intermediates A and B were optimized. The energy profile of this step is shown in Fig. S1,† alongside the structures of the optimized intermediates. The obtained activation barrier of 21.9 kcal mol−1 is compatible with the experimental results and reaction conditions of this reaction and it is consistent with other related phosphoric-acid catalyzed transformations.35–40
In order to account for the difference in stabilization energy of TS-1-majorvs.TS-1-minor, analyses of the intermolecular interactions between the substrate and the catalyst were carried out using the Non-Covalent Interactions (NCI) index method. Default parameters were used for the calculations and plots. TS-1-major (Fig. 7) shows a strong attractive interaction between the N–H group in the substrate and an O atom from the phosphate in the catalyst (shown in blue). Then, it shows a number of weak attractive interactions (shown in green): (i) aliphatic interactions between the six membered ring in the substrate and the aromatic ring in one of the naphthyl groups in the catalyst, (ii) a halogen – aromatic interaction between one of the bromines in the substrate and the other naphthyl group in the catalyst, (iii) C–H aromatic interactions between one of the phenyl groups of the substrate and one of the naphthyl groups of the catalyst, (iv) C–H aromatic interactions between the other phenyl group from the substrate and one of the [H8]-ring from the catalyst, and (v) C–H⋯O interactions between one of the phenyl groups and the bromine substituted aromatic ring in the substrate with the phosphate in the catalyst. The corresponding NCI plot for TS-1-minor (Fig. 7) also shows a strong interaction between the N–H group and the O from the phosphate and a number of weaker interactions: (i) a halogen aromatic interaction between one bromine atom from the substrate and one [H8]-rings from the catalyst, (ii) aliphatic interactions between the six member ring from the substrate and a naphthyl ring from the catalyst, and (iii) C–H⋯O interactions between one of the phenyl groups and the bromine substituted aromatic ring in the substrate with the phosphate in the catalyst. A comparison between TS-1-major and TS-1-minor shows that the latter has a fewer number of interactions, since it does now show phenyl aromatic interactions. This is consistent with the greater stabilization energy present in TS-1-major.
 | ||
| Fig. 7 NCI plots showing intermolecular interactions between the substrate and catalyst. Strong attractive interactions shown in blue, and weak attractive interactions shown in green. Obtained with M06-2X def2-TZVPP in the gas phase. | ||
The intermolecular interactions were further investigated using a Natural Bond Order (NBO) analysis of the second order perturbation energies. The greatest interaction was found between the H atoms in the reacting six membered ring in the substrate and the O atoms in the phosphate group in the catalyst. In total, the H bonds account for a stabilization of 36.8 kcal mol−1 in TS-1-major and 34.4 kcal mol−1 in TS-1-minor. The largest interactions are shown in Fig. 8. TS-1-major shows an interaction of 10.32 kcal mol−1 between an empty N–H σ* orbital and a partially-filled P–O1 σ* orbital, an interaction of 3.88 kcal mol−1 between an empty C–H3 σ* orbital and a filled O3 lone pair n, and an interaction of 2.34 kcal mol−1 between an empty C–H2 σ* orbital and an partially-filled PO1 σ* orbital. TS-1-minor shows an interaction of 9.98 kcal mol−1 between an empty N–H1 σ* orbital and a partially-filled PO1 σ* orbital, and interaction of 1.76 kcal mol−1 between an empty C–H3 σ* orbital and a filled O3 lone pair n, and an interaction of 1.53 kcal mol−1 between an empty C–H2 σ* orbital and a filled O2 lone pair. These interaction energies are consistent with the activation modes observed for these TS structures and provide a rationale for the greater stabilization of TS-1-majorvs.TS-1-minor. These greater H bond energies, together with the greater intermolecular interactions observed using the NCI plot between the phenyl groups in the substrate and the aromatic rings in the catalyst, account for the origins of enantioselectivity of the reaction.
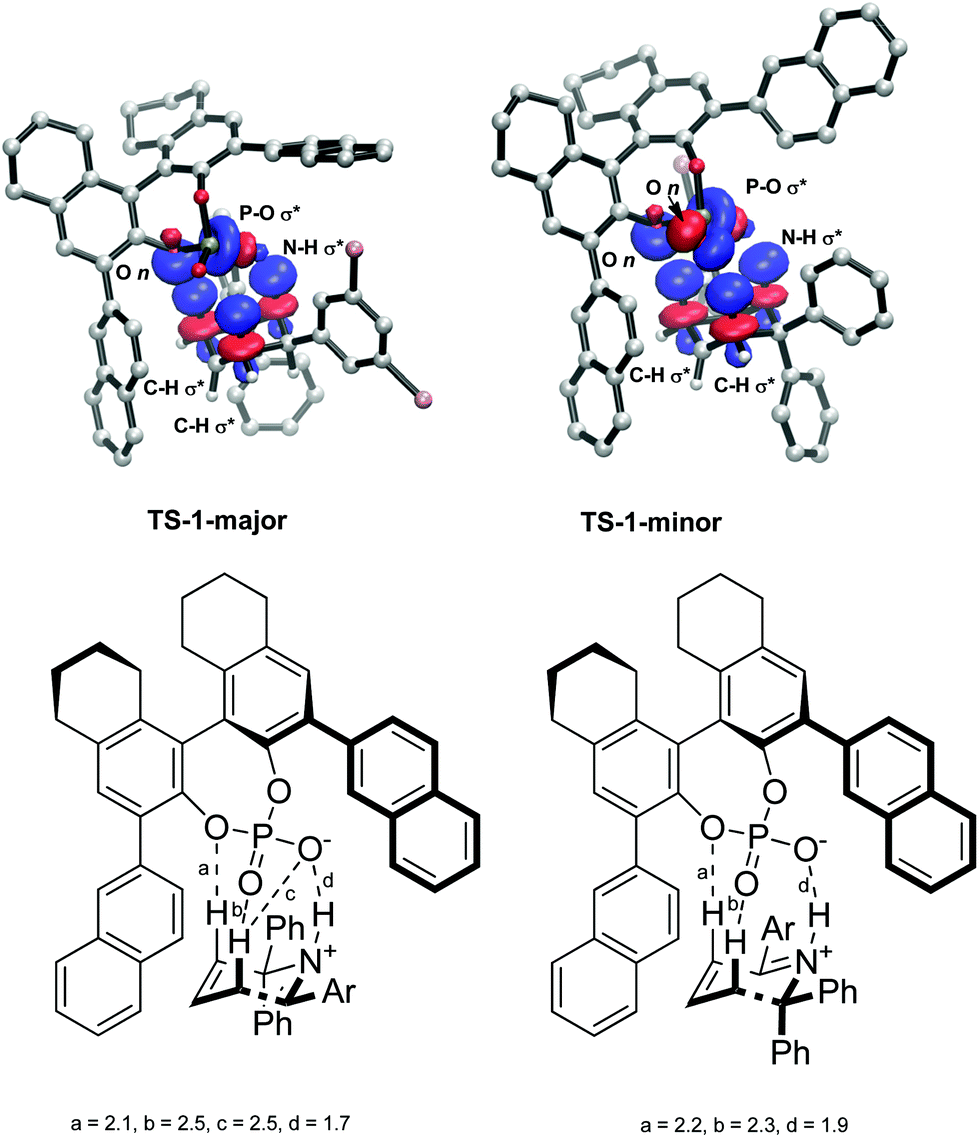 | ||
| Fig. 8 NBO analysis of hydrogen bonding interactions between the substrate and the catalyst. Obtained with M06-2X def2-TZVPP in the gas phase. H-Bond distances a, b, c and d shown in Å. | ||
Finally, we have studied the distortion41–43 of the catalyst and substrate upon the formation of the transition states, a process that has been reported as being fundamental to some related reactions, such as chiral phosphoric-acid catalyzed oxetane desymmetrizations44 and ring-openings of meso-aziridinium and episulfonium cations.33 We have calculated the single point energy of the catalyst and substrate in the TS-1-major and TS-1-minor conformations using M06-2X def2-TZVPP in dibutylether (SMD). The substrate conformation in TS-1-major is in fact 0.26 kcal mol−1 higher than in TS-1-minor, and the catalyst conformation is 0.88 kcal mol−1 higher, accounting for a total distortion energy of 1.15 kcal mol−1, so in fact the conformation of the constituents leading to TS-1-major are disfavored over TS-1-minor. These results highlight the significance of the intermolecular interactions described above, which account for the origin in the stabilization of TS-1-major over TS-1-minor, consistent with the experimental results.
Conclusions
In conclusion, DFT calculations were carried out on the chiral phosphoric-acid catalyzed 2-aza-Cope rearrangement reaction. Different activation modes of the C–C bond forming step model TS were located, and TS-A was found to be lowest in energy. It shows four H-bonds, one between N–H and a phosphate O atom, and three C–H⋯O bonds between the substrate and the phosphate group in the catalyst. Then, the origin of the enantioselectivity was evaluated by locating the TSs for the full catalyst 1h and substrate leading to product 4j. The lowest energy conformations were found to be TS-1-major and TS-1-minor. The full energy reaction profile was investigated and the obtained reaction barrier of 21.9 kcal mol−1 is consistent with the experimental results. NCI and NBO analyses were performed in order to rationalize the difference in stabilization energy between TS-1-major and TS-1-minor. The NCI plot showed that TS-1-minor has a lower number of interactions and it does not show interactions between the phenyl groups in the substrate and the aromatic rings in the catalyst. The NBO analysis showed stronger H-bond interactions between the H atoms in the substrate and the O atoms in the phosphate group from the catalyst. Finally, the distortion of the catalyst and substrate upon TS formation was explored but was not found to account for the stabilization of the preferred reaction pathway. These results provide valuable insights into the mechanism of the reaction and the different stabilization modes in the TS and can be used for the design of more effective catalysts for this and related reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Centro de Cómputos de Alto Rendimiento (CeCAR) and the Irish Centre for High-End Computing (ICHEC) for granting use of computational resources which allowed us to perform the experiments included in this work. Part of this research was funded by the Synthesis and Solid State Pharmaceutical Centre (SSPC), supported by Science Foundation Ireland (SFI) and cofunded under the European Regional Department Fund (SFI 12/RC/2275_p2/R18867). M.N.G. thanks the University of Bath for financial support. Dr. Zhongyue Yang is thanked for his helpful discussions regarding this work.References
- M. Rueping and A. P. Antonchick, Catalytic Asymmetric Aminoallylation of Aldehydes: A Catalytic Enantioselective Aza-Cope Rearrangement, Angew. Chem., Int. Ed., 2008, 47(52), 10090–10093 CrossRef CAS PubMed.
- R. M. Horowitz and T. A. Geissman, A Cleavage Reaction of α-Allylbenzylamines, J. Am. Chem. Soc., 1950, 72(4), 1518–1522 CrossRef CAS.
- U. Nubbemeyer, Recent Advances in Asymmetric [3,3]-Sigmatropic Rearrangements, Synthesis, 2003,(07), 0961–1008 CAS.
- C. Martín and A. M. Claisen, Rearrangement over the Past Nine Decades, Chem. Rev., 2004, 104(6), 2939–3002 CrossRef PubMed.
- C. G. Nasveschuk and T. Rovis, The [1,3] O-to-C Rearrangement: Opportunities for Stereoselective Synthesis, Org. Biomol. Chem., 2008, 6(2), 240–254 RSC.
- K. C. Majumdar, S. Alam and B. Chattopadhyay, Catalysis of the Claisen Rearrangement, Tetrahedron, 2008, 64(4), 597–643 CrossRef CAS.
- M. Sugiura, C. Mori and S. Kobayashi, Enantioselective Transfer Aminoallylation: Synthesis of Optically Active Homoallylic Primary Amines, J. Am. Chem. Soc., 2006, 128(34), 11038–11039 CrossRef CAS PubMed.
- L. E. Overman, Charge as a Key Component in Reaction Design. The Invention of Cationic Cyclization Reactions of Importance in Synthesis, Acc. Chem. Res., 1992, 25(8), 352–359 CrossRef CAS.
- C. O. Puentes and V. Kouznetsov, Recent Advancements in the Homoallylamine Chemistry, J. Heterocycl. Chem., 2002, 39(4), 595–614 CrossRef CAS.
- S. Stas, K. Abbaspour Tehrani and G. Laus, Carbon-Carbon Bond Formation via a Tandem Cationic 2-Aza-Cope Rearrangement-Lewis Acid Promoted Petasis Reaction, Tetrahedron, 2008, 64(16), 3457–3463 CrossRef CAS.
- Y. Nishikawa, M. Kitajima and H. Takayama, First Asymmetric Total Syntheses of Cernuane-Type Lycopodium Alkaloids, Cernuine, and Cermizine D, Org. Lett., 2008, 10(10), 1987–1990 CrossRef CAS PubMed.
- D. Kampen, C. M. Reisinger and B. List, Chiral Brønsted Acids for Asymmetric Organocatalysis, in Asymmetric Organocatalysis, ed. B. List, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp 1–37 Search PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114(18), 9047–9153 CrossRef CAS PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chiral Phosphoric Acid Catalysis: From Numbers to Insights, Chem. Soc. Rev., 2018, 47(4), 1142–1158 RSC.
- M. Terada, K. Soga and N. Momiyama, Enantioselective Activation of Aldehydes by Chiral Phosphoric Acid Catalysts in an Aza-Ene-Type Reaction between Glyoxylate and Enecarbamate, Angew. Chem., Int. Ed., 2008, 47(22), 4122–4125 CrossRef CAS PubMed.
- T. Marcelli, P. Hammar and F. Himo, Phosphoric Acid Catalyzed Enantioselective Transfer Hydrogenation of Imines: A Density Functional Theory Study of Reaction Mechanism and the Origins of Enantioselectivity, Chemistry, 2008, 14(28), 8562–8571 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, Theoretical Study of the Mechanism of Hantzsch Ester Hydrogenation of Imines Catalyzed by Chiral BINOL-Phosphoric Acids, J. Am. Chem. Soc., 2008, 130(27), 8741–8747 CrossRef PubMed.
- L. Simón and J. M. Goodman, A Model for the Enantioselectivity of Imine Reactions Catalyzed by BINOL−Phosphoric Acid Catalysts, J. Org. Chem., 2011, 76(6), 1775–1788 CrossRef PubMed.
- G.-Q. Li, H. Gao, C. Keene, M. Devonas, D. H. Ess and L. Kürti, Organocatalytic Aryl-Aryl Bond Formation: An Atroposelective [3,3]-Rearrangement Approach to BINAM Derivatives, J. Am. Chem. Soc., 2013, 135(20), 7414–7417 CrossRef CAS PubMed.
- R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Computational Ligand Design in Enantio- and Diastereoselective Ynamide [5 + 2] Cycloisomerization, Nat. Commun., 2016, 7(1), 10109 CrossRef CAS PubMed.
- D. M. Sedgwick, M. N. Grayson, S. Fustero and P. Barrio, Recent Developments and Applications of the Chiral Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds, Synthesis, 2018, 50(10), 1935–1957 CrossRef CAS.
- B. N. Falcone, M. N. Grayson and J. B. Rodriguez, Mechanistic Insights into a Chiral Phosphoric Acid-Catalyzed Asymmetric Pinacol Rearrangement, J. Org. Chem., 2018, 83(23), 14683–14687 CrossRef CAS PubMed.
- M. N. Grayson and J. M. Goodman, Understanding the Mechanism of the Asymmetric Propargylation of Aldehydes Promoted by 1,1′-Bi-2-Naphthol-Derived Catalysts, J. Am. Chem. Soc., 2013, 135(16), 6142–6148 CrossRef CAS PubMed.
- A. Lerchen, N. Gandhamsetty, E. H. E. Farrar, N. Winter, J. Platzek, M. N. Grayson and V. K. Aggarwal, Enantioselective Total Synthesis of (−)–Finerenone Using Asymmetric Transfer Hydrogenation, Angew. Chem., Int. Ed., 2020, 59(51), 23107–23111 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41(2), 157–167 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, How Reliable Are DFT Transition Structures? Comparison of GGA, Hybrid-Meta-GGA and Meta-GGA Functionals, Org. Biomol. Chem., 2011, 9(3), 689–700 RSC.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- L. Simón and R. S. Paton, QM/MM Study on the Enantioselectivity of Spiroacetalization Catalysed by an Imidodiphosphoric Acid Catalyst: How Confinement Works, Org. Biomol. Chem., 2016, 14(11), 3031–3039 RSC.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, Macromodel—an Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics, J. Comput. Chem., 1990, 11(4), 440–467 CrossRef CAS.
- R. J. Doedens, G. P. Meier and L. E. Overman, Transition-State Geometry of [3,3]-Sigmatropic Rearrangements of Iminium Ions, J. Org. Chem., 1988, 53(3), 685–690 CrossRef CAS.
- F. Duarte and R. S. Paton, Molecular Recognition in Asymmetric Counteranion Catalysis: Understanding Chiral Phosphate-Mediated Desymmetrization, J. Am. Chem. Soc., 2017, 139(26), 8886–8896 CrossRef CAS PubMed.
- M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, Mechanistic Insights into the BINOL-Derived Phosphoric Acid-Catalyzed Asymmetric Allylboration of Aldehydes, J. Am. Chem. Soc., 2012, 134(5), 2716–2722 CrossRef CAS PubMed.
- M. Yamanaka, J. Itoh, K. Fuchibe and T. Akiyama, Chiral Brønsted Acid Catalyzed Enantioselective Mannich-Type Reaction, J. Am. Chem. Soc., 2007, 129(21), 6756–6764 CrossRef CAS PubMed.
- T. Hirata and M. Yamanaka, DFT Study of Chiral-Phosphoric-Acid-Catalyzed Enantioselective Friedel-Crafts Reaction of Indole with Nitroalkene: Bifunctionality and Substituent Effect of Phosphoric Acid, Chem. – Asian J., 2011, 6(2), 510–516 CrossRef CAS PubMed.
- H. Wang, P. Jain, J. C. Antilla and K. N. Houk, Origins of Stereoselectivities in Chiral Phosphoric Acid Catalyzed Allylborations and Propargylations of Aldehydes, J. Org. Chem., 2013, 78(3), 1208–1215 CrossRef CAS PubMed.
- A. Fu, W. Meng, H. Li, J. Nie and J.-A. Ma, A Density Functional Study of Chiral Phosphoric Acid-Catalyzed Direct Arylation of Trifluoromethyl Ketone and Diarylation of Methyl Ketone: Reaction Mechanism and the Important Role of the CF3 Group, Org. Biomol. Chem., 2014, 12(12), 1908–1918 RSC.
- J. Calleja, A. B. González-Pérez, Á. R de Lera, R. Álvarez, F. J. Fañanás and F. Rodríguez, Enantioselective Synthesis of Hexahydrofuro[3,2-c] Quinolines through a Multicatalytic and Multicomponent Process. A New “Aromatic Sandwich” Model for BINOL-Phosphoric Acid Catalyzed Reactions, Chem. Sci., 2014, 5(3), 996–1007 RSC.
- M. N. Grayson and K. N. Houk, Cinchona Alkaloid-Catalyzed Asymmetric Conjugate Additions: The Bifunctional Brønsted Acid-Hydrogen Bonding Model, J. Am. Chem. Soc., 2016, 138(4), 1170–1173 CrossRef CAS PubMed.
- K. N. Houk, R. W. Gandour, R. W. Strozier, N. G. Rondan and L. A. Paquette, Barriers to Thermally Allowed Reactions and the Elusiveness of Neutral Homoaromaticity, J. Am. Chem. Soc., 1979, 101(23), 6797–6802 CrossRef CAS.
- R. D. J. Froese, J. M. Coxon, S. C. West and K. Morokuma, Theoretical Studies of Diels-Alder Reactions of Acetylenic Compounds, J. Org. Chem., 1997, 62(20), 6991–6996 CrossRef CAS.
- D. H. Ess and K. N. Houk, Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity, J. Am. Chem. Soc., 2007, 129(35), 10646–10647 CrossRef CAS PubMed.
- P. A. Champagne and K. N. Houk, Origins of Selectivity and General Model for Chiral Phosphoric Acid-Catalyzed Oxetane Desymmetrizations, J. Am. Chem. Soc., 2016, 138(38), 12356–12359 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob02458a |
| This journal is © The Royal Society of Chemistry 2021 |