
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the anomeric effect and mechanism of isomerization of oxazinane rings by DFT methods†
Constantinos A.
Tsipis
*,
Evangelos G.
Bakalbassis
,
Stavroula A.
Zisopoulou
and
John K.
Gallos

School of Chemistry, Faculty of Sciences, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: tsipis@chem.auth.gr; Fax: (+30)2310 997679; Tel: (+30)2310 997714
First published on 11th January 2021
Abstract
Mechanistic studies of the thermal amine-promoted isomerization of oxazinane rings by DFT methods showed that the isomerization proceeds through abstraction of the C-3 hydrogen atom by the amine nitrogen atom followed by its re-recruitment from C-3 that helps the oxazinane ring to avoid breaking, leading to the same or an isomeric conformer. Calculations also provided evidence that steric effects are responsible for the breaking of the O–N bond in the transition state of the thermal amine-promoted transformations of oxazinane rings, leading to the transformation of the 6-membered ring to a 5-membered ring. Extensive computational studies of the origin of the anomeric effect in the di-substituted oxazinane rings, bearing the EtO substituent at C-6 and CO2Et at C-3, and a series of analogous tetrahydro-2H-pyran ring conformers, revealed that the conformational preferences in both series of compounds are tuned by the balance of non-covalent (weak vDW, dipole–dipole, electrostatic forces, hydrogen bonding) steric effects and hyperconjugative interactions.
Introduction
5,6-Dihydro-4H-1,2-oxazines are useful intermediates in organic synthesis.1 Among them, 6-alkoxy-5,6-dihydro-4H-1,2-oxazine-3-carboxylates have previously been synthesized in our lab through the reaction of ethyl 2-nitroso-acrylate with enol ethers and several interesting products, such as non-natural amino acids,2–5 aza-sugars6–9 and alkaloids,10 are isolated and fully characterized.A first major key transformation of these hetero-Diels–Alder adducts is their C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond reduction with NaCNBH3, which proceeds non-stereoselectively to yield the respective oxazinanes.11 For example, the NaCNBH3 reduction of the parent ethyl 6-ethoxy-5,6-dihydro-4H-1,2-oxazine-3-carboxylate affords oxazinanes 1 and 2 (Scheme 1). However, further treatment of the mixture of these products with Et3N induces complete conversion of 1 to 2,6 achieving thus an indirect highly diastereoselective reduction of the CN bond in the 5,6-dihydro-4H-1,2-oxazine ring. It was postulated that the cis-isomer 2, having the CO2Et group at C-3 in an equatorial position, is thermodynamically more stable than the trans-isomer 1, whereas the OEt group at C-6 has an axial position, stabilized by the anomeric effect.
N bond reduction with NaCNBH3, which proceeds non-stereoselectively to yield the respective oxazinanes.11 For example, the NaCNBH3 reduction of the parent ethyl 6-ethoxy-5,6-dihydro-4H-1,2-oxazine-3-carboxylate affords oxazinanes 1 and 2 (Scheme 1). However, further treatment of the mixture of these products with Et3N induces complete conversion of 1 to 2,6 achieving thus an indirect highly diastereoselective reduction of the CN bond in the 5,6-dihydro-4H-1,2-oxazine ring. It was postulated that the cis-isomer 2, having the CO2Et group at C-3 in an equatorial position, is thermodynamically more stable than the trans-isomer 1, whereas the OEt group at C-6 has an axial position, stabilized by the anomeric effect.
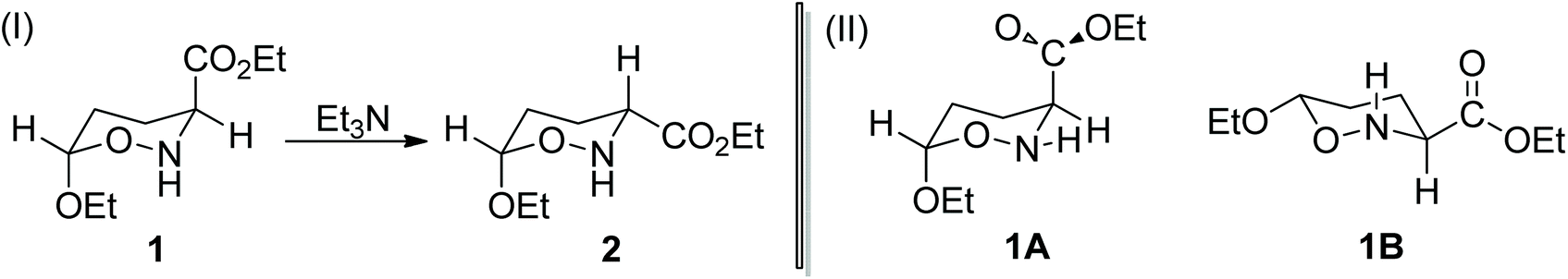 | ||
| Scheme 1 Thermal amine-promoted isomerization of trans-oxazinane, 1, to cis-oxazinane, 2 (I). The two model conformers 1A and 1B selected for the study of the thermal amine-promoted isomerization mechanism of the oxazinane ring (II). | ||
It was generally proposed6,7,11 that the mechanism of the thermal amine-promoted isomerization of oxazinane 1 to 2 (Scheme 1), adopting chair-like conformations, proceeds through the abstraction of the 3-H proton (systematic numbering), which next is bonded to the amine nitrogen; its re-recruitment from C-3 of the generated enolate affords the same or the isomeric structure.
The origin of the anomeric effect (AE), a fundamental concept in organic chemistry, defined as the thermodynamic preference for polar substituents X to occupy the axial position in the chair conformation of various heterocycles, has been the subject of intense debate. Two different explanations for the AE have been proposed: the first one is stereoelectronic in nature, based on favorable/unfavorable dipole–dipole interactions between the lone electron pairs of the ring heteroatom and the anomeric X substituent (minimization of dipole–dipole interactions).12–18 The second explanation is based on favorable/unfavorable donor–acceptor orbital interactions (hyperconjugative interactions) between the ring heteroatom and the anomeric X substituent (delocalization of the endocyclic oxygen lone pair into the antibonding σ*(C–X) orbital).16–29 However, there is no general consensus about the actual origin of the AE.25,26
Yirong Mo17 investigated the origin of the anomeric effect and concluded that it arises from electrostatic interaction associated with dipole–dipole interactions. Liu and coworkers30 pointed out that electron delocalization and electrostatic interactions alone could not explain the general validity of the AE. Bauerfeldt et al.31 pointed out that the anomeric effect has no electrostatic origin. Ferro-Costas and Mosquera32 performing population analysis in diverse anomeric compounds by combining ELF and QTAIM basins (ELF∩QTAIM scheme) provided a deeper understanding of the conformational preferences independent of hyperconjugative effects.
Having in mind the proposed thermal amine-promoted isomerization of trans-oxazinane, and the peculiarity of the oxazinane rings, bearing neighboring N and O heteroatoms, both being capable of promoting the anomeric effect, we thoroughly explored (i) the possible reaction pathways and (ii) the anomeric effect in the oxazinane and related tetrahydro-2H-pyran rings employing DFT computational protocols.
Computational details
All stationary points (reactants, transition states, and products) located on the potential energy surfaces (PES) were fully optimized at the ωB97XD/Def2-TZVP level of theory as implemented in the Gaussian 09 W suite of programs.33 The ωB97XD functional34–37 containing empirical dispersion terms and long-range corrections provides good descriptions of reaction profiles, including geometries, heats of reactions, and barrier heights,38 and has been successfully used to describe: (i) the AE for 2-substituted tetrahydropyrans and piperidines,25 (ii) the geometries and dissociation energies of halogen bonds,39 (iii) the geometries, ionization energies (IPs), electron affinities (EAs), and excitation energies of neutral and oxidized polyenes, thiophene, and furan oligomers,40 and (iv) the anion–π interaction in heterocyclic calix complexes.41 The Def2-TZVP basis set42,43 was used for all elements of the conformers. To reduce the execution time, NH3 instead of NMe3 was used for the thermal amine-promoted isomerization of 1 to 2. Analytical frequencies were calculated at the same level of theory, and the nature of the stationary points was determined in each case according to the number of negative eigenvalues of the Hessian matrix. Unless otherwise stated, Gibbs free energies are used to construct the energetic reaction profiles. Moreover, the correct transition states have been confirmed by intrinsic reaction coordinate (IRC) calculations, while intrinsic reaction paths (IRPs) were traced from the various transition structures to make sure that no further intermediates exist.44 The SMD implicit solvation model,45 recommended choice for computing ΔG of solvation, was used to account for solvation effects of chloroform solvent; still temperature corrected energy values at the boiling point (61.2 °C) of chloroform were also calculated. All energies reported throughout the text are in kcal mol−1, and the bond lengths are in angstroms (Å). Natural Bond Orbital (NBO) population analysis was performed using Weinhold's methodology46 to probe the AE for the oxazinane rings with a chair-like conformation. The NBO population analysis approach allows for a quantitative assessment of stereoelectronic effects, and the stabilization energy ΔE(2) associated with the charge transfer (CT) interactions between the relevant donor–acceptor orbitals computed from the second-order perturbative estimates of the Fock matrix in the NBO analysis according to the equationwas successfully applied to analyze the classical AE.24,25 This equation evaluates the magnitude of the donor–acceptor interaction in terms of the spatial overlap of the i and j NBOs, using the off-diagonal Fock-matrix elements Fij and the difference in energy εi − εj, weighted by the occupancy of the donor NBO, qi. The electronic delocalization contributions to the AE, ΔΔEdeloc, are calculated by the equation proposed by Cuevas and co-workers.47 The NOSTAR keyword was used in the NBO input to determine the energy of the hypothetical Lewis molecules. Calculations of the reduced density gradients, RDG, and steric energies, Es, are performed and their plots are obtained employing the Multiwfn 3.5 software package.48,49
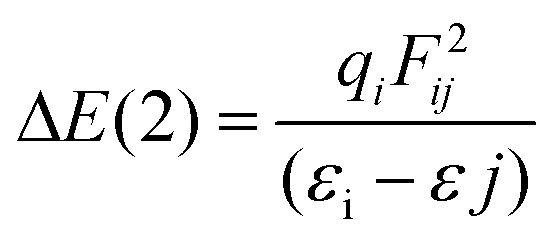
Results and discussion
Mechanism of the thermal amine-promoted transformations of oxazinane rings
In an attempt to gain a comprehensive understanding of the proposed mechanism for the thermal amine-promoted transformations of oxazinane rings, we thoroughly explored possible reaction pathways employing DFT computational protocols at the ωB97XD/Def2-TZVP level of theory. It should be stressed at this point that the compounds under study present a variety of conformers, differing mainly in the orientation (axial/equatorial) of their (a) OEt substituents, (b) H atom bonded to the N heteroatom and (c) CO2Et ester groups, and/or in the number and strength of the intramolecular hydrogen bonds. The conformers studied computationally are given in the ESI (Fig. S1†). Among them, the oxazinane ring conformers 1A and 1B (numbers 4 and 9 in Fig. S1†), differing in all three above cases of orientation, were selected as model compounds (Scheme 1) to study the mechanism of the amine-promoted oxazinane ring transformations. The reaction steps involved in the entire thermal amine-promoted isomerization mechanism of 1A and 1B have been scrutinized, and the transition states have been fully identified by monitoring the corresponding geometric and energetic reaction profiles.The geometric and free energy reaction profiles calculated by the ωB97XD/Def2-TZVP computational protocol in the gas phase are shown in Fig. 1. The calculated geometric and free energy reaction profiles in chloroform solution are given in the ESI (Fig. S2†). In the first step, the amine molecule interacts with 1A and/or 1B conformers yielding loose associations 2A and 2B, respectively, with the estimated interaction energies for the gas (solution) phases being −4.1 (−6.1) and −4.1 (−5.7) kcal mol−1 respectively, indicative of non-covalent interactions in 2A and 2B loose associations. The transformation of the reactants 1A and 1B to their isomeric products proceeds via the transition states TS1 and TS2, surmounting activation barriers of 54.0 (44.5) and 42.4 (39.2) kcal mol−1, respectively. The estimated relatively high activation barriers reveal that the transformation of 1A and 1B to their isomeric forms requires heating in solution, in excellent agreement with the experiment.6,7,11
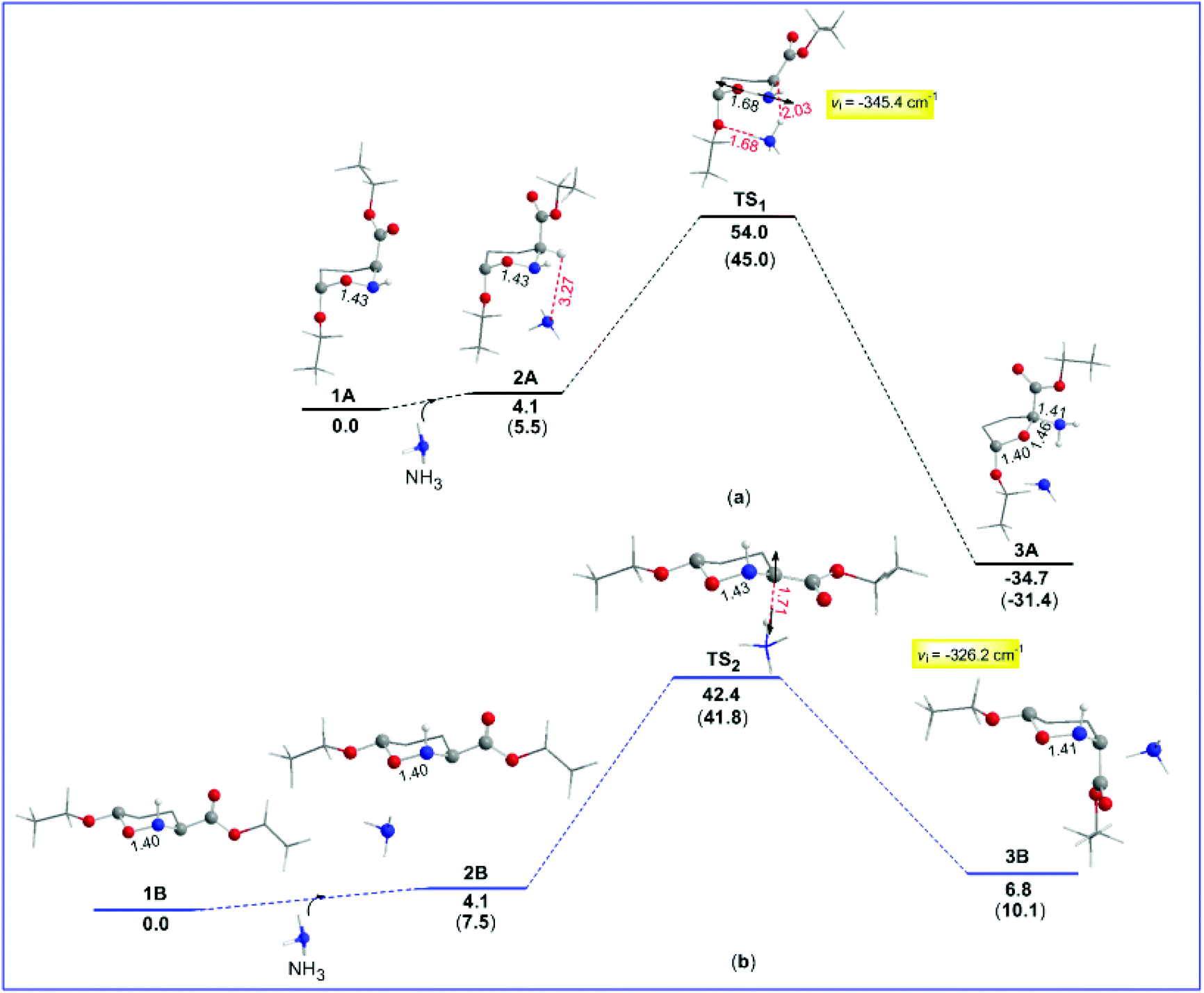 | ||
| Fig. 1 Gas-phase geometric and gas- and SMD liquid-phase temperature corrected (parentheses) free energy, ΔG (in kcal mol−1), reaction profiles of the thermal amine-promoted transformations of oxazinane rings of 1A (a) and 1B (b) conformers calculated by the ωB97XD/Def2-TZVP computational protocol. | ||
It is noteworthy that the normal coordinate vectors (arrows) of the vibrational modes, corresponding to the imaginary frequencies of TS1 and TS2, are completely different from each other. In particular, the normal coordinate vectors of TS1 at −345.4 (−65.8) cm−1 show that the dominant motions involve the lengthening of the O–N intramolecular bond to 1.68 Å. Interestingly, the O–N bond lengthening could be associated with the ring opening that yields the corresponding product 3A, being a 5-membered ring, resulting from the six-membered one with the N heteroatom and the O atom of the OEt group forming H-bridges with NH3. The thermal amine-promoted transformation of the oxazinane ring in 1A corresponds to a moderate exergonic process, and the estimated ΔRH values are −34.7 and −32.7 kcal mol−1 respectively, in the gas phase and in solution. The above breaking of the 6-membered ring leading to a 5-membered one was unknown until now.
In a recent publication, Del Valle and co-workers50 reported a synthesis of enantiopure ε-oxapipecolic acid. They found that treatment of the oxazinane ring conformer (enantiopure tetrahydrooxazine) with hydroxide bases resulted in full conversion to the 5-membered ring tetrahydrofuran derivative, an excellent analogue of 3A. It is very likely that the use of a strong base in this case leads to an irreversible abstraction of the 3-H proton in the more stable conformer 1A (see the ESI†) to give the 5-membered 3A, hence blocking (preventing) the interconversion between the two conformers 1A and 1B, and thus the generation of 2B and its equilibrium with 3B. The experimental results are also in excellent agreement with the theoretical findings (vide infra), which showed that the rupture of the O1–N2 bond and ring opening might be due to an increase of the steric energy at the middle of the O–N bond in TS1.
Moreover, as shown in Fig. 1, in contrast to 1A, the normal coordinate vectors of TS2 of 1B at −326.2 (−128.6) cm−1 show that the dominant motions involve the abstraction of the C-3 hydrogen atom by the amine nitrogen atom. Hence, TS2 corresponds to the interconversion between 2B and isomeric 3B; still, the activation energy of TS2 higher than 30.0 kcal mol−1 (vide supra) clearly shows that the interconversion between 2B and 3B requires heating in solution, in excellent agreement with the experimental findings of these compounds.6,7,11 In contrast to the experiment,6,7,11 where an interconversion between the two isomeric structures takes place upon boiling in CHCl3, calculations clearly show that there will be a breaking of the six-membered rings in 1A, too. More specifically, they show that the final product depends on the type of conformer selected. Indeed, the trans-oxazinane 1A conformer resulted in ring cleavage, while the respective trans-1B conformer resulted in the interconversion between 2B and isomeric 3B, which is a conformer of compound 2 (Scheme 1).
The question now at hand is: how could this ring system avoid breaking? 1A could adopt a variety of oxazinane ring conformations, some of which are shown in Fig. S1.† Yet, at the CHCl3 boiling temperature of 61.2 °C, all possible ring conformers exist in equilibrium in solution and therefore the conformer participating in the transformation process cannot be identified. Nevertheless, calculations showed that, despite the different products obtained, TS2 exhibits a change in the hybridization at C-3 from sp3 to sp2, as a result of the 3-H proton abstraction. Hence, it is very likely that, upon boiling of the conformer's mixture, the ammonium cation NH4+ formed could attack C-3 from either one of the two possible opposite directions at TS2, affording either 2B or isomeric 3B, respectively. Consequently, the 3-H proton re-recruitment from C-3, leading to either one of the two isomeric structures, renders the oxazinane ring stable enough to avoid breaking. It should be stressed here that, as shown above, the isomerization process requires a smaller activation energy than the one leading to the ring breaking. Therefore, the former process is thermodynamically more favorable than the latter one.
To gain insights into the factors responsible for the breaking of the intramolecular O1–N2 bond associated with ring opening we calculated the steric energy, Es, at the C-3 and C-6 centers and at the middle point (centroid, cd) of the intramolecular O1–N2 bond (Table 1). The Es distributions on the entire molecular framework are visualized49,51 by the shaded surface maps with projection shown in Fig. 2.
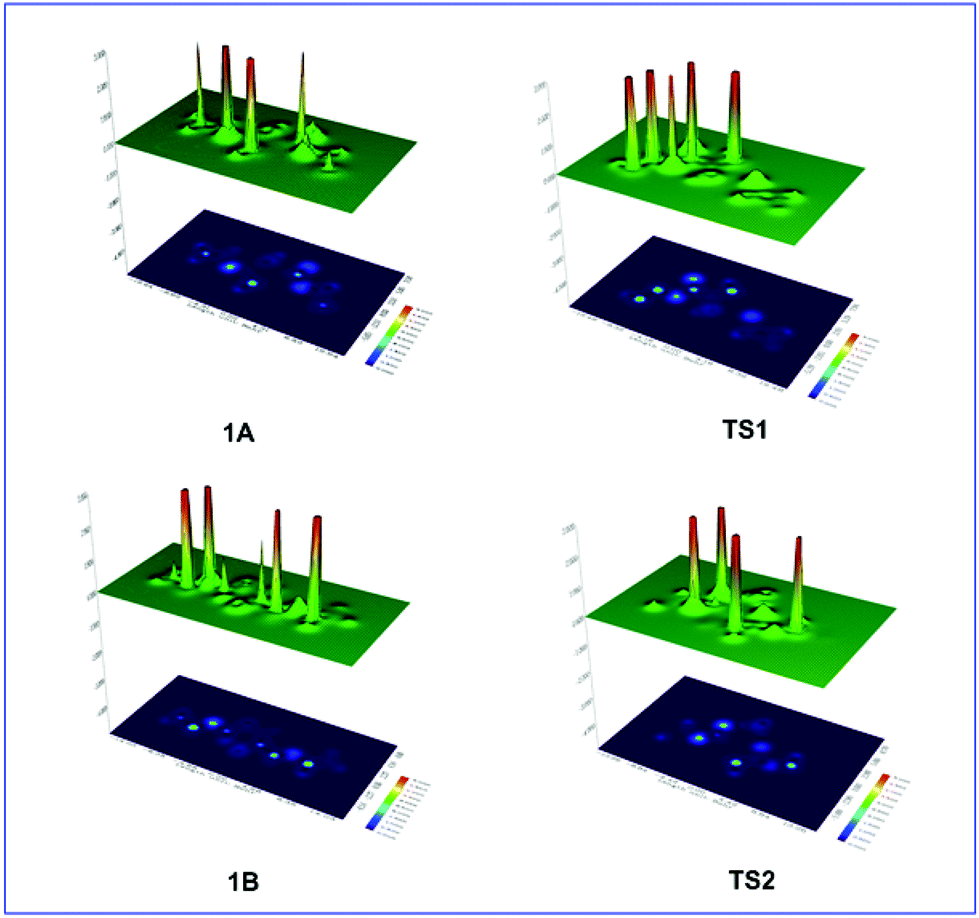 | ||
| Fig. 2 Shaded surface maps with projection of the Es distribution in 1A and 1B reactants and in TS1 and TS2 transition states involved in the thermal amine-promoted transformations of oxazinane rings. | ||
|
|
|||
|---|---|---|---|
| Compound | C-6 | C-3 | cd (O1–N2) |
| 1A | 0.82 | 0.13 | 20.08 |
| TS1 | 0.88 | 0.38 | 35.77 |
| 1B | 0.82 | 0.19 | 42.67 |
| TS2 | 0.82 | 0.25 | 8.79 |
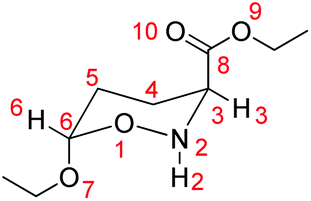
Table 1 and Fig. 2 show a dramatic increase of the steric energy at the cd point of the O1–N2 bond in TS1 upon detachment of the 3-H proton attacked by the amine molecule, which promotes the rupture of the O1–N2 bond and ring opening. In contrast, the Es at the cd point of the O1–N2 bond in TS2 is lower than that in TS1 indicating the resistance of the O1–N2 bond to break, being also in excellent agreement with the corresponding reaction profile data. Interestingly, the higher Es indicator for the anomeric C-6 atoms than for the C-3 carbon atoms accounts well for the abstraction of the 3-H proton upon interaction with the NH3 molecule.
The steric energy is a measure (indicator) of molecular stability in the absence of stereoelectronic interactions. However, in the oxazinane and tetrahydro-2H-pyran ring conformers, stereoelectronic, electrostatic and weak non-covalent (dipole–dipole, hydrogen bond and vDW) interactions strongly contribute to the stability of the conformers. Therefore, all these interactions related to the well-established concept of AE have to be taken into account. In this context, calculations are performed to probe the AE in mono- and di-substituted tetrahydro-2H-pyran and oxazinane ring conformers involving OEt and esteric CO2Et substituents and explore whether and how the endocyclic N heteroatom in the oxazinane ring affects the AE.
Probing the anomeric effect in tetrahydro-2H-pyran and oxazinane rings
To probe the anomeric effect in the oxazinane and tetrahydro-2H-pyran ring conformers, we followed the widely accepted explanations of the anomeric effect, namely the so-called electrostatic and delocalization (hyperconjugation) models. In this context, both non-covalent (dipole–dipole, hydrogen bond, steric and vDW) and hyperconjugative interactions involving the lone pair of oxygen LP(O) and nitrogen LP(N) ring heteroatoms and the antibonding σ*(C–O), σ*(C–N) and σ*(C–C) NBOs as acceptors are thoroughly explored by DFT in conjunction with Natural Bond Orbital (NBO) population analysis methods. From the NBO analysis, the delocalization contribution, ΔΔEdeloc, to the AE can be estimated using the equation47 ΔΔEdeloc = (Etotal − ELewis)eq − (Etotal − ELewis)ax, where Etotal is the total SCF energy for the equatorial and axial conformers and ELewis is the energy of the hypothetical Lewis structures of the equatorial and axial conformers involving localized bonds. It should be noted that the difference Etotal − ELewis measures the loss of stabilization (ELewis > Etotal) associated with the deletion of all possible hyperconjugative interactions.Considering the chemical equilibria, shown in Scheme 2, positive ΔEtotal and ΔELewis energies are expected to favor the axial conformation, while negative ΔEtotal and ΔELewis energies characterize the equatorial conformation. On the other hand, positive ΔEtotal and negative ΔELewis energies give positive ΔΔEdeloc delocalization energies favoring the axial conformation when electronic delocalization is dominant and the equatorial conformation when the steric effect predominates.46
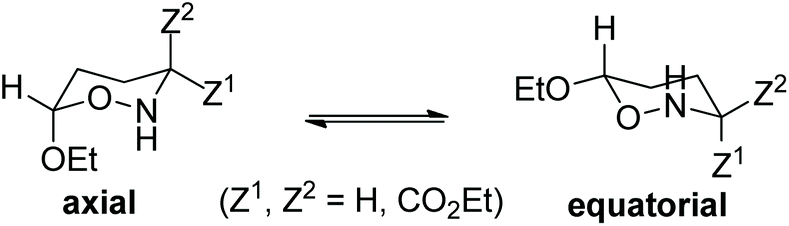 | ||
| Scheme 2 Chemical equilibria between the axial and equatorial conformers. | ||
The anomeric effect in tetrahydro-2H-pyran conformers
The equilibrium geometries with selected structural parameters (bond lengths in Å) and relative stability (in kcal mol−1, marked in red) of the cyclohexane and tetrahydro-2H-pyran conformers bearing OEt and/or OEt and esteric CO2Et substituents calculated at the ωB97XD/Def2-TZVP level of theory in the gas phase are given in Fig. 3.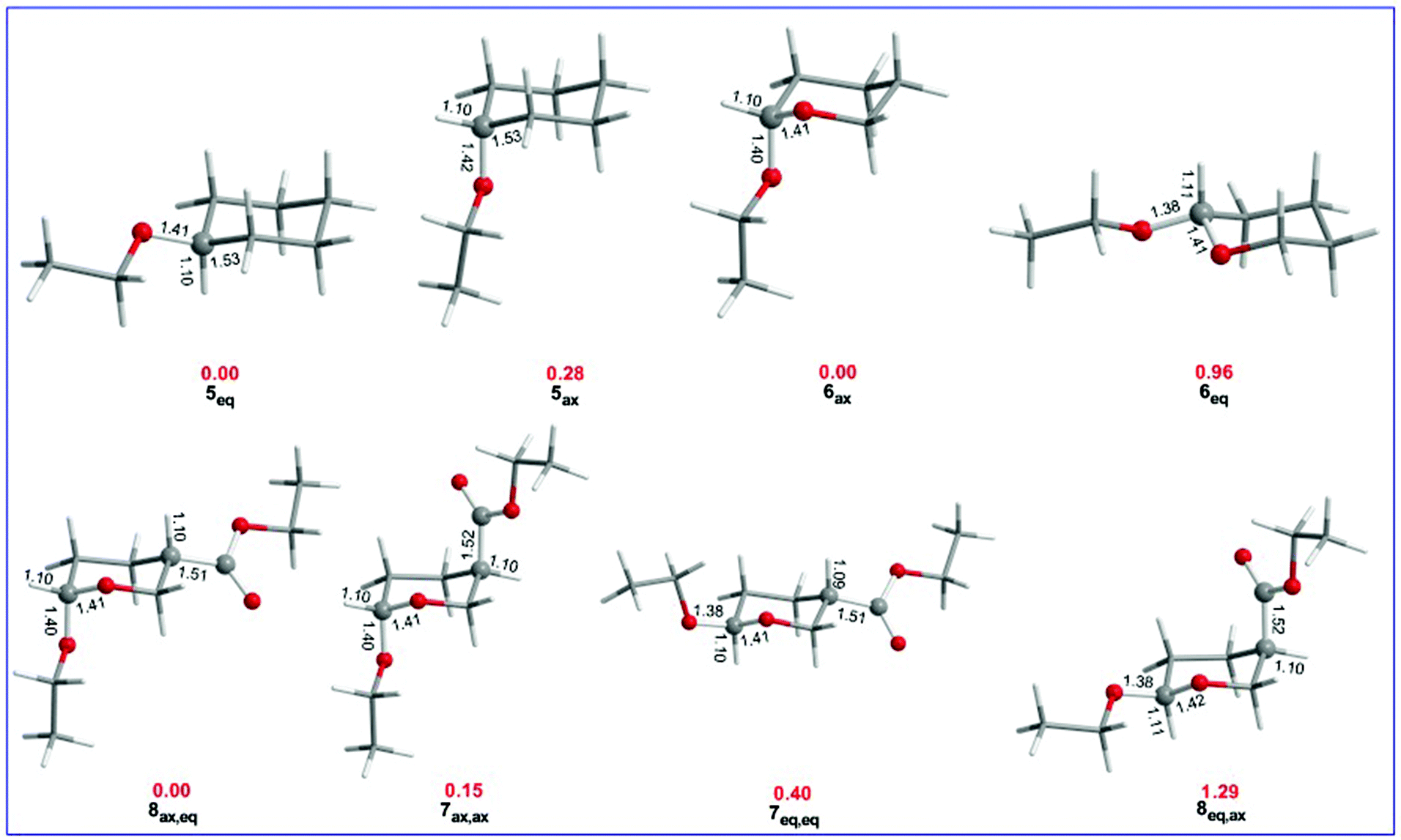 | ||
| Fig. 3 Equilibrium geometries with selected structural parameters (bond lengths in Å) and relative stability (in kcal mol−1, marked in red) of the cyclohexane and tetrahydro-2H-pyran conformers bearing OEt and/or OEt and esteric CO2Et substituents, calculated at the ωB97XD/Def2-TZVP level of theory in the gas phase. | ||
Fig. 3 shows that the R(C-OEt) bond lengths in the cyclohexane derivatives are slightly longer in 5ax (1.42 Å) than in 5eq (1.41 Å) conformers. However, in the analogous tetrahydro-2H-pyran conformers, the R(C-OEt) bond length in 6ax (1.40 Å) is much longer than that in 6eq (1.38 Å). Note that the endocyclic C–O bond length is longer (1.41 Å) in 6eq than in 6ax (1.406 Å). Similarly, in the di-substituted conformers, the R(C-OEt) bonds are longer in the axial orientation (1.40 Å) than in the equatorial orientation (1.38 Å). The same holds true for the R(C-CO2Et) bonds which are longer in the axial orientation (1.52 Å) than in the equatorial orientation (1.51 Å). It is evident that the OEt and CO2Et substituents form stronger bonds when adopting the equatorial orientation compared to the axial orientation. It is noteworthy that the endocyclic C–O bond lengths in the disubstituted tetrahydro-2H-pyran conformers follow the trend 8ax,eq < 7ax,ax < 7eq,eq < 8eq,ax which is parallel to the trend followed by the relative stabilities of the conformers. The larger R(C–X) bond lengths in the axial anomers compared to the equatorial anomers indicate that the anomeric effect in the set of conformers studied might have a predominant hyperconjugative character. The hyperconjugative interactions are maximized when the lone pair at the endocyclic oxygen atom is aligned in an antiparallel geometry with the antibonding σ*(C–X) orbital. In such an orientation, the C-OEt bond is elongated and the endocyclic C–O bond is shortened.52,53
The total energies, Lewis energies and electronic delocalization contributions ΔΔEdeloc of substituted cyclohexane and tetrahydro-2H-pyran conformers bearing OEt and esteric CO2Et substituents calculated by the ωB97XD/Def2-TZVP/NBO computational protocol are presented in Table 2.
| Conformer | Total energy | Lewis energy | ΔEtotal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
ΔELewisb |
ΔΔEdeloc |
|---|---|---|---|---|---|
| a ΔEtotal = (Etotal)eq − (Etotal)ax. b ΔELewis = (ELewis)eq − (ELewis)ax. | |||||
| 5ax | −389.745044 | −389.074716 | |||
| 5eq | −389.745298 | −389.071730 | 0.16 | −1.87 | 2.03 |
| 6ax | −425.653647 | −424.892233 | |||
| 6eq | −425.651720 | −424.884327 | 1.21 | 4.96 | −3.75 |
| 7ax,ax | −692.871109 | −691.460613 | |||
| 7eq,eq | −692.865258 | −691.456352 | 3.67 | 2.67 | 1.00 |
| 8ax,eq | −692.871077 | −691.467298 | 0.02 | −4.19 | 4.21 |
| 8eq,ax | −692.868757 | −691.448274 | 1.48 | 7.74 | −6.26 |
Perusal of Table 2 manifests the AE in the substituted tetrahydro-2H-pyran conformers bearing OEt and/or OEt and esteric CO2Et substituents. In the ethoxy substituted cyclohexane ring conformers 5ax and 5eq, the ethoxy group prefers the equatorial position. The 5eq conformer is predicted to be slightly more stable than the 5ax one by only 0.28 kcal mol−1 at the ωB97XD/Def2-TZVP level of theory being very close to the value of 0.21 kcal mol−1 calculated at the more sophisticated CCSD(T) complete basis set (CBS) limit for the analogous OMe substituted cyclohexane conformers.54 The negative ΔELewis energy corroborates the higher stability of 5eq relative to the 5ax conformer, while the positive ΔΔEdeloc delocalization energy indicates the predominance of the steric effect (gauche interactions originating from non-bonded steric interactions). These data indicate that the anomeric effect does not exist in the ethoxy substituted cyclohexane ring conformers 5ax and 5eq manifesting the key role of the endocyclic O heteroatom in the anomeric phenomenon.
In the ethoxy substituted tetrahydro-2H-pyran 6ax and 6eq conformers, the ethoxy group prefers the axial position. 6ax is predicted to be slightly more stable than 6eq by 0.96 kcal mol−1. The preference of the axial conformation of 6 is further corroborated by the positive ΔEtotal and negative ΔELewis energies that give positive ΔΔEdeloc delocalization energies indicating strong electronic delocalization due to hyperconjugative interactions.
The tetrahydro-2H-pyran system bearing EtO and ester CO2Et substituents at the C-6 and C-3 positions, respectively, could adopt four possible configurations, namely the ax,ax, eq,eq, ax,eq and eq,ax configurations which constitute two diastereomeric pairs. The more stable conformers of 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax are shown in Table 2. The 8ax,eq conformer corresponds to the global minimum in the potential energy surface (PES) while the 7ax,ax, 7eq,eq and 8eq,ax conformers are local minima at 0.15, 0.40 and 1.29 kcal mol−1 higher in energy at the ωB97XD/Def2-TZVP level of theory in the gas phase. Keeping the esteric substituent at C-3 in the axial position, the estimated delocalization energy ΔΔEdeloc for the 7ax,ax and 8eq,ax pair of conformers is negative (−6.26 kcal mol−1). The positive ΔEtotal and positive ΔELewis energies favor the axial conformation. For the 7ax,ax and 8ax,eq pair of conformers, the estimated delocalization energy is positive (4.21 kcal mol−1) indicating the predominance of steric effects, while the positive ΔEtotal and negative ΔELewis energies in combination with positive ΔΔEdeloc delocalization energies favor the axial conformation when electronic delocalization is dominant and the equatorial conformation when the steric effect predominates. For the 7ax,ax and 7eq,eq pair of conformers, the estimated delocalization energy is positive (1.00 kcal mol−1) indicating the predominance of steric effects, while the positive ΔEtotal and negative ΔELewis energies giving positive ΔΔEdeloc delocalization energies favor again the axial conformation when electronic delocalization is dominant in line with the estimated relative stabilities of the conformers.
The steric energies, Es, on various nuclear centers in the substituted cyclohexane and tetrahydro-2H-pyran conformers bearing the OEt and/or OEt and esteric CO2Et substituents calculated at the ωB97XD/Def2-TZVP level of theory are given in Table 3.
|
|
||||
|---|---|---|---|---|
| Conformer | O-7 | Y-1 | C-6 | C-3 |
| 5ax | 20.52 | 0.56 | ||
| 5eq | 20.80 | 0.54 | ||
| 6ax | 20.08 | 21.34 | 0.88 | |
| 6eq | 20.71 | 20.71 | 0.88 | |
| 7ax,ax | 20.08 | 21.34 | 0.94 | 0.44 |
| 7eq,eq | 21.34 | 20.71 | 0.88 | 0.06 |
| 8ax,eq | 20.08 | 21.34 | 0.88 | 0.63 |
| 8eq,ax | 21.34 | 20.71 | 0.94 | 0.04 |
The estimated Es at O-7 for all conformers under investigation is slightly higher when the OEt substituent adopts the equatorial orientation independently of the orientation of the CO2Et substituent. Es is slightly higher for 5eq, 7eq,eq and 8eq,ax by about 0.3, 0.6 and 1.26 kcal mol−1, respectively. Note that Es is practically negligible at the anomeric C-6 and C-3 carbon atoms, around 0.45–0.94 kcal mol−1 and 0.06–0.63 kcal mol−1, respectively, in all conformers.
Shaded surface maps with projection of the Es distribution in 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers are shown in Fig. 4.
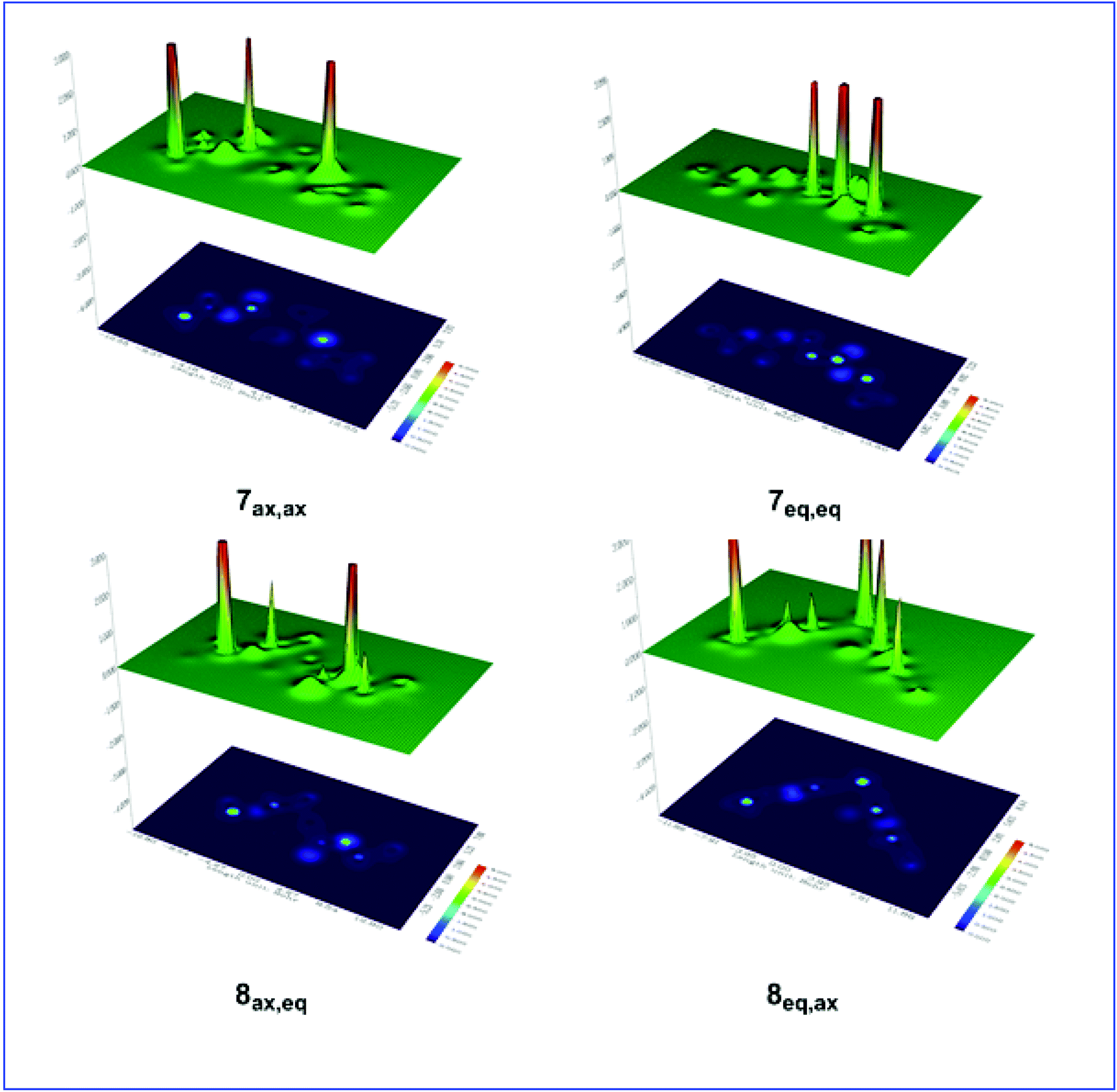 | ||
| Fig. 4 Shaded surface maps with projection of the Es distribution in 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers. | ||
The predominant hyperconjugative interactions contributing to the stabilization energy ΔE(2) in the substituted cyclohexane and tetrahydro-2H-pyran conformers bearing OEt and/or OEt and esteric CO2Et substituents calculated by the ωB97XD/Def2-TZVP/NBO computational protocol are presented in Table 4.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Conformer | LP(O1) → σ*(C6–O7) | LP(O1) → σ*(C5–C6) | LP(O1) → σ*(C6–H6) | LP(O7) → σ*(C6–O1) | LP(O7) → σ*(C5–C6) | LP(O7) → σ*(C6–H6) | LP(O10) → σ*(C3–C8) |
| 5ax | 10.90 | 9.79 | |||||
| 5eq | 11.03 | 9.84 | |||||
| 6ax | 18.29 | 9.49 | 3.38 | 18.53 | 1.37 | 9.85 | |
| 6eq | 4.10 | 9.41 | 10.37 | 19.52 | 1.61 | 11.32 | |
| 7ax,ax | 17.35 | 9.03 | 3.31 | 18.97 | 1.38 | 9.77 | 29.71 |
| 7eq,eq | 4.03 | 9.46 | 10.02 | 19.41 | 11.24 | 4.09 | 28.82 |
| 8ax,eq | 17.95 | 9.51 | 3.40 | 18.83 | 1.43 | 10.08 | 28.88 |
| 8eq,ax | 3.96 | 8.95 | 9.82 | 20.14 | 1.61 | 11.38 | 30.04 |
Inspection of Table 4 reveals that hyperconjugative interactions operate even in the mother cyclohexane 5ax and 5eq conformers having no heteroatom in the ring. These interactions involve the oxygen LP(O) lone pairs of the ethoxy substituent as donors and the antibonding σ*(C6–C5) and σ*(C6–H6) NBOs as acceptors. The 3D plots of the relevant donor and acceptor NBOs are shown in Scheme 3.
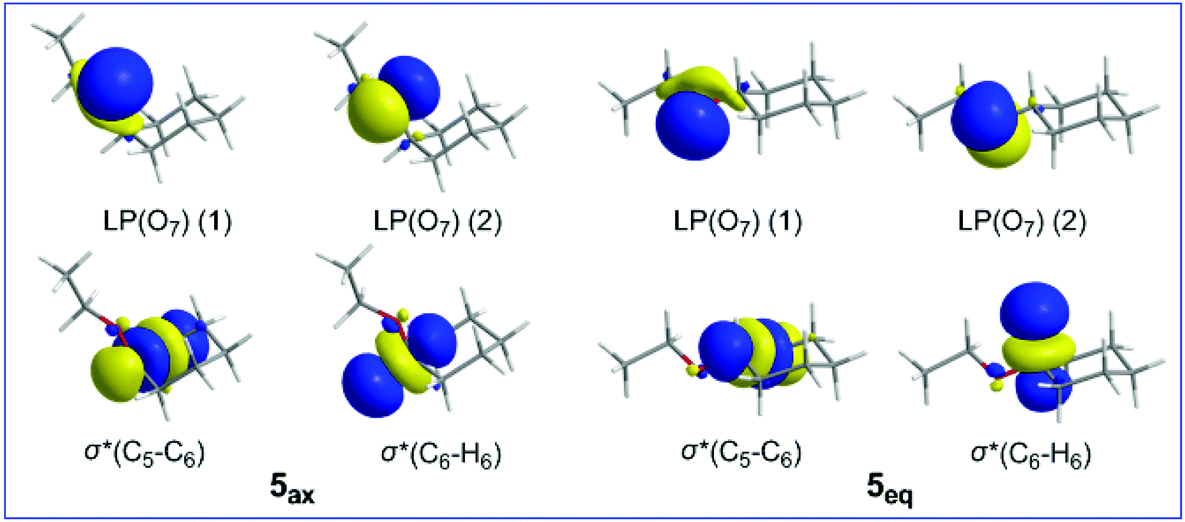 | ||
| Scheme 3 3D plots of the relevant NBOs involved in hyperconjugative interactions in 5ax and 5eq conformers. | ||
It can be seen that both hyperconjugative interactions make a slightly larger contribution to the equatorial 5eq conformation than to the axial 5ax conformation, amounting to 0.13 and 0.08 kcal mol−1, respectively, indicating that the conformational preferences in substituted cyclohexane rings are governed by a balance of steric and hyperconjugative interactions.
The axial conformation of 6 is further corroborated by considering the strong electronic delocalization due to hyperconjugative interactions (Table 4). These interactions involve the oxygen LP(O1) lone pairs of the O heteroatom in the tetrahydro-2H-pyran ring and the oxygen LP(O7) lone pairs of the ethoxy substituent as donors and the antibonding σ*(C6–O7), σ*(C6–C5), σ*(C6–H6) and σ*(C6–O1) NBOs as acceptors. The 3D plots of the relevant donor NBOs and acceptor NBOs for 6ax and 6eq are shown in Scheme 4.
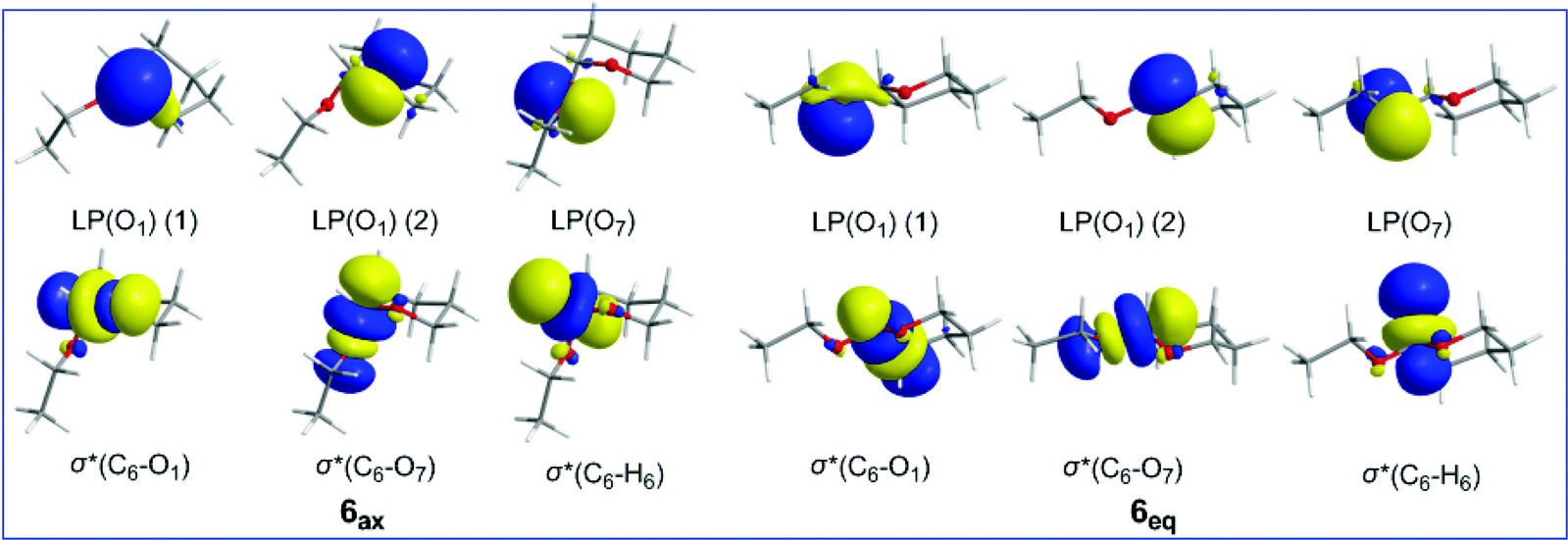 | ||
| Scheme 4 3D plots of the relevant NBOs involved in hyperconjugative interactions in 6ax and 6eq conformers. | ||
It is noteworthy that all hyperconjugative interactions, except the LP(O1) → σ*(C6–O7) one, make larger contributions to the equatorial conformation than to the axial conformation, totally amounting to 9.61 kcal mol−1. However, the contribution of the LP(O1) → σ*(C6–O7) hyperconjugative interaction to stabilization of the conformers is higher by 14.19 kcal mol−1 for 6ax than for 6eq.
Table 4 shows that the predominant hyperconjugative interactions contributing to the AE in 7ax,ax, 7eq,eq, 8ax,eq and 8eq,ax conformers follow the trend:
| 8ax,eq > 7ax,ax > 7eq,eq > 8eq,ax |
in excellent agreement with the trend of the relative stability of the 7ax,ax, 7eq,eq, 8ax,eq and 8eq,ax conformers confirming the key role of hyperconjugation in tuning the AE.
It is worth mentioning that the LP(O1) → σ*(C6–O7) hyperconjugative interactions induce a much larger contribution to the stabilization energies in the 7ax,ax and 8ax,eq conformers than in the 7eq,eq and 8eq,ax conformers, whereas the LP(O1) → σ*(C6–H6) hyperconjugative interactions induce larger contributions to the stabilization energy in the 7eq,eq and 8ax,eq conformers than in the 7ax,ax and 8ax,eq conformers. The contributions to the stabilization energies due to LP(O7) → σ*(C6–O1) and LP(O7) → σ*(C6–H6) hyperconjugative interactions are comparable for all conformers. The 3D plots of the relevant donor and acceptor NBOs for the 7ax,ax and 8eq,ax conformers given in the ESI (Scheme S1†) are analogous to the 3D plots of the monosubstituted tetrahydro-2H-pyran ring conformers.
Next, attempts have been made to identify the noncovalent interactions (dispersion forces, dipole–dipole and steric interactions) in the 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers. In this context, we calculated the reduced density gradient, 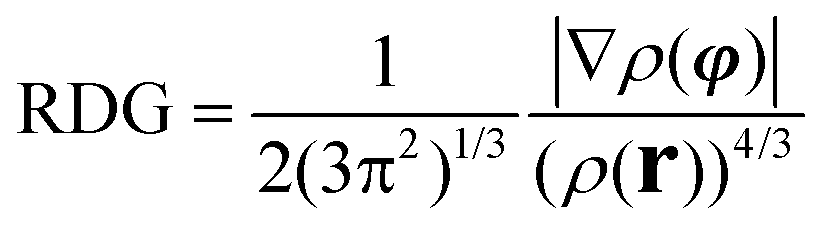 , where ρ(r) represents the electron density and |∇ρ(r)| stands for the norm of the electron density gradient vector. The Laplacian of the electron density, |∇2ρ(r)|, is usually decomposed into three components (∇2ρ(r) = λ1 + λ2 + λ3), which correspond to the three eigenvalues λi of the electron-density Hessian matrix. Analysis of the components can be used as a good descriptor of the types of intra- and intermolecular interactions in a molecular system.55–58 For covalent interactions, the Laplacian is negative, while for weaker noncovalent interactions, it is positive. It should be noted that the sign of the λ2 component allows discerning between different types of noncovalent interactions, while the electron density itself provides information about their strength.58
, where ρ(r) represents the electron density and |∇ρ(r)| stands for the norm of the electron density gradient vector. The Laplacian of the electron density, |∇2ρ(r)|, is usually decomposed into three components (∇2ρ(r) = λ1 + λ2 + λ3), which correspond to the three eigenvalues λi of the electron-density Hessian matrix. Analysis of the components can be used as a good descriptor of the types of intra- and intermolecular interactions in a molecular system.55–58 For covalent interactions, the Laplacian is negative, while for weaker noncovalent interactions, it is positive. It should be noted that the sign of the λ2 component allows discerning between different types of noncovalent interactions, while the electron density itself provides information about their strength.58
Fig. 5 shows the plots of the RDG versus the electron density multiplied by the sign of the λ2 Hessian eigenvalue, sign(λ2)ρ(r), along with the 3D plots of RDG isosurfaces (isosurface = 0.75 au) for the 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers calculated at the ωB97XD/Def2-TZVP level of theory.
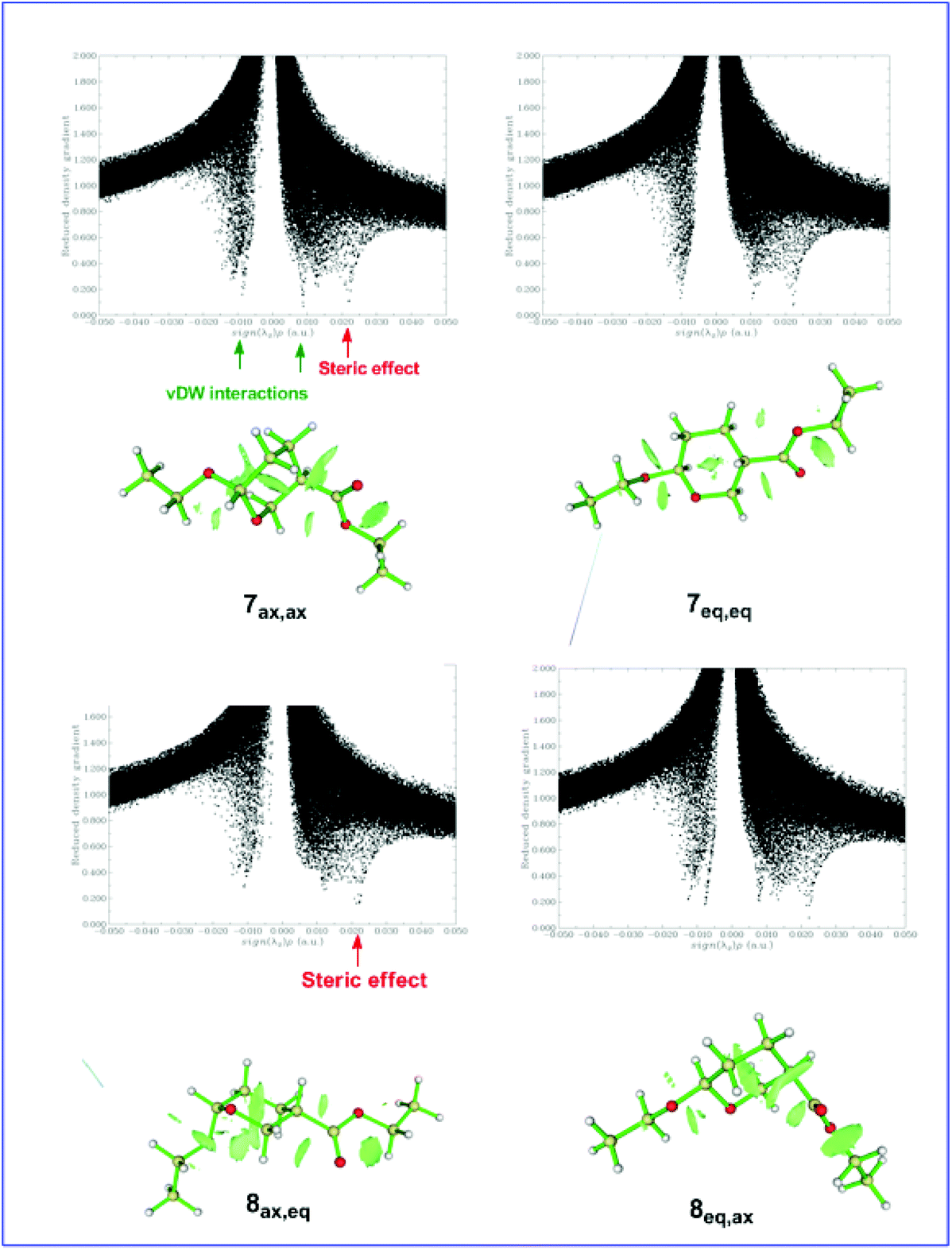 | ||
| Fig. 5 Scatter graphs of the RDG vs. sign(λ2)ρ(r) correlations along with the 3D RDG isosurfaces (isosurface = 0.75 au) for the 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers calculated at the ωB97XD/Def2-TZVP level of theory (scatter graphs obtained using promolecular electron density). | ||
It can be seen that all scatter graphs of RDG vs. sign(λ2)ρ(r) correlations exhibit one or more spikes in the low-density, low-gradient region, a signature of noncovalent interactions. Note that large negative values of sign(λ2)ρ(r) indicate attractive interactions (dipole–dipole or hydrogen bonding), large positive values of sign(λ2)ρ(r) indicate nonbonding interactions and values near zero indicate very weak van der Waals interactions. All non-covalent interactions are also clearly visualized as broad regions of real space in the molecular structures of the 7ax,ax, 7eq,eq, 8ax,eq, and 8eq,ax conformers by the RDG isosurfaces at low electron density values (colored in green). In summary, the conformational preferences in di-substituted tetrahydro-2H-pyran rings bearing the EtO substituent at C-6 and the CO2Et substituent at C-3 are tuned by the balance of non-covalent (weak vDW, dipole–dipole, hydrogen bonding) steric effects and hyperconjugative interactions.
The anomeric effect in oxazinane conformers
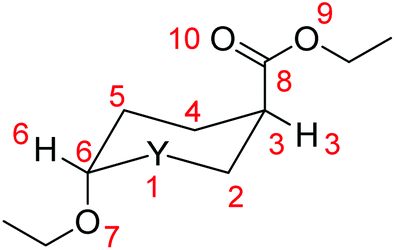
We investigated the AE for the set of substituted oxazinane conformers bearing OEt and/or OEt and esteric CO2Et substituents shown in Scheme 5.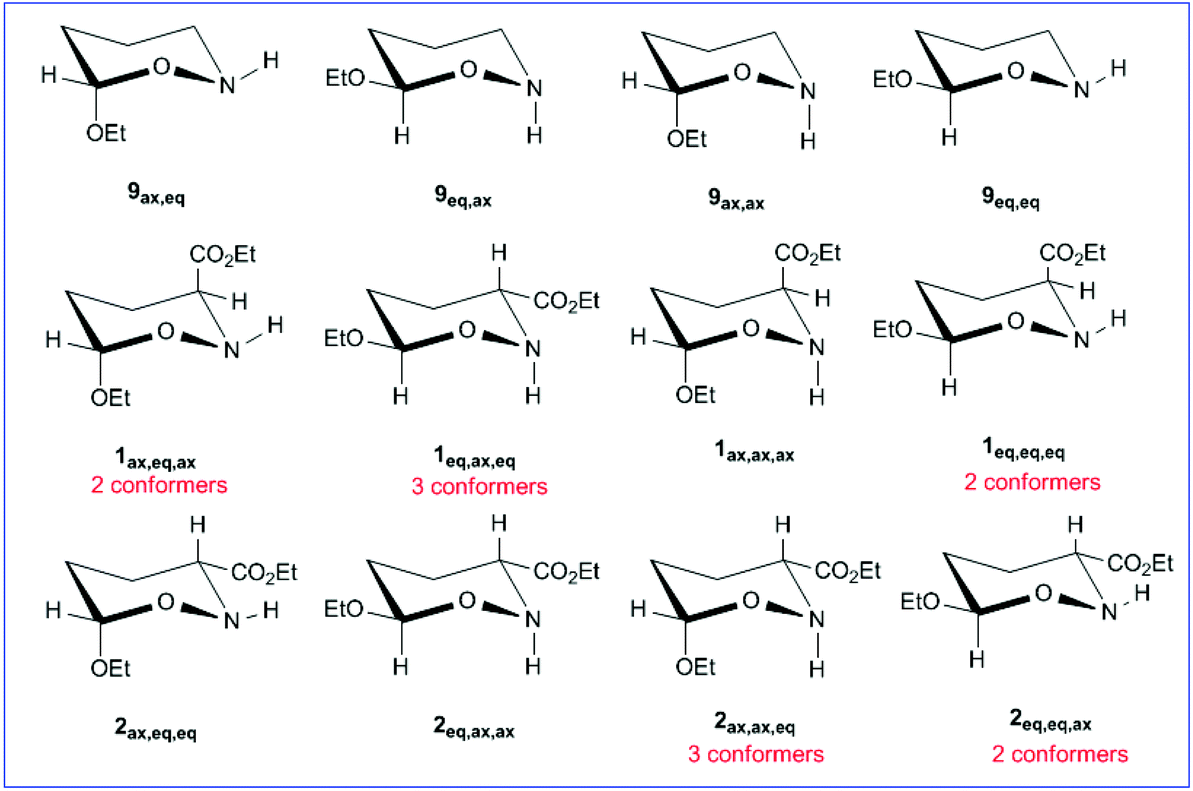 | ||
| Scheme 5 Oxazinane conformers studied (the first subscript is referred to the anomeric center, the second one shows the N–H bond orientation and the third one the CO2Et group orientation). The additional conformers are due to (C-3)-(CO) bond rotation. | ||
The equilibrium geometries of the oxazinane conformers with selected structural parameters optimized at the ωB97XD/Def2-TZVP level of theory in the gas phase along with the relative stability (numbers in red) are given in Fig. 6.
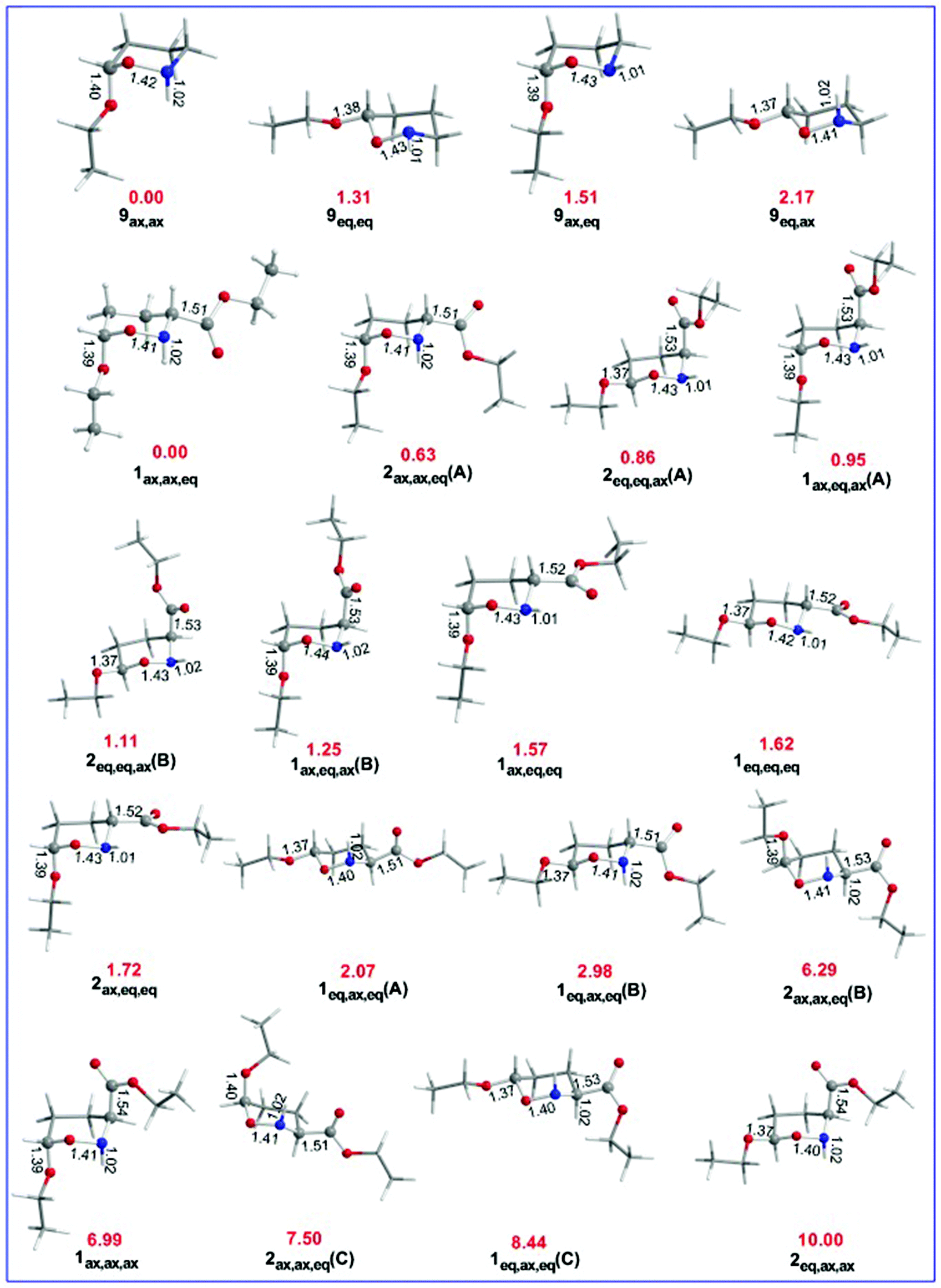 | ||
| Fig. 6 Equilibrium geometries with selected structural parameters (bond lengths in Å) and relative stability (in kcal mol−1, marked in red) of the oxazinane conformers calculated at the ωB97XD/Def2-TZVP level of theory in the gas phase. | ||
Inspection of Fig. 6 reveals that the R(C-OEt) bond lengths in the axial orientation (1.39–1.40 Å) are longer than the R(C-OEt) bond lengths in the equatorial orientation (1.37 Å). Similarly, the R(C-CO2Et) bonds are longer in the axial orientation (1.53–1.54 Å) than in the equatorial orientation (1.51–1.53 Å) indicating that the OEt and CO2Et substituents form stronger bonds when they are in the equatorial orientation than in the axial orientation. The larger R(C–X) bond lengths in axial anomers compared to equatorial anomers indicate that the anomeric effect in the set of conformers studied might have a predominant hyperconjugative character as it will be discussed later on. The R(O–N) bond lengths are also significantly affected by the substituent's orientation. In conformers 9, the R(O–N) bond lengths for the axial anomers (1.42 and 1.43 Å) are longer than those for the equatorial anomers (1.41 and 1.43 Å). In conformers 1 and 2, the R(O–N) bond lengths for all combinations of axial and equatorial orientations of the X substituents (X = OEt, CO2Et) range from 1.40 to 1.44 Å. The large width of the R(O–N) bond lengths for all combinations of axial and equatorial orientations of the X substituents is due to the orientation of the H atom bonded to the nitrogen heteroatom. Independently of the orientation of the H atom bonded to the nitrogen, the R(O–N) bond lengths follow the trend: ax,ax > ax,eq > eq,ax > eq,eq. The estimated R(N–H) bond lengths for the axial orientation of the H atom are slightly longer (average = 1.018 Å) than for the equatorial orientation (average = 1.02 Å).
The 9ax,ax conformer is predicted to be the global minimum in the potential energy surface (PES) while the 9eq,eq, 9ax,eq and 9eq,ax conformers are local minima at 1.31, 1.51 and 2.17 kcal mol−1 higher in energy at the ωB97XD/Def2-TZVP level of theory. For the set of 1 and 2 conformers, 1ax,ax,eq is predicted to be the global minimum, while 2eq,ax,ax is the conformer with the higher relative stability (10.00 kcal mol−1). The relative stability of 1 and 2 conformers is shown schematically in the form of a ladder in Fig. 7.
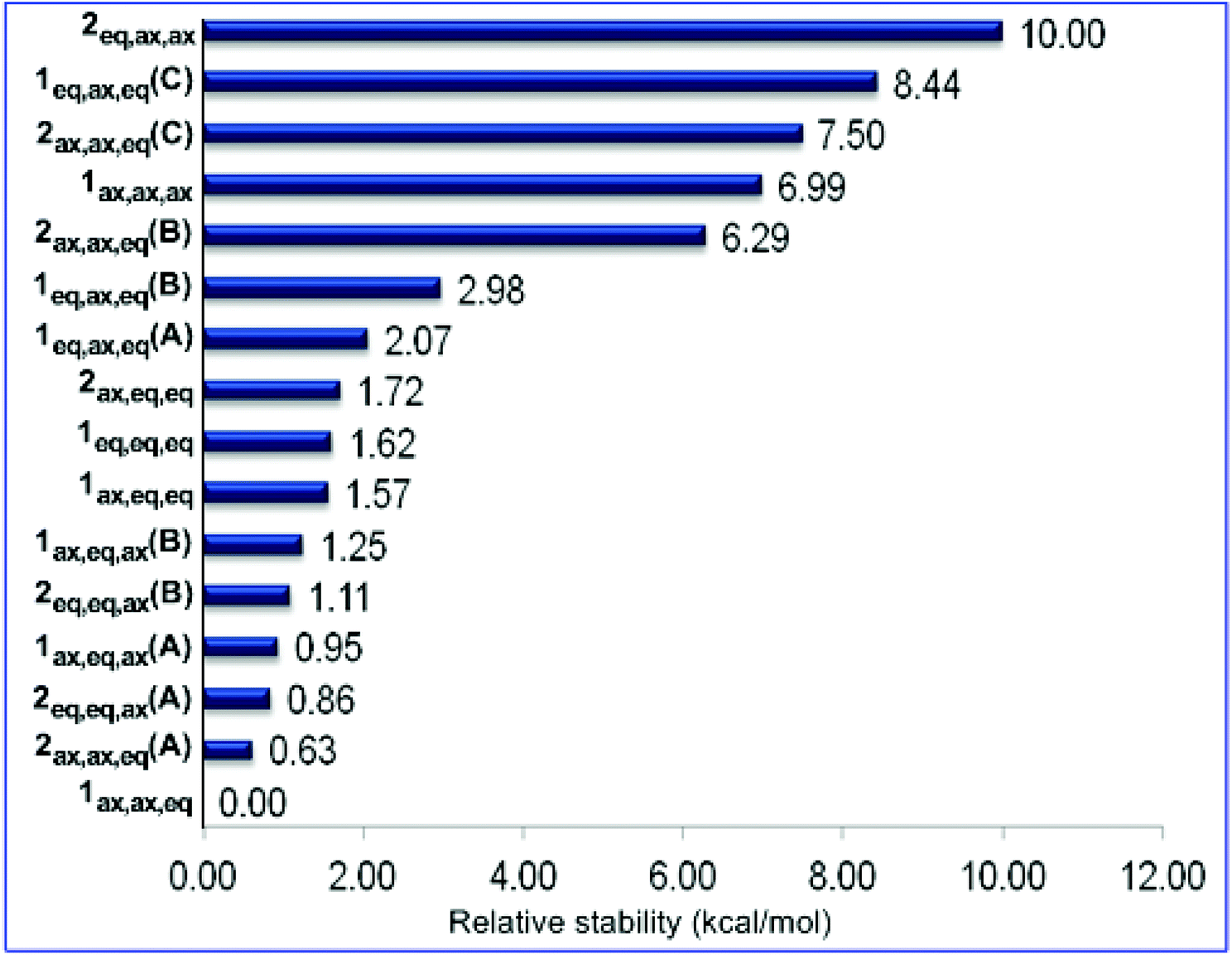 | ||
| Fig. 7 Ladder of relative stability of the oxazinane conformers calculated at the ωB97XD/Def2-TZVP level of theory in the gas phase. | ||
The total energies, Lewis energies, and electronic delocalization contributions to the AE (ΔΔEdeloc) for selected conformer pairs of substituted oxazinane ring conformers with OEt and/or OEt and esteric CO2Et substituents calculated by the ωB97XD/Def2-TZVP/NBO computational protocol are given in the ESI (Table S5†). Herein we will present and analyze the results only for some representative conformer pairs with different orientations of OEt, CO2Et and H, namely 9eq,eq–9ax,ax, 9ax,eq–9ax,ax, 9eq,ax–9ax,ax, 1eq,ax,eq(A)–1ax,ax,eq, 2ax,eq,eq–2ax,ax,eq(A), 1ax,eq,eq–1ax,eq,ax(B), 1eq,ax,eq(C)–1ax,eq,eq and 1eq,eq,eq–1ax,ax,ax. In the 9eq,eq–9ax,ax pair, both OEt and H change the orientation from equatorial to axial. The negative ΔELewis energy (−13.07 kcal mol−1) in combination with the positive ΔΔEdeloc delocalization energy (14.47 kcal mol−1) implies stereoelectronic origin for the AE. In the 9ax,eq–9ax,ax pair where only the H atom changes the orientation from equatorial to axial, the negative ΔELewis energy (−6.37 kcal mol−1) in combination with the positive ΔΔEdeloc delocalization energy (7.88 kcal mol−1) implies also a stereoelectronic origin for the AE. In the 9eq,ax–9ax,ax pair where only the OEt substituent changes the orientation from equatorial to axial, the positive ΔELewis energy (6.48 kcal mol−1) in combination with the negative ΔΔEdeloc delocalization energy (−4.00 kcal mol−1) implies a non-stereoelectronic origin for the AE and therefore electrostatic and steric effects are the key factors in tuning the conformational preference for this pair. The same holds true for the 1eq,ax,eq(A)–1ax,ax,eq pair where only the OEt substituent changes the orientation from equatorial to axial (ΔELewis = 2.52 kcal mol−1; ΔΔEdeloc = −0.15 kcal mol−1). In the 2ax,eq,eq–2ax,ax,eq(A) pair where only the H orientation changes, negative ΔELewis (−10.44 kcal mol−1) in combination with positive ΔΔEdeloc (12.21 kcal mol−1) reflects the stereoelectronic origin for the AE. In the 1ax,eq,eq–1ax,eq,ax(B) pair where only the orientation of the CO2Et substituent changes, negative ΔELewis (−2.48 kcal mol−1) in combination with positive ΔΔEdeloc (2.94 kcal mol−1) reflects the stereoelectronic origin for the AE. In the 1eq,ax,eq(C)–1ax,eq,eq pair where both OEt and H change the orientation, positive ΔELewis (18.11 kcal mol−1) in combination with negative ΔΔEdeloc (−11.12 kcal mol−1) implies a non-stereoelectronic origin for the AE. In the 1eq,eq,eq–1ax,ax,ax pair where all substituents change the orientation from equatorial to axial, negative ΔELewis (−26.04 kcal mol−1) in combination with positive ΔΔEdeloc (20.80 kcal mol−1) is in accordance with the stereoelectronic origin for the AE. Generally, in the complete set of conformers studied, the AE exhibits a stereoelectronic origin.
We then probed whether favorable/unfavorable hyperconjugative interactions (donor–acceptor orbital interactions between the ring heteroatoms and the anomeric X substituents) contribute to the AE (hyperconjugation model of the AE) in the set of conformers studied. The predominant hyperconjugative interactions contributing to the AE in the substituted oxazinane ring conformers bearing the OEt and/or the OEt and CO2Et substituents calculated at the ωB97XD/Def2-TZVP level of theory are given in the ESI (Table S6†). The numbering of the oxazinane ring is shown in Scheme 6.
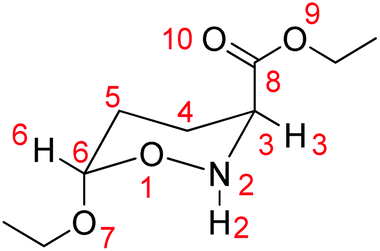 | ||
| Scheme 6 Numbering of the disubstituted oxazinane ring. | ||
Perusal of Table S6† reveals that among the predominant hyperconjugative interactions identified, the LP(O1) → σ*(C6–O7), LP(O1) → σ*(C6–H6), LP(O1) → σ*(N2–H2) and LP(N2) → σ*(C3–C8) interactions make different contributions to the conformational preferences. All other hyperconjugative interactions contribute almost equally to the stabilization energy of all conformers. The LP(O1) → σ*(C6–O7) interactions contribute to ΔE(2) about 15.8–16.8 kcal mol−1 in the conformers with the OEt substituent at the axial position and only 2.6–2.8 kcal mol−1 in the conformers with the OEt substituent at the equatorial position. It is noteworthy that the higher contribution of the LP(O1) → σ*(C6–O7) interactions to ΔE(2) (about 25.5 kcal mol−1) occurs in the 1ax,ax,ax conformer having OEt, CO2Et and H in the axial positions, while the lowest contribution (about 2.6 kcal mol−1) occurs in 1eq,eq,eq having OEt, CO2Et and H in the equatorial positions. The LP(O1) → σ*(C6–H6) hyperconjugative interactions contribute to ΔE(2) about 2.1–2.3 kcal mol−1 in conformers having the X substituents in the axial orientation, but the contribution is larger (7.5–9.4 kcal mol−1) when the X substituents are in the equatorial orientation. In contrast, the contributions to ΔE(2) from the LP(O1) → σ*(N2–H2) interactions are higher (5.6–7.5 kcal mol−1) in conformers with axial H than in conformers with equatorial H (1.10–1.7 kcal mol−1). Furthermore, the contribution to ΔE(2) from the LP(N2) → σ*(C3–C8) interactions is higher (9.0–10.5 kcal mol−1) in conformers with the axial CO2Et substituent than in conformers with equatorial CO2Et (0.6–3.3 kcal mol−1). The higher contributions to ΔE(2) result from the LP(O9) → σ*(C8–O10) and LP(O10) → σ*(C8–O9) hyperconjugative interactions localized on the esteric CO2Et substituent amounting to 70.2–85.2 kcal mol−1 and 43.2–47.6 kcal mol−1 respectively. Note that the (Z)-conformation of the CO2Et substituent exhibits relatively stronger LP(O10) → σ*(C8–O9) hyperconjugative interactions than the (E)-conformation.
Considering that the hyperconjugative interactions are not the only key factor tuning the AE in the oxazinane ring conformers, steric effects and possible non-covalent interactions have to be taken into account. The steric energies calculated at the C-6 and C-3 anomeric centers and the O-1 and N-2 heteroatoms in the ring as well as at the middle point of the O1–N2 bond are presented in Table S7.† Representative shaded surface maps with projection of the Es distribution in oxazinane conformers are shown in Fig. 8.
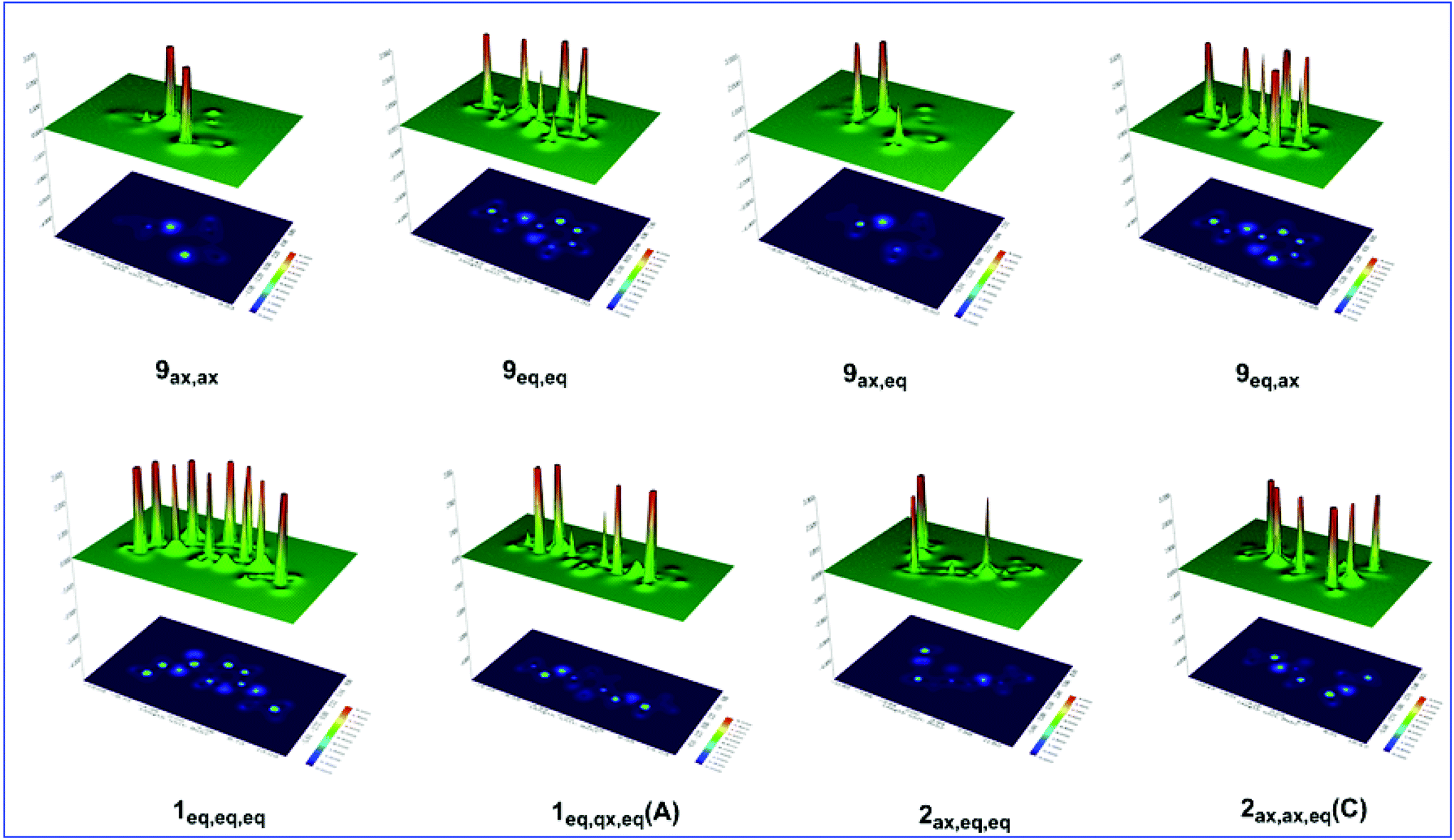 | ||
| Fig. 8 Shaded surface maps with projection of the Es distribution for selected oxazinane conformers. | ||
The estimated steric energy at the O-1 and N-2 heteroatoms is almost the same for all conformers ranging from 23.6 to 27.0 (average = 25.3 kcal mol−1) and 13.2 to 14.4 kcal mol−1 (average = 13.8 kcal mol−1), respectively. Note that the O-1 heteroatom exerts higher steric energy than the N heteroatom. Interestingly, the axial and equatorial positions of the N–H hydrogen fairly affect the steric energies at the O-1 and N-2 heteroatoms. For example, in the 9ax,eq and 9ax,ax conformers, O-1 exerts a steric energy of 23.9 and 26.4 kcal mol−1 respectively, while N-2 exerts a steric energy of 13.8 and 14.4 kcal mol−1, respectively. The same holds true for the 9eq,ax and 9eq,eq conformers, where O exerts a steric energy of 25.7 and 23.9 kcal mol−1, respectively. It is clear that the N–H hydrogen atom induces higher steric energy at both O-1 and N-2 heteroatoms when it is axial and not equatorial. This pattern is followed by the conformational pairs having the EtO substituent in the axial and equatorial positions; that is the axial EtO induces higher steric energy at both the O-1 and N-2 heteroatoms than the equatorial EtO. It should be noted that the steric energy at the C-6 and C-3 anomeric centers is relatively small, amounting to 0.9 and 0.2 kcal mol−1, respectively.
It is noteworthy that the steric energy at the middle point (centroid, cd) of the O1–N2 bond has higher values (20.0–151.2 kcal mol−1) and strongly depends on the configuration of the conformers. Conformers 9eq,eq and 9eq,ax exhibit the highest values of 136.2 and 151.2 kcal mol−1, respectively, followed by the 2ax,eq,eq conformer (112.3 kcal mol−1). Conformers 2eq,eq,ax(B) and 1eq,ax,eq(C) exhibit high steric energy values of 101.3 and 92.9 kcal mol−1, respectively. On the other hand, the lowest values of 20.1, 21.3, 26.4, 28.2 and 31.4 kcal mol−1 correspond to 1ax,eq,ax(A), 1eq,eq,eq, 1ax,eq,ax(B), 1ax,ax,ax and 9ax,ax conformers, respectively. For all other conformers. the steric energy ranges from 43.3 up to 74.7 kcal mol−1. It should be noted that the increase of steric energy at the centroid of the O1–N2 bond in the transition states of the thermal amine-promoted transformations of the oxazinane ring favors the rupture of the O1–N2 bond and the ring opening. Obviously, to avoid ring opening in the thermal amine-promoted transformations of the oxazinane ring, conformers with low values of steric energy at the centroid of the O1–N2 bond should be chosen.
Fig. 9 shows the plots of RDG versus the electron density multiplied by the sign of the λ2 Hessian eigenvalue, sign(λ2)ρ(r), along with the 3D plots of RDG isosurfaces (isosurface = 0.75 au) for representative oxazinane ring conformers, calculated at the ωB97XD/Def2-TZVP level of theory.
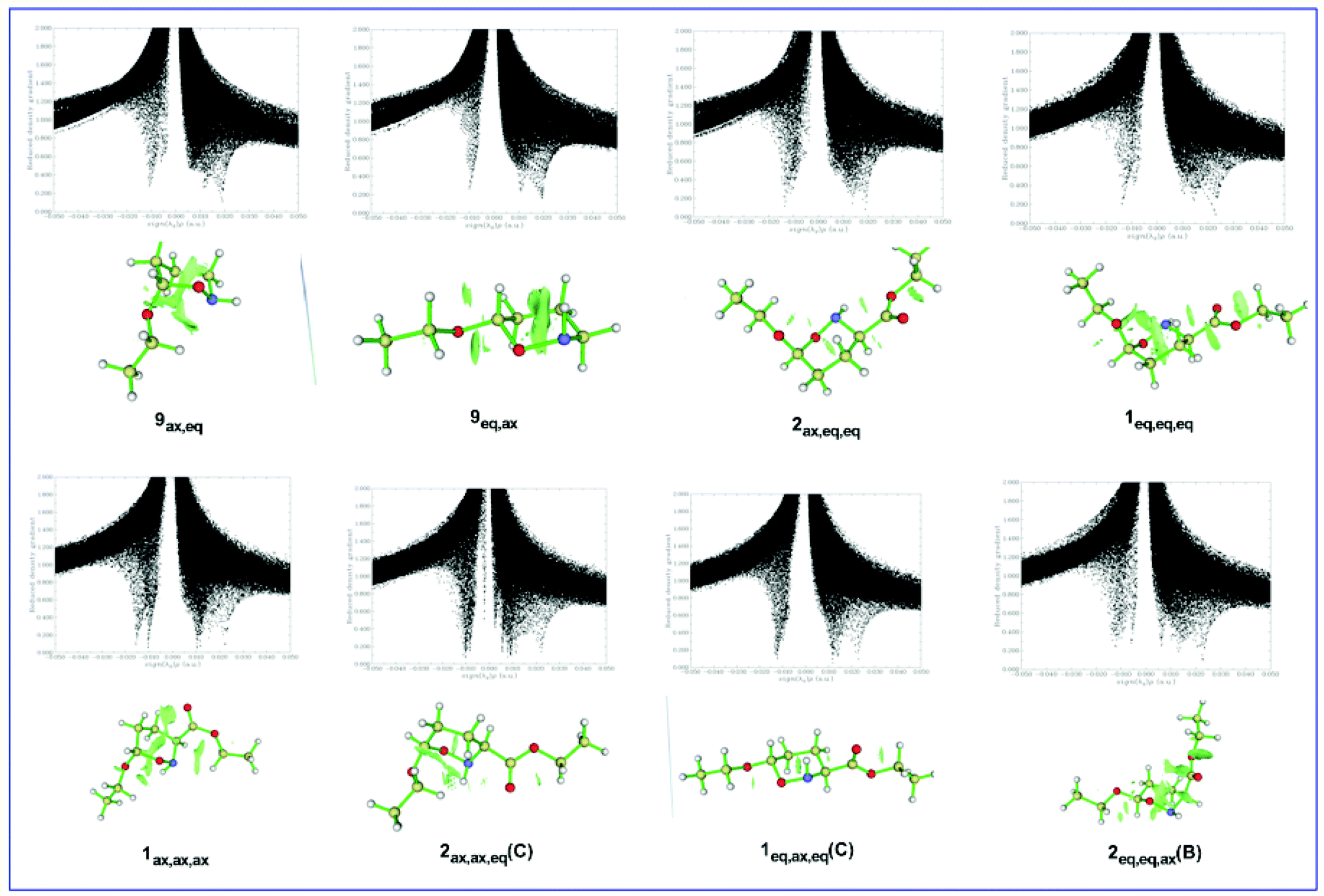 | ||
| Fig. 9 Scatter graphs of the RDG vs. sign(λ2)ρ(r) correlations along with the 3D RDG isosurfaces (isosurface = 0.75 au) for selected oxazinane ring conformers calculated at the ωB97XD/Def2-TZVP level of theory (scatter graphs obtained using SCF electron density). | ||
It can be seen that the low-density, low-gradient spike lying at negative values are characteristic of stabilizing interactions. On the other hand, the low-density, low-gradient spike for the sterically crowded oxazinane ring remains at positive values indicating the lack of bonding in the central area of the ring. The green isosurfaces located at the center of the oxazinane rings characterize the steric repulsions between the ring atoms, while the green isosurfaces located at the real space between non-bonded atoms characterize non-covalent electrostatic, dipole–dipole, weak vDW and steric interactions. The stronger non-covalent interactions in the 9eq,ax conformer than in the 9ax,eq conformer reflect the stereoelectronic origin of the AE in these conformers. The same holds true for the 2ax,eq,eq and 1eq,eq,eq(A) conformers. Similarly, the stronger non-covalent interactions in the 1ax,ax,ax conformer than in the 1eq,eq,eq(B) conformer and in the 2ax,ax,eq(D) conformer than in the 1eq,ax,eq(D) conformer are in line with the stereoelectronic nature of the AE in these conformer pairs.
In summary, the conformational preferences in di-substituted oxazinane rings bearing the EtO substituent at C-6 and the CO2Et substituent at C-3, as in the case of the tetrahydro-2H-pyran ring conformers, are tuned by the balance of non-covalent (weak vDW, dipole–dipole, hydrogen bonding, steric effects) and hyperconjugative interactions.
Conclusions
The mechanism of the thermal amine-promoted transformations of oxazinane ring conformers, adopting chair-like conformations, has been scrutinized employing DFT computational protocols. The first step of the reaction mechanism involves the abstraction of the C-3 hydrogen, upon attack by the amine nitrogen, leading to the same or the isomeric conformer. Depending on the reactant's structure and reaction conditions, breaking of the O–N bond could occur in the transition state leading to ring opening. NBO population analysis in conjunction with calculations of steric effects and non-covalent interactions illustrated the origin of the anomeric effect in tetrahydro-2H-pyran and oxazinane ring conformers. The origin of the anomeric effect in the tetrahydro-2H-pyran and oxazinane ring conformers is complex. It is the balance of non-covalent (weak vDW, dipole–dipole, electrostatic forces, hydrogen bonding) steric effects and hyperconjugative interactions that tune the AE. The breaking of the O–N bond in the transition state leading to ring opening is forced by the steric effects (steric energy, steric forces).Conflicts of interest
There are no conflicts to declare.Notes and references
- S. M. M. Lopes, A. L. Cardoso, A. Lemos and T. M. V. D. Pinho e Melo, Recent Advances in the Chemistry of Conjugated Nitrosoalkenes and Azoalkenes, Chem. Rev., 2018, 118, 11324–11352 CrossRef CAS and references therein.
- J. K. Gallos, V. C. Sarli, A. C. Varvogli, C. Z. Papadoyanni, S. D. Papaspyrou and N. G. Argyropoulos, The hetero-Diels–Alder addition of ethyl 2-nitrosoacrylate to electron-rich alkenes as a route to unnatural α-amino acids, Tetrahedron Lett., 2003, 44, 3905–3909 CrossRef CAS.
- J. K. Gallos, V. C. Sarli, Z. S. Massen, A. C. Varvogli, C. Z. Papadoyanni, S. D. Papaspyrou and N. G. Argyropoulos, A new strategy for the stereoselective synthesis of unnatural α-amino acids, Tetrahedron, 2005, 61, 565–574 CrossRef CAS.
- Z. S. Massen, V. C. Sarli, E. Coutouli-Argyropoulou and J. K. Gallos, Synthesis of C-Glycosylated Amino Acids by Hetero-Diels−Alder Addition of Ethyl 2-Nitrosoacrylate to exo-Glycals, Carbohydr. Res., 2011, 346, 230–237 CrossRef CAS.
- Z. S. Massen and J. K. Gallos, Asymmetric Synthesis of Both Enantiomers of Protected 5-Hydroxynorvaline by Hetero-Diels-Alder Addition of ethyl 2-Nitrosoacrylate to (R)- and (S)-1-Phenylbutyl Vinyl Ether, J. Heterocycl. Chem., 2012, 49, 1214–1217 CrossRef CAS.
- J. K. Gallos, V. C. Sarli, T. V. Koftis and E. Coutouli-Argyropoulou, New Entry to Hydroxylated Pyrrolizidines, Tetrahedron Lett., 2000, 41, 4819–4822 CrossRef CAS.
- J. K. Gallos, V. C. Sarli, C. I. Stathakis, T. V. Koftis, V. R. Nachmia and E. Coutouli-Argyropoulou, Hetero-Diels-Alder Additions to Pent-4-enofuranosides: Concise Synthesis of Hydroxylated Pyrrolizidines, Tetrahedron, 2002, 58, 9351–9357 CrossRef CAS.
- Z. S. Massen, E. Coutouli-Argyropoulou, V. C. Sarli and J. K. Gallos, Synthesis of a Protected Trihydroxyindolizidine 3-Carboxylate via a Hetero-Diels−Alder Addition of Ethyl 2-Nitrosoacrylate to a D-Ribose-derived exo-Glycal, Carbohydr. Res., 2011, 346, 508–511 CrossRef CAS.
- A.-T. Serafidou, E. G. Yioti and J. K. Gallos, A Protection-Free Synthetic Access to (±)-1-Deoxy-6-epi-castanospermine and (±)-1Deoxy-6,8a-di-epi-castanospermine, Eur. J. Org. Chem., 2013, 939–943 CrossRef CAS.
- E. G. Yioti, I. K. Mati, A. G. Arvanitidis, Z. S. Massen, E. S. Alexandraki and J. K. Gallos, Synthesis of (±)-Crispine A via a Nitrosoalkene Hetero-Diels-Alder Addition to Ethyl Vinyl Ether, Synthesis, 2011, 142–146 CAS.
- R. Zimmer, T. Arnold, K. Homann and H.-U. Reissig, An Efficient and Simple Synthesis of 3,4,5,6-Tetrahydro-2H-1,2-oxazines by Sodium Cyanoborohydride Reduction of 5,6-Dihydro-4H-1,2-oxazines, Synthesis, 1994, 1050–1056 CrossRef CAS.
- J. T. Edward, Stability of glycosides to acid hydrolysis, Chem. Ind., 1955, 1102–1104 CAS.
- C. B. Anderson and D. T. Sepp, Conformation and the anomeric effect in 2-halotetrahydropyrans, J. Org. Chem., 1967, 32, 607–611 CrossRef CAS.
- E. A. C. Lucken, Chemical applications of nuclear quadrupole resonance spectroscopy. Part III. The inductive effect of substituents belonging to the first row of the periodic table, J. Chem. Soc., 1959, 2954 RSC.
- C. L. Perrin, K. B. Armstrong and M. A. Fabian, The origin of the anomeric effect: conformational analysis of 2-methoxy-1,3-dimethylhexahydropyrimidine, J. Am. Chem. Soc., 1994, 116, 715–722 CrossRef CAS.
- C. M. Filloux, The Problem of Origins and Origins of the Problem: Influence of Language on Studies Concerning the Anomeric Effect, Angew. Chem., Int. Ed., 2015, 54, 8880–8894 CrossRef CAS.
- Y. Mo, Computational evidence that hyperconjugative interactions are not responsible for the anomeric effect, Nat. Chem., 2010, 2, 666–671 CrossRef CAS.
- I. V. Alabugin, Stereoelectronic Effects: A Bridge between Structure and Reactivity, John Wiley & Sons, 2016 Search PubMed.
- B. Fuchs, A. Ellencweig, E. Tartakovsky and P. Aped, Solvent polarity and the anomeric effect, Angew. Chem., Int. Ed. Engl., 1986, 25, 287–289 CrossRef.
- S. Wolfe, B. M. Pinto, V. Varma and R. Y. N. Leung, The Perlin Effect: bond lengths, bond strengths, and the origins of stereoelectronic effects upon one-bond C-H coupling constants, Can. J. Chem., 1990, 68, 1051–1062 CrossRef CAS.
- E. Juaristi and G. Cuevas, Recent studies of the anomeric effect, Tetrahedron, 1992, 48, 5019–5087 CrossRef CAS.
- A. Vila and R. A. Mosquera, Atoms in Molecules Interpretation of the Anomeric Effect in the O-C-O Unit, J. Comput. Chem., 2007, 28, 1516–1530 CrossRef CAS.
- K. Eskandari and R. A. Mosquera, Interpretation of Anomeric Effect in the N-C-N Unit with the Quantum Theory of Atoms in Molecules, J. Phys. Chem. A, 2007, 111, 8491–8499 CrossRef CAS.
- U. Salzner and P. v. R. Schleyer, Ab Initio Examination of Anomeric Effects in Tetrahydropyrans, 1,3-Dioxanes, and Glucose, J. Org. Chem., 1994, 59, 2138–2155 CrossRef CAS.
- M. P. Freitas, The anomeric effect on the basis of natural bond orbital analysis, Org. Biomol. Chem., 2013, 11, 2885–2890 RSC.
- N. Hasanzadeh, D. Nori-Shargh, M. Farzipour and B. Ahmadi, On the origin of the anomeric effect. Probing the impacts of the stereoelectronic interactions and steric exchange components on the structural and configurational behaviors of 2,3- and 2,5-dihalo-1,4-oxathianes, Org. Biomol. Chem., 2015, 13, 6965–6976 RSC.
- P. Ghanbarpour and D. Nori-Shargh, Exploring the origin of the anomeric relationships in 2-cyanooxane, 2-cyanothiane, 2-cyanoselenane and their corresponding iso-cyano isomers. Correlations between hyperconjugative anomeric effect, hardness and electrostatic interactions, RSC Adv., 2016, 6, 46406–46420 RSC.
- F. A. Martins, J. M. Silla and M. P. Freitas, Theoretical study on the anomeric effect in a-substituted tetrahydropyrans and piperidines, Carbohydr. Res., 2017, 451, 29–35 CrossRef CAS.
- M. Nishio and Y. Kohno, The Orbital-Effect-Myth, Chem-Bio Inf. J., 2018, 18, 86–95 CAS.
- Y. Huang, A.-G. Zhong, O. Yang and S. B. Liu, Origin of anomeric effect: A density functional steric analysis, J. Chem. Phys., 2011, 134, 084103–084112 CrossRef.
- F. Glauco, G. F. Bauerfeldt, T. M. Cardozo, M. S. Pereira and C. O. da Silva, The anomeric effect: the dominance of exchange effects in closed-shell systems, Org. Biomol. Chem., 2013, 11, 299–308 RSC.
- D. Ferro-Costas and R. A. Mosquera, Complementarity of QTAIM and ELF Partitions: Deeper Understanding of the Anomeric Effect, J. Chem. Theory Comput., 2013, 9, 4816–4824 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- S. Grimme, Semiempirical GGA–type density functional constructed with a long–range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- D. Becke, Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals, J. Chem. Phys., 1997, 107, 8554 CrossRef.
- D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Q. Wu and W. T. Yang, Empirical correction to density functional theory for van der Waals interactions, J. Chem. Phys., 2002, 116, 515 CrossRef CAS.
- Y. Minenkov, Å. Singstad, O. Occhipinti and V. Jensen, The accuracy of DFT-optimized geometries of functional transition metal compounds: a validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase, Dalton Trans., 2012, 41, 5526–5541 RSC.
- S. Kozuch and J. M. L. Martin, Halogen Bonds: Benchmarks and Theoretical Analysis, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS.
- U. Salzner and A. Aydin, Improved Prediction of Properties of π-Conjugated Oligomers with Range-Separated Hybrid Density Functionals, J. Chem. Theory Comput., 2011, 7, 2568–2583 CrossRef CAS.
- K. Wang, J. Lv and J. Miao, Assessment of density functionals and force field methods on anion–π interaction in heterocyclic calix complexes, Theor. Chem. Acc., 2015, 134, 5–10 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- EMSL basis set exchange, https://bse.pnl.gov/bse/portal, accessed 26-06-2019. .
- (a) C. Gonzalez and H. B. Schlegel, An improved algorithm for reaction path following, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, Reaction path following in mass-weighted internal coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (b) F. Weinhold, in The Encyclopedia of Computational Chemistry, ed. P. v. R. Schleyer, John Wiley & Sons, Chichester, U.K., 1998 Search PubMed.
- F. Cortés, J. Tenorio, O. Collera and G. Cuevas, Electronic delocalization contribution to the anomeric effect evaluated by computational methods, J. Org. Chem., 2001, 66, 2918–2924 CrossRef.
- Multiwfn Version 3.5. http://sobereva.com/multiwfn.
- (a) T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS; (b) T. Lu and F. Chen, Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS; (c) T. Lu and F. Chen, Calculation of Molecular Orbital Composition, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- E. H. Howard, C. F. Cain, C. Kang and J. R. Del Valle, Synthesis of Enantiopure ε-Oxapipecolic Acid, J. Org. Chem., 2020, 85, 1680–1686 CrossRef CAS.
- S. Liu, L. Liu, D. Yu, C. Rong and T. Lu, Steric charge, Phys. Chem. Chem. Phys., 2018, 20, 1408–1420 RSC.
- S. Pérez and I. Tvaroška, Carbohydrate–Protein Interactions: Molecular Modeling Insights, Adv. Carbohydr. Chem. Biochem., 2014, 71, 9–136 CrossRef.
- P. A. Petillo and L. E. Lerner, Origin and Quantitative Modeling of Anomeric Effect, in The Anomeric Effect and Associated Stereoelectronic Effects, ed. G. R. J. Thatcher, ACS Symposium Series, American Chemical Society, Washington, DC, 1993, ch. 9 Search PubMed.
- A. J. Weldon, T. L. Vickrey and G. S. Tschumper, Intrinsic Conformational Preferences of Substituted Cyclohexanes and Tetrahydropyrans Evaluated at the CCSD(T) Complete Basis Set Limit: Implications for the Anomeric Effect, J. Phys. Chem. A, 2005, 109, 11073–11079 CrossRef.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory. International Series of Monographs on Chemistry 22, Oxford Science Publications, Oxford, 1990 Search PubMed.
- R. F. W. Bader and H. Essen, The characterization of atom interactions, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- R. F. W. Bader, Bond Path: A Universal Indicator of Bonded Interactions, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interaction, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed computational data. See DOI: 10.1039/d0ob02453h |
| This journal is © The Royal Society of Chemistry 2021 |