
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot, three-component Fischer indolisation–N-alkylation for rapid synthesis of 1,2,3-trisubstituted indoles†
Christopher A.
Hughes-Whiffing
and
Alexis
Perry
 *
*
Biosciences, University of Exeter, Stocker Road, Exeter EX4 4QD, UK. E-mail: A.Perry@Exeter.ac.uk
First published on 17th December 2020
Abstract
A one-pot, three-component protocol for the synthesis of 1,2,3-trisubstituted indoles has been developed, based upon a Fischer indolisation–indole N-alkylation sequence. This procedure is very rapid (total reaction time under 30 minutes), operationally straightforward, generally high yielding and draws upon readily available building blocks (aryl hydrazines, ketones, alkyl halides) to generate densely substituted indole products. We have demonstrated the utility of this process in the synthesis of 23 indoles, benzoindoles and tetrahydrocarbazoles bearing varied and useful functionality.
Introduction
Indoles are prominent in natural and non-natural products of biological and pharmaceutical importance, and are often considered as a “privileged scaffold” within the drug discovery arena.1,2 Furthermore, they are versatile building blocks in synthesis, providing access to diverse heterocycles (e.g. tryptolines,3 spiropyrans,4 indolines,5 oxindoles6 and spirocycles7), substrates for asymmetric dearomatisation,8,9 and polymers and composite materials with energy storage and biomedical applications.10–12 Accordingly, indole synthesis has a long and distinguished history, and the many published protocols for generating and elaborating the indole core ensure that few indole structures are beyond the reach of the present-day synthetic chemist.13–17 Our recent studies into structurally-diverse spiropyrans4,18 required a broad range of 1,2,3-trisubstituted indole building blocks and, to meet this demand, we have developed a rapid, cheap and concise one-pot, three-component sequence for their synthesis, based upon Fischer indolisation–N-alkylation, and the evolution of this process is described herein.19 One-pot, multistep regimens can provide efficiency in terms of time, cost, yield, labour, energy and consumables,20–22 and the combination of Fischer indole synthesis and indole N-alkylation is inherently geared towards successful application as a rapid one-pot process for two principal reasons: (i) Fischer indolisation and indole N-alkylation are robust, clean, high-yielding processes which generate minimal quantities of by-products or leftovers, hence are ideal within one-pot, multicomponent reaction cascades; (ii) indole N-alkylation is rapid (commonly < 1 hour) and whilst Fischer indolisation displays more varied reaction rate, use of microwave irradiation often leads to short reaction times (<10 minutes).23 A further benefit to this Fischer indolisation–N-alkylation approach is that a broad range of structurally-diverse starting materials are commercially available for each of the aryl hydrazine, ketone and alkyl halide components, hence the process is amenable to rapid generation of trisubstituted indole libraries.There is considerable precedence for the use of multicomponent reaction cascades in indole synthesis, often combining aryl hydrazone formation with Fischer indolisation (e.g. via addition of metalloimines to aryl hydrazines,24via addition of diazonium salts to acid chlorides25 or via Heck isomerisation26 (see Scheme 1a–c respectively)), although non-hydrazone-based methodologies – e.g. Sonagashira coupling preceding intramolecular hydroamination27 (Scheme 1d) – have also proved successful. Generally, simple 1,2,3-trisubstituted indoles have been obtained by stepwise Fischer indolisation–N-alkylation,18 whilst modern techniques have accessed related indoles bearing more complex functionality (e.g. via rhodium carbene insertions,28 transition metal catalysed C–H/N–H functionalisation,29 Cu(I)-catalysed imine cyclisations30 and on a solid support).31,32
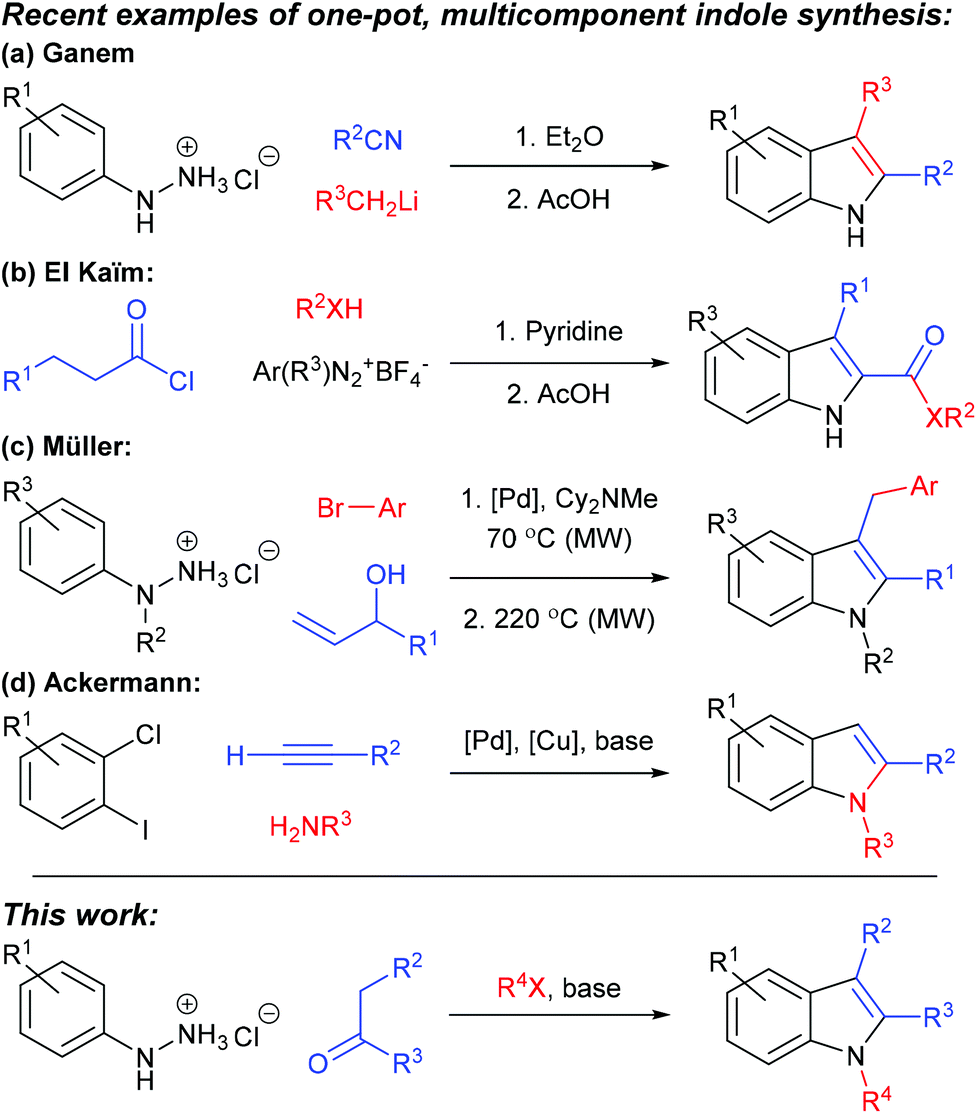 | ||
| Scheme 1 Precedence for multicomponent reactions in indole synthesis (a–d; ref. 19–22 respectively), and (lower) the methodology described in this publication. | ||
Despite the congested nature of this field of research, we consider that the one-pot Fischer indolisation–N-alkylation process documented herein is complementary to current methods, especially given its speed and simplicity, and the commercial availability of diverse substrates. This is fundamentally useful, robust methodology and we envisage that it will be adopted for rapid, straightforward access to a broad range of 1,2,3-trisubstituted indole structures.
Results and discussion
Our strategy to identify optimal conditions for Fischer indolisation–indole N-alkylation was based on the premise that N-alkylation would present limited tolerance for varied reaction conditions, hence the more adaptable conditions for Fischer indolisation should be directed to accommodate the subsequent alkylation as much as possible. Classical conditions for indole N-alkylation employ indole, sodium hydride and alkyl halide in DMF or THF, and often proceed in excellent yield and with high selectivity for N- over C-alkylation, though with caveats around reagent toxicity, limitations in substrate scope through use of strong base, and the sometimes unpredictable behaviour of hydride in DMF on a large scale.33 Whilst various alternative N-alkylation processes have been documented (e.g. alkylation with alcohols via Mitsunobu conditions34 or transition metal catalysis;35 Brønsted acid catalysed aza-Michael addition;36 transition metal catalysed allylation37) all have their strengths and weaknesses, and the simplicity and off-the-shelf nature of the classical method was more consistent with our approach, hence we considered this as the optimum alkylation to explore within this one-pot sequence.In adopting alkylation methodology based around generation of a sodium indole salt using sodium hydride, we necessarily constrained conditions for the preceding Fischer indolisation – i.e. a minimum of acid should be employed and only sodium hydride-tolerant solvents would be appropriate – without compromising reaction rate, yield or practical convenience. To achieve this, we employed microwave-promoted Fischer indole synthesis (several studies have documented considerable rate acceleration over standard thermal heating for this process),23,38,39 using 1 equivalent of acid (most conveniently through use of hydrazine hydrochloride salts) and proceeding in ethereal solvents (DMF is unstable under microwave heating).40 Our initial investigations briefly explored the influence of solvent on the reaction between phenylhydrazine hydrochloride (1) (1 eq.) and butanone (2) (1.05 eq.), to give 2,3-dimethylindole (3), at 150 °C for 15 minutes (max 300 W) (see Table 1). In THF, 2-MeTHF and cyclopentyl methyl ether (CPME) (0.63 M), Fischer indolisation was clean and high-yielding (Table 1, entries 1–3), whereas reaction in the absence of solvent41 was less effective (entry 4). To reduce solvent usage, the reaction could be performed with equal success at concentrations of up to 2.1 M in THF; further increase in concentration caused erosion of yield (entries 5 and 6). Subsequently, we optimised reaction time and temperature in THF: (i) reaction at 100 °C proved ineffective (entry 7), whereas reaction at 125 °C took only marginally more time to reach completion than at 150 °C (conversion 97% vs. >99% after 15 min); (ii) at 125 °C or above, the reaction reached 95% conversion within 5 minutes (entries 9 and 10); (iii) at 150 °C, the reaction was complete after 10 minutes (entry 11). Given that we were identifying conditions for the first step of a one-pot, two-step sequence, our main priority was that the reaction should reach completion to avoid unreacted starting materials interfering with subsequent N-alkylation. Correspondingly, we adopted 10 minutes at 150 °C in THF as our optimal conditions, whilst acknowledging that, for given specific substrates, milder conditions may be attainable. Finally, in order to explore the effect of microwave irradiation in promoting this reaction, a control experiment was performed using standard thermal heating at 125 °C in a sealed tube for 15 minutes. In this case, we observed conversion of 64%, whilst the corresponding microwave-irradiated replicate reached 97% (entry 8), thus it seems likely that microwave irradiation has a role in rate acceleration for this reaction beyond a straightforward thermal effect.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | T/°C | t/min | Solvent | Conc./M | Conv.b/% | Yieldc/% |
a Reactions were conducted using phenylhydrazine·HCl (1) (99 mg, 0.682 mmol, 1 eq.), butanone (2) (64 μL, 0.716 mmol, 1.05 eq.) and solvent in thick-walled microwave vials sealed with septa, and heated to the specified temperature using microwave irradiation (maximum of 300 W). Reactions were then held at the specified temperature for the specified time.
b Based on ratio of phenylhydrazine·HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethylindole, estimated by analysis of crude reaction mixtures using 1H NMR.
c Isolated yield.
d Heated using an oil bath, all other parameters as previously described. :dimethylindole, estimated by analysis of crude reaction mixtures using 1H NMR.
c Isolated yield.
d Heated using an oil bath, all other parameters as previously described.
|
||||||
| 1 | 150 | 15 | THF | 0.63 | >99 | 96 |
| 2 | 150 | 15 | 2-MeTHF | 0.63 | 94 | 92 |
| 3 | 150 | 15 | CPME | 0.63 | 93 | 92 |
| 4 | 150 | 15 | None | — | 71 | 64 |
| 5 | 150 | 15 | THF | 2.1 | >99 | — |
| 6 | 150 | 15 | THF | 6.3 | 75 | — |
| 7 | 100 | 15 | THF | 2.1 | 42 | — |
| 8 | 125 | 15 | THF | 2.1 | 97 | — |
| 9 | 125 | 5 | THF | 2.1 | 95 | — |
| 10 | 150 | 5 | THF | 2.1 | 98 | — |
| 11 | 150 | 10 | THF | 2.1 | >99 | 96 |
| 12d | 125 | 15 | THF | 2.1 | 64 | — |
We then explored conditions for the indole N-alkylation step of this two-step process. 2,3-Dimethyl indole was generated using the optimum conditions as described above, then deprotonation (with sodium hydride) and alkylation (with benzyl bromide) were investigated directly on this crude reaction mixture (which we assumed to consist of 2,3-dimethylindole (1 eq.), water (1 eq.), ammonium chloride (1 eq.) and THF (300 μL)). Our previous experience of indole N-alkylation (as an isolated reaction) identified DMF as a superior solvent to THF and 2-MeTHF,18 hence we were keen to employ DMF to promote the alkylation step in this one-pot sequence. Furthermore, in our hands, N-alkylation performs similarly in anhydrous or non-dried DMF, thereby enabling development of an operationally straightforward process without requirement for rigorously anhydrous reaction conditions. The instability of DMF towards aqueous acid defined the order of addition of reagents in this process: sodium hydride was added first (to quench the ammonium chloride and water, and deprotonate the indole), then DMF was added, then benzyl bromide. Our initial foray employed 3 eq. NaH then 1 mL of DMF, then allowed 5 minutes for deprotonation prior to introduction of benzyl bromide. After 1 h, the quenched reaction mixture contained a 20:48:32 mixture of unalkylated 2,3-dimethylindole (3), the desired 1-benzyl-2,3-dimethylindole (4a) and 2,3-dimethyl-3-benzylindolenine (5) (i.e. the product of competing C-3 alkylation) (Table 2, entry 1), and this ratio remained unchanged with longer reaction times or an increase in benzyl bromide quantity (entry 2). Complete consumption of dimethylindole 3 was achieved through use of 4 eq. of sodium hydride, however, this did not suppress reaction through C-3 (entry 3). Use of ethereal solvents THF, CPME and 2-MeTHF in place of DMF resulted in poorer regioselectivity and precipitation of (presumably) the indole sodium salt; in each case, 2,3-dimethylindole was the predominant product within complex product mixtures (entries 4–6). With respect to competing C-3 alkylation, our suspicion was that incomplete deprotonation was allowing direct reaction of 2,3-dimethylindole with benzyl bromide (via C-3), and this was due to poor solubility of hydride and the indole anion in solvent mixtures containing THF. This hypothesis was supported by the observation that alkylation in media containing an increased proportion of DMF displayed a slight but significant shift towards reaction through nitrogen (compare entries 3 and 7). It is also plausible that reaction of the indole anion itself through C-3 was further compromising regioselectivity, despite the kinetic and thermodynamic favourability of N-alkylation.42,43 In either case, there is an argument to suggest that higher reaction temperatures should favour alkylation through nitrogen; accordingly, we repeated our protocol (hydride addition, DMF addition, stir 5 minutes then BnBr addition) with heating applied to the reaction vessel following DMF addition. Whilst reactions at 50 °C and 65 °C offered modest improvements in regioselectivity, we were delighted to observe complete N-alkylation at 80 °C, and this corresponded to 91% isolated yield (96% per step) of the desired 1,2,3-trisubstituted indole 4a. Further development of this process identified that the DMF volume could be halved without detriment at this elevated temperature (entry 11) (further reduction resulted in reduced regioselectivity) and that alkylation was complete in under 15 minutes (entries 12 and 13).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | NaH/eq.b | BnBr/eq.b | Solventc | Volume/mL | T /°C | t /min |
3:4a:5f |
Yield 4ag/% |
| a Benzylations were performed directly on the crude reaction mixture of Fischer indolisation, derived using the conditions defined by Table 1, entry 11 (i.e. microwave promoted reaction of phenylhydrazine·HCl (99 mg, 0.682 mmol, 1 eq.) and butanone (64 μL, 0.716 mmol, 1.05 eq.) in THF (0.3 mL) at 150 °C for 10 minutes). After cooling to room temperature, NaH was added to this crude mixture, then solvent, and the mixture was stirred for 5 minutes before addition of BnBr. b Molar equivalents based upon phenylhydrazine·HCl (0.682 mmol, 1 eq.). c Standard “bench” non-anhydrous solvents were used. d Heating commenced following solvent addition. e Reaction time following addition of BnBr. f Ratio estimated by analysis of crude reaction mixtures using 1H NMR. g Isolated yield from phenylhydrazine·HCl. | ||||||||
| 1 | 3 | 1.05 | DMF | 1 | 22 | 60 | 20:48:32 |
42 |
| 2 | 3 | 2.1 | DMF | 1 | 22 | 60 | 20:48:32 |
— |
| 3 | 4 | 1.05 | DMF | 1 | 22 | 60 | 0:63:37 |
59 |
| 4 | 4 | 1.05 | THF | 1 | 22 | 60 | 52:17:31 |
— |
| 5 | 4 | 1.05 | 2-MeTHF | 1 | 22 | 60 | 56:14:30 |
— |
| 6 | 4 | 1.05 | CPME | 1 | 22 | 60 | 55:22:23 |
— |
| 7 | 4 | 1.05 | DMF | 2 | 22 | 60 | 0:72:28 |
66 |
| 8 | 4 | 1.05 | DMF | 1 | 50 | 60 | 0:78:22 |
74 |
| 9 | 4 | 1.05 | DMF | 1 | 65 | 60 | 0:80:20 |
75 |
| 10 | 4 | 1.05 | DMF | 1 | 80 | 60 | 0:100:0 |
91 |
| 11 | 4 | 1.05 | DMF | 0.5 | 80 | 60 | 0:100:0 |
90 |
| 12 | 4 | 1.05 | DMF | 0.5 | 80 | 30 | 0:100:0 |
91 |
| 13 | 4 | 1.05 | DMF | 0.5 | 80 | 15 | 0:100:0 |
89 |
In order to validate this one-pot approach, we performed a control experiment evaluating the analogous stepwise pathway as a point of comparison for our optimum conditions (defined in Table 2, entry 13), in terms of yield, time, material and energy costs (a full breakdown of which is available in the ESI†). For Fischer indolisation, we employed our microwave promoted conditions (Table 1, entry 11) followed by standard aqueous work-up/organic extraction and purification using silica gel chromatography, whilst N-alkylation was performed using standard published conditions (1.0 eq. indole, 1.2 eq. NaH, 1.1 eq. BnBr, DMF (0.4 M), RT, 1 hour), followed by a methanol quench and direct silica gel chromatography. In terms of yield, the one-pot approach offered a modest advantage over its stepwise analogue (one-pot, 91%; stepwise, 84%), however, more stark contrasts were evident in terms of time and consumable consumption. The total time, from empty reaction vessel to pure indole product, for the one-pot method was 103 minutes, whereas the stepwise approach required 268 minutes. Whilst it is harder to quantify working hours (given the variable efficiencies of laboratory workers and differing intensities of laboratory tasks), the extra chromatography, aqueous work-up and washing up in the stepwise protocol contribute to a more labour intensive process. These same additional processes were also instrumental in the use of an extra 971 mL of organic solvent over the one-pot route (one-pot: 723 mL total, 1.16 L solvent per mmol product; stepwise: 1694 mL total, 2.96 L solvent per mmol product), and silica and TLC plate usage doubled. On the other hand, the one-pot route carries two disadvantages: (i) a requirement for 3.3× more sodium hydride; (ii) 15 minutes of heating at 80 °C is necessary, corresponding to an extra 0.02 kW h over the stepwise protocol.
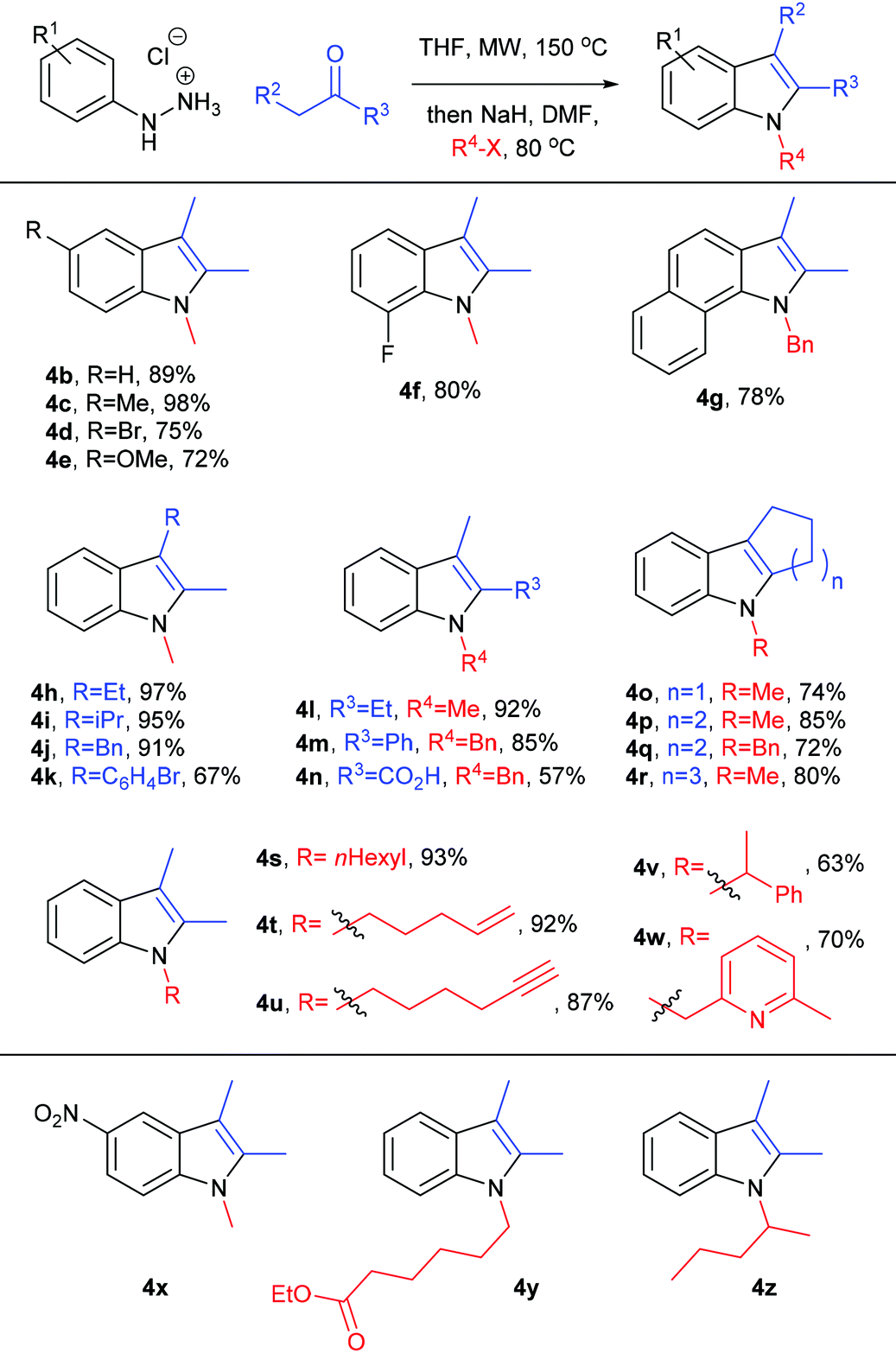
We then investigated the substrate scope of this one-pot, two-step process in terms of its hydrazine, ketone and alkyl halide components, using the optimised conditions identified above (see Table 2, entry 12). In general, a wide variety of functionality could be tolerated – derived from each of these three components – rapidly generating 1,2,3-trisubstituted indoles in high yield. We successfully incorporated useful functional groups for further elaboration, including alkenes, alkynes, heteroaromatics, carboxylates and halides, and we were able to access related heterocyclic structures such as tetrahydrocarbazoles, tetrahydrocyclopentaindoles and benzoindoles, and hindered indoles such as 4i and 4k. Carboxylic acid 4n was derived from in situ basic hydrolysis of the corresponding ester (the reaction sequence generates 1 eq. of hydroxide) and we suspect that the modest yield of 4n can be attributed to partial decarboxylation: indole 2-carboxylic acids are susceptible to acidic, basic, thermal and microwave-promoted decarboxylation (all evident conditions in this reaction sequence),44–46 and 1H NMR analysis of the crude reaction mixture revealed the presence of decarboxylated indole. The reaction sequence was unsuccessful with nitrophenylhydrazine (electron poor hydrazones are often unwilling substrates in Fischer indolisation),47,48 with enolisable ester-based alkylating agents, or with unactivated secondary alkylating agents (see products 4x–z respectively). Given that hindered, secondary electrophiles are documented as challenging substrates for indole alkylation,37 it is encouraging that a secondary benzyl halide was tolerated in this study, to give indole 4v, albeit in somewhat modest yield. Somewhat surprisingly, iodomethane was a less effective substrate in this reaction cascade and 2.5 eq. were required to achieve satisfactory yields. Presumably this can be attributed to the volatility of this reagent at 80 °C, despite the reaction occurring in a sealed tube (Table 3).
|
Conclusions
In summary, we have developed methodology that enables rapid, straightforward and generally high yielding synthesis of 1,2,3-trisubstituted indoles via one-pot, three-component Fischer indolisation–indole N-alkylation, in a process that requires neither anhydrous nor inert reaction conditions. The reaction sequence draws upon cheap, widely available substrates to incorporate diverse and useful functionality onto the indole nucleus and provides a fundamentally practical and robust approach to these valued heterocycles. Finally, we have attempted to quantify the benefits of our one-pot, two-reaction approach over its stepwise counterpart in terms of economy and efficiency, and this has provided concrete evidence for the effectiveness of this one-pot strategy.Experimental
General experimental information
Solvents and reagents were used as commercially supplied and no anhydrous solvents were employed. Analytical thin layer chromatography was carried out using Merck Kieselgel 60 F254, coated on aluminium plates, with visualisation of spots where necessary by quenching of UV (254 nm) fluorescence. Silica gel with particle size 40–63 mm was used for flash chromatography. Microwave reactions were performed in a CEM Discover microwave in 10 mL, thick-walled microwave tubes, sealed with septum caps and were magnetically stirred. Infrared spectra were recorded as thin films using attenuated total reflectance with a Nicolet iS5 FTIR spectrometer. Mass spectra were recorded on a QToF 6520 mass spectrometer (Agilent Technologies, Palo Alto, USA). 1H NMR and 13C NMR spectra were recorded at 400 MHz and 100 MHz respectively, using a Bruker Avance III HD400 spectrometer. Chemical shifts are quoted in ppm relative to tetramethylsilane, the residual solvent peak being used for referencing purposes. Coupling constants are quoted to the nearest 0.1 Hz with peak multiplicities for single resonances being labelled as: s, singlet; d, doublet; t, triplet; q, quartet; m, unresolved multiplet. Complete and unambiguous assignment of 1H and 13C NMR spectra was achieved through use of COSY, NOESY, HSQC and HMBC experiments.Synthetic methods and characterisation data
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 135.9 (7a), 135.9 (2), 128.7 (Bn3,5), 128.5 (Bn1), 128.4 (Bn4), 128.3 (Bn2,6), 128.3 (3), 125.8 (6), 123.0 (3a), 120.8 (4), 120.0 (5), 111.6 (7), 66.4 (CH2) and 10.1 (CH3); ES (m/z): 288 (55%, MNa+). This compound has been synthesised previously, but without publication of characterisation data.54,55
O), 135.9 (7a), 135.9 (2), 128.7 (Bn3,5), 128.5 (Bn1), 128.4 (Bn4), 128.3 (Bn2,6), 128.3 (3), 125.8 (6), 123.0 (3a), 120.8 (4), 120.0 (5), 111.6 (7), 66.4 (CH2) and 10.1 (CH3); ES (m/z): 288 (55%, MNa+). This compound has been synthesised previously, but without publication of characterisation data.54,55
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Debbie Salmon (Mass Spectrometry, University of Exeter) for recording of mass spectra of novel compounds presented in this work.References
- F. R. de Sá Alves, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2009, 9, 782–793 CrossRef.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS.
- M. T. Rahman and J. M. Cook, Eur. J. Org. Chem., 2018, 3224–3229 CrossRef CAS.
- H. Swinson and A. Perry, Tetrahedron, 2020, 76, 131219 CrossRef CAS.
- C. Yu, J. Wang and Y. Zhou, Org. Chem. Front., 2018, 5, 2805–2809 RSC.
- S. V. Shelar and N. P. Argade, Org. Biomol. Chem., 2019, 17, 6671–6677 RSC.
- J. T. R. Liddon, M. J. James, A. K. Clarke, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. – Eur. J., 2016, 22, 8777–8780 CrossRef CAS.
- C. Zheng and S. You, Nat. Prod. Rep., 2019, 36, 1589–1605 RSC.
- C. Zhuo, W. Zhang and S. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef.
- H. Mudila, P. Prasher, M. Kumar, A. Kumar, M. G. H. Zaidi and A. Kumar, Mater. Renewable Sustainable Energy, 2019, 8, 9 CrossRef.
- M. Elango, M. Deep, R. Subramanian and A. M. Musthafa, J. Alloys Compd., 2017, 696, 391–401 CrossRef CAS.
- M. Z. Bin Hussein and C. W. Long, Mater. Chem. Phys., 2004, 85, 427–431 CrossRef CAS.
- G. W. Gribble, Indole Ring Synthesis: From Natural Products to Drug Discovery (1st Edition), John Wiley & Sons, Ltd., New York, 2016 Search PubMed.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS.
- R. Mancuso and R. Dalpozzo, Catalysts, 2018, 8, 458 CrossRef.
- D. I. Bugaenko, A. V. Karchava and M. A. Yurovskaya, Russ. Chem. Rev., 2019, 88, 99–159 CrossRef CAS.
- A. Perry, K. Davis and L. West, Org. Biol. Chem., 2018, 16, 7245–7254 RSC.
- Recent advances in Fischer indolisation methodolgy: (a) H. Vuong, S. Duarte and D. A. Klumpp, Top. Catal., 2018, 61, 7–8 CrossRef; (b) A. Kaga, T. Fukushima, J. Shimokawa and M. Kitamura, Synthesis, 2019, 51, 3214–3220 CrossRef CAS; (c) M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedia, RSC Adv., 2017, 7, 52852–52887 RSC.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- Y. Liu, Y. Meng, X. Huang, F. Qin, D. Wu, Q. Shao, Z. Guo, Q. Li and W. Wei, Green Chem., 2020, 22, 4593–4596 RSC.
- X. Huang, F. Qin, Y. Liu, S. Wu, Q. Li and W. Wei, Green Chem., 2020, 22, 3952–3955 RSC.
- E. C. Creencia, M. Tsukamoto and T. Horaguchi, J. Heterocycl. Chem., 2011, 48, 1095–1102 CrossRef CAS.
- C. A. Simoneau and B. Ganem, Nat. Protoc., 2008, 3, 1249–1252 CrossRef CAS.
- L. El Kaïm, L. Grimaud and C. Ronsseray, Synlett, 2010, 2296–2298 CrossRef.
- J. Panther and T. J. J. Müller, Synthesis, 2016, 48, 974–986 CrossRef CAS.
- L. T. Kaspar and L. Ackermann, Tetrahedron, 2005, 61, 11311–11316 CrossRef CAS.
- M. Shen, Y. Pan, Z. Jia, X. Ren, P. Zhang and H. Xu, Org. Biomol. Chem., 2015, 13, 4851–4854 RSC.
- M. Ghobrial, M. D. Mihovilovic and M. Schnürch, Beilstein J. Org. Chem., 2014, 10, 2186–2199 CrossRef.
- F. S. Melkonyan, D. E. Kuznetsov, M. A. Yurovskaya and A. V. Karchava, RSC Adv., 2013, 3, 8388–8397 RSC.
- T. Yao, D. Yue and R. C. Larock, J. Comb. Chem., 2005, 7, 809–812 CrossRef CAS.
- S. A. Worlikar, B. Neuenswander, G. H. Lushington and R. C. Larock, J. Comb. Chem., 2009, 11, 875–879 CrossRef CAS.
- Q. Yang, M. Sheng, J. J. Henkelis, S. Tu, E. Wiensch, H. Zhang, Y. Zhang, C. Tucker and D. E. Ejeh, Org. Process Res. Dev., 2019, 23, 2210–2217 CrossRef CAS.
- A. Bombrun and G. Casi, Tetrahedron Lett., 2002, 43, 2187–2190 CrossRef CAS.
- S. M. A. Hakim Siddiki, K. Kon and K. Shimizu, Green Chem., 2015, 17, 173–177 RSC.
- S. Bahn, S. Imm, K. Mevius, L. Neubert, A. Tillack, J. M. J. Williams and M. Beller, Chem. – Eur. J., 2010, 16, 3590–3593 CrossRef.
- A. V. Karchava, F. S. Melkonyan and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 391–407 CrossRef CAS.
- O. Algul, A. Kaessler, Y. Apcin, A. Yilmaz and J. Jose, Molecules, 2008, 13, 736–748 CrossRef CAS.
- S. V. Karthikeyan, S. Perumal, K. A. Shetty, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2009, 19, 3006–3009 CrossRef CAS.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS.
- K. Matsumoto, A. Tanaka, I. Yukio, N. Hayashi, M. Toda and R. A. Bulman, Heterocycl. Commun., 2003, 9, 9–12 CAS.
- A. V. Karchava, I. S. Shuleva, A. A. Ovcharenko and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2010, 46, 291–301 CrossRef CAS.
- Q. Cai, C. Zheng and S. You, Angew. Chem., Int. Ed., 2010, 49, 8666–8669 CrossRef CAS.
- A. A. Vandersteen, S. O. C. Mundle and R. Kluger, J. Org. Chem., 2012, 77, 6505–6509 CrossRef CAS.
- E. Piers and R. K. Brown, Can. J. Chem., 1962, 40, 559–561 CrossRef CAS.
- G. B. Jones and B. J. Chapman, J. Org. Chem., 1993, 58, 5558–5559 CrossRef CAS.
- H. N. Rydon and S. Siddappa, J. Chem. Soc., 1951, 2462–2467 RSC.
- B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef.
- S. Basak, A. Alvarez-Montoya, L. Winfrey, R. L. Melen, L. C. Morrill and A. P. Pulis, ACS Catal., 2020, 10, 4835–4840 CrossRef CAS.
- J. Zhang, S. T. Kohlbouni and B. Borhan, Org. Lett., 2019, 21, 14–17 CrossRef CAS.
- A. Saito, S. Oda, H. Fukaya and Y. Hanzawa, J. Org. Chem., 2009, 74, 1517–1524 CrossRef CAS.
- S. Zhou, J. Wang, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2427–2430 CrossRef CAS.
- W. Wei, X. Dong, S. Nie, Y. Chen, X. Zhang and M. Yan, Org. Lett., 2013, 15, 6018–6021 CrossRef CAS.
- X. Li, B. Zhou, R. Yang, F. Yang, R. Liang, R. Liu and Y. Jia, J. Am. Chem. Soc., 2018, 140, 13945–13951 CrossRef CAS.
- M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. Murali Dhar, J. Am. Chem. Soc., 2020, 142, 3094–3103 CrossRef CAS.
- Y. Li, R. Wang, T. Wang, X. Cheng, X. Zhou, F. Fei and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 15436–15440 CrossRef CAS.
- M. C. Willis, G. N. Brace and I. P. Holmes, Angew. Chem., Int. Ed., 2005, 44, 403–406 CrossRef CAS.
- T. R. Pradhan, H. W. Kim and J. K. Park, Org. Lett., 2018, 20, 5286–5290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra for 4a–4w and 13C NMR spectra for 4n and 4s–4w. Full details of time, energy and consumables analysis for one-pot and stepwise syntheses of 4a. See DOI: 10.1039/d0ob02185g |
| This journal is © The Royal Society of Chemistry 2021 |