
Construction of an IMiD-based azide library as a kit for PROTAC research†
Haixia
Liu‡
abc,
Renhong
Sun‡
a,
Chaowei
Ren
acd,
Xing
Qiu
e,
Xiaobao
Yang
 *a and
Biao
Jiang
*ae
*a and
Biao
Jiang
*ae
aShanghai Institute for Advanced Immunochemical Studies, ShanghaiTech University, 393 Middle Huaxia Road, Shanghai 201210, China. E-mail: jiangbiao@shanghaitech.edu.cn; yangxb@shanghaitech.edu.cn
bSchool of Physical Science and Technology, ShanghaiTech University, 393 Middle Huaxia Road, Shanghai 201210, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dSchool of Life Science and Technology, ShanghaiTech University, 393 Middle Huaxia Road, Shanghai 201210, China
eCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 23rd November 2020
Abstract
As a promising protein degradation strategy, PROTAC technology is increasingly becoming a new star in cancer treatment. Here we report the efficient construction of an IMiD-based azide library via a quick one-step conversion of the existing IMiD-based amine library. This new azide library can act as a kit to endow PROTAC libraries with triazole moieties for various POIs through a highly effective ‘click reaction’ and then help to rapidly screen out lead degraders that are valuable for drug development. Its power in fleetly identifying potent degraders has been verified on two oncogenic proteins, BCR-ABL and BET, the degraders of which showed comparable potency to or even higher potency than the reported PROTACs in degrading target proteins and effectively inhibiting cancer cell proliferation.
Although multiple treatment methods, such as the use of small molecule inhibitors,1 monoclonal antibodies2 and oligonucleotide agents,3 have been developed over the past few decades, cancer is still the major lethal disease threatening human health nowadays.4,5 All these methods have limited applications resulting from their inherent defects and it is difficult for them to meet the demand.6 In recent years, a novel and promising strategy called the Proteolysis Targeting Chimera (PROTAC) technology,7 which uses the cellular ubiquitin–proteasome system (UPS) to regulate the levels of oncogenic proteins, has emerged and aroused extraordinary research interest in academia and the pharmaceutical industry.8
PROTACs usually consist of a warhead binding to the protein of interest (POI), a ligand binding to E3 ligase and an interval linker to bridge these two parts. Such a heterobifunctional small molecule, acting as a catalyst, is capable of artificially recruiting E3 ligase to the target protein, inducing its polyubiquitination and subsequent degradation by the proteasome. It has been proved that in addition to the two binders at the ends, the length, hydrophilicity and rigidity of the coupling linkers also have a great influence on the activity of PROTACs.9 Linkers with triazole moieties, common scaffolds in pharmaceutical molecules, that have the appropriate hydrophilicity, rigidity and potential to vary their lengths at will are widely used in the design and development of promising PROTACs.10 Rapid synthesis of triazole moieties through a 1,3-dipolar cycloaddition reaction, referred to as the ‘click reaction’,11 allows the production of PROTACs as fast as possible, thereby greatly increasing the efficiency of creating a successful degrader for a specific target. The click reaction with an almost stoichiometric yield under mild reaction conditions involves two substrates: azide and alkyne. Immunomodulatory drugs (IMiDs), composed of thalidomide and its derivatives pomalidomide and lenalidomide, are a class of well-studied E3 ligands and can hijack E3 ubiquitin ligase CRL4CRBN.7e,12 Many reported PROTACs based on IMiDs display high potency. In the present study, using a safe and effective method, we develop a library of IMiD-based azides, which can act as a kit for PROTAC research by reacting with diverse alkyne-tagged protein ligands to construct libraries of PROTACs targeting different POIs and screen out degraders with high efficacy swiftly (Scheme 1).
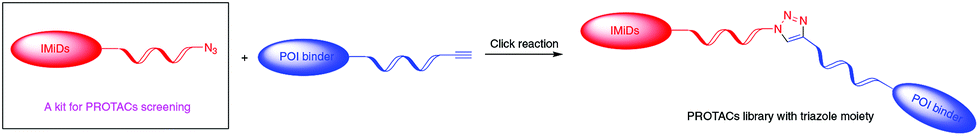 | ||
| Scheme 1 Mechanism for constructing a PROTAC library via an IMiD-based azide library as a kit. | ||
To date, IMiD-derived azides are commonly synthesized in several steps from chain aliphatic azides (e.g., Scheme 2),10b,13 which are commercially available with a limited variety and expensive price or prepared in-house via an SN2 reaction using halides and NaN3 as substrates.14 The current method used involves multiple steps and high cost or is dangerous and is not suitable for the mass production of IMiD-derived azides and further PROTACs.
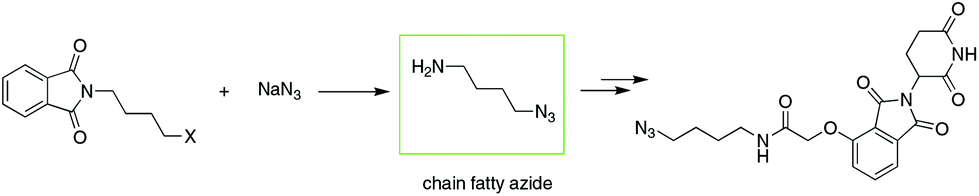 | ||
| Scheme 2 Common method to synthesize IMiD-based azides. | ||
Actually, we have developed a library of IMiD-based amines in the previous work to promote the research of PROTACs in our lab (Scheme 3a).15 Since this library is also necessary for the diversity of linkers to develop PROTACs, direct conversion from the existing amine library to the azide library will achieve twice the result with half the effort. Recently, Dong and colleagues discovered a reliable method to transform primary amines into the corresponding azides with high yields and broad substrate scopes under mild conditions by using a much safer and commercially available diazotizing reagent, fluorosulfuryl azide (FSO2N3), as the N3-source (Scheme 3b).16 Therefore, we plan to apply this method to construct a new IMiD-based azide library in one step (Scheme 3c).
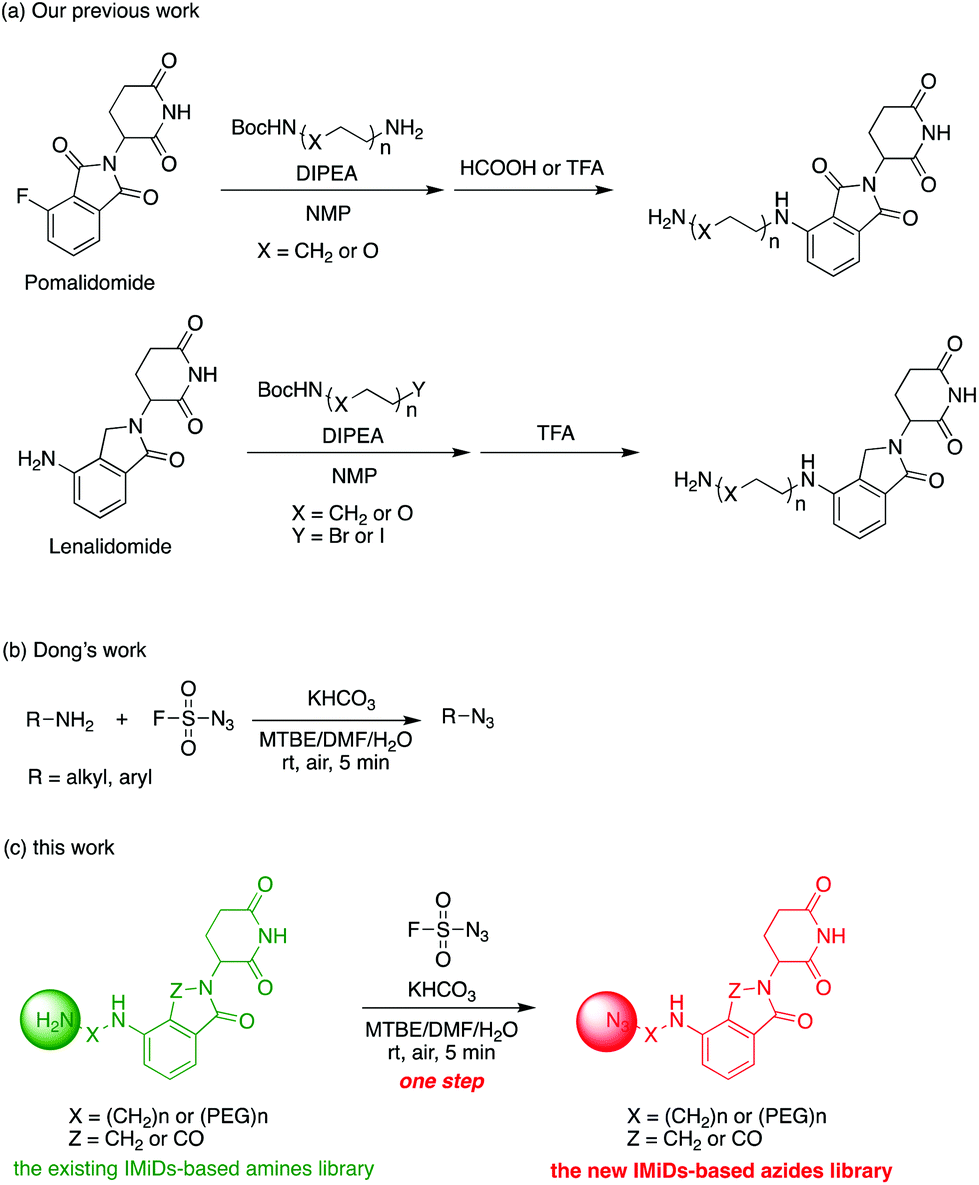 | ||
| Scheme 3 Direct conversion from the existing amine library to an azide library. | ||
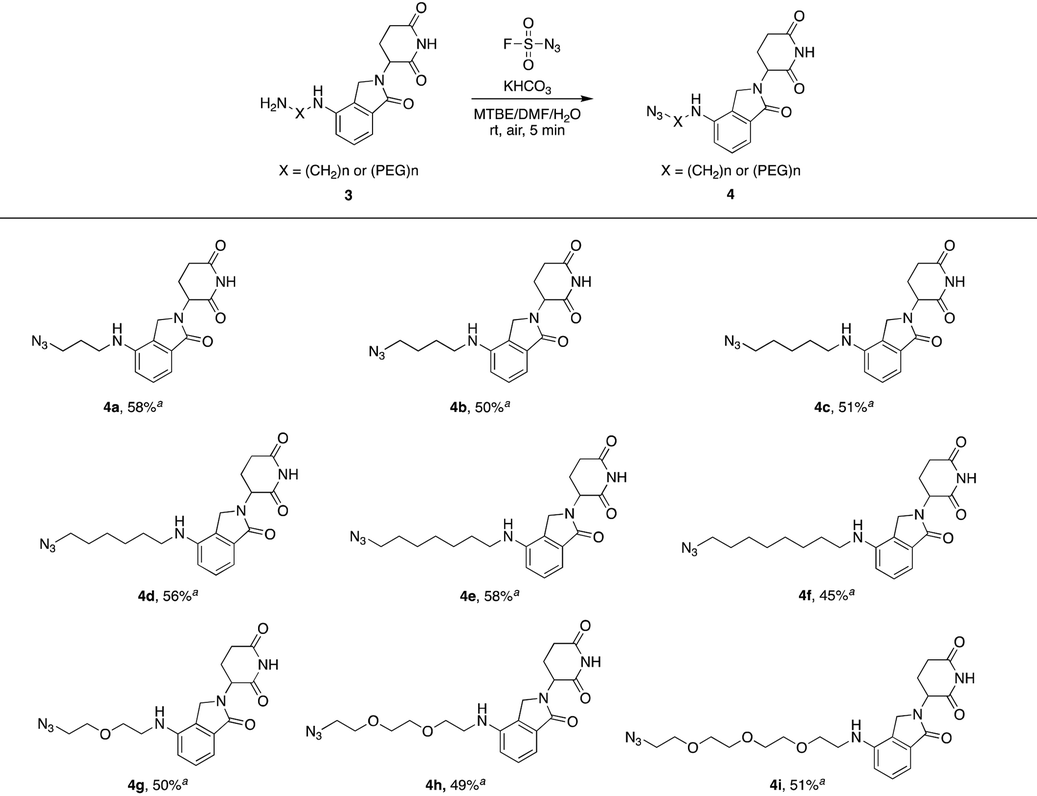
We first utilized compound 1a to perform this reaction and obtained the desired azide 2a with a yield of 89%, indicating that our hypothesis is feasible (Table 1). Thus, as shown in Table 1, we extended this reaction to a series of pomalidomide derived amines with increasing lengths of carbon or PEG chains. The corresponding azides were acquired in moderate or high yields to form an azide library based on pomalidomide.
| a Isolated yield. |
|---|
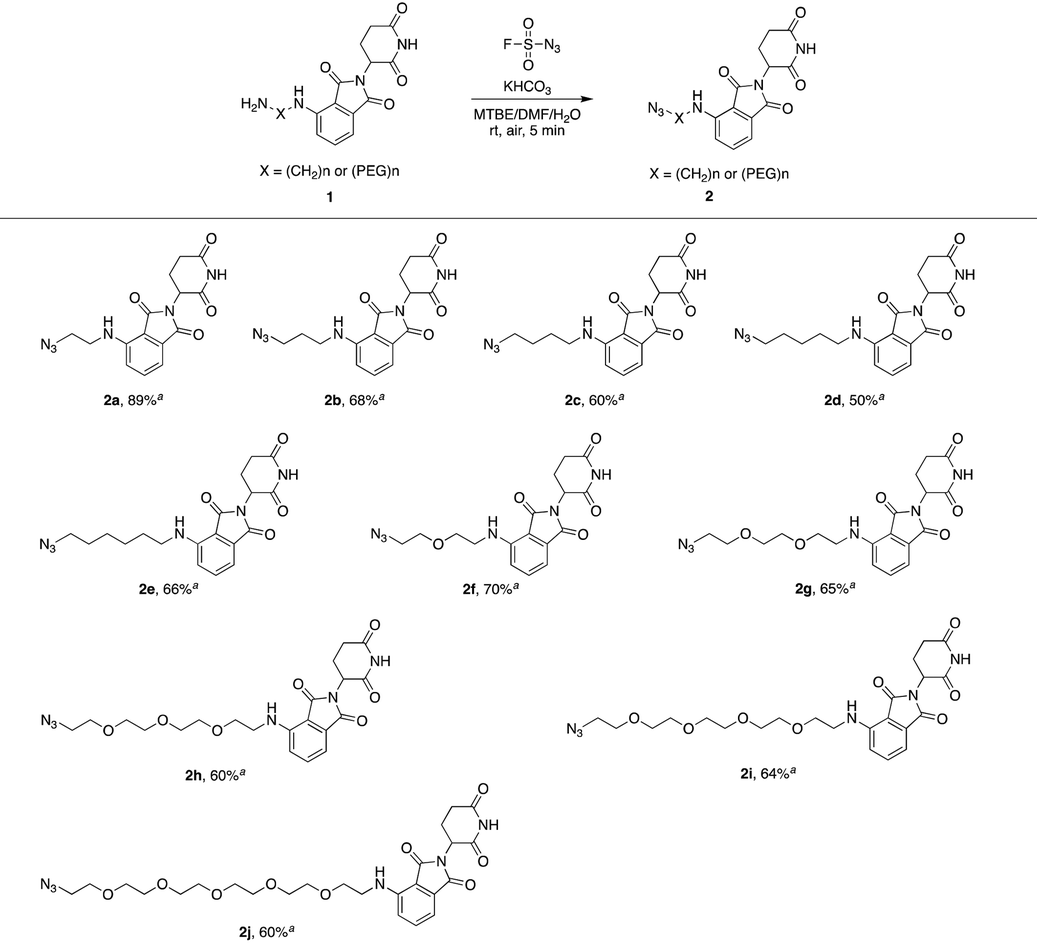
|
Lenalidomide, another kind of IMiD, has demonstrated comparable potency to or even higher potency than pomalidomide as a CRL4CRBN recruiter in many PROTACs. Hence, it is necessary to explore azides based on lenalidomide. In Table 2, a range of lenalidomide-derived azides containing different lengths of full carbon or PEG chains were prepared in moderate yields via this functional method.
| a Isolated yield. |
|---|
|
In order to verify whether this IMiD-based azide library is a practical kit for PROTAC research, we applied it to two common targets, BCR-ABL and BET (bromodomain and extra-terminal domain), both of which possessed known potent PROTAC degraders.
The BCR-ABL fusion protein is generally responsible for chronic myelogenous leukemia (CML) by constitutive activation and causing unregulated cell division.17 Crews’ group has developed a potent BCR-ABL PROTAC DAS-6-2-2-6-CRBN by conjugating a tyrosine kinase inhibitor dasatinib to pomalidomide using a long and complex linker (Fig. 1).18
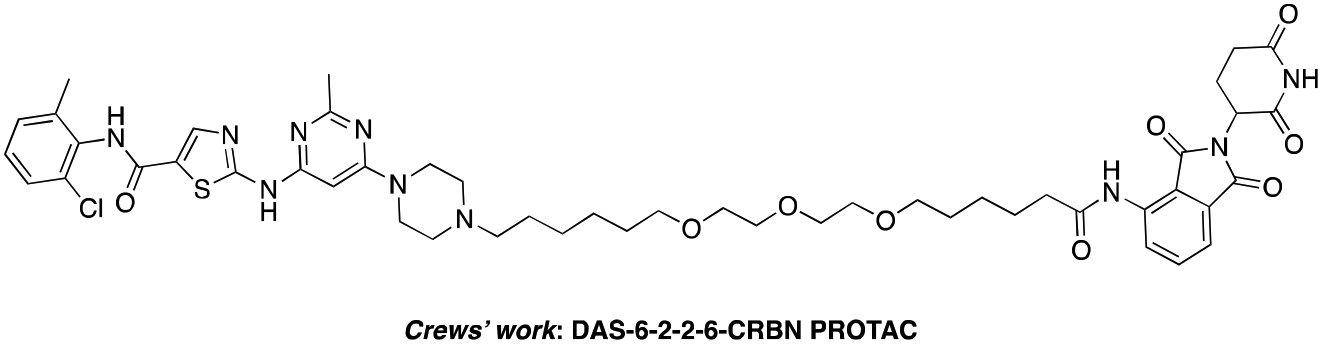 | ||
| Fig. 1 Structure of the reported DAS-6-2-2-6-CRBN PROTAC. | ||
This molecule is effective against the BCR-ABL-driven human CML cell line K562 and can mediate the degradation of the BCR-ABL protein. Here, we produced two new BCR-ABL PROTACs SIAIS629050 and SIAIS629051, which can be easily afforded by the click reactions of pomalidomide-based azide 2c and dasatinib derivatives with an alkyne handle. To optimize the synthesis, we explored a three-step one-pot approach using N-Boc protected pomalidomide-based amine as the starting material: (1) removal of the N-Boc protection group; (2) key conversion of the amine to the azide and (3) click reactions with two different dasatinib derivatives linked with a terminal alkynyl group (Scheme 4). These two PROTACs presented comparable antiproliferation activities in K562 cells to DAS-6-2-2-6-CRBN (Table 3). Furthermore, like DAS-6-2-2-6-CRBN, SIAIS629050 and SIAIS629051 degraded c-ABL and BCR-ABL very efficiently in a dose-dependent manner (Fig. 2). In particular, they displayed even stronger activity than the reported degrader DAS-6-2-2-6-CRBN when the concentration was as low as 10 nM. These results hint that simple triazole linkers, which are easily provided by azides, can also produce potent PROTACs compared with complex linkers. Such a one-pot approach will make it much easier to screen out highly efficient degraders.
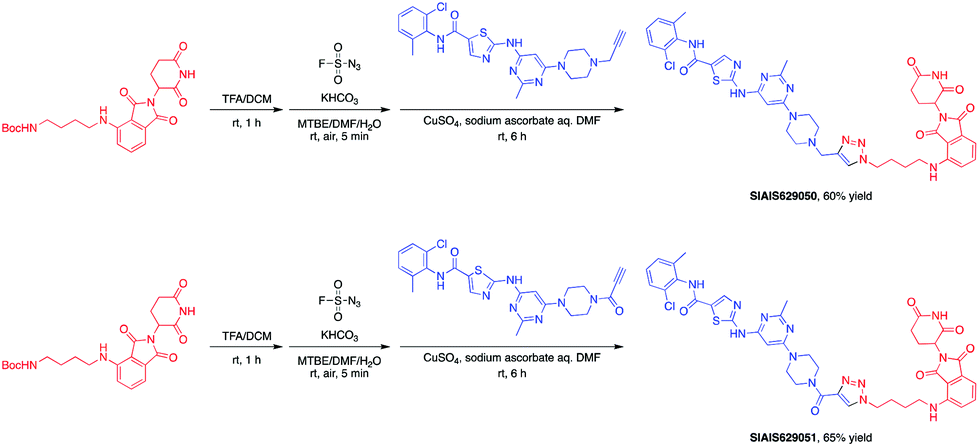 | ||
| Scheme 4 One-pot synthesis of new BCR-ABL PROTACs. | ||
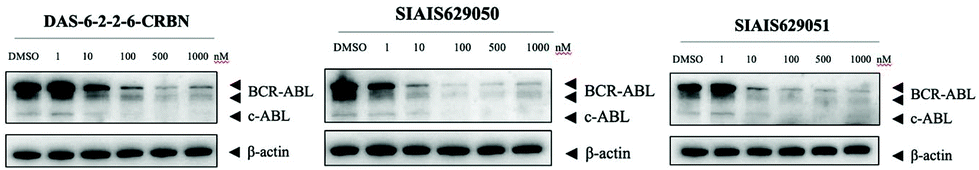 | ||
| Fig. 2 Western blotting analysis of the BCR-ABL protein in K562 cells treated with DAS-6-2-2-6-CRBN, SIAIS629050 and SIAIS629051. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
The BET family, the “reader” of lysine acetylation, has been unveiled as a key player in cancer biology and implicated as a target in multiple human cancers.19dBET1, one of the known BET degraders with high efficacy, which is documented by Bradner and colleagues, consists of an eight-atom N-butyl-2-hydroxyacetamide as the linker, JQ1 as the warhead and thalidomide as the CRBN recruiter (Fig. 3).20 In this study, two new BET PROTACs SIAIS629048 and SIAIS629049 were generated from two pomalidomide-based amines that have linkers of different lengths by using the same one-pot strategy (Scheme 5), and then tested together with dBET1. As shown in Table 4, these two new degraders displayed high antiproliferative effects, 8- or 11-fold better than dBET1 in the leukemia cell line MV-4-11. Compared to dBET1, SIAIS629048 showed stronger ability to degrade BET proteins at 5 nM concentration. Unexpectedly, SIAIS629049 equipped with a longer linker can selectively downregulate BRD2 and BRD3 while another BET protein member BRD4 remained (Fig. 4). Taken together, similar to the BCR-ABL protein target, these results indicate that PROTACs rapidly prepared by the domino method have the potential to act as efficient degraders, and also prove that the azide library is indeed a practical kit for PROTAC research.
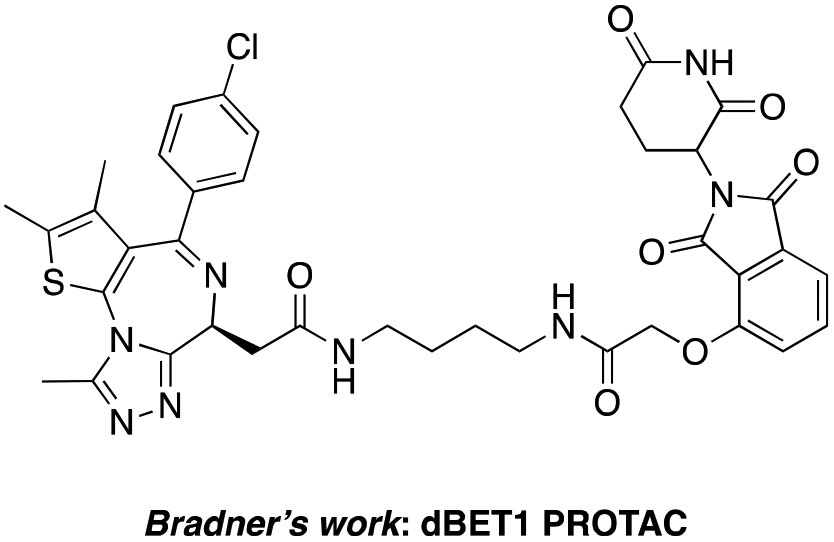 | ||
| Fig. 3 Structure of the reported dBET1 PROTAC. | ||
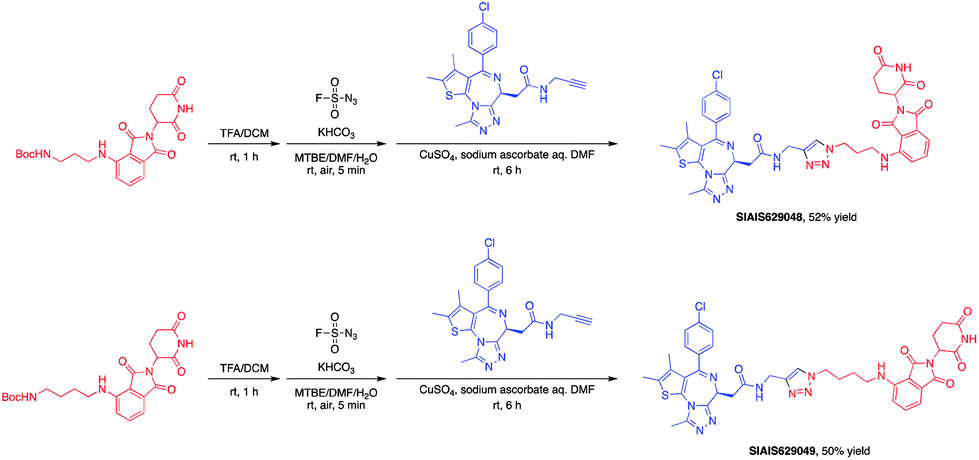 | ||
| Scheme 5 One-pot synthesis of new BET PROTACs. | ||
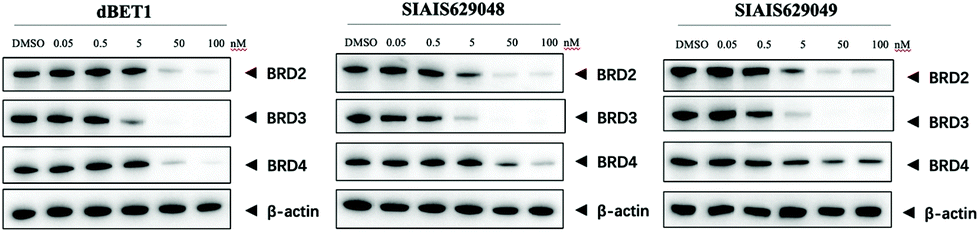 | ||
| Fig. 4 Western blotting analysis of BET proteins in MV-4-11 cells treated with dBET1, SIAIS629048 and SIAIS629049. | ||
In conclusion, we reported a highly efficient protocol to construct a new IMiD-based azide library through one-step direct conversion from the existing CRBN recruiter amine library. The azide library can provide rapid access to multiple PROTAC libraries for various POIs by reacting with the corresponding alkynyl-tagged POI ligands in the ‘click reaction’. Good application results on two targets, BCR-ABL protein and BET protein, confirmed the effectiveness of the azide library in developing PROTAC degraders. Today, the competition in the field of PROTAC development is becoming more and more fierce. For pharmaceutical companies and research groups, how to screen out potent degraders more quickly has become the key to success. In spite of the minor limitation of the need for preparing alkynyl-functionalized POI ligands, the azide library developed in this paper, as a kit, allows speedy construction of unlimited PROTAC libraries and just meets this urgent need. Further application of the azide library on other new targets is ongoing in our laboratory to develop better degraders.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21502114 to B. J.), the Shanghai Natural Science Foundation (Grant No. 19ZR1433600 to X. Y.) and the China Postdoctoral Science Foundation (Grant No. 2019M651609 to R. S.) for financial support.Notes and references
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301 CrossRef CAS.
- J. Tol and C. J. A. Punt, Clin. Ther., 2010, 32, 437 CrossRef CAS.
- Y. Deng, C. C. Wang and K. W. Choy, et al. , Gene, 2014, 538, 217 CrossRef CAS.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69 CrossRef.
- D. M. Parkin, P. Pisani and J. Ferlay, Ca-Cancer J. Clin., 1999, 49, 33 CrossRef CAS.
- (a) L. Xie and P. E. Bourne, Curr. Opin. Struct. Biol., 2011, 21, 189 CrossRef CAS; (b) Y. Mei and B. Yang, Future Med. Chem., 2018, 10, 1835 CrossRef CAS; (c) G. P. Adams and L. M. Weiner, Nat. Biotechnol., 2005, 23, 1147 CrossRef CAS; (d) A. L. Nelson;, E. Dhimolea and J. M. Reichert, Nat. Rev. Drug Discovery, 2010, 9, 767 CrossRef.
- (a) S. An and L. Fu, EBioMedicine, 2018, 36, 553 CrossRef; (b) I. Churcher, J. Med. Chem., 2018, 61, 444 CrossRef CAS; (c) Y. Zou;, D. Ma and Y. Wang, Cell Biochem. Funct., 2019, 37, 21 CrossRef; (d) D. P. Bondeson, B. E. Smith; and G. M. Burslem, et al. , Cell Chem. Biol., 2018, 25, 78 CrossRef CAS; (e) M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966 CrossRef CAS; (f) J. Qi and G. Zhang, Future Med. Chem., 2019, 11, 723 CrossRef CAS.
- For reviews, see: (a) A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101 CrossRef CAS; (b) T. K. Neklesa, J. D. Winkler and C. M. Crews, Pharmacol. Ther., 2017, 174, 138 CrossRef CAS; (c) P. Ottis and C. M. Crews, ACS Chem. Biol., 2017, 12, 892 CrossRef CAS. For papers, see: (d) K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554 CrossRef CAS; (e) L. Bai;, H. Zhou;, R. Xu; and S. Wang, et al. , Cancer Cell, 2019, 36, 498 CrossRef.
- (a) S. Wang, L. Han, J. Han and Y. Yu, et al. , Nat. Chem. Biol., 2019, 15, 1223 CrossRef CAS; (b) X. Han, C. Wang, C. Qin and S. Wang, et al. , J. Med. Chem., 2019, 62, 941 CrossRef CAS.
- (a) R. P. Wurz, K. Dellamaggiore and H. Dou, et al. , J. Med. Chem., 2018, 61, 453 CrossRef CAS; (b) M. Schiedel, D. Herp and S. Hammelmann, et al. , J. Med. Chem., 2018, 61, 482 CrossRef CAS; (c) Z. An, W. Lv, S. Su, W. Wu and Y. Rao, Protein Cell, 2019, 10, 606 CrossRef CAS; (d) Y. Sun, X. Zhao, N. Ding and Y. Rao, et al. , Cell Res., 2018, 28, 779 CrossRef CAS; (e) Y. Sun;, N. Ding; and Y. Song, et al. , Leukemia, 2019, 33, 2105 CrossRef; (f) C. P. Tinworth, H. Lithgow and L. Dittus, et al. , ACS Chem. Biol., 2019, 14, 342 CrossRef CAS; (g) J. L. Bian, J. Ren and Y. R. Li, et al. , Bioorg. Chem., 2018, 81, 373 CrossRef CAS; (h) Q. Zhao, T. Lan, S. Su and Y. Rao, Chem. Commun., 2019, 55, 369 RSC; (i) H. Lebraud, D. J. Wright, C. N. Johnson and T. D. Heightman, ACS Cent. Sci., 2016, 2, 927 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra, A. S. Singh and X. Chen, et al. , Chem. Rev., 2016, 116, 3086 CrossRef CAS.
- (a) E. S. Fischer, K. Bohm, J. L. Jenkins and N. H. Thomä, et al. , Nature, 2014, 512, 49 CrossRef CAS; (b) P. P. Chamberlain, A. Lopez-Girona and K. Miller, et al. , Nat. Struct. Mol. Biol., 2014, 21, 803 CrossRef CAS; (c) T. Ito, H. Ando and T. Suzuki, et al. , Science, 2010, 327, 1345 CrossRef CAS; (d) A. Broyl, R. Kuiper and M. van Duin, et al. , Blood, 2013, 122, 624 CrossRef.
- S. Su, Z. Yang, H. Gao and Y. Rao, et al. , J. Med. Chem., 2019, 62, 7575 CrossRef CAS.
- J. Zhang, P. Chen, P. Zhu, J. Zhou and H. Zhang, et al. , Bioorg. Chem., 2020, 99, 103817 CrossRef CAS.
- (a) X. Qiu, N. Sun, Y. Kong, Y. Li, X. Yang and B. Jiang, Org. Lett., 2019, 21, 3838 CrossRef CAS; (b) For another example of an IMID-based amine library, see: J. Lohbeck and A. K. Miller, Bioorg. Med. Chem. Lett., 2016, 26, 5260 CrossRef CAS.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86 CrossRef CAS.
- (a) O. Hantschel and G. Superti-Furga, Nat. Rev. Mol. Cell Biol., 2004, 5, 33 CrossRef CAS; (b) O. Hantschel, W. Warsch, E. Echelhart, I. Kaupe and F. Grebien, et al. , Nat. Chem. Biol., 2012, 8, 285 CrossRef CAS.
- A. Lai, M. Toure, D. Hellerschmied, J. Salami and C. M. Crews, et al. , Angew. Chem., Int. Ed., 2016, 55, 807 CrossRef CAS.
- (a) D. J. Patel and Z. Wang, Annu. Rev. Biochem., 2013, 82, 81 CrossRef CAS; (b) A. C. Belkina and G. V. Denis, Nat. Rev. Cancer, 2012, 12, 465 CrossRef CAS.
- G. E. Winter, D. L. Buckley, J. Paulk and J. E. Bradner, et al. , Science, 2015, 348, 1376 CrossRef CAS . For other known dBET1 degraders, see: (a) J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755 CrossRef CAS; (b) M. Zengerle, K. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob02120b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |