
Enantioselective N-heterocyclic carbene-catalysed intermolecular crossed benzoin condensations: improved catalyst design and the role of in situ racemisation†
Eoghan G.
Delany
and
Stephen J.
Connon
 *
*
Centre for Synthesis and Chemical Biology, Trinity Biomedical Sciences Institute, School of Chemistry, The University of Dublin, Trinity College, Dublin 2, Ireland. E-mail: delanye@tcd.ie; e.delany@latrobe.edu.au; Fax: +353 16712826
First published on 8th December 2020
Abstract
The enantioselective intermolecular crossed-benzoin condensation mediated by novel chiral N-heterocyclic carbenes derived from pyroglutamic acid has been investigated. A small library of chiral triazolium ions were synthesised. Each possessed a tertiary alcohol H-bond donor and a variable N-aryl substituent. It was found that increasing both the steric requirement and the electron-withdrawing characteristics of the N-aryl ring led to more chemoselective, efficient and enantioselective chemistry, however both quenching the reaction at different times and deuterium incorporation experiments involving the product revealed that this is complicated by product racemisation in situ (except in the case of benzoin itself), which explains the dependence of enantioselectivity on the electrophilicity of the reacting aldehydes common in the literature. Subsequent protocol optimisation, where one reacting partner was an o-substituted benzaldehyde, allowed a range of crossed-benzoins to be synthesised in moderate-good yields with moderate to excellent enantioselectivity.
Introduction
Since its discovery in 1832,1 the benzoin condensation between two molecules of benzaldehyde is acknowledged as one of the oldest carbon–carbon bond forming reactions in organic synthesis. Initially catalysed by the cyanide anion,2 over 100 years later Ukai and co-workers successfully employed thiamine (vitamin B1) and base to mediate the reaction.3 It was through this work that the thiazolium ring came to be recognised as the catalytically relevant unit, which led to Breslow's subsequent elucidation of the associated mechanism in 1958 (Scheme 1).4,5 | ||
| Scheme 1 Mechanism of the thiazolium ion-derived carbene-catalysed benzoin condensation. | ||
He proposed that initial deprotonation of 1 leads to in situ production of 1a, which may also be formally considered a carbene. Attack upon a molecule of benzaldehyde (2) forms adduct 3 and, following a proton exchange, generates the key nucleophilic enolamine intermediate 4 (often referred to as the Breslow intermediate). This attacks a second molecule of benzaldehyde to furnish adduct 5. A final proton exchange allows the elimination of 1a, which re-enters the catalytic cycle, with concurrent formation of benzoin (6).
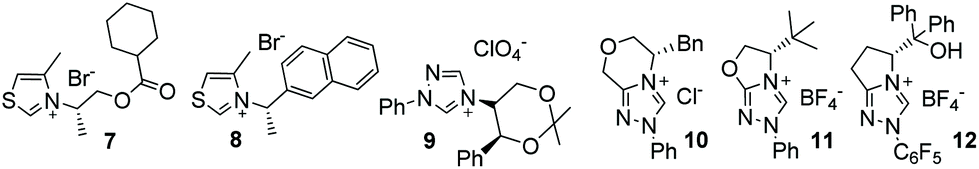
In the intervening years, the use of N-heterocyclic carbenes derived from the deprotonation of thiazolium and triazolium ion precatalysts in homobenzoin/acyloin reactions condensations has been explored extensively.6 These reactions produce synthetically malleable7,8 chiral α-hydroxy ketone products. Consequently, many attempts have been made to design chiral precatalysts capable of bringing about highly enantioselective benzoin condensations. In 1966, seminal work from Sheehan and Hunnemann detailed the synthesis of the chiral thiazolium salt 7 (Fig. 1).9 The carbene derived from this material could promote the formation of 6 with 21% ee. Subsequent design of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 several years later allowed the generation of 6 with 52% ee but in just 6% yield.11 Although thiazolium-derived NHCs of various design were utilised by other groups in later years, no efforts succeeded in substantially improving enantiocontrol.12
10 several years later allowed the generation of 6 with 52% ee but in just 6% yield.11 Although thiazolium-derived NHCs of various design were utilised by other groups in later years, no efforts succeeded in substantially improving enantiocontrol.12
 | ||
| Fig. 1 Early chiral precatalysts employed in the benzoin condensation. | ||
Enders reported the use of the first chiral triazolium ion precatalyst in 1996.13 Under basic conditions, use of 9 allowed the production of 6 in 66% yield and 75% ee, although both product yields and optical purities varied considerably when employing other aromatic aldehydes; the use of activated benzaldehydes tended to negatively impact the enantioselectivity of the reaction, while electron-rich benzaldehydes induced the opposite effect, albeit at the expense of reaction rates. Similar observations were reported by Leeper et al. in 1998 using precatalyst 10,14 which produced (R)-benzoin in 80% ee. Enders’ later developed 11, which allowed the isolation of benzoin in 90% ee,15,16 while You and co-workers showed that bis-triazolylidene carbenes could also mediate various homobenzoin condensations with high-excellent enantioselectivity (84–95% ee).17,18 In 2009 – as part of a programme aimed as using hydrogen-bond donation as a control element in acyloin condensations19 – we devised the NHC precatalyst 12 which, in the presence of Rb2CO3, could catalyse the benzoin condensation in 90% yield with almost perfect enantioselectivity (>99%).20 Again, however, significant variations in both yield and ee were observed depending upon the identity of the aromatic aldehyde employed – with trends paralleling those outlined above.21
Despite the advances made in recent years in the homobenzoin condensation (which has dominated the field until recently), the lack of an efficient protocol for the NHC-catalysed intermolecular cross-condensation22–26 between two non-identical aromatic aldehydes has hampered the synthetic utility of the reaction.
To the best of our knowledge, there exists in the literature no study dedicated exclusively to the development of an efficient protocol for the chemoselective and asymmetric intermolecular crossed-benzoin condensation between two aryl aldehydes catalysed by an NHC. As part of a programme aimed at the development of highly efficient crossed acyloin reactions,23c,27,28 we recently reported the synthesis of a novel triazolium precatalyst capable of directing the racemic intermolecular crossed-benzoin condensation with excellent chemoselectivity if one of the partners incorporated an o-halo substituent.29 The steric and electronic characteristics of the catalyst's N-aryl substituent proved to be key: steric bulk near the carbene centre is required to attain high selectivity, yet the aryl unit needs to incorporate electron-withdrawing functionality to allow useful efficacy. Given the dearth of NHC-mediated systems for the asymmetric synthesis of crossed-benzoins, we sought to extend this methodology through the design of chiral analogues capable of representing a reliable and reproducible solution to this problem.
Results and discussion
Synthesis of chiral triazolium salts (12, 21–26)
Having previously designed precatalyst 12, capable of mediating a high-yielding and enantioselective homobenzoin condensation under basic conditions, we set about the synthesis of a family of chiral triazolium salts (21–26, Scheme 2) possessing the same general structure but also incorporating the structural elements previously discerned29 to be sine qua non for promoting chemoselective intermolecular crossed-benzoin chemistry. Precatalysts 12 and 22–26 were prepared according to a general procedure: esterification of (L)-pyroglutamic acid 13 provides 14, which was reacted with excess phenylmagnesium bromide to afford alcohol 15. Subsequent silylation provided the TMS-protected lactam 16 which, when treated with Meerwein salt overnight followed by addition of the relevant aryl hydrazine, yielded the crude hydrazone intermediate 18via17. Ring closure with triethyl orthoformate at elevated temperature and deprotection of the silylated triazolium salt 19 with HBr (generated catalytically through use of bromotrimethylsilane in MeOH) provides the desired triazolium salt of general structure 20. The relevant hydrazine precursors, if not commercially available, were derived from the parent aniline through diazotisation and subsequent reduction in situ using SnCl2·H2O. An important note is that the 19F NMR spectra of catalysts of general type 20 are silent, so full or partial anion exchange (most likely with bromide derived from the last step) may occur.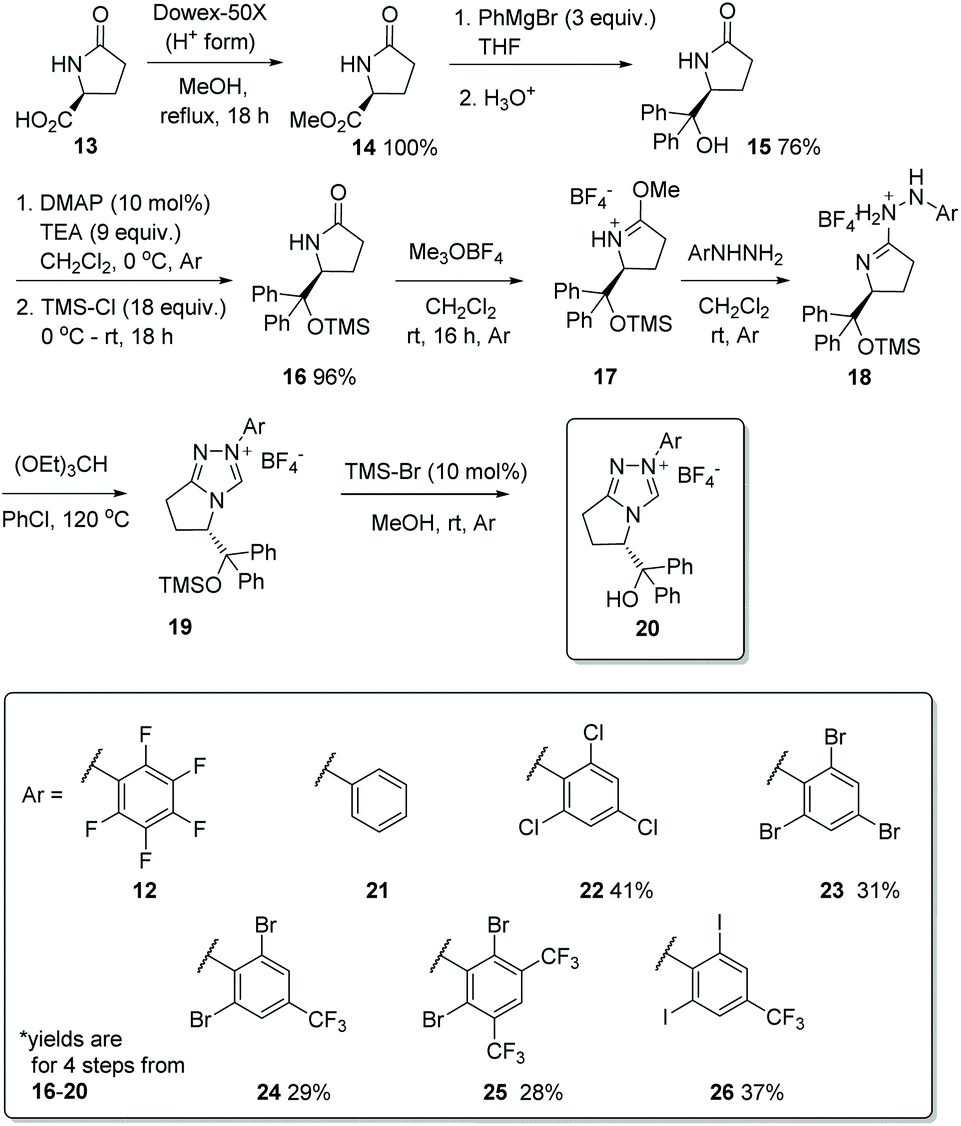 | ||
| Scheme 2 General synthesis of chiral triazolium salts. | ||
Preliminary studies
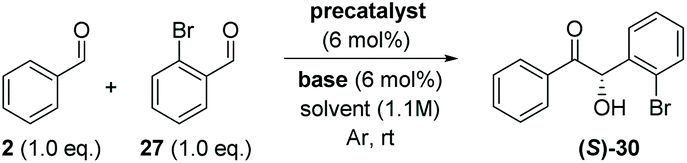
With the necessary precatalysts in hand, we set about establishing a set of conditions amenable to an asymmetric crossed-benzoin condensation between two non-identical aromatic aldehydes. Previous studies have shown that, in crossed acyloin condensations where one coupling partner is an ortho-substituted benzaldehyde, use of an NHC catalyst typically results in higher yields of crossed acyloins derived from initial attack of the NHC upon the non-ortho-substituted coupling partner.27,29–32 With this in mind, we focused upon the potential cross-condensation between benzaldehyde 2 and 2-bromobenzaldehyde 27 and the effect of various precatalysts on selectivity under basic conditions (Table 1).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Precatalyst | Base | Time (h) | Yield 28 (%) | Yield 6a (%) | Yield 29a (%) | Yield 30a,b (%) | ee 30c (%) |
| a Determined via1H NMR spectroscopic analysis of the crude reaction mixture using styrene (0.25 equiv.) as an internal standard. b Value in parentheses represents isolated yield. c Determined via CSP-HPLC analysis. d Reaction carried out on a 3.3 mmol scale as opposed to 1.1 mmol scale. | ||||||||
| 1 | 21 | K2CO3 | 48 | 0 | 0 | 0 | 0 | n/a |
| 2 | 22 | K2CO3 | 48 | 0 | 8 | 0 | 79 (71) | 7 |
| 3 | 22 | K2CO3 | 17 | 0 | 9 | 0 | 65 (60) | 64 |
| 4d | 22 | K2CO3 | 2 | 0 | 4 | 0 | 87 (85) | 15 |
| 5 | 22 | DABCO | 1 | 0 | 14 | 0 | 30 (25) | 67 |
| 6 | 22 | DABCO | 2 | 0 | 19 | 0 | 49 (45) | 67 |
| 7 | 22 | DABCO | 3 | 0 | 20 | 0 | 59 (55) | 67 |
| 8 | 22 | DABCO | 4 | 0 | 20 | 0 | 60 (57) | 67 |
| 9 | 22 | DIPEA | 1 | 0 | 7 | 0 | 20 (14) | 67 |
| 10 | 22 | DIPEA | 2 | 0 | 9 | 0 | 34 (30) | 67 |
| 11 | 22 | DIPEA | 3 | 0 | 11 | 0 | 48 (45) | 67 |
| 12 | 22 | DIPEA | 4 | 0 | 14 | 0 | 65 (63) | 67 |
| 13 | 22 | DMAP | 4 | 0 | 1 | 0 | 16 (9) | 67 |
| 14 | 22 | NMI | 20 | 0 | 0 | 0 | 6 (n/d) | n/d |
| 15 | 22 | DBU | 1 | 0 | 17 | 0 | 48 (n/d) | n/d |
| 16 | 22 | DBU | 2 | 0 | 18 | 0 | 59 (55) | 57 |
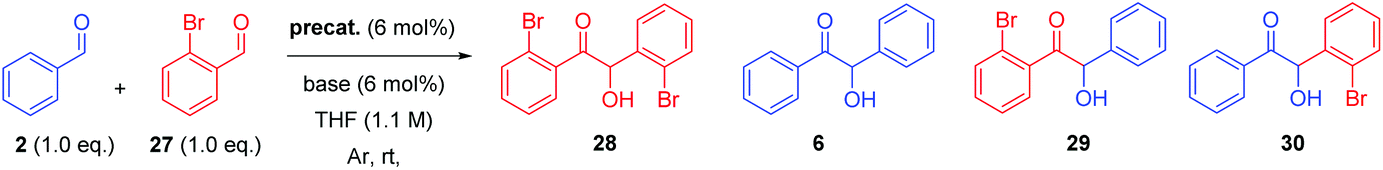
In the presence of 6 mol% of 21 and an equimolar loading of K2CO3, we observed no conversion to any of the four benzoin products 6, 28, 29 or 30 (entry 1) after 48 h; which underscores the need for the N-aryl substituent to be electron deficient in these systems.33 Subsequent use of the N-2,4,6-trichlorophenyl analogue 22 under identical conditions produced benzoin (6) in 15% yield (via1H NMR spectroscopic analysis of the crude reaction mixture) and allowed isolation of 30 in 71% yield, albeit with very poor enantiocontrol (entry 2). Conscious of potential racemisation through base-mediated enolisation, the reaction time was reduced to 17 h; resulting in improved enantioselectivity of 64% ee (entry 3). However, repetition of the same experiment on threefold scale for 2 h resulted in the generation of 30 in high yield but with just 15% ee (entry 4). Concerned that this irreproducibility could be due to the poor solubility of K2CO3 in THF, we screened a series of soluble nitrogenous bases of varying basicities to evaluate their effect upon the stereochemical outcome. Use of 1,4-diazabicyclo[2.2.2]octane (DABCO, pKaH conj. acid at 25 °C = 8.8) afforded 30 in low yield after just 1 h in 67% ee (entry 5).
Gratifyingly, we observed no change in the ee of the isolated product over the course of a 2–4 h reaction time when DABCO was employed as the base, while yields increase in an approximately time-dependent linear fashion; with 1H NMR spectroscopic analysis indicating a 65% yield of 30 after 4 hours (entries 6–8). Similar results were obtained employing N,N-diisopropylethylamine (pKaH 10.8, (entries 9–12). Surprisingly, employing N,N-dimethylaminopyridine (DMAP, pKaH 9.7) resulted in a slower reaction rate, despite its increased basicity relative to DABCO (entry 13); the cause of this observation was not elucidated but may relate to the less hindered (and thus more nucleophilic) nature of DMAP.
Use of the considerably less basic N-methylimidazole (NMI, pKaH 7.1) led to slower product formation – with just 6% of 30 observed after 20 h (entry 14). Finally, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKaHca. 13) mediated significantly faster coupling than observed using other bases. Quenching the reaction after only 2 h allowed the isolation of 36 in 55% yield but with reduced ee (entry 15).
It is noteworthy that in these reactions, none of either the homodimer of 27 (i.e.28) or the product 29 (derived from formation of the Breslow intermediate from 27 followed by attack on 2) were formed. Significant (albeit as a minor product) levels of 6 were usually observed.
Catalyst screening
Confident that – although the enantio- and chemoselectivity of the reaction remained suboptimal – we had established a reaction timeframe and suitable base (DIPEA) amenable to the generation of reproducible data, we then set about evaluating the other members of the triazolium precatalyst library in the coupling between 2 and 27 (Table 2). As previously detailed, in the presence of DIPEA, 6 mol% of the carbene derived from 22 promotes the reaction in 63% yield and 67% ee after 4 h in THF (entry 1). To our surprise, reducing the nucleophilicity of the carbene through the use of precatalyst 12 under the same conditions provided 30 in just 26% isolated yield in racemic form (entry 2). In addition, benzoin (6) was isolated as the major product in 74% yield and >99% ee. Use of triazolium salt 23 – possessing a bulkier N-aryl ring but a more electron-rich carbene centre – led to an improvement in the ee of 30 albeit stemming from slow, inefficient reaction (entry 3). Intrigued by this observation, we postulated that the presence of a sterically demanding substituent in the ortho-positions of the N-aryl ring may serve to improve the catalyst's ability to distinguish between different faces of the aromatic aldehydes. However, an increase in the pKa of the triazolium precatalyst may consequently impact upon the levels of carbene generated in situ and thus, the isolated yields of the desired cross-product.
The precatalyst 24 (in which the p-bromo atom of the N-aryl substituent has been replaced with the more electron withdrawing CF3 unit) mediated the formation of 29 with an ee of 75% ee but just 28% isolated yield (entry 4). We have previously shown28 that under basic conditions an achiral analogue of 25 has been shown to catalyse the crossed-benzoin condensation between benzaldehyde and 2-bromobenzaldehyde with unprecedented levels of chemoselectivity. It was therefore disappointing that under our reaction conditions, utilisation of precatalyst 25 led to isolation of 30 in 38% yield and 68% ee; although accelerated reaction rates relative to 24 were observed, (entry 5). The iodo-substituted salt 26 offered no improvement: the influence of the reduced electronegativity but greater steric bulk of iodine relative to bromine resulted in sluggish coupling and moderate product ee (entry 6).
The observation that the carbene derived from precatalyst 12 – containing the smaller but more electronegative N-pentafluorophenyl ring (relative to 23–26) – exhibits a marked preference for promoting the formation of benzoin 6 in good yields and excellent enantioselectivity (see Table 2, entry 2) with concurrent production of crossed-benzoin 30 in racemic form in the same pot is remarkable. Catalyst 22 promotes the formation of 30 as the major reaction product with modest enantiocontrol (see Table 2, entry 1) via attack on 27 from the opposite face to that attacked on benzaldehyde during the formation of 6.
After crossed-benzoin reactions between 2 and 27 in the presence of either 12, 22 or 23, hydrogenolysis of 27 using a palladium catalyst under a hydrogen atmosphere and subsequent CSP-HPLC analysis revealed that the benzoin is formed as the (R)-enantiomer, whereas crossed-benzoin 30 is produced in enantioenriched (S)-form (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Precatalyst | Yield 6a (%) | ee 6b (%) | Yield 30a (%) | ee 30c,d (%) |
| a Isolated yields. b Determined via CSP-HPLC analysis. c Determined through hydrogenolysis of the brominated crossed-benzoin adduct and subjection to CSP-HPLC analysis. d No racemisation of 30 is assumed. | |||||
| 1 | 12 | 74 | >99 | 36 | 0 |
| 2 | 22 | 12 | 80 (R) | 63 | 67 (S) |
| 3 | 23 | 2 | 78 (R) | 23 | 72 (S) |
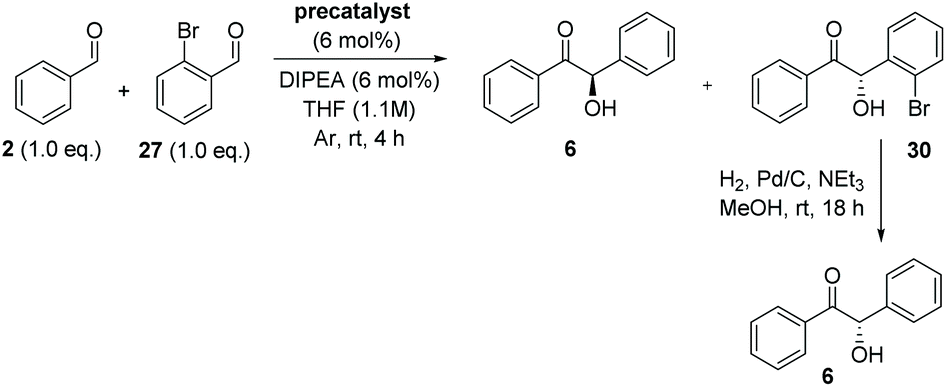
As previously noted, precatalyst 12 catalyses the formation of 6 in >99% ee while 30 is concurrently synthesised as a racemate (entry 1). However, in addition to promoting the production of (S)-30 in 63% yield and 67% ee, precatalyst 22 allows the formation of (R)-6 in 12% yield and 80% ee (entry 2). Similar results were observed using the bromo-substituted 23 (entry 3).
We next endeavoured to determine whether this trend was reliant solely on the steric characteristics of the catalyst or if the identity of the ortho-substituted aldehyde also influenced the enantioselectivity of the reaction. We therefore evaluated the effect selected precatalysts had on both the chemo- and enantioselectivity of the crossed-condensation between 2 and 2-chlorobenzaldehyde (31) and 2-iodobenzaldehyde (32, Table 4). In the presence of 6 mol% of precatalyst 12 and DIPEA, the cross-coupling between 2 and 31 proceeds with adduct (R)-33 formed in moderate yield and ee. However, once again, (R)-6 represented the major reaction product (entry 1). Use of 22 as the carbene precursor results in a change in the absolute configuration of the crossed-benzoin product, with (S)-37 produced also in moderate yield and ee. Formation of (R)-6 under these conditions is less pronounced (entry 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Precatalyst | R | Yield 33/34a (%) | Yield 6b,c (%) | Yield 35/36a (%) | Yield 37/38b (%) | ee 37/38d (%) |
| a Determined via1H NMR spectroscopic analysis of the crude reaction mixture using styrene (0.25 equiv.) as an internal standard. b Isolated yields. c Value in parentheses represents enantiomeric excess of isolated product, for (R)-benzoin, as determined via CSP-HPLC analysis. d Determined via CSP-HPLC analysis. | |||||||
| 1 | 12 | Cl | 8 | 52 (>99) | 2 | 44 | 44 (R) |
| 2 | 22 | Cl | 5 | 32 (80) | 1 | 49 | 40 (S) |
| 3 | 23 | Cl | 2 | 24 (78) | 1 | 28 | 53 (S) |
| 4 | 12 | I | 0 | 65 (n/d) | 0 | 35 | 58 (S) |
| 5 | 22 | I | 0 | 42 (n/d) | 0 | 43 | 72 (S) |
| 6 | 23 | I | 0 | 32 (n/d) | 0 | 27 | 74 (S) |

Consistent with previous observations; employment of the N-2,4,6-tribromophenyl precatalyst 23 resulted in a further improvement in enantioselectivity, alongside a diminution in chemoselectivity and crossed-product yield (entry 3). The observation of the formation of 35 may be rationalised through the reduced steric bulk of the ortho-chlorine substituent (relative to the bromine atom in 27), which results in a greater propensity of the catalyst to attack 31 initially relative to 27, which also produces a less hindered, less discerning Breslow intermediate. This is reflected in the production of both 33 and 35 in trace amounts (entries 1–3), a feature not observed in the data detailed in Table 1.
Utilising 32 as the ortho-substituted coupling partner results in similarly low levels of chemoselectivity; when the reaction is mediated by the carbene derived from 12, (R)-6 is again the primary reaction product, whereas the desired crossed-benzoin adduct 38 is produced in 35% isolated yield (entry 4). However, in contrast to the analogous use of 31 under these conditions, (S)-38 was formed in 58% ee (compare with entry 1). Subsequent use of 22 resulted in improved enantioselectivity (entry 5). Distinct chemoselectivity issues arise here; 6 and 38 were generated with almost equal facility. Increasing the size of the precatalyst's N-aryl ring negatively impacts chemoselectivity further, to the extent that the carbene derived from 23 will preferentially induce the formation of (R)-6 over the crossed-benzoin (S)-38 (entry 6), albeit with an increase in the cross-product ee to 74%.
The results obtained in Tables 3 and 4 are indicative of a clear trend – that, in general, the identity of the ortho-halo-benzaldehyde substituent does influence the stereochemical outcome of cross-benzoins with 2 mediated by the novel chiral triazolium ion-based precatalysts under basic conditions. In general, the bigger the halogen, the higher the crossed product ee. Furthermore, as the steric bulk of the ortho-halogen atoms on the carbene's N-aryl ring increase, the enantiomeric excess of the crossed adducts also improves.
Despite these findings, our efforts to develop an efficient asymmetric variant of the intermolecular crossed-benzoin condensation could not have been considered successful at this point. While utilisation of o-iodobenzaldehyde (32) offered the greatest potential for achieving a highly enantioselective crossed-benzoin condensation, we were discouraged by the lack of chemoselectivity observed using this substrate. Conversely, use of 27 gave rise to a comparable process from an enantioselectivity standpoint (see Table 1, entry 12 vs. Table 4, entry 6) but also provided the desired crossed-benzoin adduct as the main reaction product (albeit with sub-optimal chemoselectivity). The o-bromo aldehyde 27 was therefore chosen as the best substrate for further study.
In order to develop a truly enantioselective protocol, we first had to understand why, if the size of the catalyst N-aryl ring has a direct effect on facial selectivity, the design of precatalysts 24–26 did not result in a significantly improved methodology. We reasoned that because – for instance – the steric bulk around the carbene centre in precatalyst 23 relative to 24 and 25 were identical (owing to the presence of the N-aryl ortho-bromine atoms), the answer must lie in the electronic characteristics of the materials. The additional trifluoromethyl moieties incorporated into salts 24 and 25 would allow for more facile deprotonation and thus a greater concentration of carbene in solution relative to 23. We further posited that crossed-benzoin 30, possessing an electronegative o-bromine atom in close proximity to the α-carbon chiral centre, would have a pKa lower than benzoin and so, under our reaction conditions, may therefore be subject to racemisation via either base-mediated enolisation or a catalyst-mediated addition/elimination mechanism (Scheme 3).
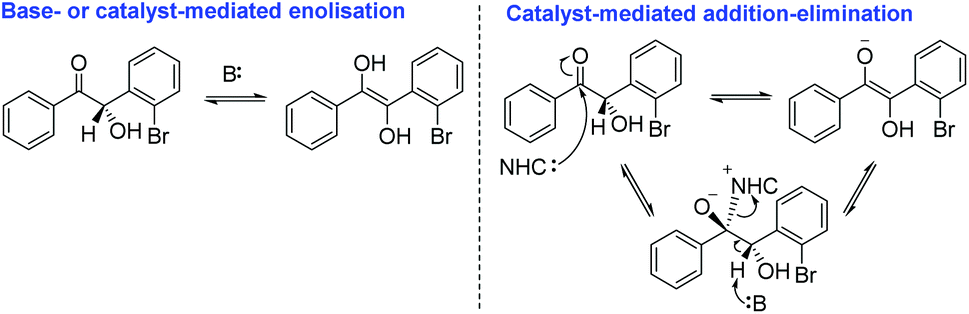 | ||
| Scheme 3 Potential racemisation pathways. | ||
In order to investigate these possibilities, we initially sought to expose an enantioenriched sample of 30 to a solution of pre-formed carbene in the presence of 5 equivalents of CD3OD as solvent and monitor the corresponding deuterium incorporation into 30via1H NMR spectroscopy. However, the excess of CD3OD resulted in deactivation of the carbene in situ and we failed to observe any reaction.
Therefore, we instead exposed both 30 and 6 to a 6 mol% loading of the non-nucleophilic base DIPEA in THF in the presence of 10 equivalents of CD3OD (but the absence of carbene) and monitored the rate of deuterium incorporation as a function of time (Table 5). For accuracy and reproducibility, it was necessary to operate at a lower concentration (0.45 M) to that which we had used previously – as benzoin exhibits poor solubility in THF at 1.1 M concentration (which were the previously typical reaction conditions).
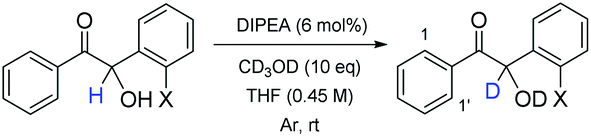
As expected, at t0 there is no incorporation of deuterium observed (entry 1). However, at t = 2 h we were able to detect trace levels of deuteration (entry 2). This deuterium uptake continued – with 12% incorporation after 44 h (entries 3–5). In contrast, under identical conditions and sampling timepoints, 6 displays no inclination for D-uptake at all (entries 6–9). With these results, we were confident that in parallel with inducing a change in the cross-product's absolute configuration, the presence of the ortho-bromine atom also increases the acidity of the α-proton of (S)-30, resulting in base-mediated racemisation to which 6 is not susceptible under the same conditions. This provides an explanation for how 6 and 30 can be formed in the same pot (by the same catalyst) but with such contrasting degrees of enantiocontrol.
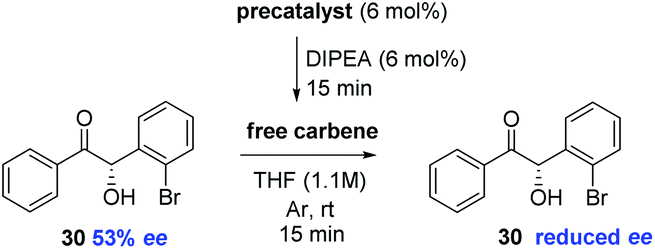
We sought to further support this hypothesis by studying the effect of exposure of enantioenriched 30 to various carbenes (Table 6). After stirring 6 mol% of precatalyst 12 in the presence of DIPEA in THF for 15 minutes; addition of (S)-30 (53% ee) leads to rapid onset of racemisation: just 15 min following exposure, the ee of the sample was depleted (entry 1); similarly, but in a less pronounced manner, the use of precatalyst 22 under identical conditions returned (S)-30 in 47% ee after 15 minutes reaction time (entry 2). The use of the less acidic and more hindered precatalyst 23 allowed the re-isolation of (S)-30 without any observable loss of ee after 15 minutes (entry 3). Finally, the carbene derived from 24 appears to represent a median between its N-pentafluorophenyl and N-trichlorophenyl analogues; with (S)-30 recovered in 43% ee. It appears that the size of the N-aryl ring and the pKa of the triazolium ring (and thus the concentration of carbene generated in solution) are the key factors influencing the racemisation of 30 under these reaction conditions.
In an attempt to ameliorate the racemisation problem, we examined the effect of various mixed solvent systems (Table 7) upon the chemo- and enantioselective outcome of the cross-condensation between 2 and 27 in the presence of chiral triazolium precatalysts and base. In a 4:1 THF:n-hexane solution, (S)-30 is produced in low isolated yield and 54% ee after 18 h employing 6 mol% of precatalyst 22.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Precat. | Base | Solvent | Time (h) | Yield 36a (%) | ee 36b (%) |
| a Isolated yield. b Determined via CSP-HPLC analysis. c 1.25 equiv. of 27 employed. d Reaction carried out using 12 mol% of both precatalyst and base. e Reaction performed at −40 °C. f Reaction performed at 0 °C. g Reaction carried out using 12 mol% KHMDS. h 1.5 equiv. of 27 employed. | ||||||
| 1 | 22 | DIPEA | THF:n-hex. (4:1) |
18 | 19 | 54 |
| 2 | 22 | DIPEA | THF:MTBE (4:1) |
18 | 56 | 33 |
| 3 | 22 | DIPEA | PhMe:CH2Cl2 (10:1) |
18 | 54 | 66 |
| 4c | 22 | DIPEA | PhMe:THF (7.5:1) |
20 | 63 | 75 |
| 5 | 22 | DIPEA | PhMe:THF (7.5:1) |
4 | 40 | 75 |
| 6 | 23 | KHMDS | PhMe:THF (7.5:1) |
18 | 27 | 85 |
| 7d | 23 | KHMDS | PhMe:THF (7.5:1) |
20 | 52 | 57 |
| 8e | 22 | KHMDS | PhMe:THF (7.5:1) |
72 | 2 | 72 |
| 9e | 23 | KHMDS | PhMe:THF (7.5:1) |
72 | 5 | 80 |
| 10f | 22 | KHMDS | PhMe:THF (7.5:1) |
24 | 6 | 79 |
| 11g,h | 22 | KHMDS | PhMe:THF (7.5:1) |
18 | 94 | 0 |
Alternatively, conducting the reaction in a 4:1 THF:MTBE solvent system increased the yield significantly but was accompanied by a marked reduction in ee (entry 2). A 10:1 mixture of PhMe:CH2Cl2 allowed isolation of (S)-30 in 54% yield and higher ee (entry 3). While the solubility of 22 in conjunction with DIPEA was too low in toluene alone to allow the reaction to be carried out efficiently, a 7.5:1 PhMe:THF mixture resulted in a moderate 63% isolated yield and 75% ee after 20 h when 1.25 equivalents of 27 were employed (entry 4). Gratifyingly, under these conditions, concurrent formation of 6 was suppressed to just 5% levels (determined via1H NMR spectroscopic analysis of the crude reaction mixture). Reduction of the reaction time still allowed us to obtain (S)-30 in 40% yield but no improvement in enantioselectivity was detected (entry 5) – suggesting that while the use of non-polar solvents succeeds in partially alleviating the problems associated with product racemisation, there is an inherent lack of facial selectivity displayed by the carbene derived from precatalyst 22. Seeking to overcome the slow reaction rate, we decided to employ precatalyst 23 in toluene alone and utilised KHMDS (6 mol%) as base – to generate a higher concentration of carbene in situ while concurrently avoiding solubility issues. Under these conditions (S)-30 was formed in 85% ee, albeit in poor yield (entry 6).
Further modifications to this system proved unsuccessful – doubling the catalyst and base loading was accompanied by a corresponding improvement in yield after 20 h but a reduction in ee (entry 7). While lowering the reaction temperature caused the reaction rate to drop sharply (entry 8). Under these conditions, use of precatalyst 23 offered slight improvements: producing (S)-30 in 5% yield and 80% ee (entry 9) at −40 °C. Increasing the temperature to 0 °C was not effective (entry 10). Conversely, doubling the loading of KHMDS and employing 1.5 equivalents of 27 enables chemoselective production of 30 in 94% isolated yield but as a racemate (entry 11).
It seemed clear that in these systems the factors which favour high efficiency are detrimental to achieving high enantioselectivity. For instance; the optimal reaction conditions to maximise yields of (S)-30 also encourage racemisation, while a system promoting a more enantioselective crossed-condensation relies on a non-polar environment and a bulky, less active carbene catalyst, which reduces rate and yield. Acknowledging the above, we decided to strike a compromise between the two extremes and screen a range of substituted benzaldehydes in the intermolecular cross-condensation with 27 using the best conditions identified (Table 8).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yielda (%) | eeb(%) |
| a Isolated yield. b Determined via CSP-HPLC analysis. c Reaction carried out at 0.5 M. d Reaction time reduced to 2 h. e Subsequent recrystallisation returned the product in racemic form. | ||||
| 1 |

|

|
63 | 75 (S) |
| 2c |

|
|
40 | 80 (S) |
| 3 |

|

|
76 | 0 |
| 4d |
|
|
62 | 84e |
| 5 |
|
|
66 | 83 |
| 6 |
|
|
57 | 77 |
| 7 |
|
|
47 | 66 |
| 8 |
|

|
48 | 45 |
| 9 |
|
|
79 | 52 |
| 10 |
|
|
48 | 83 |
| 11 |
|
|
42 | 76 |
| 12 |
|
|
56 | 67 |
| 13 |
|

|
76 | 71 |
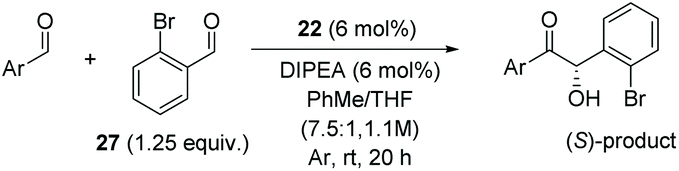
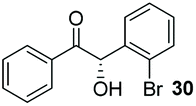
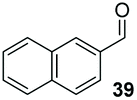
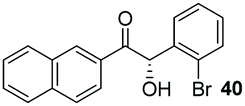
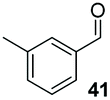
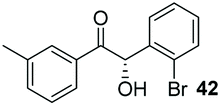
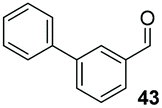
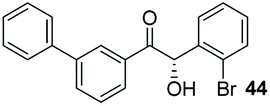
Precatalyst 22 was employed in the presence of DIPEA in a 7.5:1 PhMe:THF solvent system (see Table 7, entry 4) using 1.25 equivalents of 27: conditions which we felt enabled moderate chemoselective production of the desired cross-product without significantly impacting the enantioselectivity of the reaction. Under these conditions, benzaldehyde (2) can be coupled with 27 in 63% isolated yield and 75% ee (entry 1). A lower concentration furnished 30 in 80% ee, although as expected, the yield was negatively impacted (entry 2). Interestingly, 2-naphthaldehyde (39) proved an exceptionally active substrate: 40 precipitated from solution after just 1 h; however, the product is present in racemic form after 20 h (entry 3). Quenching the reaction after 2 h enabled the isolation of 40 in moderate yield and with good ee (entry 4). Electron-neutral benzaldehydes proved the most suitable coupling partners in our hands, with 3-tolualdehyde (41) undergoing asymmetric condensation with 27 to form 42 with high ee (entry 5). Biphenyl-3-carbaldehyde (43) reacts more slowly, undoubtedly due to the increased steric requirement, but still allows access to 44 in 57% yield and appreciable enantiopurity (77% ee, entry 6).
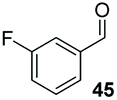
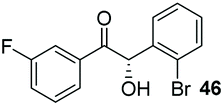
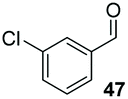
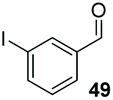
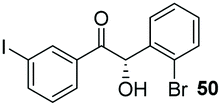
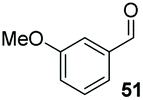
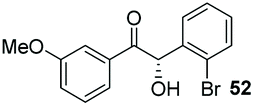
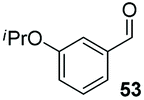
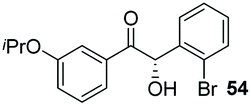
Consistent with observations disclosed in previous studies,19,20 enantioselectivity in the presence of electron-deficient benzaldehydes 45, 47 and 49 is reduced (entries 7–9). This may be attributable to increased acidity of the α-proton in products 46, 48 and 50. In addition, the chemoselectivity of the process is reduced when employing 45 and 47 due to a greater proclivity for the formation of the corresponding homobenzoin products derived from these aldehydes; resulting in diminished yields. Conversely, electron-rich benzaldehydes tend to participate in highly chemoselective but slower reactions: 52 can be obtained in 48% yield through cross-coupling between 3-anisaldehyde (51) and 27 with 83% ee after 20 hours (entry 10), while 3-isopropoxybenzaldehyde (53) participates in the condensation with isolation of 54 with marginally lower enantiocontrol (entry 11).
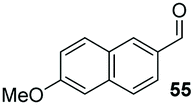
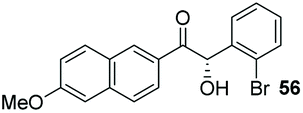
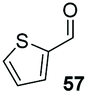
The methoxy-substituted naphthaldehyde 55 proved less adequate as a substrate, however 56 could still be obtained in 67% ee (entry 12). Finally, we were pleased to find that heterocyclic aldehyde 2-thiophenecarbaldehyde (57) was also a serviceable substrate (entry 13). We attempted to recrystallise enantioenriched 40 (84% ee) from hexane: however, it appears that enol formation in these adducts is so facile that even the mild heating required to dissolve the material promotes complete racemisation.
Since our efforts to marry both high yields and enantioselectivity had reached an impasse, we dedicated our efforts to maximising enantiocontrol, irrespective of yield. Having already extensively evaluated a range of reaction conditions, we focused our attention instead on our choice of ortho-substituted benzaldehyde. Conscious of the fact that the increased acidity of the crossed-benzoin α-proton was hindering our efforts, we postulated that installation of an ortho-substituent capable of deterring deprotonation might be a viable strategy to overcome this obstacle. In this regard, we took inspiration from the pKa values of the ortho-substituted phenol series where, despite the superior electronegativity of fluorine, the pKa of 2-fluorophenol (8.73) is actually greater than that of 2-chlorophenol (8.51) or 2-bromophenol (8.39). This is primarily attributed to the fact that fluorine can favourably hydrogen-bond with the acidic proton (O–H–F) to form a chelating intramolecular five-membered ring to a greater extent than chlorine, while the larger atomic radius of bromine prevents it from participating in this way (Fig. 2).34
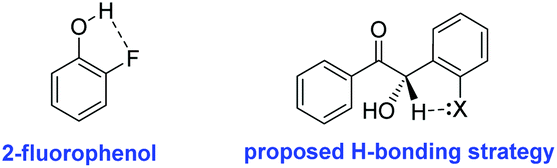 | ||
| Fig. 2 2-Fluorophenol and a potential strategy for reducing product acidity in a crossed-benzoin product using an o-fluoro substituent. | ||
We postulated that such an interaction would be potentially achievable in a crossed-benzoin product and, in an analogous manner, could lead to decrease in the acidity of the α-proton. Thus, under our previously established conditions, we attempted to cross-couple 2 and 2-fluorobenzaldehyde (59) (Table 9, entry 1). All four possible benzoin products were observed at approximately 20% levels each via1H NMR spectroscopic analysis of the crude material. Attempts to separate these using flash chromatography were unsuccessful. It seems obvious, a posteriori, that the fluorine atom is not of sufficient steric bulk to control both the formation and reactivity of the required Breslow intermediate. However, we noted how a similar trend in pKa emerges when one considers the pKa of both 2-fluoromphenol and 2-(trifluoromethyl)phenol (8.95 and 8.68 respectively). Thus, we repeated the attempted cross-condensation with 2 as before, employing 2-trifluoromethylbenzaldehyde (61).

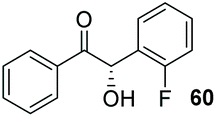
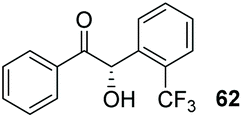

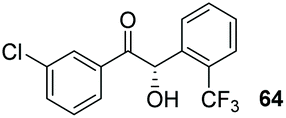
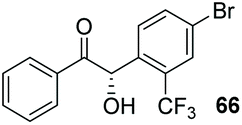

Despite the slow reaction rate, we were able to isolate 62 in 34% yield and 92% ee after 20 hours (entry 2), which, to the best of our knowledge, represents the first ever highly enantioselective intermolecular crossed-benzoin condensation between two non-identical aromatic aldehydes as catalysed by a chiral NHC catalyst. Aldehyde 62 participates in the reaction quite slowly: reaction with the methoxybenzalehyde 51 in the presence of the carbene provided 63 in very low yield but again, with excellent enantiocontrol (entry 3). Use of the more activated aromatic aldehyde 47 allowed the preparation of 64 in marginally improved yield and 88% ee after 20 hours (entry 4). In a final attempt to improve efficacy, we synthesised aldehyde 65 – this retains the CF3 unit which contributes to high chemoselectivity and enantiocontrol, yet also possesses a (removable) p-bromo moiety to increase the activity of the electrophile. Use of 65 in conjunction with 2 was unsuccessful: the product 66 was obtained in 58% yield, but as a near racemate. Shortening the reaction time to 4 h produced just 10% yield of 66 – albeit with improved enantiocontrol (entry 6). Evidently, the installation of the para-bromine atom again lowers the pKa of the α-proton to the extent that our proposed H-bonding protective effect to alleviate product racemisation is obviated under these conditions.
Conclusions
In conclusion, we have carried out the first extensive study into the asymmetric intermolecular crossed benzoin condensation between two non-identical aromatic aldehydes catalysed by chiral NHCs. The presence of at least one ortho-substituted benzaldehyde (usually chosen so that the o-substituent could be removed after reaction if required) proved crucial in enabling the NHC to direct a chemoselective reaction. Using a catalytic loading of a novel chiral triazolium precatalyst salt and base, our initial efforts to develop a highly enantioselective version of the reaction met with failure: deuterium exchange experiments revealed how – in the presence of carbene and base – these crossed-benzoin adducts are enolisable and can undergo racemisation via deprotonation at the aryloin α-carbon atom (or possibly an addition–elimination process involving the carbene). Susceptibility to enolisation is enhanced in the presence of electron-withdrawing substituents; which could rationalise in part why previous studies investigating homobenzoin condensations have observed fluctuating enantioselectivity when employing activated benzaldehydes. Benzoin itself appears resistant to such racemisation in the presence of catalyst and base (and sometimes precipitates from solution at higher concentrations) and so we propose that the archetypal benzoin condensation can no longer be considered a suitable model reaction for the meaningful evaluation of the potential catalytic utility of a chiral carbene in mediating crossed acyloin condensations.After investigation of these pathways, reaction conditions could be modified to minimise the impact of racemisation, thus allowing the synthesis of a range of crossed-benzoins in moderate-good yields with moderate-high enantioselectivities for the first time. Furthermore, careful planning and choice of the substrate aldehyde coupling partners employed allowed the first highly enantioselective intermolecular crossed-benzoin condensation using an NHC. It was also shown that the enantioselectivity of the reaction is heavily dependent upon the identity of the ortho-substituted benzaldehyde, with general trends suggesting that the larger the substituent, the greater the enantiocontrol. A similar trend was observed when considering the size of the precatalyst N-aryl ring.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Irish Research Council for financial support.Notes and references
- F. Wöhler and J. Liebig, Ann. Pharm., 1832, 3, 249 CrossRef.
- (a) N. Zinin, Ann. Pharm., 1839, 31, 329 CrossRef; (b) N. Zinin, Ann. Pharm., 1840, 34, 329 Search PubMed.
- T. Ukai, R. Tanaka and T. Dokawa, J. Pharm. Soc. Jpn., 1943, 63, 269 Search PubMed.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- For a more recent kinetic study see: M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124 CrossRef CAS.
- (a) J. L. Moore and T. Rovis, Top. Curr. Chem., 2009, 291, 77 CrossRef; (b) D. Enders, J. Org. Chem., 2008, 73, 7857 CrossRef CAS; (c) T. Rovis, Chem. Lett., 2008, 37, 1 CrossRef; (d) K. Zeitler, Ernst Schering Found. Symp. Proc., 2007, 2, 183 Search PubMed; (e) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5506 CrossRef; (f) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (g) K. Zeitler, Angew. Chem., Int. Ed., 2005, 44, 7506 CrossRef CAS; (h) M. Christmann, Angew. Chem., Int. Ed., 2005, 2632 CrossRef CAS; (i) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS; (j) J. S. Johnson, Angew. Chem., Int. Ed., 2004, 43, 1326 CrossRef CAS; (k) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (l) R. S. Menon, A. T. Biju and V. Nair, Beilstein J. Org. Chem., 2016, 12, 444 CrossRef CAS; (m) X.-Y. Chen, Z.-H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690 CrossRef CAS.
- P. Hoyos, J.-V. Sinisterra, F. Molinari;, A. R. Alcántara and P. Domínguez de María, Acc. Chem. Res., 2010, 43, 288 CrossRef CAS.
- B. Rosche, V. Sandford, M. Breuer, B. Hauer and P. L. Rogers, J. Mol. Catal. B: Enzym., 2002, 19, 109 CrossRef.
- J. Sheehan and D. H. Hunnemann, J. Am. Chem. Soc., 1966, 88, 3666 CrossRef CAS.
- J. Sheehan and T. Hara, J. Org. Chem., 1974, 39, 1196 CrossRef CAS.
- Rawal later improved the yield: C. A. Dvorak and V. H. Rawal, Tetrahedron Lett., 1998, 39, 2925 CrossRef CAS.
- (a) W. Takagi, Y. Tamura and Y. Yano, Bull. Chem. Soc. Jpn., 1980, 53, 478 CrossRef; (b) J. Marti, J. Castells and F. Lopez-Calahora, Tetrahedron Lett., 1993, 34, 521 CrossRef CAS; (c) R. L. Knight and F. J. Leeper, Tetrahedron Lett., 1997, 38, 3611 CrossRef CAS; (d) A. U. Gerhard and F. J. Leeper, Tetrahedron Lett., 1997, 38, 3615 CrossRef CAS; (e) R. L. Knight and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 1998, 1891 RSC; (f) Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438 CrossRef CAS.
- D. Enders, K. Breuer and J. H. Teles, Helv. Chim. Acta, 1996, 79, 1217 CrossRef CAS.
- R. L. Knight and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 1998, 1891 RSC.
- D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743 CrossRef CAS.
- See also: D. Enders and J. Han, Tetrahedron: Asymmetry, 2008, 19, 1367 CrossRef CAS.
- Y. Ma, S. Wei, J. Wu, F. Yang, B. Liu, J. Lan, S. Yang and J. You, Adv. Synth. Catal., 2008, 350, 2645 CrossRef CAS.
- For representative other NHC-systems for catalysis of the asymmetric homobenzoin condensation see: (a) Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438 CrossRef CAS; (b) J. P. Brand, J. I. O. Siles and J. Waser, Synlett, 2010, 881 CAS; (c) T. Soeta, Y. Tabatake, K. Inomata and Y. Ukaji, Tetrahedron, 2012, 68, 894 CrossRef CAS; (d) Z. Rafiński, A. Kozakiewicz and K. Rafińska, Tetrahedron, 2014, 70, 5739 CrossRef.
- S. E. O'Toole and S. J. Connon, Org. Biomol. Chem., 2009, 7, 3584 RSC.
- L. Baragwanath, C. A. Rose, K. Zeitler and S. J. Connon, J. Org. Chem., 2009, 74, 9214 CrossRef CAS.
- Very recently Milo et al. have shown that product ee can be improved through the simple step of adding a small excess (relative to the catalyst) of aryl boronic acids, see: V. Dhayalan, S. C. Gadekar, Z. Alassad and A. Milo, Nat. Chem., 2019, 11, 543 CrossRef CAS.
- For an approach involving a chiral metallophosphite catalyst (and acylsilanes as one coupling partner instead of aldehydes) see: X. Linghu, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070 CrossRef.
- For the use of activated ketones as coupling partners see: (a) D. Enders and A. Henseler, Adv. Synth. Catal., 2009, 351, 1749 CrossRef CAS; (b) D. Enders, A. Grossmann, J. Fronert and G. Raabe, Chem. Commun., 2010, 46, 6282 RSC; (c) C. A. Rose, S. Gundala, C.-L. Fagan, J. F. Franz, S. J. Connon and K. Zeitler, Chem. Sci., 2012, 3, 735 RSC; (d) K. Thai, S. M. Langdon, F. Bilodeau and M. Gravel, Org. Lett., 2013, 15, 2214 CrossRef CAS; (e) C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2014, 136, 14698 CrossRef CAS; (f) J. Xu, J. Peng, C. He and H. Ren, Org. Chem. Front., 2019, 6, 172 RSC.
- For a representative enzymatic approach see: A. S. Demir, Ö. Şeşenoglu, E. Eren, B. Hosrik, M. Pohl, E. Janzen, D. Kolter, R. Feldmann, P. Dünkelmann and M. Müller, Adv. Synth. Catal., 2002, 344, 96 CrossRef CAS.
- For a recent review of the aza-benzoin reaction see: D. C. M. Albanese and N. Gaggero, Catalysts, 2019, 8, 181 CrossRef.
- For representative examples of NHC-catalysed aza-benzoin condensations see: (a) Q. Peng, D. Guo, J. Bie and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 3767 CrossRef CAS; (b) M. D. Wilde and M. Gravel, Org. Lett., 2014, 16, 5308 CrossRef CAS; (c) J. Xu, C. Mou, T. Zhu, B.-A. Song and Y.-R. Chi, Org. Lett., 2014, 16, 3272 CrossRef CAS; (d) D. A. DiRocco and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 5904 CrossRef CAS; (e) K.-J. Wu, G.-Q. Li, Y. Li, L.-X. Dai and S.-L. You, Chem. Commun., 2011, 47, 493 RSC; (f) M. He and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 418 CrossRef CAS; (g) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654 CrossRef CAS.
- (a) S. E. O'Toole, C. A. Rose, S. Gundala, K. Zeitler and S. J. Connon, J. Org. Chem., 2011, 76, 347 CrossRef; (b) C. A. Rose, S. Gundala, K. Zeitler and S. J. Connon, Synthesis, 2011, 190 CAS.
- For representative studies from other groups on crossed acyloins involving aromatic and aliphatic aldehydes see: (a) M. Y. Jin, S. M. Kim, H. Han, D. H. Ryu and J. W. Yang, Org. Lett., 2011, 13, 880 CrossRef CAS; (b) M. Y. Jin, S. M. Kim, H. Mao, D. H. Ryan, C. E. Song and J. W. Yang, Org. Biomol. Chem., 2014, 12, 1547 RSC; (c) S. M. Langdon, M. M. D. Wilde, K. Thai and M. Gravel, J. Am. Chem. Soc., 2014, 136, 7539 CrossRef CAS; (d) C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2014, 136, 14698 CrossRef CAS; (e) B. T. Ramanjaneyulu, S. Mahesh and R. V. Anand, Org. Lett., 2015, 17, 6 CrossRef CAS.
- E. G. Delany and S. J. Connon, Org. Biomol. Chem., 2018, 16, 780 RSC.
- I. Piel, M. D. Pawelczyk, K. Hirano, R. Fröhlich and F. Glorius, Eur. J. Org. Chem., 2011, 5475 CrossRef CAS.
- C. J. Collett, R. S. Massey, J. E. Taylor, O. R. Maguire, A. C. O'Donoghue and A. D. Smith, Angew. Chem., Int. Ed., 2015, 54, 6887 CrossRef CAS.
- This is despite convincing evidence (see ref. 31) that benzaldehydes equipped with an o-heteroatom are attacked by NHCs faster than benzaldehyde itself. It is clear that in such (reversible) crossed benzoin condensations chemoselectivity is not determined by the relative rates of the first attack of the carbene on the two aldehydes. For a more in depth discussion see ref. 27.
- For discussions regarding the influence of the N-aryl substituent on selectivity in NHC reactions see: (a) T. Rovis, Chem. Lett., 2008, 37, 2 CrossRef CAS; (b) C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514 RSC.
- (a) M. H. Abraham, R. J. Abraham, A. E. Aliev and C. F. Tormena, Phys. Chem. Chem. Phys., 2015, 17, 25151 RSC; (b) S. W. Dietrich, E. C. Jorgensen, P. A. Kollman and S. Rothenburg, J. Am. Chem. Soc., 1976, 98, 8310 CrossRef CAS; (c) J. Jover, R. Bosque and J. Sales, QSAR Comb. Sci., 2007, 26, 385 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob02017f |
| This journal is © The Royal Society of Chemistry 2021 |