
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProfiling of Haemophilus influenzae strain R2866 with carbohydrate-based covalent probes†
Camille
Metier
a,
Jennifer
Dow
b,
Hayley
Wootton
a,
Steven
Lynham
c,
Brendan
Wren
b and
Gerd K.
Wagner
 *d
*d
aKing's College London, Department of Chemistry, Britannia House, 7 Trinity Street, London, SE1 1DB, UK
bLondon School of Hygiene and Tropical Medicine, Department of Infection Biology, Keppel Street, London, WC1E, 7HT, UK
cProteomics Facility, Centre of Excellence for Mass Spectrometry, King's College London, The James Black Centre, 125 Coldharbour Lane, London, SE5 9NU, UK
dQueen's University Belfast, School of Pharmacy, Medical Biology Centre, 97 Lisburn Road, Belfast, BT9 7BL, UK. E-mail: G.Wagner@qub.ac.uk
First published on 23rd December 2020
Abstract
We demonstrate the application of four covalent probes based on anomerically pure D-galactosamine and D-glucosamine scaffolds for the profiling of Haemophilus influenzae strain R2866. The probes have been used successfully for the labelling of target proteins not only in cell lysates, but also in intact cells. Differences in the labelling patterns between lysates and intact cells indicate that the probes can penetrate into the periplasm, but not the cytoplasm of H. influenzae. Analysis of selected target proteins by LC-MS/MS suggests predominant labelling of nucleotide-binding proteins, including several known antibacterial drug targets. Our protocols will aid the identification of molecular determinants of bacterial pathogenicity in Haemophilus influenzae and other bacterial pathogens.
Introduction
The profiling of pathogenic bacteria with chemical probes is a powerful strategy to uncover molecular determinants of bacterial pathogenicity, virulence, and antimicrobial resistance.1–3 Typically, such probes contain a reactive moiety for the formation of a covalent linkage, enabling the irreversible labelling of target proteins. Both targeted4 and target-agnostic5 approaches have been explored for such applications, with natural products being a particularly common scaffold for probe development.6–8 For target deconvolution in complex systems, such covalent probes can be conveniently interfaced with bioanalytical methods such as mass spectrometry and gel electrophoresis.9Carbohydrates are an attractive scaffold for the development of covalent probes for the profiling of bacterial pathogens, both from a chemistry and microbiology perspective. Even a simple monosaccharide allows the facile installation of both an electrophilic warhead for covalent labelling, and a reporter group for detection (Fig. 1). Moreover, most sugars are readily water soluble, which is a considerable advantage for applications in biological media. Sugars such as D-glucose and α-lactose are preferred bacterial carbon sources10 and, together with other sugars, serve as building blocks for complex glycans of the bacterial cell wall (e.g., lipopolysaccharide, peptidoglycan).11 Carbohydrates and carbohydrate-recognising proteins are often directly involved in multiple aspects of bacterial pathogenicity,12 including adhesion,12 immune evasion,12 virulence,13,14 and biofilm formation.15 Carbohydrate-based covalent probes are therefore excellent tools for the profiling of bacterial cells and the labelling of proteins involved in bacterial pathogenicity.
 | ||
| Fig. 1 General design of carbohydrate-based covalent probes. | ||
Several examples for the labelling of bacterial proteins with covalent probes based on a carbohydrate, or carbohydrate-like, scaffold have previously been reported. Usually, these probes are designed to specifically recognise a single target protein. Thus, a glucosamine-based probe has been used successfully to capture the β-N-acetylhexosaminidase NagZ, which is involved in the induction of resistance to β-lactams, from non-purified lysates of P. aeruginosa.16 More recently, Titz et al. have described a galactose-derived probe for the labelling and in vitro imaging of the lectin LecA, a virulence factor and biofilm building block from P. aeruginosa.17 A probe based on the cyclitol mimic cyclophellitol aziridine has been used for the labelling of the retaining GH29 α-L-fucosidase from Bacteroides thetaiotaomicron, both in recombinant form and in lysates from an E. coli culture overexpressing the target enzyme.18
We have recently used a covalent probe based on a D-glucosamine scaffold for the profiling of K. pneumoniae lysates.19 In the present study, we have extended our general approach to a different scaffold based on D-galactosamine, and another bacterial pathogen, Haemophilus influenzae. H. influenzae is a Gram-negative coccobacillus responsible for life-threatening invasive diseases such as pneumonia and meningitis.20 A cytoplasmic glycosylation system of H. influenzae has been reported, and many carbohydrate binding proteins21 involved in the pathogenicity of the bacterium, including lectins22 and glycosyltranferases,23 have been identified. H. influenzae strain R2866 is an unusually virulent non-typeable strain first isolated from the blood of children with meningitis infection.24,25 Although lacking a capsular polysaccharide structure, H. influenzae R2866 displays elevated serum resistance and a virulence level approaching that of encapsulated type b H. influenzae,24 and these traits have been closely related to the presence of a terminal galactoside epitope of the outer-membrane lipooligosaccharide (LOS) structure.24,26
In this study, we sought to assess the capacity of different carbohydrate-based probes to simultaneously label multiple target proteins in H. influenzae R2866. We reasoned that such a protocol may aid not only the profiling of this strain, but also the identification of antibacterial targets. We were particularly interested in the application of our probes not only with cell lysates, but also, for the first time, with intact cells. Our results show that probes derived from D-glucosamine and D-galactosamine are indeed suitable for such applications. Moreover, notable differences between the profiles obtained from lysates and intact cells suggest, that our protocols can also provide additional information about the localisation of target proteins within the bacterial cell.
Results and discussion
Chemical synthesis of carbohydrate-based probes
The two anomers 7-α and 7-β of D-galactosamine-based target molecule 7 were synthesised in six steps from D-galactosamine 1 (Scheme 1A). We reasoned that O-acetylated probes were more likely than free sugars to penetrate the bacterial membrane and were therefore more suited for labelling experiments with intact bacterial cells. | ||
Scheme 1 (A) Synthesis of D-galactosamine-based probes 7-α and 7-β. Reagents and conditions: (i) p-Anisaldehyde (1 equiv.), NaOH (1 M), 0 °C; (ii) acetic anhydride (8 equiv.), pyridine (excess), rt, 16 hours; (iii) acetone, HCl (4 M, 1.2 equiv.), 56 °C, 5 min; (iv) chloroacetyl chloride (3 equiv.) TEA (2 equiv.), DCM, rt, 2 hours; (v) propynol ethoxylate (3 equiv.), BF3 × Et2O (4 equiv.), DCM, 40 °C, 8 hours; (vi) dansyl azide 9 (1 equiv.), CuSO4–5H2O (1 equiv.), sodium ascorbate (1.5 equiv.), DIPEA (3 equiv.), THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (10:1), rt, 3 hours. (B) D-Glucosamine-based probes 8-α, 8-β, 11-α and 11-β.19 (C) Control compounds 9 and 10 (see ESI† for synthesis). :water (10:1), rt, 3 hours. (B) D-Glucosamine-based probes 8-α, 8-β, 11-α and 11-β.19 (C) Control compounds 9 and 10 (see ESI† for synthesis). | ||
First, O-selective acetylation of 1 was achieved in three steps via the strategy of Wulffen et al.271 was converted into the corresponding imine 2 by reaction with p-anisaldehyde, followed by quantitative O-acetylation of 2. Imine hydrolysis of 3 under acidic conditions gave O-acetylated D-galactosamine 4. Next, the electrophilic warhead was installed in position 2 by reaction of 4 with chloroacetyl chloride, to give the 2-(2-chloroacetamido) derivative 5. Finally, the reporter group was attached at the anomeric position via a short linker. 5 was reacted with propynol ethoxylate under Fischer glycosylation conditions to give glycoside 6 as a mixture of both anomers, which were separated by normal phase chromatography. Each anomer was reacted separately in a Cu-catalysed 1,3-dipolar cycloaddition with azide 9,28 introducing a dansyl fluorophore, to give the D-galactosamine-based target molecules 7-α and 7-β. The successful cycloaddition was evidenced by 1H NMR (CDCl3), which showed the diagnostic triazole signal at 7.61 and 7.67 ppm for 7-α and 7-β respectively. The corresponding D-glucosamine derivatives 8-α and 8-β as well as their deacetylated parent sugars 11-α and 11-β (Scheme 1B) were prepared as previously reported.19
Assignment of anomers
The identity of the individual anomers was unambiguously assigned for both series by 1D and 2D NMR analysis. The weak orbital overlap between H1 and H2 protons in the α-anomer is associated with a small coupling constant 3J1,2, as observed for 7-α (3J1,2 3.5 Hz). A large coupling constant 3J1,2 on the other hand, is characteristic of the β-anomer (7-β: 3J1,2 8.5 Hz). These assignments are further supported by ROESY spectra of each anomer. The 1,3-diaxial orientation of the H1, H3, and H5 protons in the β-anomer leads to diagnostic H1–H3 and H1–H5 crosspeaks, as observed in the ROESY spectrum of 7-β (ESI Fig. S1†). In contrast, these crosspeaks are absent from the ROESY spectrum of 7-α (ESI Fig. S2†) due to the equatorial orientation of H1 in the α-anomer. Assignments for 8-α and 8-β were made as previously reported.19Labelling of a bacterial glycosyltransferase in cell lysates
In order to assess the capability of our probes to label a carbohydrate-recognising protein, we carried out labelling experiments with the retaining α-1,4-galactosyltransferase LgtC from Neisseria meningitidis, which catalyses the transfer of D-galactose from a UDP α-D-galactose donor to a lactose acceptor.29 LgtC was overexpressed in BL21* E. coli cells. Cells were lysed, and cell lysates were incubated for 30 min at 30 °C with the D-glucosamine derivatives 8-α and 8-β, or the fluorophore control 10. To assess the potential effect of the acetate protecting groups on labelling, we also included the deacetylated sugars 11-α and 11-β (Scheme 1B) in these experiments. Samples were separated on a 12% SDS-page gel under denaturing conditions and analysed by in-gel fluorescence emission and Coomassie staining (Fig. 2).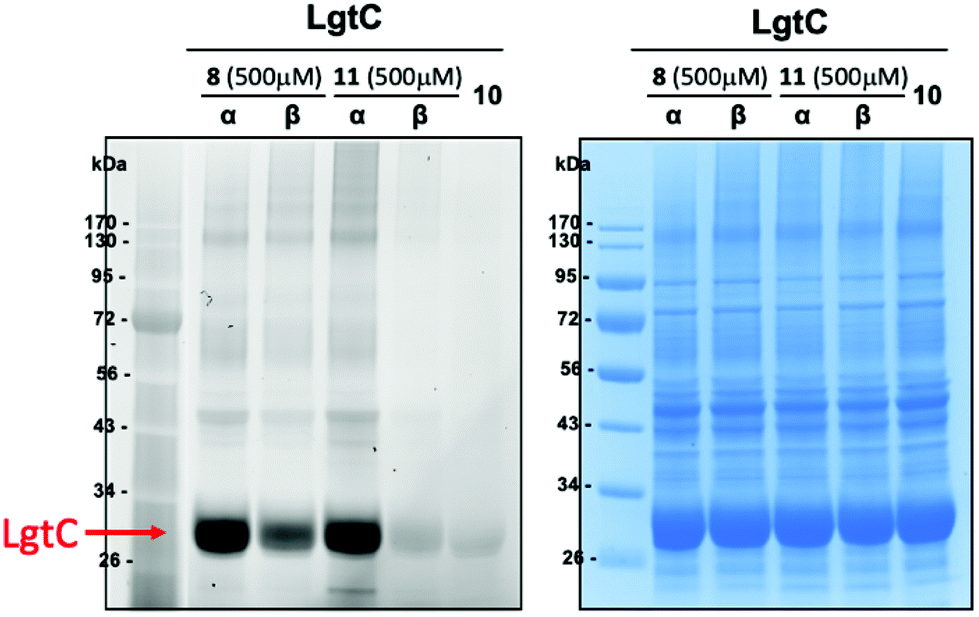 | ||
| Fig. 2 Labelling of recombinant LgtC with 8-α, 8-β, 11-α and 11-β in E. coli lysates. General conditions: E. coli cells overexpressing LgtC were lysed (8 h after IPTC induction) by mixing with BugBuster (100 μL for 100 mg of cell pellet, prepared from 10× stock in HEPES buffer) for 30 min at rt. Following centrifugation, the supernatant was divided into fractions (9 μL) and incubated with stock solutions (1 μL, 5 mM stock in DMSO) of 8-α, 8-β, 11-α, 11-β or 10 (fluorophore control) for 1 hour at 30 °C. Loading buffer (2.5 μL) was added to each fraction (10 μL). Samples were incubated at 50 °C for 10 min and loaded onto a 12% SDS-page gel. The gel was run with MES running buffer at 160 V for 70 min. Protein bands were detected by fluorescence scanning (left) and Coomassie staining (right). | ||
In the case of the acetylated probes, both anomers 8-α and 8-β produced a fluorescently labelled band of LgtC, although this band was weaker for the β-anomer. Interestingly, in the case of the deacetylated probes, strong fluorescent labelling was only observed with the α-anomer 11-α. As expected, no labelling was observed with the fluorophore control 10. These results demonstrate that 8-α and 8-β are capable of labelling target proteins in complex biological media. They also suggest that the presence of the acetate protecting groups, in conjunction with the anomeric configuration, may affect labelling efficacy and/or target recognition.
Profiling of H. influenzae R2866 lysates
Next, we set out to assess the suitability of the pure anomers 7-α, 7-β, 8-α and 8-β for the profiling of H. influenzae R2866. First, we investigated all four probes for the labelling of target proteins in cell lysates. To identify optimum growth conditions for labelling experiments, growth kinetics of R2866 were established over 24 hours. We observed a doubling time of 60 min, with the stationary phase reached at 8–9 hours, and a maximum OD590 of 1.9 (ESI Fig. S3†). For labelling experiments, cell cultures of H. influenzae R2866 were grown in Brain Heart infusion (BHI) broth until mid-logarithmic phase was reached (OD590 0.9; 5 hours). The cells were then harvested by centrifugation and pellets equivalent to 1 mL of cell culture were lysed with BugBuster protein extraction reagent. Cell debris was removed by centrifugation, and the supernatant was incubated with probes 7-α, 7-β, 8-α or 8-β (0.5 mM) or controls. Controls included a DMSO-only sample, as well as the dansyl fluorophore 10 (0.5 mM), which is lacking both the sugar scaffold and the electrophilic warhead (Scheme 1C). Lysate fractions treated with either probe or control were separated on a 4–12% SDS-page gel, and analysed by in-gel fluorescence emission and Coomassie staining (Fig. 3).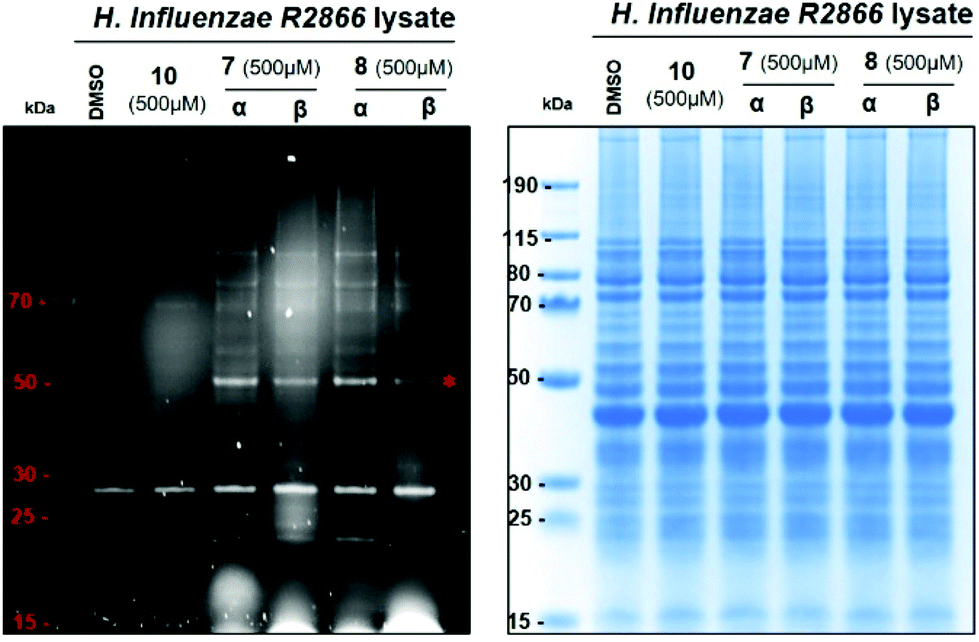 | ||
| Fig. 3 Protein labelling with 7-α, 7-β, 8-α and 8-β in H. influenzae R2866 cell lysates. General conditions: Cells were grown to OD590 0.9 and lysed by mixing with BugBuster (100 μL, ×1 in PBS, for pellet of 4 mL of cell culture) for 30 min at rt. Following centrifugation, the supernatant was divided into fractions (9 μL) and incubated with stock solutions (1 μL, 5 mM stock in DMSO) of 7-α, 7-β, 8-α, 8-β or 10 (fluorophore control), or DMSO (1 μL, control) for 1 hour at 30 °C. Loading buffer (2.5 μL) was added to each fraction (10 μL) and loaded onto a 4–12% SDS-page gel. The gel was run with MES running buffer at 150 V for 70 min. Protein bands were detected by fluorescence scanning (left) and Coomassie staining (right). The asterisk denotes the low-abundance protein at 50 kDa labelled by all four probes (see text). | ||
For all four probes, a defined labelling pattern of proteins over a wide MW range (∼20–100 kDa) was observed (Fig. 3). This pattern was absent from the control lanes, with the exception of a strong band of an autofluorescent protein at ∼28 kDa. There was no significant difference in the labelling profile between the four different probes. This suggests that the configuration at position 4 (D-gluco vs.D-gala series) or position 1 (α vs. β anomer) of the sugar scaffold has only a minor influence on the target profile. The similar labelling intensity observed for both anomers, both for 7 and 8, is reminiscent of the observation in the labelling experiments with LgtC, and suggest that the acetate groups remain intact under these conditions.
Particularly strong fluorescence was observed for a band at ∼50 kDa (Fig. 3, asterisk). The corresponding band in the Coomassie stain was relatively weak, indicating that this is a low-abundance protein. In contrast, several high-abundance proteins that give strong Coomassie bands were not, or only weakly, labelled by the fluorescent probes. Taken together, this suggests that protein abundance is not the determining factor for labelling by these probes.
Under these experimental conditions, labelling efficiency was dependent on probe concentration, as expected (Fig. 4). The resolution of the labelling pattern is significantly reduced at probe concentrations lower than 250 μM, although individual bands with strong fluorescent labelling, such as the low-abundance protein at 50 kDa, can still be detected at probe concentrations as low as 50 μM (Fig. 4).
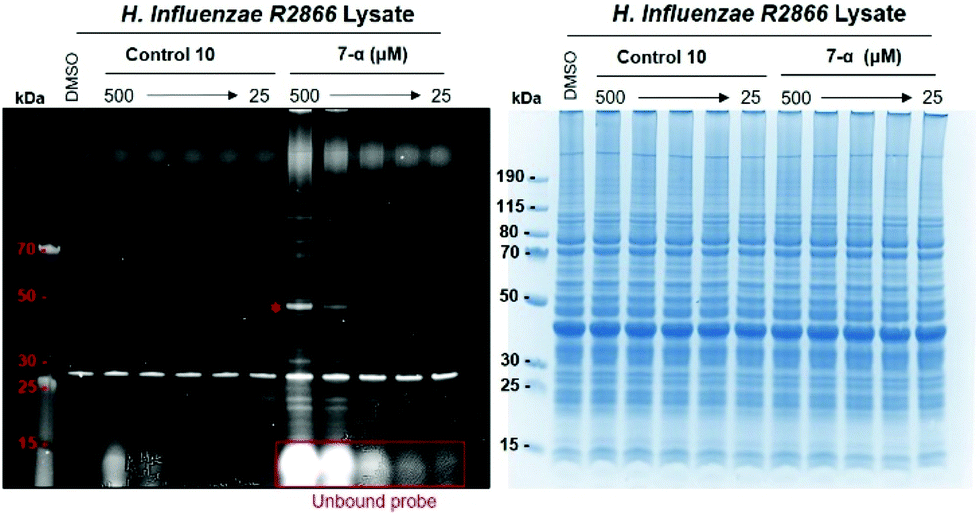 | ||
| Fig. 4 Effect of probe concentration on the labelling of H. influenzae R2866 lysates. General conditions as in Fig. 2. Following cell lysis and centrifugation, the supernatant was divided into fractions (9 μL) and incubated with stock solutions (1 μL, 5–0.25 mM stock in DMSO) of 7-α or 10 (control). Protein bands were detected by fluorescence scanning (left) and Coomassie staining (right). The asterisk denotes the low-abundance protein at 50 kDa labelled by all four probes (see text). | ||
Protein labelling in intact H. influenzae R2866 cells
Following the successful profiling of H. influenzae R2866 lysates, we next tested the suitability of probes 7-α, 7-β, 8-α and 8-β for the labelling of proteins in intact cells. First, we carried out bacterial growth assays to ensure that the probes did not compromise cell viability. No effect on bacterial growth was observed, suggesting that the probes were suited for profiling experiments with intact cells (ESI Fig. S4†). Next, we assessed the capacity of the probes to penetrate the bacterial membrane. Cells were grown as described and harvested by centrifugation. Pellets equivalent to 4 mL of cell cultures were incubated for 2 hours with 7-α, 7-β, 8-α, 8-β, 10 (fluorophore control, Scheme 1), or DMSO (control). After incubation, unbound probe was removed by consecutive washes (5% DMSO in PBS) and centrifugation.After four washes, no fluorescence was detected in the supernatant (Fig. 5A). Cell pellets that had been incubated with probes or fluorophore control remained fluorescent, while cells incubated with DMSO control showed no fluorescence (Fig. 5B). Cell pellets were collected, lysed, and centrifuged. The fluorescence of samples that had been incubated with probes or fluorophore control persisted in the soluble fraction after cell lysis (Fig. 5C). These results provided a first indication that all four probes as well as the fluorophore control can penetrate into the periplasm and/or cytoplasm of H. influenzae R2866, and hence that the carbohydrate scaffold in 7 and 8 does not impede penetration of the bacterial membrane.
 | ||
| Fig. 5 Protein labelling in intact H. influenzae R2866 cells. Cell pellets were incubated for 2 hours with 7-α, 7-β, 8-α, 8-β or 10 (fluorophore control) at 1 mM, or DMSO (control). (A) Supernatant after repeated washes (5% DMSO in PBS) of pellets that had been incubated with probe (results shown for 7-α) or DMSO (control); (B) cell pellets that had been incubated with probe, DMSO (control) or 10 (fluorophore control) after four washes (5% DMSO in PBS); (C) soluble fraction of the pellets from (B) after lysis and centrifugation. Fluorescence emission at 365 nm. | ||
To further confirm these results, we next investigated the labelling of intracellular proteins by our probes. Cell lysates were separated on a 4–12% SDS-page gel and analysed by fluorescence emission and Coomassie staining (Fig. 6). As for the application of 7 and 8 with cell lysates, a defined labelling pattern of proteins over a wide MW range (∼13–100 kDa) was observed for all four probes under these conditions (Fig. 6). Labelling efficiency was again concentration-dependent (ESI, Fig. S5†). While the only strong fluorescent band in the control lanes for DMSO and 10 resulted from the unknown autofluorescent protein at ∼28 kDa, a number of additional bands were observed for control 9. Most likely, these are due to protein labelling by the reactive azide group in 9 (Scheme 1C).
 | ||
| Fig. 6 Protein labelling with 7-α, 7-β, 8-α and 8-β in intact H. influenzae R2866 cells. General conditions: Cells were grown to OD590 0.9. Cell pellets were suspended in PBS (900 μL) and incubated with stock solutions (100 μL, 10 mM in DMSO) of probes 7-α, 7-β, 8-α, 8-β or fluorophore controls 9 or 10, or with DMSO (control), for two hours at 30 °C. Cells were washed (4×, 5% DMSO in PBS), lysed with BugBuster (100 μL, ×1 in PBS), and shaken for 30 min at rt, and centrifuged. Loading buffer (2.5 μL) was added to the clear supernatant (10 μL) and loaded onto a 4–12% SDS-page gel. Gels were run with MES running buffer at 150 V for 70 min. Protein bands were detected by fluorescence scanning (left) and Coomassie staining (right). | ||
While the labelling pattern of intact cells was very similar for all four probes, there were notable differences between this profile (Fig. 6), and the profile obtained from labelling of cell lysates (Fig. 3). Most significantly, the strongly fluorescent band at ∼50 kDa that had been observed with the cell lysate labelling protocol was absent from the intact cell experiment. To further investigate these intriguing differences, we decided to compare both labelling protocols side by side in a single experiment.
Direct comparison of H. influenzae R2866 profiling in intact cells and cell lysates
A bacterial culture was grown as described, half of which was used for protein labelling through cells (1 mM of 7-α/β and 8-α/β, 2 hours) while the other half was lysed and subsequently incubated with the glucosamine-based probe (0.5 mM, 1 hour). The lysate samples from both experiments were loaded on a single 4–12% SDS-page gel to enable the direct comparison of the labelling profiles (Fig. 7). | ||
| Fig. 7 Direct comparison of protein labelling with 7-α, 7-β, 8-α and 8-β in intact cells and cell lysates from H. influenzae R2866. See Fig. 3 and 5 for general conditions. The asterisk denotes the low-abundance protein at 50 kDa which is detected, by all four probes, in cell lysates only. | ||
The labelling profiles obtained with the two different protocols showed a number of differences, confirming our observations in the previous, separate experiments. Several targets were labelled exclusively with one protocol, but not the other (Fig. 7). The most notable difference was the presence of a strongly fluorescent band at ∼50 kDa, which is detected only by labelling of cell lysates, but not of intact cells.
A possible explanation for these differences is, that the probes penetrate the outer bacterial membrane and reach the periplasm of H. influenzae R2866, but cannot cross the inner membrane to reach the cytoplasmic space. The probes would therefore be able to only capture periplasmic proteins in the labelling experiment with intact cells, but both periplasmic and cytoplasmic proteins in the protocol with cell lysates. We therefore hypothesised that the proteins labelled exclusively in the bacterial cell lysate experiment may be located in the bacterial cytoplasm.
Analysis of selected target proteins by LC-MS/MS
To further assess this hypothesis, we sought to establish the identity of selected target proteins. Selected bands were excised from the requisite gel and analysed by LC-MS/MS following in-gel reduction, alkylation and digestion with trypsin. The resulting peptide fragments were matched against the current H. influenzae taxonomy in the Uniprot database (ESI†).This analysis suggests two probable assignments for the band at ∼50 kDa that is detected selectively by labelling of cell lysates: FtsZ (45 kDa), a cytoplasmic GTPase that is central for the bacterial cell division machinery;30 and Elongation Factor Tu (43 kDa), another cytoplasmic GTPase involved in polypeptide chain elongation in protein biosynthesis.31 Interestingly, both proteins have been identified as attractive targets for small-molecule antibacterial drug discovery.30,31 These results are in keeping with our hypothesis that the protein labelled exclusively with the cell lysate protocol is located in the cytoplasm.
High-probability assignments for other target proteins (ESI, Fig. S6†) include many nucleotide-binding proteins, including the chaperone protein DnaK (68 kDa), periplasmic NAD nucleotidase (66 kDa), and DNA-directed RNA polymerase subunit beta, a nucleotidyl-transferase (157 kD). DnaK is an outer membrane protein known to mediate sulfatide recognition32 and a biofilm protein in non-typeable Haemophilus Influenzae.33 These results suggest that probes 7 and 8 may act as nucleotide mimics, possibly because of the combination of a simple sugar with the dansyl fluorophore, which may be reminiscent of a nucleobase.
Conclusion
We have synthesised both anomers of a D-galactosamine-based fluorescent probe in six steps from D-galactosamine with an overall yield of 22%. These probes, alongside their D-glucosamine congeners, were used successfully for the labelling of multiple target proteins in both cell lysates and intact cells of Haemophilus influenzae R2866. In both the D-galactosamine and D-glucosamine series, the labelling profiles and intensities for the fully acetylated probes were practically identical for both anomers. This is consistent with observations for the labelling of the recombinant galactosyltransferase LgtC, where labelling was comparable between both anomers of the fully acetylated D-glucosamine-based probes 8-α and 8-β, but significantly different between both anomers of the corresponding free sugars 11-α and 11-β. This suggests that the fully acetylated probes are also the active species in the labelling experiments with Haemophilus lysates and intact cells, and that the acetate groups remain intact under these conditions.While the labelling profile observed with all four probes was practically identical, notable differences were observed between profiles from the lysate and intact cell experiments. The exclusive labelling of a cytoplasmic protein in lysate samples, but not intact cells, suggests that these probes can indeed penetrate the outer, if probably not the inner, bacterial membrane.
The cell envelope of Gram-negative bacteria represents a formidable permeability barrier, and considerable efforts are being made to identify small molecules that can cross this barrier.34–36 The ability of these probes to penetrate the bacterial membrane, as reported in this study, is therefore of considerable interest. Initial target identification experiments suggest that these probes preferentially label nucleotide-binding proteins. This is consistent with the labelling of LgtC, where labelling may occur at the sugar-nucleotide donor binding site. Interestingly, potential target proteins include several known antibacterial drug targets. These probes therefore represent exciting new tool compounds to study the role of such nucleotide-binding proteins for bacterial pathogenicity of Haemophilus and, potentially, other Gram-negative pathogens.
Experimental
Chemistry
Compound 2. Galactosamine hydrochloride 1 (9.25 mmol) was dissolved in aq. NaOH (1 M, 9.3 mL) at 0 °C. p-Anisaldehyde (1 equiv.) was added dropwise, and the mixture was shaken vigorously. Upon formation of a white solid, the reaction was placed in the freezer (−20 °C) for 1 h to complete precipitation. The solid was collected by filtration, washed dropwise with ice-cold H2O (10 mL) and ice-cold EtOH
:Et2O (1:1, 10 mL), and dried under vacuum to give 2 as a white powder in 56% yield. 1H NMR (400 MHz, DMSO-d6) δ 3.09 (dd, 3J 9.5 Hz, 3J 7.7 Hz, 1H, H-2 sugar), 3.46 (t, 3J 6.2 Hz, 1H, H-5 sugar), 3.50–3.62 (m, 3H, H-3, H-6 sugar), 3.67 (t, 3J 3.5 Hz, 1H, H-4 sugar), 3.80 (s, 3H, H-methoxy), 4.39 (d, 3J 4.4 Hz, 1H, H-hydroxyl), 4.50 (d, 3J 7.0 Hz, 1H, H-hydroxyl), 4.58–4.64 (m, 2H, H-1 anomeric, H-hydroxyl), 6.42 (d, 3J 6.3 Hz, 1H, H-hydroxyl), 6.98 (d, 3J 8.8 Hz, 2H), 7.67 (d, 3J 8.8 Hz, 2H), 8.13 (s, 1H, H-imine). 13C NMR (100 MHz, DMSO-d6) δ 55.3 (C-methoxy), 60.7 (C-6 sugar), 67.2 (C-4 sugar), 71.6 (C-3 sugar), 74.5 (C-2 sugar), 75.1 (C-5 sugar), 96.1 (C-1 sugar), 113.9 (×2), 129.2, 129.5 (×2), 161.0, 161.2 (C-imine).
Compound 3. 2 (3.20 mmol) was dissolved in pyridine (10 mL) at 0 °C. Acetic anhydride (8 equiv.) was added dropwise, the reaction was warmed to room temperature and stirred overnight (16 h). The reaction mixture was poured into ice (50 mL) under stirring. The precipitate was collected by filtration, washed with ice-cold H2O (200 mL) and ice-cold EtOH (2 mL), and dried under vacuum, to give 3 as a white powder in 99% yield. 1H NMR (400 MHz, DMSO-d6) δ 1.82, 1.97, 2.00, 2.12 (4s, 12H, CH3CO), 3.52 (dd, 3JH2–H3 10.3 Hz, 3JH2–H1 8.2 Hz, 1H, H-2 sugar), 3.79 (s, 3H, H-methoxy), 4.04–4.09 (m, 2H, H-6 sugar), 4.46 (t, 3JH5–H6 6.5 Hz, 1H, H-5 sugar), 5.27 (d, 3JH4–H3 3.3 Hz, 1H, H-4 sugar), 5.35 (dd, 3JH3–H2 10.4 Hz, 3JH3–H4 3.4 Hz, 1H, H-3 sugar), 5.98 (d, 3JH1–H2 8.2 Hz, 1H, H-1 anomeric), 6.99 (d, 3J 8.8 Hz, 2H), 7.67 (d, 3J 8.8 Hz, 2H), 8.31 (s, 1H, H-imine). 13C NMR (100 MHz, DMSO-d6) δ 20.3, 20.4, 20.5, 20.5, (4C, CH3CO), 55.4 (C-methoxy), 61.4 (C-6 sugar), 66.0 (C-4 sugar), 68.4 (C-2 sugar), 70.9 (C-5 sugar), 70.9 (C-3 sugar), 92.8 (C-1 sugar), 114.2, 128.3, 129.9, 161.8, 164.7 (C-imine), 168.6, 169.2, 169.9, 170.0 (4C, CH3CO).
Compound 4. 3 (2.70 mmol) was dissolved in acetone (11 mL) and the solution was heated to reflux. Aq. HCl (4 M, 1.2 equiv.) was added dropwise under stirring. After about 5 min, a white precipitate formed. Heating was continued for a further 2 min. The precipitate was collected by filtration and washed with ice-cold acetone (50 mL), to give 4 as a white powder in 88% yield. 1H NMR (400 MHz, DMSO-d6) δ 1.99, 2.00, 2.12, 2.16 (4s, 12H, CH3CO), 3.39 (dd, 3JH2–H1 8.2 Hz, 3JH2–H3 6.2 Hz, 1H, H-2 sugar), 4.98–4.09 (m, 2H, H-6 sugar), 4.29 (t, 3J 6.3 Hz, 1H, H-5 sugar), 5.24–5.32 (m, 2H, H-3, H-4 sugar), 5.89 (d, 3JH1–H2 8.7 Hz, 1H, H-1 anomeric), 8.71 (s, 3H, NH). 13C NMR (100 MHz, DMSO-d6) δ 20.3, 20.5, 20.7, 20.8 (4C, CH3CO), 49.4 (C-2 sugar), 61.2 (C-6 sugar), 68.8, 65.8 (C-3, C-4 sugar), 71.1 (C-5 sugar), 90.3 (C-1 sugar), 168.6, 169.3, 169.9, 169.9 (4C, CH3CO).
Compound 5. To a suspension of 4 (0.4 mmol) in DCM (2 mL), triethylamine (2 equiv.) was added. The reaction mixture was stirred at room temperature until all solid was dissolved (approx. 2 min). The solution was cooled to 0 °C, and chloroacetyl chloride (3 equiv.) was added dropwise. The reaction mixture was stirred at 0 °C for 10 min, and at room temperature until TLC (8
:1 EtOAc:hexane) showed full conversion (1 h). The reaction was quenched by addition of saturated aq. NaHCO3 solution (5 mL). DCM (10 mL) was added and the two layers were separated. The aqueous phase was extracted with DCM (3 × 10 mL). The organic layers were combined and dried over MgSO4. After filtration the mixture was concentrated under vacuum. The residue was purified by normal phase column chromatography (EtOAc/hexane) to afford the title compound as an off-white crystalline product in 85% yield. 1H NMR (400 MHz, CDCl3) δ 2.02, 2.05, 2.13, 2.17 (4s, 12H, CH3CO), 4.00 (s, 2H, H-7), 4.06 (td, 3J 6.5 Hz, 3J 1.0 Hz, 1H, H-5), 4.09–4.20 (m, 2H, H-6), 4.38 (dt, 3J2–3 11.3 Hz, 3J2-NH 9.0 Hz, 1H, H-2), 5.24 (dd, 3J3–2 11.3 Hz, 3J3.4 3.3 Hz, 1H, H-3), 5.38–5.42 (m, 1H, H-4), 5.82 (d, 3J1–2 8.8 Hz, 1H, H-1), 6.48 (d, 3J 9.2 Hz, 1H, NH). 13C NMR (100 MHz, CDCl3): δ 20.7, 20.7, 20.8, 21.0 (4C, CH3CO), 42.5 (C-7), 50.5 (C-2), 61.5 (C-6), 66.6 (C-4), 69.9 (C-3), 71.9 (C-5), 92.5 (C-1), 166.9 (C-8), 169.5, 170.2, 170.6, 170.6 (CH3CO). LC-MS (m/z) 446.3 [M + Na+] (calculated 446.2).
Compounds 6-α and 6-β. 5 (1.06 mmol) was dissolved in DCM (3 mL), and the solution was cooled to 0 °C. Borontrifluoride diethyletherate (4 equiv.) and propynol ethoxylate (3 equiv.) were added dropwise, and the reaction was stirred at 40 °C for 8 h. The reaction mixture was diluted with DCM (7 mL) and washed with saturated aq. K2CO3 (2 × 10 mL) and brine (2 × 10 mL). The organic layer was separated, dried over MgSO4, and concentrated under vacuum. The crude residue was purified by normal phase column chromatography (0–20% acetone in toluene) to give two products. Each product was further purified by reverse phase column chromatography (10–30% EtOAc in hexane) to give the title compounds as pale yellow oils in 34% (6-α) and 36% (6-β) yield. Cmpd 6-α: 1H NMR (400 MHz, CDCl3) δ 1.99, 2.05, 2.16 (3s, 9H, CH3CO), 2.47 (t, 3J 2.4 Hz, 1H, H-13), 3.67–3.74 (m, 3H, H-9, H-10), 3.83–3.90 (m, 1H, H-9), 4.01 (d, 3J 3.2 Hz, 2H, H-7), 4.11 (m, 2H, H-6), 4.19 (d, 3J 2.4 Hz, 1H, H-11), 4.28 (t, 3JH5–H6 6.6 Hz, 1H, H-5), 4.57 (ddd, 3JH2–H3 11.2 Hz, 3JH2–NH 9.7 Hz, 3JH2–H1 3.7 Hz, 1H, H-2), 4.95 (d, 3JH1–H2 3.7 Hz, 1H, H-1), 5.24 (dd, 3JH3–H2 11.3 Hz, 3JH3–H4 3.3 Hz, 1H, H-3), 5.41 (dd, 3JH4–H3 3.2 Hz, 3JH4–H5 1.2 Hz, 1H, H-4), 6.71 (d, 3JNH–H2 9.7 Hz, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 20.9 (3 × CH3CO), 42.6 (C-7), 48.3 (C-2), 58.5 (C-11), 62.0 (C-6), 67.0 (C-5), 67.5 (C-4), 67.7 (C-9), 68.6 (C-3), 68.6 (C-10), 75.1 (C-13), 79.5 (C-12), 97.9 (C-1), 166.4 (C-8), 170.4, 170.6, 170.8 (3C, CH3CO). LC-MS (m/z) 486.3 [M + Na+] (calculated 486.1). Cmpd6-β: 1H NMR (400 MHz, CDCl3) δ 2.01, 2.05, 2.15 (3s, 9H, CH3CO), 2.45 (t, 3J 2.3 Hz, 1H, H-13), 3.65–3.73 (m, 2H, H-10), 3.76–3.84 (m, 1H, H-9), 3.93 (t, 3J 6.7 Hz, 1H, H-5), 3.97–4.08 (m, 4H, H-2, H-9, H-7), 4.12–4.23 (m, 4H, H-6, H-11), 4.84 (d, 3JH1–H2 8.4 Hz, 1H, H-1), 5.30 (dd, 3JH3–H2 11.3 Hz, 3JH3–H4 3.4 Hz, 1H, H-3), 5.38 (d, 3JH4–H3 3.4 Hz, 1H, H-4), 6.49 (d, 3JNH–H2 8.6 Hz, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 20.8, 20.8, 20.8 (3C, CH3CO), 42.7 (C-7), 52.1 (C-2), 58.6 (C-11), 61.6 (C-6), 66.8 (C-4), 68.9 (C-9), 69.3 (C-10), 69.8 (C-3), 71.0 (C-5), 74.9 (C-13), 79.6 (C-12), 101.2 (C-1), 166.6 (C-8), 170.3, 170.5, 170.6 (3C, CH3CO). LC-MS (m/z) 486.3 [M + Na+] (calculated 486.1).
Compounds 7-α and 7-β. Under stirring, 6-α or 6-β (0.16 mmol) and 10 (1 equiv.) were dissolved in THF (1 mL). A solution of sodium ascorbate (2 equiv.) and copper sulfate pentahydrate (1.2 equiv.) in H2O (110 μL) was added, followed by DIPEA (5 equiv.). The reaction was covered with aluminium foil and stirred at room temperature for 3 h. The reaction was extracted with EtOAc (15 mL). The organic extract was washed with H2O (5 mL) and brine (5 mL), and concentrated under vacuum. The crude residue was purified by normal phase column chromatography twice (first solvent system: 50–100% EtOAc in hexane. Second solvent system: 0–70% acetone in toluene) to give the title compounds as powders in 74% (7-α) and 82% (7-β) yield. Cmpd7-α: 1H NMR (400 MHz, CDCl3) δ 1.98, 2.00, 2.11 (3s, 9H, CH3CO), 2.89 (s, 6H), 3.37–3.43 (m, 2H), 3.70–3.76 (m, 3H), 3.80–3.88 (m, 1H), 3.98 (s, 2H, H-7), 4.03–4.15 (m, 2H, H-6 sugar), 4.29 (td, 3JH5–H6 6.4 Hz, 3JH5–H4 1.4 Hz 1H, H-5 sugar), 4.39–4.52 (m, 2H), 4.56–4.70 (m, 3H, H-2 sugar, H-aliphatic), 5.01 (d, 3JH1–H2 3.6 Hz, 1H, H-1 anomeric), 5.20 (dd, 3JH3–H2 11.3 Hz, 3JH3–H4 3.3 Hz, 1H, H-3 sugar), 5.43 (dd, 3JH4–H3 3.3 Hz, 3JH4–H5 1.4 Hz, 1H, H-4 sugar), 6.14 (t, 3JNH–H15 5.6 Hz, 1H, NH-sulfonamide), 7.14 (d, 3JNH–H2 9.6 Hz, 1H, NH-amide), 7.18 (d, 3J 7.4 Hz, 1H), 7.50–7.57 (m, 2H), 7.61 (s, 1H, H-triazole), 8.19–8.26 (m, 2H), 8.56 (d, 3J 8.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 20.8, 20.9, 21 (3C, CH3CO), 42.7 (C-7), 42.8, 45.6 (×2), 48.3 (C-2 sugar), 50.4, 62.0 (C-6 sugar), 64.4, 66.9 (C-5 sugar), 67.6, 67.6 (C-4 sugar), 68.7 (C-3 sugar), 69.3, 98.0 (C-1 sugar), 115.5, 118.9, 123.3, 124.2 (C-triazole), 128.6, 129.6, 129.6, 130.1, 130.8, 134.7, 144.8 (C-triazole), 152.2, 166.9 (C-amide), 170.6, 170.7, 171.3 (3C, CH3CO). LC-MS (m/z) 784.3 [M + H+] (calculated 784.2). Cmpd7-β: 1H NMR (400 MHz, CDCl3) δ 1.97, 1.99, 2.14 (3s, 9H, CH3CO), 2.89 (s, 6H), 3.33–3.50 (m, 2H), 3.67–3.81 (m, 2H), 3.84–3.99 (m, 3H, H-5 sugar, H-aliphatic), 4.02 (d, 3J 6.9 Hz, 2H, H-7), 4.05–4.20 (m, 3H, H-2, H-6 sugar), 4.40–4.49 (m, 1H), 4.50–4.59 (m, 1H), 4.63 (s, 2H), 4.89 (d, 3JH1–H2 8.5 Hz, 1H, H-1 anomeric), 5.21 (dd, 3JH3–H2 11.2 Hz, 3JH3–H4 3.4 Hz, 1H, H-3 sugar), 5.36 (d, 3J 2.9 Hz, 1H, H-4 sugar), 6.07 (t, 3JNH–H15 5.8 Hz, 1H, NH-sulfonamide), 7.09 (d, 3JNH–H2 8.8 Hz, 1H, NH-amide), 7.19 (d, 3J 7.6 Hz, 1H), 7.61–7.50 (m, 2H), 7.67 (s, 1H, H-triazole), 8.19 (d, 3J 8.7 Hz, 1H), 8.24 (dd, 3J 7.3 Hz, 1.1 Hz, 1H), 8.56 (d, 3J 8.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 20.8, 20.8, 20.9 (3C, CH3CO), 42.8 (C-7), 45.6 (×2), 50.4, 51.8 (C-2 sugar), 61.6 (C-6 sugar), 64.5, 67.0 (C-4 sugar), 68.6, 70.4 (C-3 sugar), 70.7, 70.8 (C-5 sugar), 101.3 (C-1 sugar), 115.6, 118.8, 123.6, 124.6 (C-triazole), 128.8, 129.6, 129.7, 130.2, 131.0, 134.3, 144.8 (C-triazole), 152.2, 167.3 (C-amide), 170.5, 170.6, 170.8 (3C, CH3CO). LC-MS (m/z) 784.3 [M + H+] (calculated 784.2).
Compound 9. A solution of dansyl chloride 11 (5.00 mmol), 2-bromoethylamine hydrobromide (1 equiv.) and triethylamine (2 equiv.) in DCM (25 mL) was stirred at room temperature for 4 h. The solvent was removed under vacuum. The resulting residue was dissolved in MeCN (50 mL). NaN3 (2.5 equiv.) was added, and the reaction was stirred at reflux overnight. The solvent was removed under vacuum, and the crude product was purified by flash column chromatography (solvent system: 0–50% EtOAc in Hexane) to give 9 as an oil in 87% yield. 1H NMR (400 MHz, CDCl3) δ 2.89 (s, 6H), 3.03–3.08 (m, 2H), 3.29–3.33 (m, 2H), 4.99 (t, 3J 5.9 Hz, 1H, NH), 7.21 (d, 3J 7.6 Hz, 1H), 7.53 (dd, 3J 8.5 Hz, 3J 7.4 Hz, 1H), 7.60 (dd, 3J 8.6 Hz, 3J 7.7 Hz, 1H), 8.24–8.26 (m, 2H), 8.26–8.28 (m, 2H), 8.57 (d, 3J 8.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 42.5, 45.6, 45.6, 51.1, 115.5, 118.6, 123.3, 128.8, 129.6, 129.8, 130.1, 131.0, 134.6, 152.3. HR-MS (m/z) 320.1182 [M + H]+ (calculated 320.1181).
Compound 10. The title compound was obtained as previously described28 from 9 and 1-pentyne, under the cycloaddition reaction conditions described for 7, in 87% yield. 1H NMR (400 MHz, CDCl3) 0.95 (t, 3J 7.4 Hz, 3H), 1.58–1.68 (m, 2H), 2.58–2.63 (m, 2H), 2.86 (s, 6H), 3.44 (dd, 3J 11.2 Hz, 3J 6.1 Hz, 2H), 4.30–4.36 (m, 2H), 4.41–4.48 (m, 1H, NH), 7.11 (s, 1H, H-triazole), 7.19 (d, 3J 7.1 Hz, 1H), 7.51–7.57 (m, 2H), 8.18 (dt, 3J 8.7 Hz, 3J 1 Hz, 1H) 8.25 (dd, 3J 7.3 Hz, 3J 1.2 Hz, 1H), 8.54–8.58 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 14.0, 22.8, 27.7, 42.9, 45.6 (×2), 50.0, 115.5, 118.6, 122.1 (C-triazole), 123.3, 128.9, 129.6, 129.7, 130.1, 131.0, 134.5, 148.3, 152.2.
Biology
:33 to an OD590 of 0.1 and incubated aerobically at 37 °C and 180 rpm. For growth curves, OD590 was recorded at various time points in triplicate cultures.
Materials. NuPAGE™ 4–12% bis–tris protein gels, 1.0 mm, 12-well. PageRuler™ prestained protein ladder 15–190 kDa. NuPAGE™ MES SDS running buffer (20×).
Protein labelling in intact cells. Cell cultures of H. influenzae R2866 were grown as described. Upon reaching mid log phase (OD590 0.9), 4 mL cell cultures were centrifuged at 13
000g for 1 minute. The supernatant was discarded. Cell pellets were resuspended in PBS (900 μL), and stock solutions of probe or fluorophore control (100 μL, 10× desired concentration in DMSO), or DMSO (100 μL, control) were added. Samples were incubated for 2 hours at 30 °C with regular mixing, and centrifuged (2 min, 13000g). Cell pellets were separated from the supernatant, washed with a solution of 5% DMSO in PBS, and centrifuged. This procedure was repeated four times to remove unbound fluorophore. After each cycle, the fluorescence of pellets and washes was examined at 365 nm (Fig. 4). BugBuster protein extraction reagent (Millipore, 1× in PBB, 100 μL) was added to the cell pellets. Samples were incubated for 30 min at rt with shaking, and centrifuged (10 min, 13000g). Supernatant and cell debris were separated, and the fluorescence of both was examined at 365 nm. Supernatant (10 μL) from each sample was incubated with loading buffer (no dye, 2.5 μL) for 10 min at 50 °C. Samples (10 μL) were loaded on precast SDS-page gels (NuPAGE™ 4 to 12%, bis–tris, 1.0 mm, Novex). Gels were run in 1× MES running buffer at 150 V for 70 min and analysed by fluorescence emission and Coomassie staining.
Protein labelling in cell lysates. Cell cultures of H. influenzae R2866 were grown as described above. Cell pellets were isolated by centrifugation and lysed with BugBuster protein extraction reagent as described. Following centrifugation, the supernatant from each sample was kept on ice, and the cell debris was discarded. The supernatants (9 μL) were incubated with stock solutions of probe or fluorophore control (1 μL, 10× desired concentration in DMSO), or DMSO (1 μL, control) for 1 hour at 30 °C with regular mixing. Samples (10 μL) were incubated with loading buffer (2.5 μL) for 10 min at 50 °C and analysed by SDS-page as described.
Protein mass spectrometry
Chromatographic separation was performed using a U3000 UHPLC NanoLC system (ThermoFisherScientific, UK). Peptides were resolved by reversed phase chromatography on a 75 μm C18 Pepmap column (50 cm length) using a three-step linear gradient of 80% acetonitrile in 0.1% formic acid. The gradient was delivered to elute the peptides at a flow rate of 250 nl min−1 over 60 min starting at 5% B (0–5 minutes) and increasing solvent to 40% B (5–40 minutes) prior to a wash step at 99% B (40–45 minutes) followed by an equilibration step at 5% B (45–60 minutes). The eluate was ionised by electrospray ionisation using an Orbitrap Fusion Lumos (ThermoFisherScientific, UK) operating under Xcalibur v4.1.5. The instrument was first programmed to acquire using an Orbitrap-Ion Trap method by defining a 3 s cycle time between a full MS scan and MS/MS fragmentation. Orbitrap spectra (FTMS1) were collected at a resolution of 120000 over a scan range of m/z 375–1500 with an automatic gain control (AGC) setting of 4.0 × 105 with a maximum injection time of 35 ms. Monoisotopic precursor ions were filtered using charge state (+2 to +7) with an intensity threshold set between 5.0 × 103 to 1.0 × 1020 and a dynamic exclusion window of 35 s ± 10 ppm. MS2 precursor ions were isolated in the quadrupole set to a mass width filter of 1.6 m/z. Ion trap fragmentation spectra (ITMS2) were collected with an AGC target setting of 1.0 × 104 with a maximum injection time of 35 ms with CID collision energy set at 35%. This method takes advantage of multiple analyzers in the Orbitrap Fusion Lumos and drives the system to use all available parallelizable time, resulting in decreasing dependence on method parameters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank King's College London for a studentship (to CM), the EPSRC National Mass Spectrometry Facility, Swansea, for the recording of mass spectra, and Dr Jon Cuccui (LSHTM) for helpful discussions. The LgtC-expressing clone NMC-41 was a gift from Prof Warren Wakarchuk (University of Alberta). Funding from the Biotechnology & Biological Sciences Research Council (grant BB/N001591/1, to BW) is gratefully acknowledged.Notes and references
- J. Krysiak and S. A. Sieber, in Methods in Molecular Biology, ed. H. Overkleeft and B. Florea, Humana Press, New York, 2017, vol. 1491, pp. 57–74 Search PubMed.
- S. Sharifzadeh, J. D. Shirley and E. E. Carlson, in Current Topics in Microbiology and Immunology, ed. B. F. Cravatt, K.-L. Hsu and E. Weerapana, Springer Nature, Switzerland, 2019, vol. 420, pp. 23–48 Search PubMed.
- N. C. Sadler and A. T. Wright, Curr. Opin. Chem. Biol., 2015, 24, 139–144 CrossRef CAS.
- S. Ray and A. S. Murkin, Biochemistry, 2019, 58, 5234–5244 CrossRef CAS.
- C. C. Ward, J. I. Kleinman and D. K. Nomura, ACS Chem. Biol., 2017, 12, 1478–1483 CrossRef CAS.
- R. Dandela, D. Mantin, B. F. Cravatt, J. Rayo and M. M. Meijler, Chem. Sci., 2018, 9, 2290–2294 RSC.
- J. Krysiak, M. Stahl, J. Vomacka, C. Fetzer, M. Lakemeyer, A. Fux and S. A. Sieber, J. Proteome Res., 2017, 16, 1180–1192 CrossRef CAS.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- X. Chen, Y. Wang, N. Ma, J. Tian, Y. Shao, B. Zhu, Y. K. Wong, Z. Liang, C. Zou and J. Wang, Signal Transduction Targeted Ther., 2020, 5, 72 CrossRef.
- C. L. Beisel and T. Afroz, J. Bacteriol., 2016, 198, 374–376 CrossRef CAS.
- H. L. P. Tytgat and S. Lebeer, Microbiol. Mol. Biol. Rev., 2014, 78, 372–417 CrossRef.
- J. Poole, C. J. Day, M. Von Itzstein, J. C. Paton and M. P. Jennings, Nat. Rev. Microbiol., 2018, 16, 440–452 CrossRef CAS.
- M. L. Ferrando, P. van Baarlen, G. Orrù, R. Piga, R. S. Bongers, M. Wels, A. De Greeff, H. E. Smith and J. M. Wells, PLoS One, 2014, 9, e89334 CrossRef.
- S. A. Shelburne III, D. Keith, N. Horstmann, P. Sumby, M. T. Davenport, E. A. Graviss, R. G. Brennan and J. M. Musser, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1698–1703 CrossRef.
- L. Hobley, C. Harkins, C. E. MacPhee and N. R. Stanley-Wall, FEMS Microbiol. Rev., 2015, 39, 649–669 CrossRef CAS.
- K. A. Stubbs, A. Scaffidi, A. W. Debowski, B. L. Mark, R. V. Stick and D. J. Vocadlo, J. Am. Chem. Soc., 2008, 130, 327–335 CrossRef CAS.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS.
- J. Jiang, W. W. Kallemeijn, D. W. Wright, A. M. C. H. Van Den Nieuwendijk, V. C. Rohde, E. C. Folch, H. Van Den Elst, B. I. Florea, S. Scheij, W. E. Donker-Koopman, M. Verhoek, N. Li, M. Schürmann, D. Mink, R. G. Boot, J. D. C. Codée, G. A. Van Der Marel, G. J. Davies, J. M. F. G. Aerts and H. S. Overkleeft, Chem. Sci., 2015, 6, 2782–2789 RSC.
- C. Metier, J. Peng, Y. Xu, H. Wootton, V. Riesi, S. Lynham, S. Zadi, C. Turner, M. E. Wand, J. M. Sutton and G. K. Wagner, Profiling protein expression in Klebsiella pneumoniae with a carbohydrate-based probe, Manuscript submitted Search PubMed.
- D. C. Turk, J. Med. Microbiol., 1984, 18, 1–16 CrossRef CAS.
- K. L. Fox, J. M. Atack, Y. N. Srikhanta, A. Eckert, L. A. Novotny, L. O. Bakaletz and M. P. Jennings, PLoS One, 2014, 9, e90505 CrossRef.
- J. M. Atack, C. J. Day, J. Poole, K. L. Brockman, L. O. Bakaletz, S. J. Barenkamp and M. P. Jennings, Biochem. Biophys. Res. Commun., 2018, 503, 1103–1107 CrossRef CAS.
- S. Grass, C. F. Lichti, R. R. Townsend, J. Gross and J. W. St. Geme III, PLoS Pathog., 2010, 6, e1000919 CrossRef.
- A. L. Erwin, S. Allen, D. K. Ho, P. J. Bonthius, J. Jarisch, K. L. Nelson, D. L. Tsao, W. C. T. Unrath, M. E. Watson, B. W. Gibson, M. A. Apicella and A. L. Smith, Infect. Immun., 2006, 74, 6226–6235 CrossRef CAS.
- V. Nizet, K. F. Colina, J. R. Almquist, C. E. Rubens and A. L. Smith, J. Infect. Dis., 1996, 173, 180–186 CrossRef CAS.
- R. Griffin, C. D. Bayliss, M. A. Herbert, A. D. Cox, K. Makepeace, J. C. Richards, D. W. Hood and E. R. Moxon, Infect. Immun., 2005, 73, 7022–7026 CrossRef CAS.
- B. Wulffen, C. Rudolf, M. Pofahl and A. Heckel, Photochem. Photobiol. Sci., 2012, 11, 489–492 RSC.
- R. Yan, K. Sander, E. Galante, V. Rajkumar, A. Badar, M. Robson, E. El-emir, M. F. Lythgoe, R. B. Pedley and E. Årstad, J. Am. Chem. Soc., 2013, 135, 703–709 CrossRef CAS.
- K. Persson, H. D. Ly, M. Dieckelmann, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, Nat. Struct. Biol., 2001, 8, 166–175 CrossRef CAS.
- N. R. Stokes, N. Baker, J. M. Bennett, J. Berry, I. Collins, L. G. Czaplewski, A. Logan, R. Macdonald, L. MacLeod, H. Peasley, J. P. Mitchell, N. Nayal, A. Yadav, A. Srivastava and D. J. Haydon, Antimicrob. Agents Chemother., 2013, 57, 317–325 CrossRef CAS.
- M. M. K. Jayasekera, K. Onheiber, J. Keith, H. Venkatesan, A. Santillan, E. M. Stocking, L. Tang, J. Miller, L. Gomez, B. Rhead, T. Delcamp, S. Huang, R. Wolin, E. V. Bobkova and K. J. Shaw, Antimicrob. Agents Chemother., 2005, 49, 131–136 CrossRef CAS.
- E. Hartmann, C. A. Lingwood and J. Reidl, Infect. Immun., 2001, 69, 3438–3441 CrossRef CAS.
- T. K. Gallaher, S. Wu, P. Webster and R. Aguilera, BMC Microbiol., 2006, 6, 65 CrossRef.
- B. Spangler, D. Dovala, W. S. Sawyer, K. V. Thompson, D. A. Six, F. Reck and B. Y. Feng, ACS Infect. Dis., 2018, 4, 1355–1367 CrossRef CAS.
- W. Phetsang, R. Pelingon, M. S. Butler, S. Kc, M. E. Pitt, G. Kaeslin, M. A. Cooper and M. A. T. Blaskovich, ACS Infect. Dis., 2016, 2, 688–701 CrossRef CAS.
- Z. Z. Deris, J. D. Swarbrick, K. D. Roberts, M. A. K. Azad, J. Akter, A. S. Horne, R. L. Nation, K. L. Rogers, P. E. Thompson, T. Velkov and J. Li, Bioconjugate Chem., 2014, 25, 750–760 CrossRef CAS.
- T. Pesnot, R. Jørgensen, M. M. Palcic and G. K. Wagner, Nat. Chem. Biol., 2010, 6, 321–323 CrossRef CAS.
- W. W. Wakarchuk, A. Cunningham, D. C. Watson and N. M. Young, Protein Eng., 1998, 11(4), 295–302 CrossRef CAS.
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols for fluorophore controls 9 and 10; additional Fig. S1–S6; LC-MS/MS analysis of SDS-page gel bands; spectroscopic data for compounds 2–7, 9 and 10. See DOI: 10.1039/d0ob01971b |
| This journal is © The Royal Society of Chemistry 2021 |