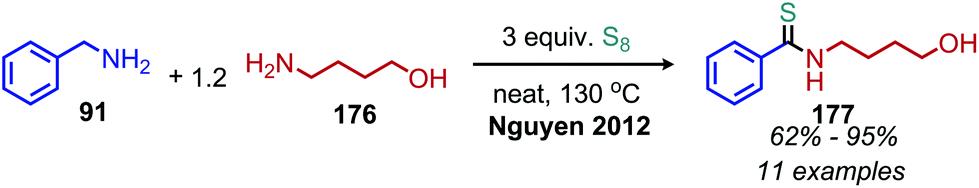DOI:
10.1039/D0OB01807D
(Review Article)
Org. Biomol. Chem., 2021,
19, 11-36
Reframing primary alkyl amines as aliphatic building blocks
Received
1st September 2020
, Accepted 7th October 2020
First published on 20th October 2020
Abstract
While primary aliphatic amines are ubiquitous in natural products, they are traditionally considered inert to substitution chemistry. This review highlights historical and recent advances in the field of aliphatic deamination chemistry which demonstrate these moieties can be harnessed as valuable C(sp3) synthons. Cross-coupling and photocatalyzed transformations proceeding through polar and radical mechanisms are compared with oxidative deamination and other transition metal catalyzed reactions.
 Kathleen J. Berger | Kate (Kathleen) Berger is from Medicine Hat in Alberta, Canada. She studied chemistry and chemical biology at the University of British Columbia, completing her undergraduate degree in 2017. At UBC, her honours thesis research focused on the synthesis and reactivity of cobalt dinitrogen complexes with Prof. Michael Fryzuk. Currently, Kate is a Ph.D. candidate at the University of Chicago under the direction of Prof. Mark Levin. Her research focuses on developing new synthetic methodologies for aliphatic deamination chemistry. |
 Mark D. Levin | Mark Levin was born and raised in Cleveland, OH and attended the University of Rochester as an undergraduate, conducting research under Prof. Alison J. Frontier. As a graduate student with Prof. F. Dean Toste at the University of California, Berkeley, Mark studied reductive elimination processes at Au(III), graduating with his Ph.D. in 2017. He conducted postdoctoral studies with Prof. Eric N. Jacobsen at Harvard University studying enantioselective fluorination, and since 2019 has been an assistant professor of chemistry at the University of Chicago, where his group develops new synthetic methods. |
1. Introduction
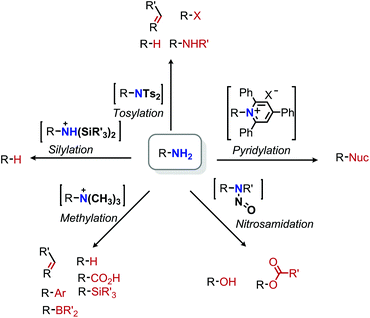
A persistent challenge in modern organic synthesis is the activation and transformation of bonds to C(sp3) carbons at a level of versatility matching their C(sp2) counterparts. In considering potential aliphatic reactants to meet this challenge, alkyl halides stand out as a prototypical choice because they can deftly serve as synthetic equivalents to alkyl cations, radicals, and anions by the appropriate choice of reaction conditions.1 However, aliphatic halides nonetheless lag aromatic halides in this regard for many reaction classes, and moreover are not often a native functionality in natural products or pharmaceuticals.2,3 Considering this deficit, advancing the chemistry of typically unreactive aliphatic bonds is critical to the pursuit of late-stage modifications of bioactive scaffolds. The field of C–H bond functionalization has (perhaps most prominently) demonstrated the concept that activating inert bonds can lend valuable synthetic tools.4–8 The sheer prevalence of C–H bonds, however, requires complex considerations of selectivity. As such, a focus on omnipresent functional groups beyond C–H moieties offers complementary advantages. Though many functional groups fit this requirement, this review specifically highlights contributions to aliphatic deaminative functionalization chemistry.
The deamination of alkyl amines may in some ways seem like an odd choice, given the synthetic effort often required for their installation and their poor leaving group character. Neither have amines been the area of greatest recent focus, with much of contemporary aliphatic activation research utilizing photochemical strategies to target functionalities such as carboxylic acids, olefins, and alcohols.9 However, primary amines are similarly ubiquitous in bioactive compounds.10 Indeed, a recent search of Pfizer's internal chemical database alone found over 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 primary aliphatic amines.2 Leveraging such a repository of compounds for late-stage modification holds substantial promise. However, the C–N bond is typically unreactive towards substitution, with N-centered reactivity dominating its chemistry.11–13 For these reasons, we have chosen to highlight deaminative functionalization reactions that convert primary aliphatic amines into other, potentially useful functional groups.
000 primary aliphatic amines.2 Leveraging such a repository of compounds for late-stage modification holds substantial promise. However, the C–N bond is typically unreactive towards substitution, with N-centered reactivity dominating its chemistry.11–13 For these reasons, we have chosen to highlight deaminative functionalization reactions that convert primary aliphatic amines into other, potentially useful functional groups.
Primary amines in particular offer retrosynthetic simplicity as terminal, peripheral groups that represent well-defined synthons. In contrast, secondary and tertiary amines are often positioned in skeletally critical positions within molecules, which complicates their implementation as reactive fragments. Indeed, transition metal catalyzed C–N bond functionalization has commonly focused on secondary and tertiary amines, with comprehensive reviews published elsewhere.14,15 Finally, diazotization has been broadly useful for activating aromatic amines (again beyond our scope) and there are several existing reviews on this topic.16–18
Within this narrow delineation, our review aims to highlight different synthetic strategies including examples from the growing fields of radical deamination, deaminative oxidation, and transition metal catalyzed pathways. These strategies are contrasted with functionalizations that proceed through polar mechanisms, including both modern applications and some historical examples which deserve recognition in a modern light. While there have also been some reports of surface supported or heterogenous transformations,19,20 we mainly focus on homogenous examples that are easily accessible to synthetic organic chemists.
2. Deamination by a polar mechanism
Due to the poor nucleofugality of primary amines, substitution reactions by polar mechanisms are challenging. A typical strategy involves a two-step procedure in which the amine is first converted to a better leaving group and undergoes subsequent substitution or elimination reactions (Scheme 1). Early work in the mid 1900s attempted to functionalize primary aliphatic amines by diazotization in an analogous manner to aromatic amines, but this typically resulted in a complex mixtures of products, likely due to the high reactivity of these intermediates.21 As a consequence, alkyl amine functionalization reactions were achieved by conversion to more stable leaving groups (e.g. bis(toluenesulfonyl)imides, vide infra). However, in 2014 Wang and coworkers reported deaminative C(sp3)–C(sp2) coupling of α-aminoacetonitriles and α-aminoesters via in situ diazotization.22 The resulting diazo compounds undergo C–B insertion with aryl boronic acids, yielding arylacetonitrile or arylacetic acid derivatives.
 |
| | Scheme 1 Scope of deamination reactions mediated by a polar functionalization mechanism. X = I, Br; Nuc = PPh3, NaOPh, NaSPh, piperidine. | |
2.1 Deamination reactions via tosylation or silylation of primary amines
Analogous to the tosylation of alcohols, the tosylation of primary amines has been pursued to enhance their leaving group ability. Seminal work by DeChristopher reported installing two sulfonamide leaving groups on primary alkyl amines. When these moieties were heated under reflux in DMF with iodide, bromide or amine nucleophiles, a mixture of substitution and elimination products were formed (Scheme 2).23 Work from the Baumgarten group demonstrated that alkenes could be produced as the sole product of the reaction and in high yield when neat N-alkyl-N,N-disulfonimides were heated to high temperatures.24 Guziec and Wei subsequently reported that treatment of N-alkyl-N-sulfonimides with chloramine and potassium hydroxide could selectively result in the production of alkanes or halides depending on the order of addition of reagents.25 Work from the Hutchins laboratory also demonstrated that N-alkyl-N,N-disulfonimides could be converted to alkanes by reaction with sodium borohydride as a nucleophile.26
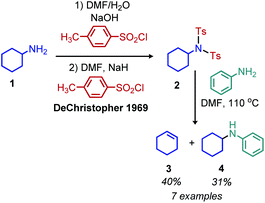 |
| | Scheme 2 Substitution reactions of N-alkyl-N,N-disulfonimides. Ts = tosyl. | |
While there has been little investigation into deaminative functionalization via tosylation since the 1990s, the Oestreich group has very recently reported a one-pot reductive deamination inspired by Hutchins’ conversion of N-alkyl-N,N-disulfonimides to alkanes. In situ dehydrogenative Si–N coupling of primary amines was performed with PhSiH3, and the Lewis acid B(C6F5)3 was used to abstract the silylated primary amine (Scheme 3).27 α-Tertiary primary amines including tritylamine, cumylamine and adamantylamine were converted to alkanes in 62% to 99% yield, which is quite remarkable considering how inert these primary amines are to other functionalization reactions (vide infra). However, primary amines without α-branching were converted in low yield. Two possible reaction pathways are proposed, a dehydrogenative pathway to form the silylated amine which is subsequently reduced, or a dissociation pathway to form a carbocation frustrated Lewis pair which then reacts with the borohydride or trihydrosilane to give the alkane product.
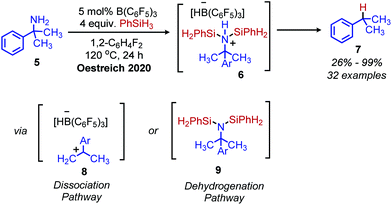 |
| | Scheme 3 One-pot conversion of amines to alkanes with trihydrosilane and tris(pentafluorophenyl)borane and proposed intermediates. | |
2.2 Deamination reactions via methylation of primary amines
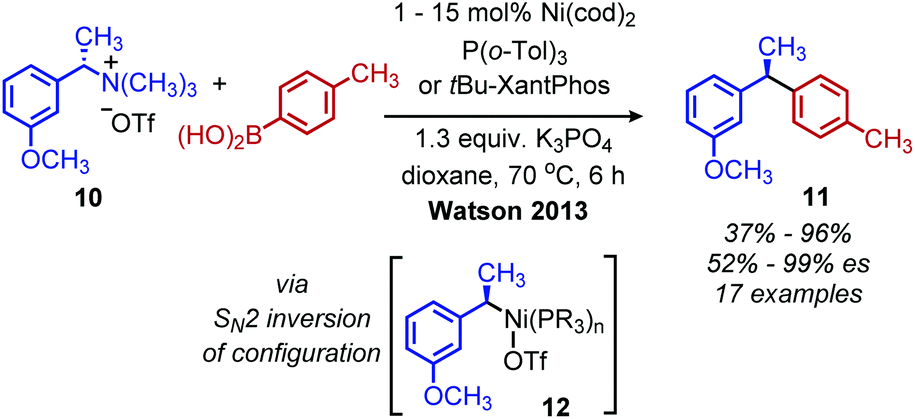
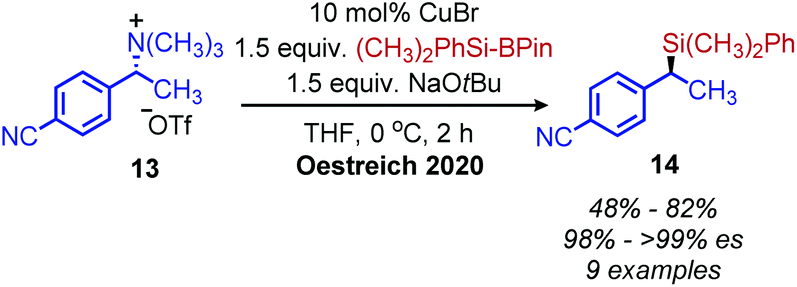
Classically, aliphatic amines can also be converted to alkenes by the Hoffman elimination reaction.28 While the leaving group is technically a quaternary amine, primary amines can be easily methylated with methyl iodide and then eliminated with anti-selective stereochemistry. A related reaction is the Cope elimination, in which a tertiary amine produced from the methylation of a primary amine reacts with m-CPBA to generate an N-oxide which undergoes syn-selective elimination.29 A modern strategy for removing methylated quaternary amines is to harness these moieties as substrates in cross-coupling reactions. The Watson group demonstrated deaminative cross-coupling of methylated quaternary amines using aryl boronic acids and a nickel catalyst.30 Enantiopure α-secondary benzylic quaternary amines were converted to diarylethanes in up to 99% es with a functional group tolerance for sulfones, esters and nitriles (Scheme 4). The enantioselectivity was rationalized by an inversion of configuration from SN2 addition of the amine substrate with the nickel catalyst.30,31 The Watson group has also reported a deaminative Miyaura borylation of methylated quaternary amines using a nickel catalyst and bis(pinacolato)diboron.32 Recent work from Oestreich and coworkers demonstrated that methylated quaternary amines can undergo deaminative silylation with silyl boronic esters and a copper catalyst (Scheme 5).33 An SN2 type inversion of configuration was also observed with enantiopure α-secondary benzylic amines, suggesting a polar mechanism is operative.
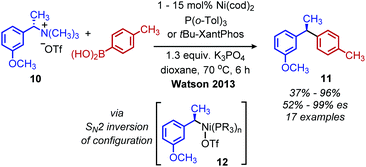 |
| | Scheme 4 Enantioselective deaminative conversion of benzylic quaternary amines to diarylethanes. Cod = cyclooctadiene; Tol = toluene; OTf = triflate; es = enantiospecificity. | |
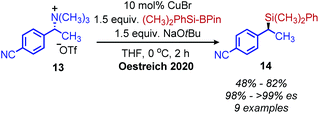 |
| | Scheme 5 Enantioselective deaminative conversion of benzylic quaternary amines to silanes. Pin = pinacolato. | |
The Martin group has also disclosed a nickel catalyzed reductive cross- coupling reaction of benzylic ammonium salts with carbon dioxide (Scheme 6).34 Unlike previous work from the same group employing alkyl halide electrophiles, this reaction displays tolerance for substrates with β-alkyl chains. The authors note that stoichiometric reactions with both nickel(I) and nickel(0) complexes successfully produce carboxylic acid product, but whether this indicates a radical oxidative addition or a non-radical disproportionation mechanism is unclear. Palladium catalyzed deaminative carbonylation of benzylic methylated quaternary amine salts has also been reported and is highlighted in a recent review by Wu and co-workers.13,35
 |
| | Scheme 6 Deaminative conversion of benzylic quaternary amines to carboxylic acids. dBuMePhen = 2,9-dibutyl-3,4,7,8-tetramethyl-1,10-phenanthroline. | |
2.3 Deamination reactions via pyridinium salts
The landscape of amine functionalization chemistry was substantially advanced by the Katritzky laboratory during the 1970s and 1980s. The facile conversion of an alkyl amino moiety into a pyridinium leaving group by reaction with a pyrylium salt allowed primary alkyl amines to be considered, in principle, as electrophiles (Scheme 7). These N-alkyl 2,4,6-triphenylpyridinium salts were initially discovered by Ziegler36 and Balaban,37 but are widely known today as Katritzky salts. These salts were shown to exhibit rich substitution chemistry, with select transformations demonstrating evidence for a polar mechanism (among other transformations that proceeded via a radical mechanism, vide infra).38 Pyrylium salts themselves have a rich history, and the interested reader is referred to a recent review highlighting their synthesis and discovery, as well as a review of the applications of pyridinium salts to radical reactions.39,40
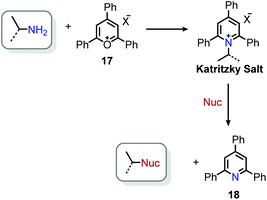 |
| | Scheme 7 Functionalization of primary amines by reaction with pyrylium salts. | |
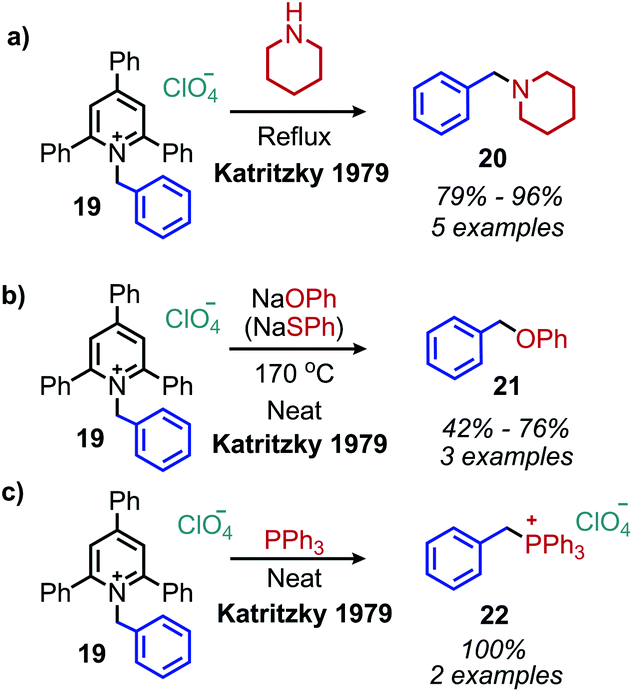
The conversion of primary amines to tertiary amines by reaction with piperidine and morpholine under reflux conditions was initially investigated with N-benzyl 2,4,6-triphenylpyridinium perchlorate and gave the product amine in 85% and 92% yield, respectively (Scheme 8a). When sodium phenoxide, sodium thiophenoxide and aniline were used as nucleophiles, the corresponding ethers, thioethers and secondary amines were produced in approximately 50% yield (Scheme 8b). Triphenylphosphine also reacts to produce benzyltriphenylphosphonium perchlorate (Scheme 8c).41
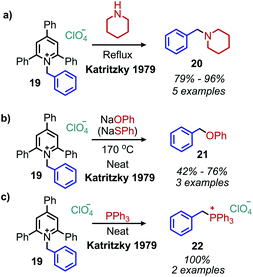 |
| | Scheme 8 Nucleophilic conversion of benzylic primary amines to (a) tertiary amines, (b) ethers and (c) phosphonium salts. | |
In detailed studies of amine alkylation with pyridinium salts, multiple mechanisms for nucleophilic transformations were shown to occur: SN2, classical SN1 and SN1 via an intimate ion–molecule pair.42 Initial kinetic studies were performed which involved piperidine and a series of N-substituted pyridinium salts. N-Methyl, -allyl and -benzyl substituents demonstrated second order kinetics. However, secondary alkyls including N-isopropyl, N-cyclohexyl and N-sec-butyl substituted pyridinium cations exhibited SN1 behavior (i.e. independent of the concentration of piperidine). Further mechanistic analysis was performed with 2-pentyl and 3-pentyl substrates by solvolysis in trifluoroacetic acid, and it was determined that both molecules produce the same mixture of 2-pentyl and 3-pentyl trifluoroacetates. This suggests that a carbocation is formed by an SN1 mechanism and has time to equilibrate. However, when morpholine was used as the solvent no rearrangement occurred suggesting that an intimate ion–molecule pair first forms. This intimate ion–molecule pair can either be intercepted by a nucleophile or drift apart to form a free carbocation before solvolysis.
2.4 Deamination by the nitration of amides to esters and alcohols
Early work by White demonstrated that primary alkyl amines can be converted to N-nitrosamides.43 These moieties undergo a rearrangement reaction to release dinitrogen and form an ester. Jakobsche and coworkers have recently reported the conversion of primary amines to N-nitrosodichloroacetamide intermediates using methyldichloroacetate and sodium nitrite (Scheme 9).44 The ester product from the rearrangement reaction is subsequently hydrolyzed by triethylamine/methanol, resulting in a net conversion of amine to alcohol in 66% yield.
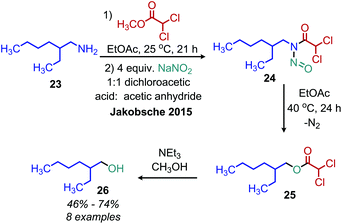 |
| | Scheme 9 Deaminative conversion of N-alkyl-N-nitrosamides to alcohols. | |
3. Radical deamination
The recent renaissance of radical chemistry in organic synthesis has demonstrated important applications of alkyl radical species. Compared to polar mechanisms, radical intermediates are not susceptible to Wagner–Meerwein rearrangements and typically suffer less from steric constraints as a result of early bond-forming transition states.45 Additionally, these reactive species can be easily intercepted in modern photoredox/cross-coupling methodologies.9 In this section, we briefly highlight historical contributions to radical aliphatic deamination reactions by Barton and Katritzky. This introduction is followed by an exploration of the recent evolution of Katritzky salts in deaminative alkyl functionalization reactions via nickel cross-coupling, photocatalysis, and Lewis base catalysis (Scheme 10).
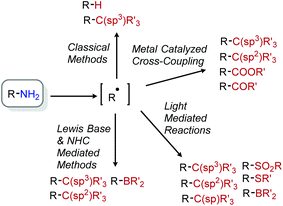 |
| | Scheme 10 Scope of radical mediated deamination reactions. | |
3.1 Classical examples of deaminative radical C–H and C–C bond formation
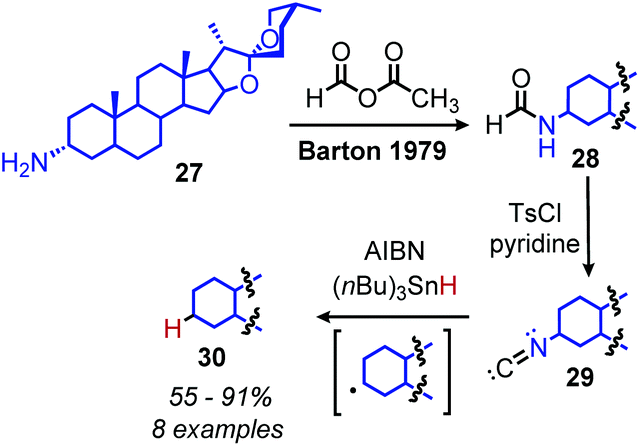
Analogous to the classic Barton–McCombie deoxygenation reaction of alcohols, Barton also developed a method for radical deamination of primary amines. Inspired by initial observations from Saegusa that isonitriles can be reduced to alkanes,46 Barton and coworkers disclosed in 1979 the radical conversion of alkyl primary amines to alkanes.47 The amine functionality can be converted to an isonitrile by formylation and subsequent dehydration with either p-toluenesulfonyl chloride or phosphorus oxychloride. In the presence of AIBN (2,2′-azobis(methylpropionitrile)) as an initiator and (nBu)3SnH (or hypophosphorus acid),48 isonitriles gave alkanes in 55–91% yield (Scheme 11).
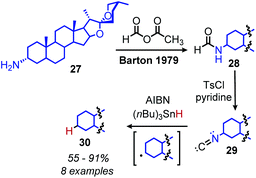 |
| | Scheme 11 Barton–Saegusa deamination of amines to alkanes. AIBN = 2,2′-azobis(methylpropionitrile). | |
Katritzky salts can also be utilized for radical deamination reactions of primary amines in addition to their polar substitution chemistry (vide supra). The earliest evidence for a radical mechanism in the chemistry of Katritzky salts came in reports of alkylation with nitronate (Scheme 12a) and malonate anions (Scheme 12b),49 resulting in C(sp3)–C(sp3) bond formation.50 These reactions demonstrated bimolecular kinetics with relative rates across N-alkyl groups consistent with a non-chain radical mechanism rather than an SN2 pathway trend (namely that secondary aliphatic substituents react faster than primary).41,51,52 This radical mechanism was further described as proceeding via a charge transfer complex (CTC), and UV-vis spectroscopy demonstrated quenching of N-benzyl-2,4,6-triphenylpyridinium tetrafluoroborate absorption upon immediate addition of sodium nitromethanide solution.51 Katritzky salts also undergo reduction with NaBH4 to 1,2-dihydropyridines, which, upon subsequent pyrolysis at 200 °C, yield alkanes (Scheme 13a).53 Labelling experiments with NaBD4 gave α-deutero-toluene as the sole product, also providing evidence for a radical mechanism (Scheme 13b).54
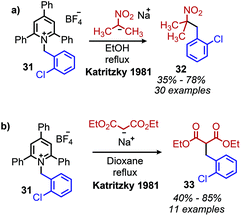 |
| | Scheme 12 Deaminative alkylation reactions with (a) nitronate and (b) malonate anions. | |
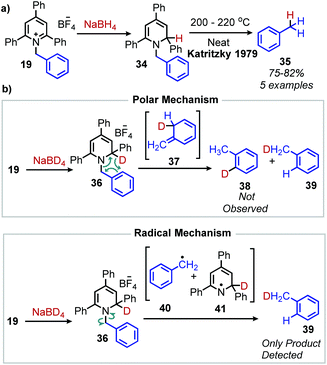 |
| | Scheme 13 a) Deaminative conversion to of amines to alkanes and (b) a labelling experiment with NaBD4 shows evidence for a radical mechanism. | |
3.2 Metal catalyzed cross-coupling reactions
The modern application of Katritzky salts to cross-coupling reactions was first disclosed by Watson and coworkers in 2017. Using catalytic nickel(II) acetate and a bathophenanthroline (BPhen) ligand, deaminative C(sp3)–C(sp2) coupling of both primary and secondary N-alkyl pyridinium tetrafluoroborate salts was achieved with aryl boronic acids (Scheme 14).55 A high functional group tolerance was demonstrated for amines containing ethers and esters, as well as heterocyclic boronic acids.55 Mechanistic studies demonstrated an alkyl radical intermediate, as evidenced by TEMPO trapped adducts and a radical clock experiment.55 The authors thus suggest a NiI/III cycle, however the exact timing of transmetallation of the aryl boronic acid is unclear. This procedure was later expanded to include the deamination of benzylic amines56 and amino acid methyl esters.57 Additionally, vinyl boronic esters could be utilized as the coupling partner,58 and in situ hydroboration of alkenes with 9-BBN allowed for a deaminative C(sp3)–C(sp3) coupling.59
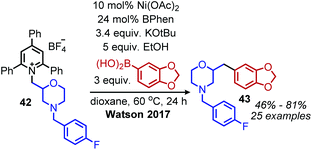 |
| | Scheme 14 Deaminative Suzuki coupling. BPhen = bathophenanthroline. | |
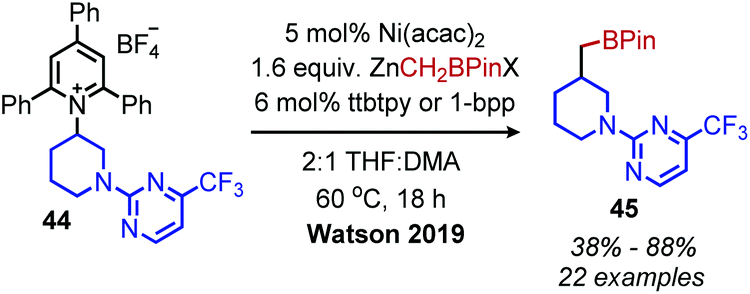
Watson and coworkers further expanded the deaminative cross-coupling to Negishi-type C(sp3)–C(sp3) bond formation with alkyl zinc halides and Ni(acac)2 catalyst (Scheme 15).2 Primary N-alkyl pyridinium salts were coupled most effectively using the ligand ttbtpy (4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine); the authors postulate this ligand was too sterically hindered for secondary N-alkyl pyridinium salts, which gave the highest yields using the 1-bpp (bis(pyrazoy-1yl)pyridine) ligand. A variety of pharmaceutical scaffolds bearing primary amines were functionalized with this methodology, including the anti-viral drug Tamiflu. Another notable application was the successful coupling of methylzinc iodide with an N-alkyl Katritzky salt, allowing primary amines to be replaced with methyl isosteres. Finally, a one-pot procedure was realized by adding triphenylpyrylium tetrafluoroborate and primary alkyl amine to the reaction mixture containing the nickel catalyst and alkyl zinc reagent.
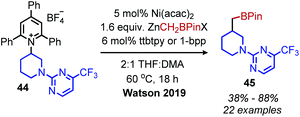 |
| | Scheme 15 Deaminative Negishi coupling. ttbtpy = 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine; 1-bpp = bis(pyrazoyl-1-yl)pyridine; Acac = acetylacetone. | |
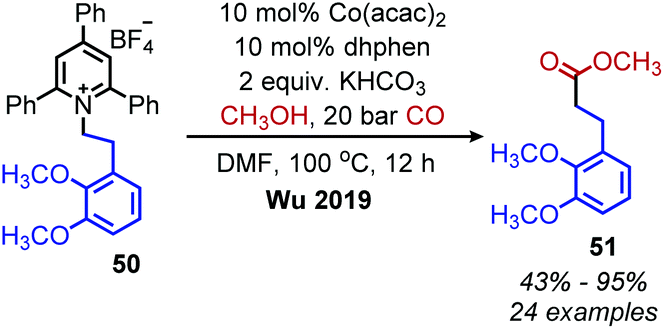
The Martin group also disclosed deaminative C(sp3)–C(sp3) bond formation by addition to non-activated alkenes (Scheme 16a). Terminal alkenes afforded anti-Markovnikov products in up to 81% yield using NiBr2·glyme catalyst, dtbpy (4,4′-di-tert-butyl-2,2′-bipyridine) ligand and (EtO)3SiH as the hydride reagent.60 α-Functionalization of internal olefins was also achieved using NiI2 and a 2-(oxazolin-2-yl)-6-methylpyridine ligand.60 Independently, Liu and co-workers further explored deaminative hydroalkylation of internal alkynes (Scheme 16b).61 Using a NiBr2·dme catalyst, tbbpy (tris-4,4′-di-tert-butyl-2,2′-bipyridine) ligand and (Me2SiH)2O, regioselectivity ratios of >20:1 could be achieved for coupling phenyl alkyl alkynes with α-secondary alkyl amines.61 Lower regioselectivity ratios of around 2:1 were observed for dialkyl alkynes, which the authors ascribe to the similar steric profiles of the alkyne substituents.61 Another transition metal catalyzed radical deamination of Katritzky salts was reported by Wu and collaborators. Using catalytic Co(acac)2 and a 4,7-dihydroxy-1,10-phenanthroline (dhphen) ligand, alkoxycarbonylation of primary amines to methyl esters was achieved with methanol under 20 bar of carbon monoxide (Scheme 17).62 The proposed mechanism involves reduction of the Katritzky salt by cobalt(0) and formation of a cobalt(II) alkyl species which undergoes CO insertion. A subsequent reaction with methanol affords the ester product and regenerates the cobalt catalyst (Scheme 18).
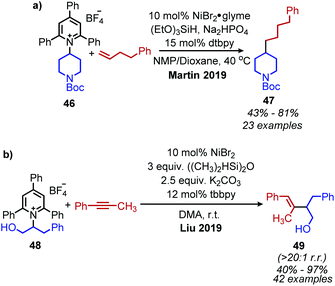 |
| | Scheme 16 a) C(sp3)–C(sp3) bond formation with non-activated alkenes and (b) C(sp3)–C(sp2) bond formation with non-activated alkynes. NMP = N-methyl-2-pyrrolidone; dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine; tbbpy = tris-4,4′-di-tert-butyl-2,2′bipyridine. | |
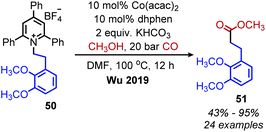 |
| | Scheme 17 Cobalt-catalyzed deaminative alkoxycarbonylation. Dhphen = 4,7-dihydroxy-1,10-phenanthroline. | |
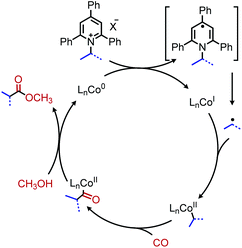 |
| | Scheme 18 Proposed catalytic cycle for deaminative alkoxycarbonylation. | |
Very recently, the Liu group has disclosed the conversion of primary amines to difluoromethane isosteres using catalytic copper and the Vicic–Mikami reagent (Scheme 19).63 Interestingly, the use of N-alkyl Katritzky salt derivates with electron-withdrawing substituents or additional ring fusions was necessary for reactivity. The reaction demonstrated tolerance for a variety of heterocyclic functional groups, as well as a α,β-unsaturated amide and was successful in derivatizing four pharmaceuticals containing primary amines.
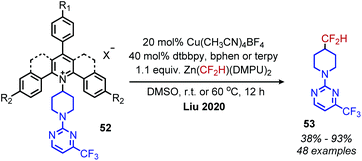 |
| | Scheme 19 Deaminative difluoromethylation with catalytic copper. Terpy = tertpyridine; Dtbbpy = 4,4′-ditertbutyl 2,2′ bipyridine; DMPU = N,N′-dimethylpropyleneurea. R1 = CF3, F or H. R2 = CF3 or H. X = BF4 or OTf. | |
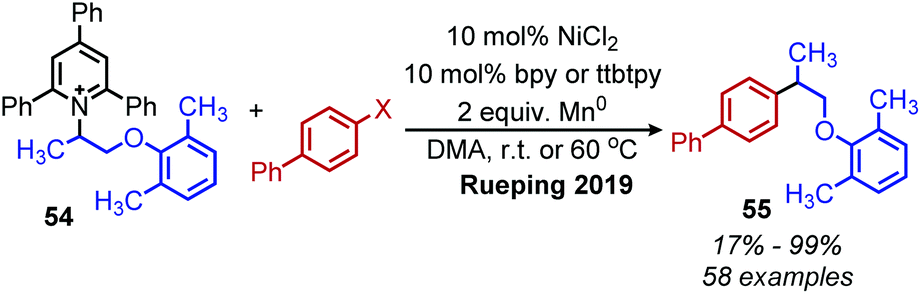
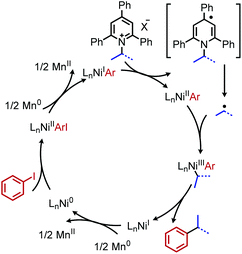
Reductive deaminative cross-coupling reactions have also been developed by several groups. Using catalytic NiCl2·dme ligated with bpy (2,2′-bipyridine) for secondary alkyl amines or ttbtpy for primary alkyl amines, Rueping and coworkers achieved C(sp3)–C(sp2) coupling with aryl iodides (and some aryl bromides) using Mn(0) as the reducing reagent (Scheme 20).64 The catalytic cycle proposed by the authors involves initial oxidative addition of the aryl iodide with nickel(0) (Scheme 21). The resulting nickel(II) species is reduced by Mn(0) to a nickel(I) aryl intermediate which reduces the Katritzky salt. A nickel(III) intermediate bearing alkyl and aryl groups is formed and undergoes reductive elimination. Mn(0) then regenerates the catalyst to nickel(0). Similar procedures were subsequently reported for reductive deamination of both aryl bromides and aryl iodides using either a Zn(0) or Mn(0) reductant.65–67 Additionally, Han and collaborators have achieved a reductive C(sp3)–C(sp3) coupling of N-alkyl Katritzky salts with alkyl bromides using a nickel catalyst and 2.5 equivalents of Zn(0).65 Notably, Watson and coworkers also disclosed the reductive coupling of 1-methylcyclopropanamine in a 35% yield, a rare example of α-tertiary primary amines in Katritzky methodology.66 This is notable as typically, α-tertiary amines are too sterically hindered to form the requisite pyridinium salts precursors. Molander and collaborators were able to use the organic chromophore 4-CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene) excited by blue light, with triethylamine as a terminal reductant in place of Mn(0) or Zn(0) to turn over the nickel catalyst.68 A reductive deaminative acylation was also reported by Matsuo using catalytic nickel, Mn(0) and N-acylsuccinimides.69
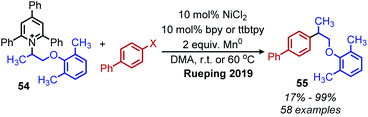 |
| | Scheme 20 Reductive deaminative cross-coupling. X = Br, I; bpy = bipyridine. | |
 |
| | Scheme 21 Proposed mechanism for deaminative reductive cross-coupling. | |
3.3 Photoredox catalyzed deaminative functionalizations
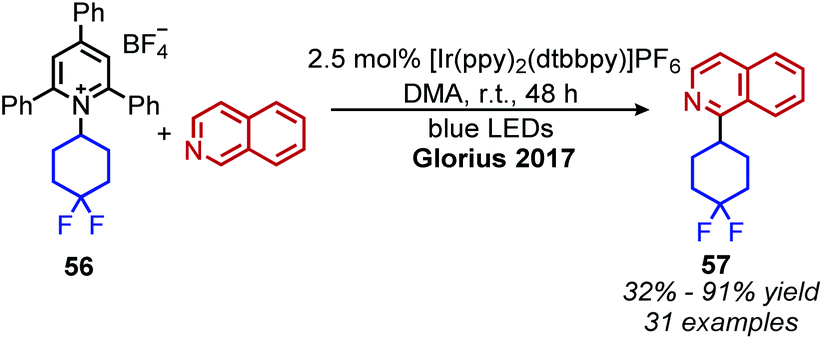
Photocatalyzed reactions have emerged in the past decade as powerful synthetic tools. As photocatalyzed deaminative alkylation reactions have recently been review elsewhere,39,70 this section is not intended to be comprehensive. Rather, we aim to provide several illustrative examples of the types deaminative transformations made possible by visible-light mediated processes. Glorius and coworkers ignited modern photoredox catalysis applications of Katritzky salts with their seminal publication in 2017, wherein the chromophore [Ir(ppy)2(dtbbpy)]PF6 was used to reduce the Katritzky salt under blue light and the resulting alkyl radical was trapped by an electron-rich heterocycle, resulting in C(sp3)–C(sp2) bond formation (Scheme 22).71 α-Secondary N-alkyl primary amines, including amino acid methyl esters were successfully coupled to indole and isoquinoline with Minisci-type selectivity. After this initial report, numerous groups reported other photoredox catalyzed C(sp3)–C(sp3) and C(sp3)–C(sp2) bond formations via a variety of alkylation methods including Heck and Giese type syntheses.72–78
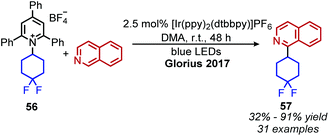 |
| | Scheme 22 Deaminative photoredox catalyzed Minisci alkylation. Ppy = 2-phenylpyridine. | |
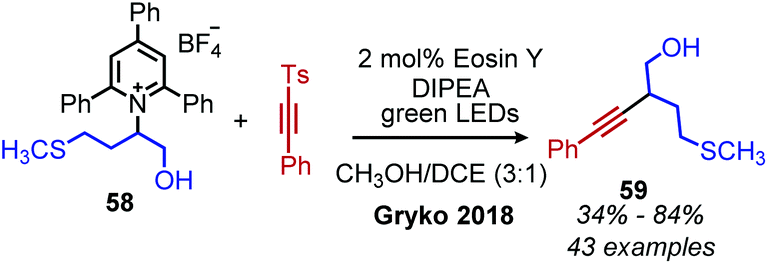
The first example of deaminative C(sp3)–C(sp) coupling was reported by Gryko and coworkers.79 Using eosin Y and green light, with DIPEA as a sacrificial reductant, α-secondary and benzylic primary Katritzky salts couple with vinyl and alkynyl sulfones (Scheme 23), in which the toluenesulfonyl group serves as a radical nucleofuge (Scheme 24). The reaction displays good functional group tolerance including halides, alcohols and heterocycles. The alkenylation reactions demonstrated an E:Z ratio of >99:1 for most substrates.
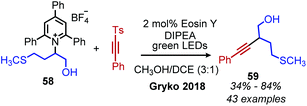 |
| | Scheme 23 Eosin Y catalyzed deaminative alkynylation. DIPEA = diisopropylethylamine. | |
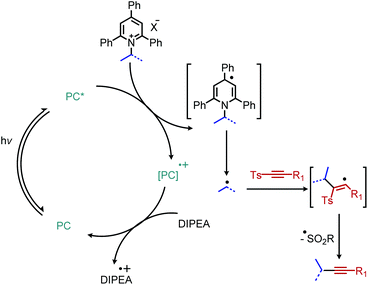 |
| | Scheme 24 Proposed catalytic cycle for eosin Y catalyzed deaminative alkynylation. PC = photocatalyst. | |
Wu and collaborators recently disclosed a deaminative photoredox-catalyzed synthesis of alkyl sulfones by coupling of N-secondary Katritzky salts with potassium metabisulfite and aryl substituted silyl enol ethers (Scheme 25).80 The proposed mechanism involves radical addition to sulfur dioxide to generate an alkylsulfonyl radical which is trapped by the silyl enol ether. Subsequent oxidation and base-induced desilylation results in the formation of a ketone (Scheme 26).
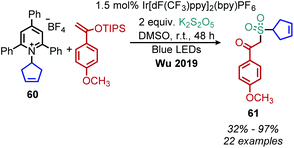 |
| | Scheme 25 Deaminative synthesis of alkyl sulfones. dF(CF3)ppy = 2,4,-difluorophenyl-5-(trifluoromethyl)pyridine. | |
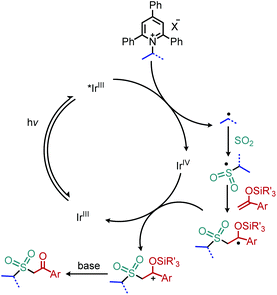 |
| | Scheme 26 Proposed catalytic cycle for the deaminative synthesis of alkyl sulfones. | |
3.4 Visible light mediated deaminative functionalization via an EDA complex
Electron–donor–acceptor (EDA) complexes involve one reaction component (the donor) that is excited by visible light causing transfer of an electron to another reaction component (the acceptor), enabled by close intermolecular association.81 Light induced EDA processes are an attractive alternative to conventional photoredox catalyzed reactions which require either precious metal or complex organic chromophores. Katrizky salts have proven exceptionally versatile in this regard, with chromophore-free deaminative borylation of alkyl amines reported concurrently by the groups of Aggarwal and Glorius in 2018 (Scheme 27).82,83 In the presence of blue light, alkyl boronic esters were successfully synthesized from N-alkyl Katritzky salts and B2cat2. Subsequent transesterification with pinacol allowed isolation of stable pinacol boronic esters.82,83 Functional group tolerance for heterocycles, sulfonamides and esters was demonstrated, and the amino acid lysine was successfully borylated at the residue nitrogen with retention of enantioselectivity at the α-carbon.83 UV-vis spectroscopy demonstrated a color change and bathochromic absorbance upon addition of B2cat2 to a DMA solution of Katritzky pyridinium salt. Job's method indicated the EDA complex is formed in a 1:1 ratio and a reaction quantum yield of greater than 1 was determined by both groups, indicating that a radical chain mechanism is operative. In the proposed mechanism (Scheme 28), the B2cat2 DMA adduct is shown to reduce the Katritzky salt. This process is analogous to the reactions originally reported by Katritzky to proceed via a charge transfer complex (vide supra).
 |
| | Scheme 27 Deaminative borylation mediated by an EDA complex. Cat = catecholato. | |
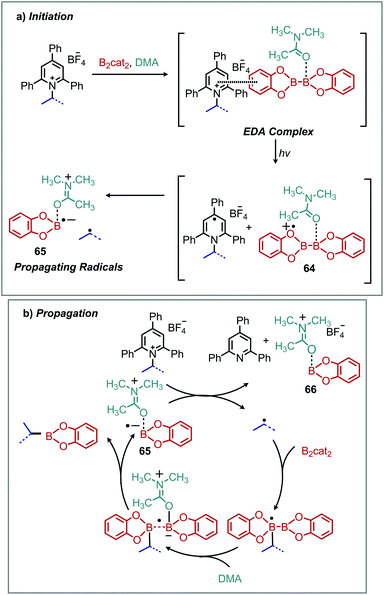 |
| | Scheme 28 Proposed mechanism for deaminative borylation with (a) EDA complexation and (b) radical chain propagation. | |
The synthesis of alkyl thioesters via an EDA complexation of a variety of amino acid derived Katritzky salts with a thiobenzoic acid anion was also reported by Liao and collaborators (Scheme 29a).84 Deprotection of the resulting thioesters resulted in the net synthesis of α-mercapto acids. Thiobenzoic acid could also be replaced with alkyl and aromatic thiols resulting in the synthesis of amino acid derived thioethers (Scheme 29b). The alkyl radical generated as a result of reduction of the Katritzky salt is proposed to either undergo radical cross-coupling with the oxidized radical thioacid species, or alternatively add to the thioacid anion to generate a radical anion species which is subsequently oxidized.
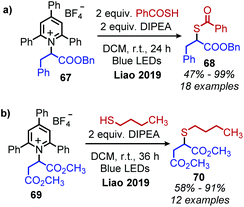 |
| | Scheme 29 a) Synthesis of alkyl thioesters and (b) thioethers. | |
Reports of photoinduced, catalyst free C(sp3)–C(sp3) and C(sp3)–C(sp2) bond formation were first reported by the groups of Fu and Aggarwal.85,86 Most recently, Xu and coworkers reported another example of deaminative C(sp3)–C(sp3) bond formation via an EDA complex of Katritzky salts and amino acid derivatives.87 In the presence of violet light, alkylation of the α-carbon of N-arylated glycine derivatives and glycine containing peptides was observed (Scheme 30). A variety of dipeptides and polypeptides of up to 6 different amino acids were successfully coupled, with selectivity for only the glycine unit. The substrate scope also included α-secondary Katritzky salts containing functional groups including an unprotected alcohol. However, primary Katritzky salts (both benzylic and non-benzylic) did not react. (This is attributed to a slower fragmentation rate of primary alkyl dihydropyridine radicals.) UV-vis spectroscopy studies indicated a new absorbance when a Katritzky salt solution was mixed with the glycine derivative ethyl 2-(phenylamino)acetate, supporting the formation of an EDA complex. The authors invoke the base DBU to deprotonate the amino radical species produced by SET (Scheme 31) but also indicate that it might participate in forming a ternary EDA complex.
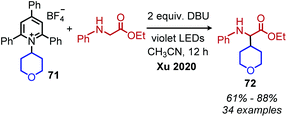 |
| | Scheme 30 Deaminative alkylation of amino acids via an EDA complex. DBU = diazobicycloundecene. | |
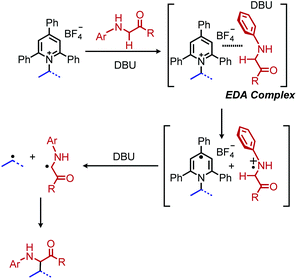 |
| | Scheme 31 Proposed mechanism for alkylation of amino acids. | |
3.5 Lewis base and NHC catalyzed deaminations
Shi and collaborators disclosed in 2018 a conceptually distinct method for deaminative borylation using a Lewis base (Scheme 32).88N-Alkyl Katritzky salts were reacted with B2cat2 in the presence of 10 mol% 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy). Control experiments indicated that the transformation is not light promoted, ruling out an EDA mechanism. Substrates bearing heterocycles, esters and halides were successfully utilized in the reaction, and an additional additive-based robustness screen was performed which demonstrated a wide range of functional groups could be tolerated. DFT studies and boron NMR experiments indicated that B2cat2 forms an adduct with dtbpy, at which point the boron–boron bond is cleaved. This intermediate is proposed to form an adduct with the solvent DMAc which has a reduction potential low enough for single electron transfer to Katritzky salts (Scheme 33). Alternatively, homolysis of this intermediate can generate radical species that are also capable of reducing the alkyl Katritzky salt.
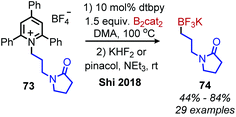 |
| | Scheme 32 Lewis base catalyzed deaminative borylation. | |
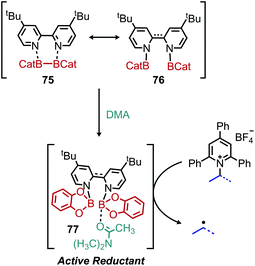 |
| | Scheme 33 Proposed mechanism for Lewis base catalyzed deamination. | |
In 2020, Hong and coworkers published the deaminative cross coupling of aldehydes using an N-heterocyclic carbene (NHC) catalyst (Scheme 34).89 The proposed mechanism involves reaction of the NHC catalyst with the aldehyde to generate a Breslow intermediate which is able to reduce N-alkyl Katritzky salts (Scheme 35). Alkyl radicals generated from variety of amino acid and peptide derived Katritzky salts were successfully coupled to different benzaldehyde derivatives and heterocyclic aldehydes.
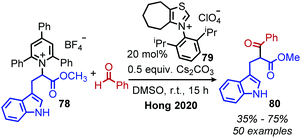 |
| | Scheme 34 NHC catalyzed deaminative cross-coupling of aldehydes. | |
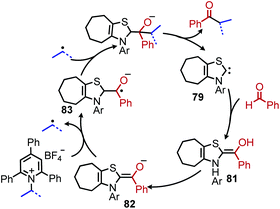 |
| | Scheme 35 Proposed mechanism for NHC catalyzed deaminative cross-coupling. | |
Wu and coworkers have also disclosed a base mediated deaminative carbonylation reaction in the absence of any catalysts. In the presence of DBU, N-alkyl Katritzky salts were shown to generate alkyl radicals which couple with CO and styrene derivatives (Scheme 36).90 Deaminative vinylation of N-alkyl Katritzky salts with DBU has also been demonstrated by the Loh group.91
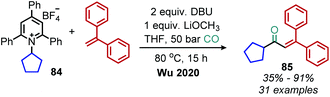 |
| | Scheme 36 Base promoted deaminative coupling with CO and styrenes. | |
4. Oxidative deamination
Carbonyl and imine moieties are extremely important and versatile functional groups, as indicated by their foundational role in introductory organic chemistry courses. Ketones, aldehydes and primary amines are abundant in natural products and thus reactions for their interconversion are of high synthetic importance. While reductive amination reactions achieve the facile conversion of carbonyls to amines,11,92 the reverse reaction involving oxidative deamination of amines to carbonyls has been historically more challenging. This has resulted in a wide variety of approaches to oxidative deamination. In this section, we begin with bio-inspired catalysts for these transformations and then explore strategies mediated by other organic and inorganic compounds. We then discuss additional synthetic applications of deaminative oxidation, including the synthesis of heterocycles and multicomponent reactions (Scheme 37).
 |
| | Scheme 37 Scope of oxidative deamination reactions. | |
4.1 Biomimetic conversion of amines to carbonyl and imine compounds by a Schiff base mechanism
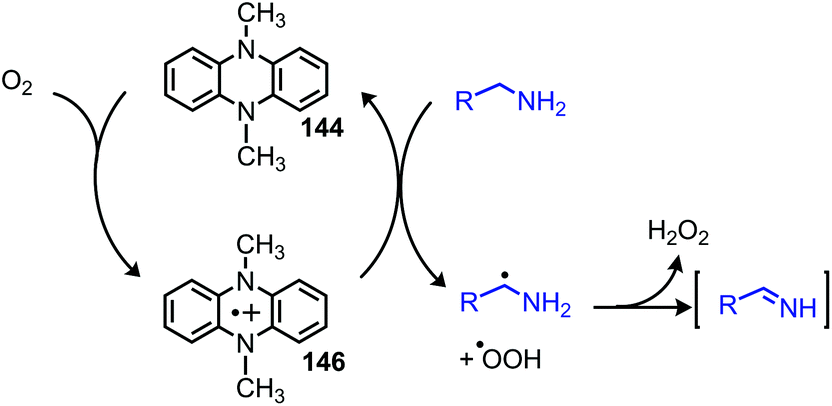
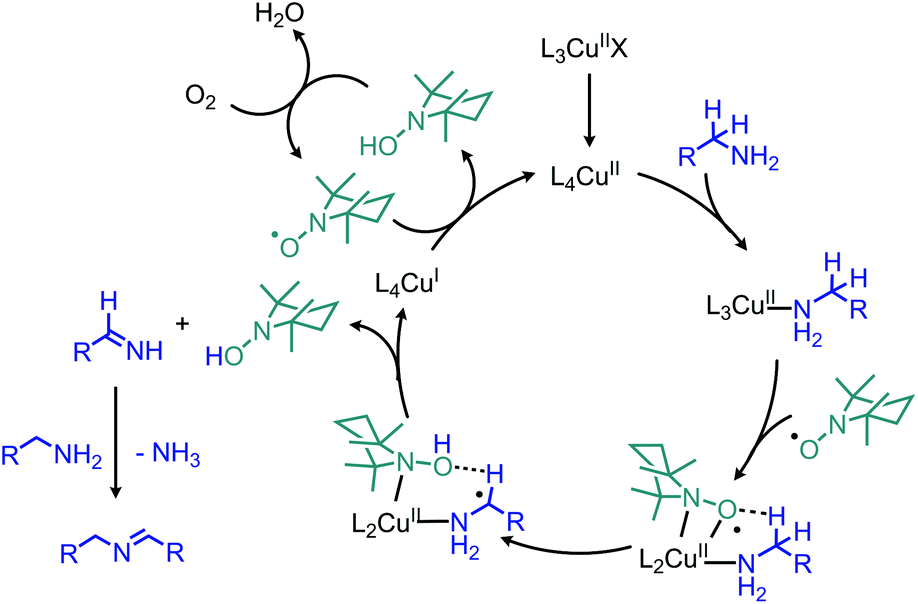
Enzyme-catalyzed deaminative oxidation of amines is critical to certain metabolic processes and has been a source of much inspiration to chemists.93 Copper-containing Amine Oxidases (CuAOs) are a family of enzymes that catalyze the degradation of amine containing neurotransmitters and histamines in animals.93,94 Work by the groups of Sayre95 and Klinman96 has identified the cofactor of CuAOs as the quinone containing derivative 2,4,5-trihydroxyphenylalaninequinone (TPQ). TPQ reacts with amines to form a Schiff base, which is converted to an aldehyde product and the reduced aminoquinol form of TPQ by reaction with water (Scheme 38a).94,95 The cofactor is then re-oxidized by the conversion of dioxygen to hydrogen peroxide. Amine dehydrogenase (ADH) is another enzyme that catalyzes the oxidative deamination of nitrogen-containing small molecules necessary for prokaryotic metabolism. ADH has been determined to contain a distinct quinone derived cofactor known as tryptophan tryptophylquinone (TTQ) and also reacts via a Schiff base mechanism (Scheme 38b).97 The cofactor is not directly re-oxidized by dioxygen and instead transfers the electrons to the blue copper proteins azurin or amicyanin.98 The majority of biomimetic strategies utilize quinone derived compounds to facilitate Schiff base formation and isomerization (Scheme 38c). Subsequent reaction with excess primary amine or hydrolysis results in the dialkyl imine or carbonyl product, respectively.
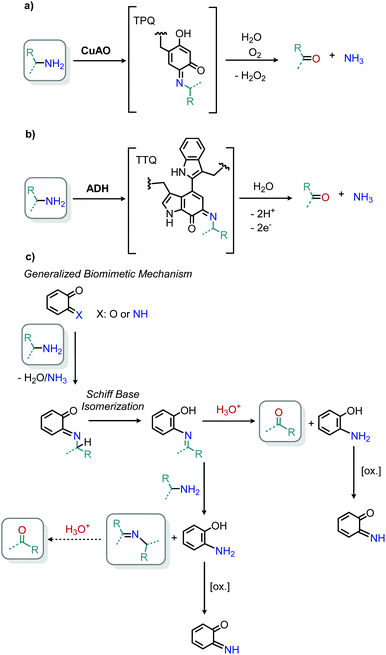 |
| | Scheme 38 Oxidative deamination in nature mediated by (a) CuAO and (b) ADH enzymes, and (c) generalized biomimetic strategy utilized by organic chemists. CuAO = copper amine oxidase; TPQ = 2,4,5-trihydroxyphenylalaninequinone; ADH = amine dehydrogenase; TTQ = tryptophan tryptophyquinone. | |
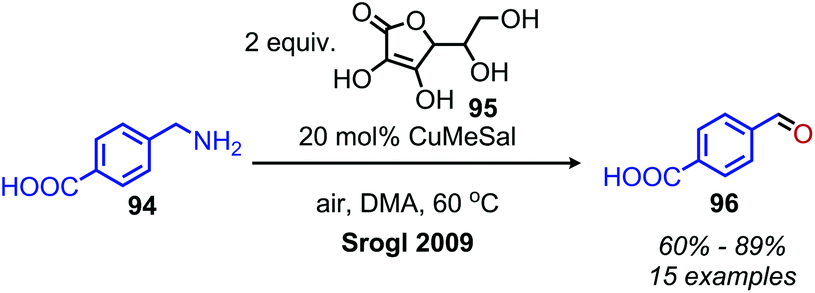
The Corey laboratory disclosed seminal work using the bio-inspired compounds 3,5-di-tert-butyl-1,2-benzoquionone and mesitylglyoxyl derivatives in 1969.99 The steric hinderance of 3,5-di-tert-butyl-1,2-benzoquionone favors Schiff base formation in the presence of primary aliphatic amines (Scheme 39a). Hydrolysis between at pH 2–4 results in the formation of ketone products. However, α-primary amines did not afford aldehydes, as the production of benzoxazoles was favored. As such, the use of mesitylglyoxyl derivatives was developed to successfully convert benzylamines to benzaldehydes (Scheme 39b). This initial work disclosed by the Corey laboratory prompted the development of a variety of additional carbonyl-containing compounds to mediate this transformation. One notable example by Voltrova and Srogl utilizes readily available ascorbic acid, which is oxidized in situ by air and catalytic copper to the active dehydroascorbic acid (Scheme 40).100 This reaction system was shown to convert a substrate scope consisting of primary benzylic amines to aldehydes with functional group tolerance including carboxylic acids.
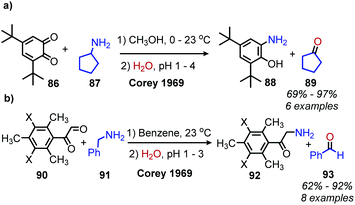 |
| | Scheme 39 Oxidative deamination with (a) benzoquinone and (b) mesitylglyoxyl derivatives. X = H, NO2. | |
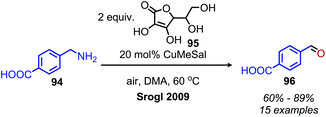 |
| | Scheme 40 Oxidative deamination with ascorbic acid. CuMeSal = copper(I) methylsalicylate. | |
An elegant recent application of this methodology was demonstrated by Lisnyak and Snyder as a key step in their enantioselective synthesis of chilocorine C, an alkaloid isolated from ladybugs with a distinctive 6/6/5 fusion of quinolizidine and two indolizidines.101 After synthesizing intermediate 97, conversion to an aldehyde moiety was pursued by the biomimetic oxidation procedure reported by Srogl and Voltrova (Scheme 41).100 The product of this transformation successfully undergoes an aldol cyclization sequence resulting in the desired natural product. This example clearly demonstrates the importance of harnessing aliphatic amines as synthons for C(sp3) bond functionalization in bioactive molecules.
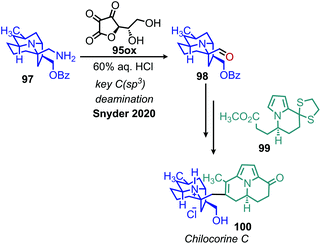 |
| | Scheme 41 Oxidative deamination in the total synthesis of chilocorine C. | |
Largeron and coworkers demonstrated oxidative deamination of a substrate scope containing mainly non-benzylic primary alkyl amines (Scheme 42).102,103 Catalytic quantities of a 3,4-iminoquinone were used for deaminative coupling of aliphatic amines to afford unstable dialkyl imines, which were subsequently isolated as aldehyde 2,4-dinitrophenylhydrazone adducts. A platinum electrode was used to re-oxidize the reduced aminoquinone species, resulting in a catalytic turnover number of up to 23. Later work demonstrated that the platinum electrode could be replaced with catalytic copper(I) and dioxygen from an air atmosphere to regenerate the quinone derived compound (Scheme 43).104 This procedure was effective for deaminative homo-coupling of benzylic primary amines. These dibenzylic imine products are more stable than their aliphatic imine counterparts and can be readily isolated. Functional group tolerance for heterocycles, including thiophene and furan was also demonstrated.
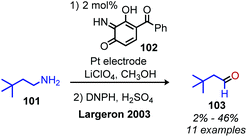 |
| | Scheme 42 Biomimetic oxidation of non-activated primary alkyl amines to aldehydes. DNPH = 2,4-dinitrophenylhydrazone. | |
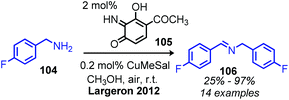 |
| | Scheme 43 Biomimetic oxidative coupling of benzylic amines using copper and air. | |
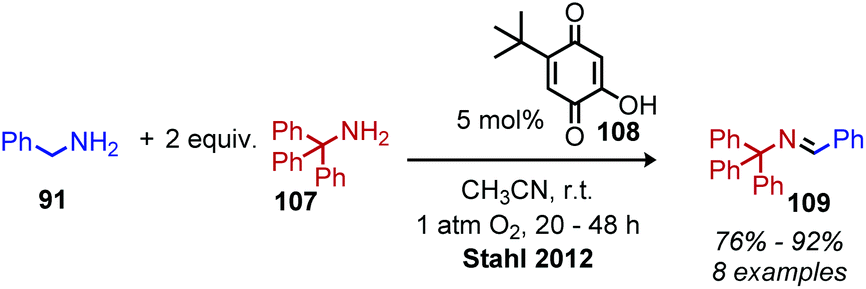
In the past 10 years, the development of biomimetic oxidative deamination procedures has rapidly evolved. An excellent review by Largeron in 2017 covers the substrate scope of these reports.93 We aim to highlight illustrative and recent examples of biomimetic conversion to carbonyl and imine compounds. In 2012, the Stahl group reported a deaminative biomimetic cross-coupling of different primary alkyl amines to a benzylic imine product using catalytic 4-tert-2-hydroxybenzoquinone and 1 atm of dioxygen.105 Notably, the α-tertiary primary amine substrate tritylamine was successfully cross-coupled with benzylamine (Scheme 44). The cross-coupling of an α-tertiary aliphatic primary amine with benzyl amine was further demonstrated by Largeron using 2,4,4-trimethylpentan-2-amine and their catalytic 3,4-iminoquinone system (Scheme 45).106 Monitoring these reactions demonstrated homo-coupled imines are initially produced and accumulate at the start of the reaction, but the selective reactivity of benzylic amines drives the equilibrium mixture quantitatively to cross-coupled products by trans-imination.105–107
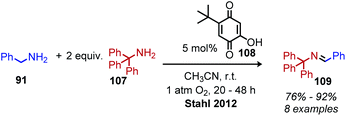 |
| | Scheme 44 Deaminative cross-coupling of primary amines using 4-tert-2-hydroxybenzoquinone. | |
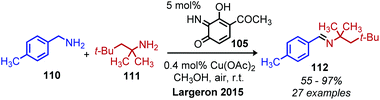 |
| | Scheme 45 Deaminative cross-coupling of primary amines using 3,4-iminoquinone. | |
In the above deaminative oxidation reactions, a second acid hydrolysis step is necessary to convert the product imines to ketones and aldehydes. In 2018, Oh and coworkers reported the first example of one-pot biomimetic conversion of amines to ketones using an ortho-naphthoquinone catalyst (Scheme 46).108 Under the optimized solvent and elevated temperature conditions, the water produced from the initial condensation of primary amines serves to hydrolyze the imine intermediates to ketone products. The substrate scope contained benzylic amines bearing halides, nitro and ester moieties, heterocyclic groups and one example of a non-benzylic secondary amine.
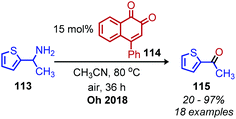 |
| | Scheme 46 One-pot conversion of primary amines to ketones. | |
Largeron and coworkers recently reported an example of ‘second-order’ biomimetic oxidative deamination, in which the catalyst purpurogallin is generated in situ from the commercially available pre-catalyst pyrogallol (Scheme 47a).109 This procedure is advantageous because most biomimetic quinone compounds are either expensive to purchase or not commercially available, requiring researchers to first synthesize the desired catalyst. In situ generated purpurogallin was shown to successfully catalyze deaminative cross-coupling of benzylic amines with primary alkyl amines (Scheme 47b).
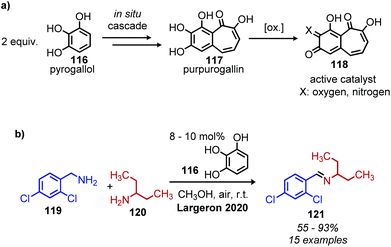 |
| | Scheme 47 Second order biomimetic deaminative coupling by (a) in situ production of purpurogallin for (b) deaminative cross-coupling of primary amines. | |
4.2 Biomimetic conversion of amines to carbonyl and imine compounds by an electron transfer mechanism
Another oxidative deamination mechanism has also been observed to occur in nature with monoamine oxidase (MAO), a mitochondrial enzyme which is responsible for the degradation of amine containing neurotransmitters.110 MAO contains a flavin cofactor which has been proposed to directly oxidize primary amines to imines (Scheme 48) instead of proceeding via a Schiff base mechanism,111 which has also been a source of inspiration for chemists. Recent work from the Carbery group inspired by flavin biomimicry demonstrated the redox mediator 126 can catalyze the oxidative deamination of benzylic primary amines to homo-coupled imines in the presence of CuCl and air.112,113 Cross-coupling of benzylic amines with aniline derivatives was also demonstrated with selectivity of greater than 25:1 for the cross coupled product when anilines bearing electron donating groups were utilized (Scheme 49).
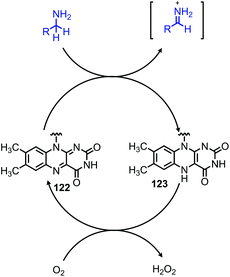 |
| | Scheme 48 Mechanism of the MAO flavin co-factor. | |
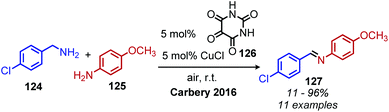 |
| | Scheme 49 Oxidative conversion of benzylic amines to cross-coupled imines with anilines. | |
4.3 Biomimetic conversion of amines to carbonyl and imine compounds by metalloporphyrin complexes
Cytochrome P450 (CYP) is an enzyme involved in biological oxidation reactions with a heme cofactor.114 While CYP does not perform oxidative deamination in nature, biomimetic metalloporphyin complexes have long been exploited by chemists for a variety of oxidative transformations.114 Ji and co-workers reported the use of manganese(III) meso-tetraphenylporphyrin chloride (MnTPPCl) for the deaminative homo-coupling of primary amines with tert-butyl hydropyroxide (t-BuOOH) as the oxidant (Scheme 50).115,116 Non-benzylic primary amines typically undergo homo-coupling in lower yields but using MnTPPCl, hexylamine was homo-coupled in 96% yield. Monitoring the reaction by UV-vis spectroscopy revealed a decrease of the MnTPPCl metalloporphyrin absorption band, replaced by a new absorbance assigned to a Mn(V) oxo complex upon the addition of t-BuOOH and benzylamine to the reaction solution. This intermediate complex subsequently oxidizes the primary amine to an imine.
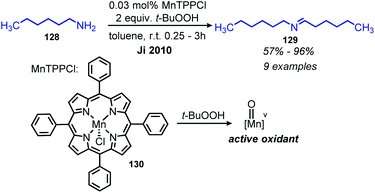 |
| | Scheme 50 Deaminative oxidative homo-coupling using biomimetic MnTPPCl. TPP = tetraphenylporphyrin. | |
The Chen group has also demonstrated that cobalt(II) β-tetrakis-(trifluoromethyl)-meso-tetraphenylporphyrin (CoTPP(CF3)4) is an effective catalyst for deaminative homo-coupling of benzylic amines in the presence of 6 atm dioxygen, excluding the need for t-BuOOH. Their proposed mechanism invokes a reaction between CoTPP(CF3)4 and dioxygen to form an active oxo intermediate species, which reacts with primary amines in an analogous manner to MnTPPCl. This cobalt oxo species is stabilized by steric protection from the trifluoromethyl groups.117
4.4 Organic oxidants for the conversion of amines to carbonyl and imine compounds
A variety of organic oxidants can be utilized for conversion of primary amines to imines, which may undergo subsequent deamination reactions. One well known oxidant, the hypervalent iodine(V) reagent o-iodoxybenzoic acid (IBX), has been shown to oxidize both primary and secondary benzylic amines to imines (Scheme 51).118,119 The substrate scope was limited to benzylic primary amines, and the resulting imines undergo in situ deaminative hydrolysis to produce carbonyl compounds. The proposed mechanism involves attack of the primary amine at iodine resulting in intermediate 134 which rearranges to give an imine and the iodine(III) by-product.
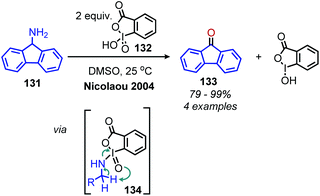 |
| | Scheme 51 Oxidative deamination with o-iodoxybenzoic acid (IBX). | |
N-tert-Butylsulfinimoyl chloride is another organic oxidant which, notably, has been applied to the oxidation of non-benzylic primary amines to imines.120 Initial studies showed the reagent was effective at oxidizing secondary aliphatic amines but was only able to oxidize primary aliphatic amines in low yield, likely due to the instability to the resulting alkyl imine species. Thus, primary amines were first mesylated with methanesulfonyl chloride to produce a more stable corresponding imine species upon oxidation (Scheme 52). However, linear mesylated primary amines remained unstable to oxidation. In order to address this, an additional procedure was developed in which primary amines were converted to secondary amines through reductive amination with cyclohexyl ketone, followed by oxidization and hydrolysis to yield aldehyde products (Scheme 53).120
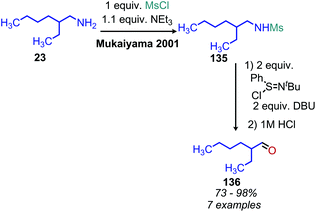 |
| | Scheme 52 Oxidative deamination with N-tert-butylsulfinimoyl chloride. Ms = methanesulfonyl. | |
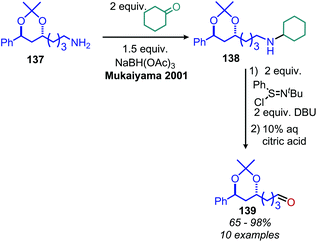 |
| | Scheme 53 Oxidative deamination with N-tert-butylsulfinimoyl chloride. Ms = methanesulfonyl. | |
The Mejia group has also demonstrated that the stable 1,4-allyl-tetramethylpyrazine radical cation species, generated from the corresponding dihydropyrazine in the presence of air, can be used for the oxidative homo-coupling of benzylic amines to dibenzylic imines (Scheme 54).121 Mejia and coworkers expanded the oxidative deamination reaction to include both homo- and cross-coupled imine products using phenazine radical cation as the oxidant (Scheme 55).122 Mechanistic studies including EPR spectroscopy demonstrated the presence of α-amino radicals, leading them to propose a mechanism involving SET oxidation of the primary amine by the phenazine radical cation followed by proton abstraction by a hydroperoxyl radical (Scheme 56).
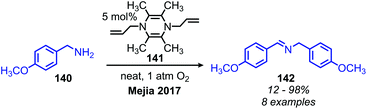 |
| | Scheme 54 Oxidative homocoupling of benzylic amines with a pyrazine derived radical cation. | |
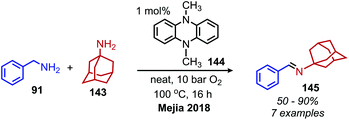 |
| | Scheme 55 Oxidative cross-coupling of benzylic amines with phenazine derived radical cation. | |
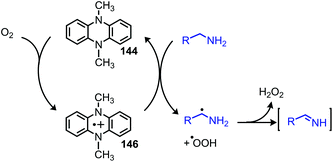 |
| | Scheme 56 Proposed mechanism for oxidative coupling with a phenazine radical cation. | |
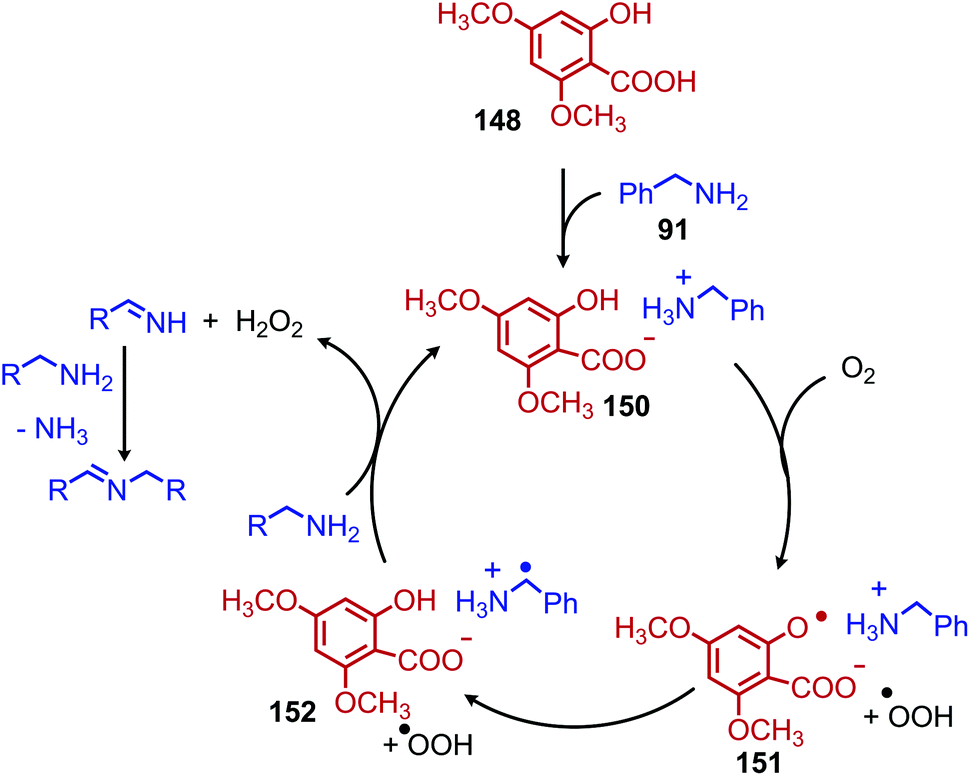
Another organocatalyst for the deaminative homo-coupling of primary benzylic amines has been disclosed by Ogawa and coworkers and is derived from salicylic acid (Scheme 57).123 The reaction demonstrated functional group tolerance for both electron withdrawing and donating functional groups as well as heterocycles. Mechanistic experiments demonstrated that the intermediate salt 150 could catalyze the reaction when it was prepared separately and added with benzylamine (1 equiv.), leading the authors to propose an organocatalytic cycle in which 150 is oxidized by dioxygen to form a phenoxy radical and a hydroperoxyl radical which then undergo subsequent hydrogen abstraction reactions (Scheme 58).
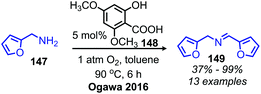 |
| | Scheme 57 Organocatalyzed oxidative deaminative homo-coupling with a salicylic acid derivative. | |
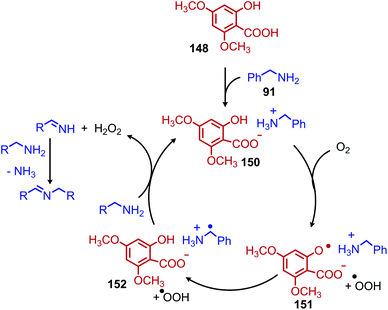 |
| | Scheme 58 Proposed organocatalytic cycle using a salicylic acid derivative. | |
4.5 Mechanical oxidants and catalytic metal compounds for the conversion of amines to carbonyl and imine compounds
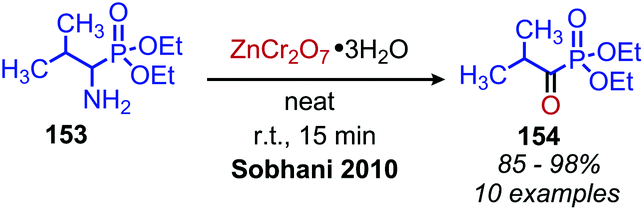
Analogous to organic oxidants, there are also a variety of metal oxidants that can be utilized for oxidative deamination. Early reports demonstrated that stoichiometric metal oxidants including KMnO4124,125 and K2FeO4126 were effective at converting aliphatic primary amines to carbonyl compounds. However, these methods have the obvious drawback of generating large quantities of metal waste and often suffered from poor yield. A more recent report by Sobhani and coworkers showed ZnCr2O7·3H2O oxidizes primary amines to ketones and aldehydes in 90% yields and aminophosphonates to ketones in 85–98% yield (Scheme 59) in the solid phase upon grinding the reagents.127
 |
| | Scheme 59 Solid phase oxidation of aminophosphonates to ketones with ZnCr2O7·3H2O. | |
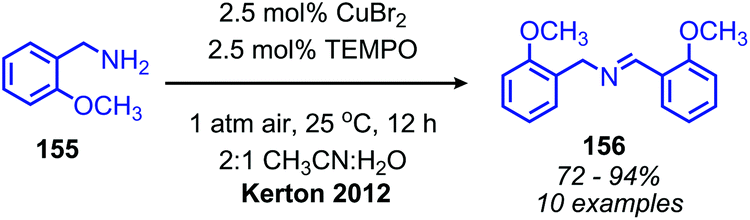
Recent research has focused heavily on eliminating stoichiometric metal oxidants in favor of utilizing readily available oxygen or hydrogen peroxide for oxidative deamination. The oxidative conversion of amines to imines can be mediated by a wide range of metal catalysts, which has been reviewed elsewhere.20,128,129 We highlight here examples involving homogenous first-row transition metal catalysts, which have garnered substantial interest recently due to their abundance and affordability. Patil and Adimurthy have shown that a 0.5 mol% loading of copper(I) chloride catalyst in neat benzylic primary amine can be used for the oxidative synthesis of homo-coupled imines at 100 °C.130 They also demonstrate deaminative cross-coupling of benzylic primary amines with substituted anilines. An example using milder temperature conditions for oxidative coupling of benzylic amines to dibenzylic imines with catalytic copper(II) bromide and co-catalytic TEMPO was disclosed by the Kerton group (Scheme 60).131 The proposed mechanism is based on a related Cu(II)-TEMPO system for aerobic alcohol oxidation and invokes coordination of the amine and TEMPO to the copper centre (Scheme 61). Subsequent hydrogen atom abstraction from the amine by TEMPO results in a stabilized radical species which dissociates to form the imine product. The Xu group has additionally found that copper(I) iodide can also be utilized as the catalyst at room temperature with co-catalytic TEMPO.132
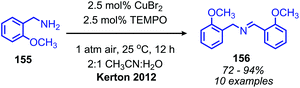 |
| | Scheme 60 Co-catalytic CuBr and TEMPO catalyzed oxidative homo-coupling. TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl. | |
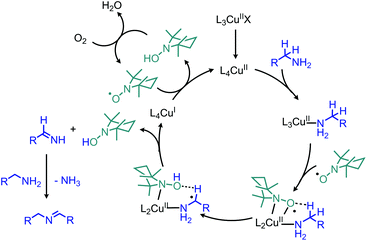 |
| | Scheme 61 Proposed mechanism for oxidative deamination with catalytic CuBr and TEMPO. X = Br; L = X, H2O, CH3CN. | |
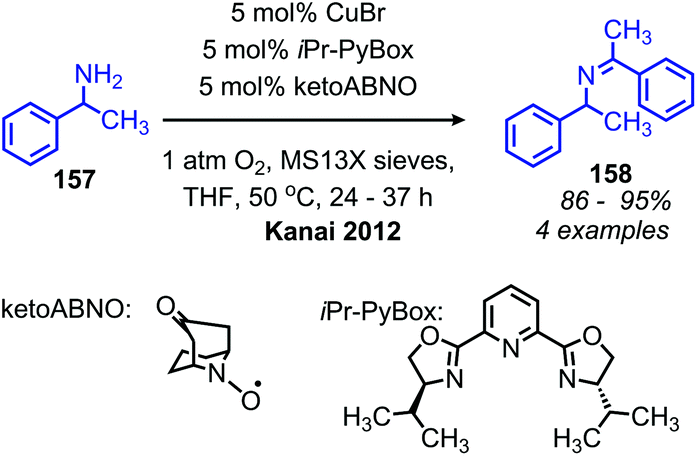
The Kanai group has also disclosed the use of a new N-oxyl radical compound ketoABNO (9-Azabicyclo[3,3,1]nonan-3-one-9-oxyl) as a co-catalyst with copper(I) bromide for the oxidative coupling of benzylic amines (Scheme 62).133 KetoABNO is inductively more electron-poor and less sterically hindered than TEMPO, which makes it a more effective mediator of C–H abstraction from the α-carbon of primary amines. Thus, oxidative homocoupling of the α-secondary primary amine substrate 1-phenylethylamine is achieved, a transformation that outside the scope of the other copper-catalyzed methods.
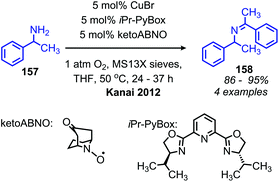 |
| | Scheme 62 Co-catalytic CuBr and ketoABNO catalyzed oxidative homo-coupling. | |
Arndtsen and collaborators have also explored the product selectivity for oxidative deamination between the formation of dibenzylic imines versus oxidation to benzylic nitriles.134 Catalytic copper(I) hexafluorophosphate in the presence of 1 atm molecular oxygen was shown to favor the formation of dibenzylic imines, while copper(I) iodide catalyst resulted in the synthesis of nitrile products (Scheme 63). The hexafluorophosphate supported imine complex is more ionic in nature and thus the electrophilicity of the copper centre favors nucleophilic attack by another amine molecule. The resulting copper(I) aminal species then liberates the dibenzylic imine product. In contrast, the iodide supported complex is more electron rich at copper and the coordinated imine may undergo an additional oxidation to form the nitrile product.
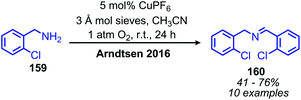 |
| | Scheme 63 CuPF6 catalyzed deaminative homo-coupling. | |
Iron is another earth-abundant metal that has been used catalytically in oxidative deamination reactions with dioxygen as the terminal oxidant. Analogous to their report utilizing catalytic copper, Xu and coworkers have demonstrated that catalytic Fe(NO3)3 and co-catalytic TEMPO can be used for the deaminative homo-coupling of benzylic amines.135 Gopalaiah and Saini reported using catalytic iron(II) bromide and molecular oxygen without TEMPO for the deaminative homo- and cross-coupling of primary amines under solvent-free conditions.136 Notably, the alkyl amine substrates cyclohexylamine and octylamine were homo-coupled in 61% and 53% yield, respectively. They also demonstrate deaminative cross-coupling of benzylic amines with alkyl amines (Scheme 64). The Minakawa group has also disclosed the conversion of benzylic primary amines to ketones and aldehydes using catalytic FeCl3·6H2O, formaldehyde and water as the solvent.137
 |
| | Scheme 64 FeBr2 catalyzed deaminative cross-coupling. | |
Two recent examples of copper and iron mediated oxidative deamination involve a catalyst which is supported by inorganic ligands, namely polyoxometalates (POMs).138 POM ligands provide a stable environment for metal catalysts to undergo aerobic oxidation.139 The POM-ligand supported copper catalyst of the form (NH4)3–4[CuMo6O18(OH)6] was demonstrated facilitate the oxidative homo-coupling of benzylic amines to imines in the presence of dioxygen.139
4.6 Photocatalyzed oxidative deamination
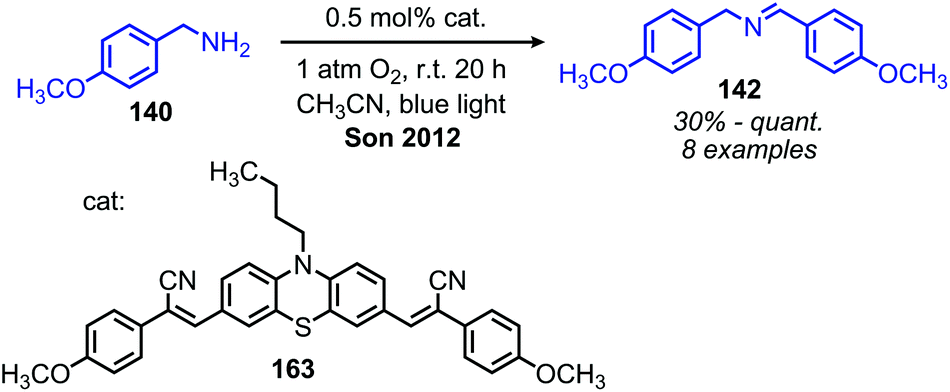
In addition to the photocatalytic radical deamination described in section 3.3, photocatalysis has also been utilized for oxidative deamination chemistry. The use of titanium dioxide and other heterogenous photocatalysts for the conversion of primary amines to imines has been reviewed by Gao and collaborators.20 We highlight here several examples which demonstrate the use of homogenous photocatalysts for oxidative deamination. Son and collaborators disclosed the use of blue-light absorbing phenothiazine dyes for the deaminative homo-coupling of benzylic amines (Scheme 65).140 The excited-state dye is proposed to reduce dioxygen to superoxide, generating a stable cationic phenothiazine radical which then oxidizes the primary amine substrate to an amino radical cation (Scheme 66). Subsequent reactions between superoxide and oxidized amine result in the production of homo-coupled imines and hydrogen peroxide, which was detected by NMR spectroscopy. The Rueping group has also reported photocatalyzed homo-coupling of benzylic primary amines using fac-Ir(ppy)3PF6 under ambient atmosphere.141
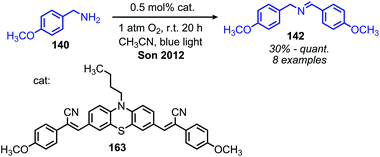 |
| | Scheme 65 Photocatalytic oxidative deamination of benzylic amines with a phenothiazine dye. | |
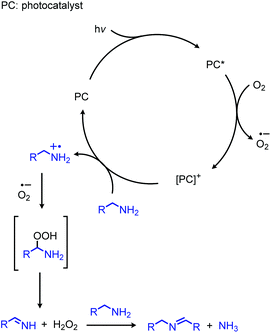 |
| | Scheme 66 Proposed mechanism for photocatalyzed oxidative deamination with phenothiazine. | |
In addition to electron transfer processes, excited-state chromophores can also be used to transfer energy to dioxygen via sensitization, resulting in singlet oxygen. Berlicka and König have demonstrated that 2,7,12,17-tetrapropylporphycene (H2TPrPc) can be used to convert primary and secondary amines to imines via a photosensitization mechanism (Scheme 67).142 The substrate scope included benzylic amines with electron donating groups and the reaction also could tolerate branching at the α-carbon, but benzylic amines with electron withdrawing groups only resulted in a trace conversion. The proposed mechanism involves the production of singlet oxygen by energy transfer from H2TPrPc which then reacts with amines, resulting in the formation of homo-coupled imines, hydrogen peroxide and ammonia (Scheme 68).
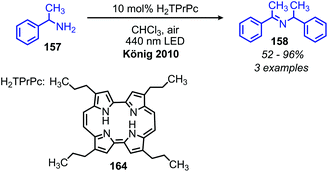 |
| | Scheme 67 Porphycene photocatalyzed deaminative oxidative coupling. H2TPrPc = 2,7,12,17-tetrapropylporphycene. | |
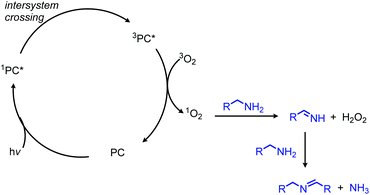 |
| | Scheme 68 Proposed mechanism for photocatalyzed deamination with a porphycene via sensitization. | |
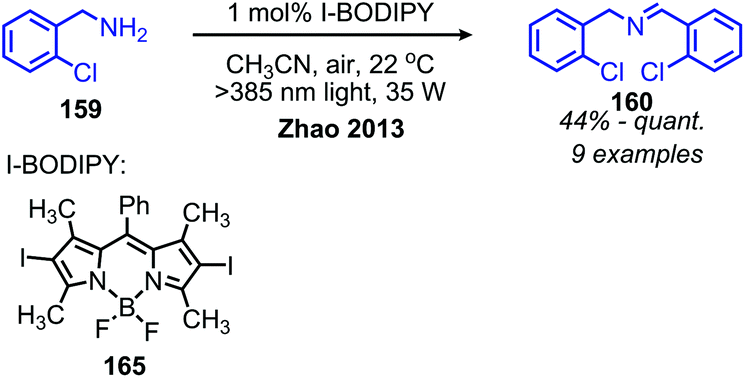
The Zhao group has also demonstrated that iodo-BODIPY derivates can be used as photosensitizers for the deaminative homo-coupling of benzylic amines (Scheme 69).143 This reaction displayed an improved functional group tolerance for benzylic primary amines bearing electron withdrawing functional groups compared to the report by Berlicka and König. Additional work by Son and collaborators disclosed the design of an iridium photosensitizer containing a bis(imidazoline selone) ligand with improved efficiency for the oxidative homo-coupling of primary amines (Scheme 70).144 This efficiency can be attributed to both the increased visible light absorbance of the complex and the presence of a selenium containing ligand which is able to enhance the excited triplet state due to the heavy atom effect, resulting in the increased production of singlet oxygen.144 The substrate scope demonstrated good tolerance for electron-withdrawing functional groups, with 4-chlorobenzylamine and 4-fluorobenzylamine coupled in 89% and 76% yield, respectively. The α-branched substrate 1-phenylethylamine was also coupled in 61% yield.
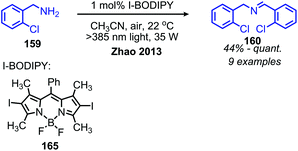 |
| | Scheme 69 Iodo-BODIPY photosensitized deaminative oxidative coupling. BODIPY = boron-dipyrromethene difluoride. | |
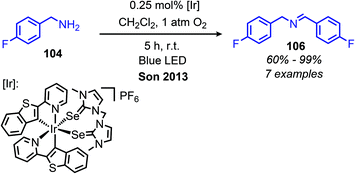 |
| | Scheme 70 Photocatalyzed oxidative homo-coupling of benzylic amines using an iridium bis-(imidazoline selone) complex. | |
4.7 Other oxidative deamination reactions using molecular oxygen or hydrogen peroxide
While catalytic metal complexes and photocatalysts have generated considerable interest for oxidative deamination reactions, several groups have demonstrated that it is possible convert primary amines to imines without these catalysts. The Fu group has shown that 10 mol% azobisisobutyronitrile (AIBN) radical initiator can be used to initiate peroxy radical reactions which facilitate the deaminative homo-coupling of primary benzylic amines.145 Functional group tolerance for both electron donating and withdrawing groups, heterocycles and the α-secondary primary amine benzhydrylamine was demonstrated. Monopoli and collaborators have demonstrated that the ionic liquids tetraethylammonium acetate and tetramethylammonium bromide (melting points 50 °C and 100 °C, respectively) can facilitate the deaminative oxidative homo- and cross-coupling of benzylic primary amines in the presence of molecular oxygen without any catalyst.146 The Wu group has also used ionic liquids to facilitate the conversion of benzylic amines to aldehydes with hydrogen peroxide as the oxidant.147,148 Fu and coworkers disclosed an additional method for catalyst-free homo- and cross-coupling of benzylic amines to dibenzylic imines by refluxing in water.149 The best conversion was observed when the reaction mixture was a biphasic with the volume of water eight times that of the volume of amine.
4.8 Applications of oxidative deamination
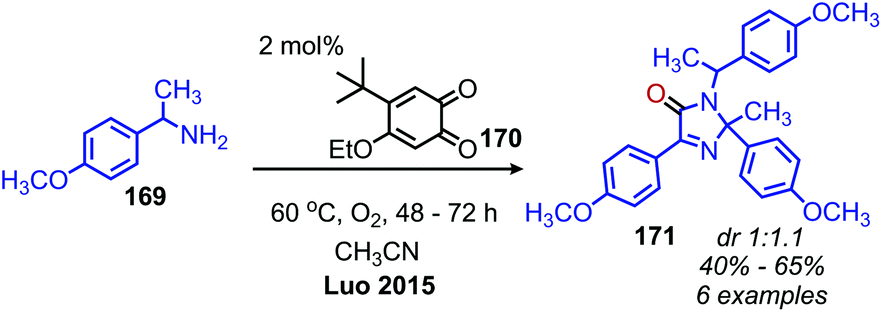
Given the numerous methods available for the oxidative conversion of amines to carbonyl and imine compounds, research has been focused on utilizing these reactions as a stepping stones in the conversion of amines to other functional groups. Chandrasekhar and coworkers demonstrated that Pd/C was an effective catalyst for the oxidative conversion of amines to aldehydes. Using this procedure, they then developed a one-pot Wittig reaction to convert the product aldehydes to α,β-unsaturated esters.150 Subsequent hydrogenation of the reaction mixture resulted in the isolation of C(sp3) hybridized product esters. The Fu group also reported extracting the dibenzylic imine product from their catalyst-free oxidative deaminative homo-coupling reaction (vide supra) and adding Danishefsky's diene, resulting in an aza Diels–Alder synthesis of an N-alkyl-4-pyridone (Scheme 71).149 Oxidative trimerization of benzylic primary amines has been demonstrated by Luo and collaborators using a biomimetic ortho-quinone catalyst (Scheme 72).151 The dibenzylic imine product was demonstrated to undergo an additional reaction to yield an imidazolinone when the temperature was increased from room temperature to 60 °C.
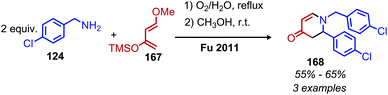 |
| | Scheme 71 Oxidative deaminative aza-Diels Alder synthesis of N-alkyl-4-pyridones. TMS = trimethylsilane. | |
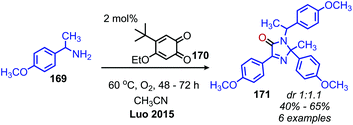 |
| | Scheme 72 Deaminative oxidative trimerization of primary amines to an imidazolinone. dr = diastereomeric ratio. | |
Lu and collaborators have demonstrated photocatalyzed oxidative deamination of 2-phenylcyclohexanamine derivatives to carboxylic acid products by ring opening and loss of formamide using rose Bengal and compact fluorescent light bulbs in dioxane (Scheme 73a).152 Interestingly, when the light sources was changed to green LEDs, lactones were observed as the products in DMF (Scheme 73b). They propose that changing the solvent and light source may result in the increased production of singlet oxygen by sensitization. This causes the enolate tautomer of the carboxylic acid product to undergo another oxidization and cyclization to the lactone by release of hydrogen peroxide.
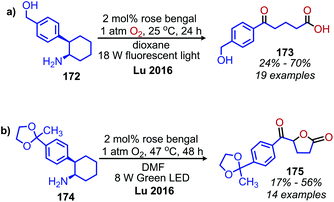 |
| | Scheme 73 Oxidative conversion of primary amines to (a) carboxylic acids and (b) lactones. | |
The synthesis of thioamides was also reported by the Nguyen group using elemental sulfur and two different primary amines, a benzylic and an aliphatic primary amine (Scheme 74).153 The substrate scope included heterocycles, an unprotected primary alcohol and one example of an α-secondary benzylic amine. The benzylic amine is preferentially oxidized over the aliphatic amine by sulfur, resulting in the release of hydrogen sulfide and the production of a benzylic imine. This imine intermediate then undergoes deaminative cross-coupling with the aliphatic primary amine to produce a disubstituted imine which reacts with elemental sulfur to form the thioamide product.
 |
| | Scheme 74 Oxidative deaminative synthesis of thioamides with elemental sulfur. | |
Several recent examples have demonstrated that imines produced by deaminative oxidation can serve as intermediates in four component Ugi reactions. Sharma and collaborators reported a one-pot oxidative deaminative homo-coupling of benzylic amines using IBX and reaction with an isocyanide and a carboxylic acid to form a bis(amide) product.154 A similar one-pot procedure has been reported by Batra and coworkers, who employed catalytic Fe(NO3)3 and co-catalytic TEMPO to facilitate the oxidative deamination step.155 The Ogawa group has shown that deaminative four component Ugi reactions can be expanded to use cross-coupled imine moieties as intermediates in a one-pot reaction using molecular oxygen and the salicylic acid derived organocatalyst 2,4,6-trihydroxybenzoic acid (Scheme 75).156
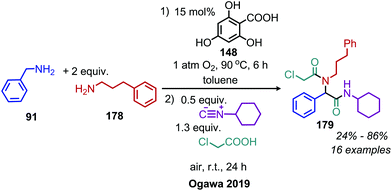 |
| | Scheme 75 One-pot oxidative deaminative four component Ugi reaction utilizing cross-coupled imines. | |
Utilizing oxidative deamination to synthesize heterocycles has generated considerable interest in recent years due to the prevalence of these motifs in bioactive compounds.157 Benzimidazoles can be produced from the deaminative coupling of primary amines with o-phenylenediamine derivatives.157 As the wide variety of deaminative oxidation procedures that have been used in the synthesis of benzimidazoles was recently reviewed by Largeron in 2018,157 we highlight here one illustrative example of the transformation reported by the Largeron group in 2016. Using a biomimetic topaquinone-like catalyst and co-catalytic CuBr, primary alkyl amines were successfully coupled with substituted o-phenylenediamine derivatives to produce 1,2-disubstituted benzimidazoles (Scheme 76).158
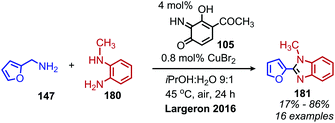 |
| | Scheme 76 Deaminative synthesis of 1,2-disubstituted benzimidazoles. | |
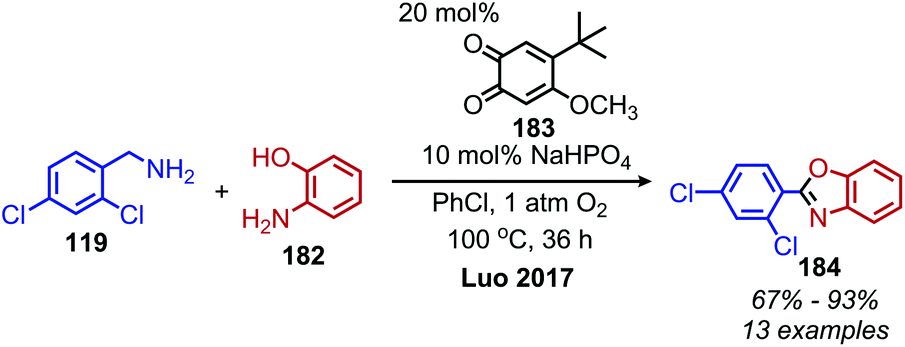
In an analogous manner, benzoxazoles can also be produced from oxidative deaminative coupling of primary amines with 2-aminophenol derivatives. Luo and collaborators synthesized benzimidazoles and benzoxazoles with primary benzylic amine substrates using a biomimetic ortho-quinone catalyst (Scheme 77).159 Additionally when 2-phenylethanamine derivatives were used as the primary amine substrate and coupled with o-phenylenediamines, they observed quinoxaline heterocycles as the product (Scheme 78).159 The Sawant group have also reported the synthesis of benzothiazole derivatives from the deaminative oxidation of benzylic primary amines and 2-aminothiophenols using the oxidant K2S2O8 (Scheme 79).160 Zhou and Yang have also demonstrated the synthesis of benzothiazoles by deaminative oxidation of primary amines using visible light and the photocatalyst BODIPY, which acts as a sensitizer for dioxygen.161 The Wu group has also reported the synthesis of quinazolinone derivatives from benzylic primary amines and o-aminobenzamides using hydrogen peroxide as the oxidant (Scheme 80).162
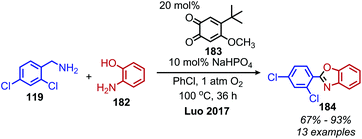 |
| | Scheme 77 Deaminative synthesis of benzoxazoles. | |
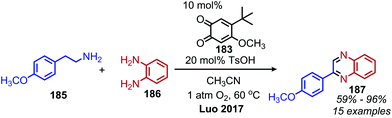 |
| | Scheme 78 Deaminative synthesis of quinoxalines. | |
 |
| | Scheme 79 Deaminative synthesis of benzothiazoles. | |
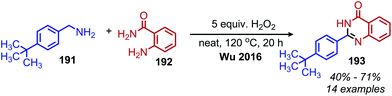 |
| | Scheme 80 Deaminative synthesis of quinazolinones. | |
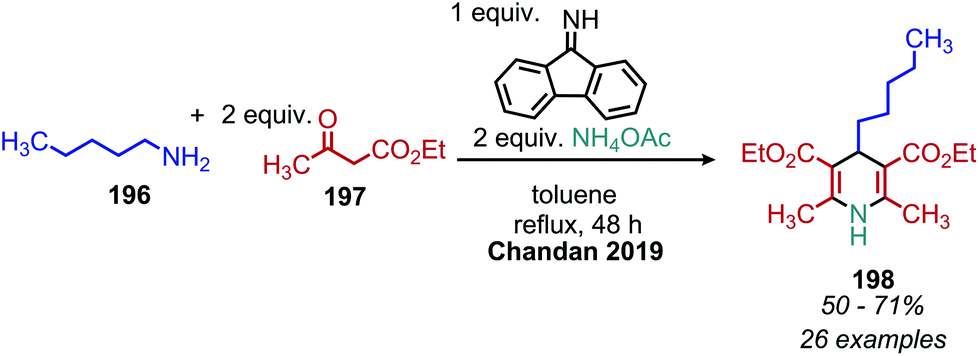
The deaminative coupling of primary amines with electron-rich C(sp3) moieties can be utilized for the synthesis of alkene products via oxidative olefination. 2-Methylquinoline derivatives are known to undergo benzylic C(sp3) activation by an aldol type mechanism.163 Wang and collaborators demonstrated the oxidative olefination of 2-methylquinoline derivatives using benzylic primary amines, catalytic NBS and the oxidant tert-butylhydroperoxide (Scheme 81). The proposed mechanism involves an aldol-type condensation between either the initially generated aldimine or its aldehyde hydrolysis product. Mao et al. also reported oxidative olefination of 2-methylquinoline using air as the oxidant to first convert the benzylic amines to aldehydes. Lanthanum pentafluorobenzoate was used as a Lewis acid catalyst to promote the aldol condensation between the benzylic aldehyde and 2-methylquinoline, resulting in the 2-alkenylquinoline products.164 Oxidative olefination of primary amines with active methylene compounds such as diethyl malonate was achieved by Ghosh and Jana who utilized a biomimetic flavin derivative 9-fluorenone imine.165 Additionally, when alkyl acetoacetates were coupled with benzylic or aliphatic primary amines in the presence of ammonium acetate, dihydropyridines were produced (Scheme 82). Oxidative olefination of primary amines with the activated methylene compounds cyanoacetamide, malononitrile and ethylcyanoacetate was also reported by the Suryavanshi group using the hypervalent iodide oxidant (diacetoxyiodo)benzene (PIDA).166
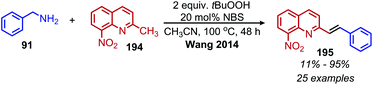 |
| | Scheme 81 Deaminative oxidative olefination of 2-methylquinoline with NBS and t-BuOOH. NBS = N-bromosuccinimide. | |
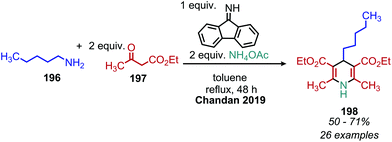 |
| | Scheme 82 Deaminative oxidative synthesis of 1,2-dihydropyridines. | |
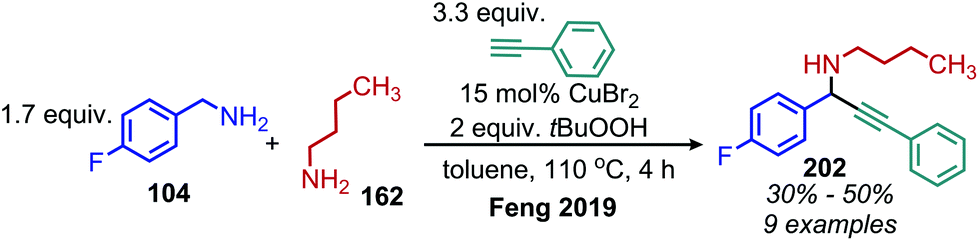
α-Cyanoepoxides are typically prepared in two steps by a Knoevenagel condensation and subsequent oxidation mediated by a hypochlorite anion.167 Work from the Seeberger group has demonstrated a one-pot synthesis of α-cyanoepoxides by deaminative oxidative olefination of primary amines and subsequent in situ oxidation of the olefin intermediate to the epoxide product (Scheme 83).167 Feng and collaborators have disclosed the synthesis of secondary propargylamines by an oxidative deamination and alkynylation reaction involving catalytic copper and tert-butylhydroperoxide (Scheme 84).168 Homo-coupled benzylic imines were successfully alkynylated with acetylenes bearing both electron-donating and withdrawing functional groups. Cross-coupled imines generated from benzylic and aliphatic amines were also alkynylated when the amount of t-BuOOH was increased from 1.2 to 2 equivalents.
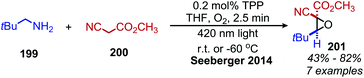 |
| | Scheme 83 One-pot oxidative deaminative synthesis of α-cyanoepoxides. | |
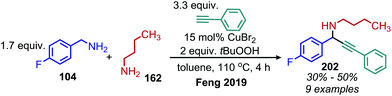 |
| | Scheme 84 Oxidative deaminative alkynylation with t-BuOOH. | |
5. Additional transition metal catalyzed deamination reactions
Transition metal catalyzed methodologies are an atom economic way to mediate difficult transformations. Despite the longstanding interest in transition metal catalysis, reports of deamination reactions remain rare. In this section, we highlight several notable ruthenium and palladium catalyzed transformations which harness primary amines without any prior functionalization reactions required to enhance their nucleofugality (Scheme 85).
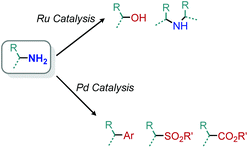 |
| | Scheme 85 Scope of transition metal catalyzed deamination reactions. | |
In 2013, the Milstein group reported an example of ruthenium pincer complex catalyzed conversion of primary aliphatic amines to primary and secondary alcohols using water and releasing ammonia (Scheme 86).169 This transformation is of significant importance because a simple nucleophilic substitution of an amine by water is highly unfavorable. The functional group tolerance for the ruthenium catalyzed reaction included α-primary and -secondary primary aliphatic amines with indole, chloride and methoxy moieties. However, α-tertiary primary amines failed to react. The ruthenium complex is thought to catalyze the dehydrogenation of amines to dihydrogen and an imine, which reacts with water to form an aldehyde that is subsequently reduced by the dihydrogen. The terminal diamino alkane substrates 1,4-diaminobutane and 1,6-diaminohexane undergo deaminative cyclization to the five- and seven-membered heterocycles pyrrolidine and azepane, respectively. They postulate that the first amine reacts with the catalyst to form an imine which then undergoes an entropically driven intramolecular attack by the second amine.
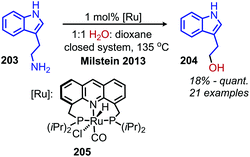 |
| | Scheme 86 Ruthenium catalyzed conversion of primary amines to alcohols using water. | |
Ruthenium complexes are known to catalyze the cleavage of secondary amines to primary amines.170 The Marciniec group has recently disclosed that benzylic and aliphatic primary amines can also be re-distributed to yield a secondary amine and ammonia using the complex [Ru(H)(Cl)(CO)(PCy3)2] (Scheme 87).171 The proposed mechanism involves β-hydride elimination from the α-CH group to yield a coordinated imine which undergoes attack by another primary amine molecule. The dialkyl imine species is consequently reduced, resulting in the C(sp3) hybridized secondary amine product (Scheme 88). A second example from Yi and co-workers using the tetranuclear ruthenium hydride complex [(PCy3)(CO)RuH]4(O)(OH)2 disclosed the synthesis of unsymmetrical secondary amines from re-distribution reactions of mixtures containing two primary amines, typically one benzylic and one non-benzylic (Scheme 89).172 The Yi group has further expanded the scope of this ruthenium catalyzed reaction include the deaminative synthesis of quinazolinones from primary benzylic amines and 2-aminobenzamides.173
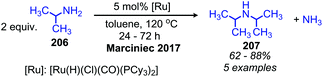 |
| | Scheme 87 Ruthenium catalyzed conversion of primary amines to secondary amines and ammonia. | |
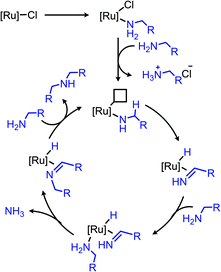 |
| | Scheme 88 Proposed catalytic cycle for the re-distribution of primary amines to secondary amines. | |
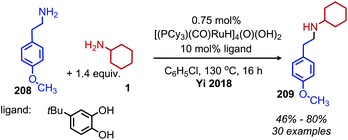 |
| | Scheme 89 Ruthenium catalyzed synthesis of unsymmetrical secondary amines. | |
While deaminative cross-coupling reactions have been successful by application of Katritzky pyridinium salts (vide supra), there are few reports of cross-coupling reactions that do not require an initial functionalization of the primary amine. One such example from Tian and coworkers demonstrates a palladium catalyzed coupling of aryl boronic acids with allylic primary amines.174 The product alkene exhibits retention of E-selectivity and an enantiospecific inversion of configuration was observed with chiral allylic primary amines when TMEDA was used as the ligand (Scheme 90). The authors propose a mechanism in which a palladium allyl intermediate is formed, analogous to the Tsuji–Trost reaction. An additional example from the Tian group demonstrated a deaminative synthesis of allylic sulfones from allylic primary amines using catalytic palladium, boric acid and sodium sulfinates.175 Addition of the ligand (R)-BINOL resulted in resulted in enantiospecific deamination with retention of configuration (Scheme 91).
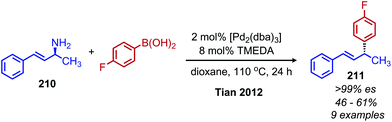 |
| | Scheme 90 Deaminative cross-coupling of allylic amines with boronic acids. Dba = dibenzylidenecaetone; TMEDA = N,N,N′,N′-tetramethylethylenediamine. | |
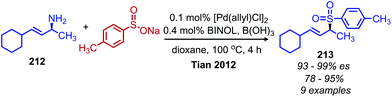 |
| | Scheme 91 Deaminative synthesis of allylic sulfones. BINOL = 1,1′-bi-2-naphthol. | |
Wu and coworkers have reported the conversion of benzylic primary amines to methyl 2-arylacetates using catalytic Pd2(dba)3, methanol and 1 bar CO (Scheme 92). This methodology was successfully used to synthesize methylphenidate (Ritalin) from a primary amine substrate.176
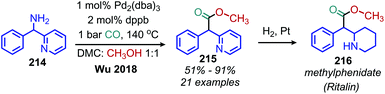 |
| | Scheme 92 Palladium catalyzed deaminative carbonylation with methanol and CO. Dppb = 1,4-bis(diphenylphosphino)butane. | |
6. Conclusions
This review has surveyed the wide-ranging and innovative strategies developed for deaminative functionalization reactions of primary alkyl amines including radical, polar, oxidative and transition metal catalyzed mechanisms. Clearly, classical reports have inspired new strategies for harnessing this relatively unreactive moiety into a new synthetic building block for the development of bioactive compounds. Recent applications of radical and polar deamination reactions have seen the catalytic conversion of primary amines into previous inaccessible functional groups including silanes, boranes, sulfides and unsaturated carbon substituents. The field of oxidative deamination has developed many different procedures for the conversion of amines to carbonyl and imine compounds with varying functional group tolerance, allowing researchers to select reaction conditions that are optimal to their compounds. Unsurprisingly, the downstream applications of these chemistries are rich and quickly growing.
Despite these recent advances, there are still challenges associated with this transformation and certainly more objectives to be realized. Namely, many methods are only or mostly effective for benzylic primary amine substrates, with decreased yields reported for aliphatic primary amines. This challenge has particularly plagued oxidative deamination reactions due to the instabilities of aliphatic imines. While recent cross-coupling and photocatalyzed reactions have been able to functionalize primary alkyl radicals, these methods suffer from the drawback of having to first functionalize the primary amine to a pyridinium salt. Ideally, more reactions for the direct conversion of primary amines without prior functionalization can be developed. Finally, sterically hindered α-tertiary primary amines remain largely intransigent, despite their prevalence in natural products. Very few methods have been able to address these moieties.
In summary, it is evident that research into aliphatic deamination chemistry will continue to develop in order to achieve the late-stage functionalization of natural products and pharmaceuticals. We hope that this review encourages the synthetic community to more seriously consider the potential of primary amines as aliphatic surrogates for an expanding host of applications.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by start-up funding from the University of Chicago. We thank Dr. Benjamin W. Rawe for helpful discussions.
References
- R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2019, 2769–2806 CrossRef CAS
 .
.
- S. Plunkett, C. H. Basch, S. O. Santana and M. P. Watson, J. Am. Chem. Soc., 2019, 141, 2257–2262 CrossRef CAS .
- B. Gál, C. Bucher and N. Z. Burns, Mar. Drugs, 2016, 14, 206 CrossRef .
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC .
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS .
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS .
- K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS .
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC .
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS .
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS .
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS .
- B. Mahjour, Y. Shen, W. Liu and T. Cernak, Nature, 2020, 580, 71–75 CrossRef CAS .
- Z. Yin, J.-X. Xu and X.-F. Wu, ACS Catal., 2020, 10, 6510–6531 CrossRef CAS .
- K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS .
- Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257–1272 RSC .
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC .
- M. E. Trusova, K. V. Kutonova, V. V. Kurtukov, V. D. Filimonov and P. S. Postnikov, Resour.-Effic. Technol., 2016, 2, 36–42 Search PubMed .
- N. Oger, M. d'Halluin, E. Le Grognec and F.-X. Felpin, Org. Process Res. Dev., 2014, 18, 1786–1801 CrossRef CAS .
- M. Bachrach, T. J. Marks and J. M. Notestein, New J. Chem., 2016, 40, 6001–6004 RSC .
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS .
- J. H. Bayless, F. D. Mendicino and L. Friedman, J. Am. Chem. Soc., 1965, 87, 5790–5791 CrossRef CAS .
- G. Wu, Y. Deng, C. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 10510–10514 CrossRef CAS .
- P. J. DeChristopher, J. P. Adamek, G. D. Lyon, J. J. Galante, H. E. Haffner, R. J. Boggio and R. J. Baumgarten, J. Am. Chem. Soc., 1969, 91, 2384–2385 CrossRef CAS .
- V. A. Curtis, F. J. Knutson and R. J. Baumgarten, Tetrahedron Lett., 1981, 22, 199–202 CrossRef CAS .
- F. S. Guziec and D. Wei, J. Org. Chem., 1992, 57, 3772–3776 CrossRef CAS .
- R. O. Hutchins, D. Kandasamy, F. Dux, C. A. Maryanoff, D. Rotstein, B. Goldsmith, W. Burgoyne, F. Cistone, J. Dalessandro and J. Puglis, J. Org. Chem., 1978, 43, 2259–2267 CrossRef CAS .
- H. Fang and M. Oestreich, Angew. Chem., 2020, 59, 11394–11398 CrossRef CAS .
- A. W. V. Hofmann and J. Clark, Philos. Trans. R. Soc. London, 1851, 141, 357–398 CrossRef .
- A. C. Cope, E. Ciganek, C. F. Howell and E. E. Schweizer, J. Am. Chem. Soc., 1960, 82, 4663–4669 CrossRef CAS .
- P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285 CrossRef CAS .
- D. M. Shacklady-McAtee, K. M. Roberts, C. H. Basch, Y.-G. Song and M. P. Watson, Tetrahedron, 2014, 70, 4257–4263 CrossRef CAS .
- C. H. Basch, K. M. Cobb and M. P. Watson, Org. Lett., 2016, 18, 136–139 CrossRef CAS .
- J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., 2020, 59, 1577–1580 CrossRef CAS .
- T. Moragas, M. Gaydou and R. Martin, Angew. Chem., Int. Ed., 2016, 55, 5053–5057 CrossRef CAS .
- W. Yu, S. Yang, F. Xiong, T. Fan, Y. Feng, Y. Huang, J. Fu and T. Wang, Org. Biomol. Chem., 2018, 16, 3099–3103 RSC .
- K. Ziegler and F. A. Fries, Ber. Dtsch. Chem. Ges., 1926, 59, 242 CrossRef .
-
A. T. Balaban, W. Schroth and G. Fischer, in Adv. Heterocycl. Chem, ed. A. R. Katritzky and A. J. Boulton, Academic Press, 1969, vol. 10, pp. 241–326 Search PubMed .
- S. L. Rössler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., 2020, 59, 9264–9280 CrossRef .
- Y. Pang, D. Moser and J. Cornella, Synthesis, 2020, 489–503 CAS .
- F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943–8960 CrossRef CAS .
- A. R. Katritzky, J. B. Bapat, R. J. Blade, B. P. Leddy, P.-L. Nie, C. A. Ramsden and S. S. Thind, J. Chem. Soc., Perkin Trans. 1, 1979, 418–425 RSC .
- A. R. Katritzky and G. Musumarra, Chem. Soc. Rev., 1984, 13, 47–68 RSC .
- E. H. White, J. Am. Chem. Soc., 1955, 77, 6011–6014 CrossRef CAS .
- N. S. MacArthur, L. Wang, B. G. McCarthy and C. E. Jakobsche, Synth. Commun., 2015, 45, 2014–2021 CrossRef CAS .
- G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS .
- T. Saegusa, S. Kobayashi, Y. Ito and N. Yasuda, J. Am. Chem. Soc., 1968, 90, 4182–4182 CrossRef CAS .
- D. H. R. Barton, G. Bringman, G. Lamotte, R. S. Hay Motherwell and W. B. Motherwell, Tetrahedron Lett., 1979, 20, 2291–2294 CrossRef .
- D. H. R. Barton, D. Ok Jang and J. C. Jaszberenyi, Tetrahedron Lett., 1992, 33, 5709–5712 CrossRef CAS .
- A. R. Katritzky and S. S. Thind, J. Chem. Soc., Perkin Trans., 1, 1981, 661–663 RSC .
- A. R. Katritzky, G. De Ville and R. C. Patel, Tetrahedron, 1981, 37, 25–30 CrossRef .
- A. R. Katritzky, M. A. Kashmiri, G. Z. De Ville and R. C. Patel, J. Am. Chem. Soc., 1983, 105, 90–96 CrossRef CAS .
- A. R. Katritzky, J.-l. Chen, C. M. Marson, A. Maia and M. A. Kashmiri, Tetrahedron, 1986, 42, 101–108 CrossRef CAS .
- A. R. Katritzky, K. Horvath and B. Plau, J. Chem. Soc., Chem. Commun., 1979, 300–301 RSC .
- A. R. Katritzky, K. Horvath and B. Plau, J. Chem. Soc., Perkin Trans. 1, 1980, 2554–2560 RSC .
- C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS .
- J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030–3033 CrossRef CAS .
- M. E. Hoerrner, K. M. Baker, C. H. Basch, E. M. Bampo and M. P. Watson, Org. Lett., 2019, 21, 7356–7360 CrossRef CAS .
- W. Guan, J. Liao and M. P. Watson, Synthesis, 2018, 3231–3237 CAS .
- K. M. Baker, D. Lucas Baca, S. Plunkett, M. E. Daneker and M. P. Watson, Org. Lett., 2019, 21, 9738–9741 CrossRef CAS .
- S.-Z. Sun, C. Romano and R. Martin, J. Am. Chem. Soc., 2019, 141, 16197–16201 CrossRef CAS .
- Z.-F. Zhu, J.-L. Tu and F. Liu, Chem. Commun., 2019, 55, 11478–11481 RSC .
- C.-L. Li, X. Jiang, L.-Q. Lu, W.-J. Xiao and X.-F. Wu, Org. Lett., 2019, 21, 6919–6923 CrossRef CAS .
- X. Zeng, W. Yan, S. B. Zacate, A. Cai, Y. Wang, D. Yang, K. Yang and W. Liu, Angew. Chem., 2020, 59, 16398–16403 CrossRef CAS .
- H. Yue, C. Zhu, L. Shen, Q. Geng, K. J. Hock, T. Yuan, L. Cavallo and M. Rueping, Chem. Sci., 2019, 10, 4430–4435 RSC .
- S. Ni, C.-X. Li, Y. Mao, J. Han, Y. Wang, H. Yan and Y. Pan, Sci. Adv., 2019, 5, eaaw9516 CrossRef CAS .
- J. Liao, C. H. Basch, M. E. Hoerrner, M. R. Talley, B. P. Boscoe, J. W. Tucker, M. R. Garnsey and M. P. Watson, Org. Lett., 2019, 21, 2941–2946 CrossRef CAS .
- R. Martin-Montero, V. R. Yatham, H. Yin, J. Davies and R. Martin, Org. Lett., 2019, 21, 2947–2951 CrossRef CAS .
- J. Yi, S. O. Badir, L. M. Kammer, M. Ribagorda and G. A. Molander, Org. Lett., 2019, 21, 3346–3351 CrossRef CAS .
- C.-G. Yu and Y. Matsuo, Org. Lett., 2020, 22, 950–955 CrossRef CAS .
- J. T. M. Correia, V. A. Fernandes, B. T. Matsuo, J. A. C. Delgado, W. C. de Souza and M. W. Paixão, Chem. Commun., 2020, 56, 503–514 RSC .
- F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS .
- X. Jiang, M.-M. Zhang, W. Xiong, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 2402–2406 CrossRef CAS .
- Z.-K. Yang, N.-X. Xu, C. Wang and M. Uchiyama, Chem. – Eur. J., 2019, 25, 5433–5439 CrossRef CAS .
- Z.-F. Zhu, M.-M. Zhang and F. Liu, Org. Biomol. Chem., 2019, 17, 1531–1534 RSC .
- M.-M. Zhang and F. Liu, Org. Chem. Front., 2018, 5, 3443–3446 RSC .
- Y. Xu, Z.-J. Xu, Z.-P. Liu and H. Lou, Org. Chem. Front., 2019, 6, 3902–3905 RSC .
- F. J. R. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2019, 9, 236–241 CrossRef CAS .
- D. Schönbauer, C. Sambiagio, T. Noël and M. Schnürch, Beilstein J. Org. Chem., 2020, 16, 809–817 CrossRef .
- M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS .
- X. Wang, Y. Kuang, S. Ye and J. Wu, Chem. Commun., 2019, 55, 14962–14964 RSC .
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS .
- F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem. – Eur. J., 2018, 24, 17210–17214 CrossRef CAS .
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS .
- M. Yang, T. Cao, T. Xu and S. Liao, Org. Lett., 2019, 21, 8673–8678 CrossRef CAS .
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS .
- J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697–5701 CrossRef CAS .
- C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Angew. Chem., 2020, 59, 7461–7466 CrossRef CAS .
- J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227–15231 CrossRef CAS .
- I. Kim, H. Im, H. Lee and S. Hong, Chem. Sci., 2020, 11, 3192–3197 RSC .
- F. Zhao, C.-L. Li and X.-F. Wu, Chem. Commun., 2020, 56, 9182–9185 RSC .
- J. Hu, B. Cheng, X. Yang and T.-P. Loh, Adv. Synth. Catal., 2019, 361, 4902–4908 CrossRef CAS .
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS .
- M. Largeron, Org. Biomol. Chem., 2017, 15, 4722–4730 RSC .
- J. P. Klinman, Chem. Rev., 1996, 96, 2541–2562 CrossRef CAS .
- Y. Lee and L. M. Sayre, J. Am. Chem. Soc., 1995, 117, 11823–11828 CrossRef CAS .
- S. M. Janes, D. Mu, D. Wemmer, A. J. Smith, S. Kaur, D. Maltby, A. L. Burlingame and J. P. Klinman, Science, 1990, 248, 981 CrossRef CAS .
- Y.-L. Hyun and V. L. Davidson, Biochemistry, 1995, 34, 816–823 CrossRef CAS .
- A. Roujeinikova, P. Hothi, L. Masgrau, M. J. Sutcliffe, N. S. Scrutton and D. Leys, J. Biol. Chem., 2007, 282, 23766–23777 CrossRef CAS .
- E. J. Corey and K. Achiwa, J. Am. Chem. Soc., 1969, 91, 1429–1432 CrossRef CAS .
- J. Srogl and S. Voltrova, Org. Lett., 2009, 11, 843–845 CrossRef CAS .
- V. G. Lisnyak and S. A. Snyder, J. Am. Chem. Soc., 2020, 142, 12027–12033 CrossRef CAS .
- M. Largeron, A. Neudorffer and M.-B. Fleury, Angew. Chem., Int. Ed., 2003, 42, 1026–1029 CrossRef CAS .
- M. Largeron, A. Chiaroni and M.-B. Fleury, Chem. – Eur. J., 2008, 14, 996–1003 CrossRef CAS .
- M. Largeron and M.-B. Fleury, Angew. Chem., Int. Ed., 2012, 51, 5409–5412 CrossRef CAS .
- A. E. Wendlandt and S. S. Stahl, Org. Lett., 2012, 14, 2850–2853 CrossRef CAS .
- M. Largeron and M.-B. Fleury, Chem. – Eur. J., 2015, 21, 3815–3820 CrossRef CAS .
- Y. Goriya, H. Y. Kim and K. Oh, Org. Lett., 2016, 18, 5174–5177 CrossRef CAS .
- G. Golime, G. Bogonda, H. Y. Kim and K. Oh, ACS Catal., 2018, 8, 4986–4990 CrossRef CAS .
- M. Largeron, P. Deschamps, K. Hammad and M.-B. Fleury, Green Chem., 2020, 22, 1894–1905 RSC .
- D. E. Edmondson, C. Binda, J. Wang, A. K. Upadhyay and A. Mattevi, Biochemistry, 2009, 48, 4220–4230 CrossRef CAS .
- H. Gaweska and P. F. Fitzpatrick, Biomol. Concepts, 2011, 2, 365–377 CAS .
- A. T. Murray, R. King, J. V. G. Donnelly, M. J. H. Dowley, F. Tuna, D. Sells, M. P. John and D. R. Carbery, ChemCatChem, 2016, 8, 510–514 CrossRef CAS .
- A. T. Murray, M. J. H. Dowley, F. Pradaux-Caggiano, A. Baldansuren, A. J. Fielding, F. Tuna, C. H. Hendon, A. Walsh, G. C. Lloyd-Jones, M. P. John and D. R. Carbery, Angew. Chem., Int. Ed., 2015, 54, 8997–9000 CrossRef CAS .
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS .
- Q.-L. Yuan, X.-T. Zhou and H.-B. Ji, Catal. Commun., 2010, 12, 202–206 CrossRef CAS .
- X.-T. Zhou, Q.-G. Ren and H.-B. Ji, Tetrahedron Lett., 2012, 53, 3369–3373 CrossRef CAS .
- S. Zhao, C. Liu, Y. Guo, J.-C. Xiao and Q.-Y. Chen, J. Org. Chem., 2014, 79, 8926–8931 CrossRef CAS .
- K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef CAS .
- K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, Angew. Chem., Int. Ed., 2003, 42, 4077–4082 CrossRef CAS .
- J.-i. Matsuo, A. Kawana, Y. Fukuda and T. Mukaiyama, Chem. Lett., 2001, 30, 712–713 CrossRef .
- R. Brisar, D. Hollmann and E. Mejia, Eur. J. Org. Chem., 2017, 5391–5398 CrossRef CAS .
- R. Brisar, F. Unglaube, D. Hollman, H. Jiao and E. Mejia, J. Org. Chem., 2018, 83, 13481–13490 CrossRef CAS .
- C.-P. Dong, Y. Higashiura, K. Marui, S. Kumazawa, A. Nomoto, M. Ueshima and A. Ogawa, ACS Omega, 2016, 1, 799–807 CrossRef CAS .
- S. S. Rawalay and H. Shechter, J. Org. Chem., 1967, 32, 3129–3131 CrossRef CAS .
- N. A. Noureldin and J. W. Bellegarde, Synthesis, 1999, 939–942 CrossRef CAS .
- R. J. Audette, J. W. Quail and P. J. Smith, Tetrahedron Lett., 1971, 12, 279–282 CrossRef .
- S. Sobhani and M. F. Maleki, Synlett, 2010, 383–386 CrossRef CAS .
- M. Largeron, Eur. J. Org. Chem., 2013, 5225–5235 CrossRef CAS .
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS .
- R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695–1700 CrossRef CAS .
- Z. Hu and F. M. Kerton, Org. Biomol. Chem., 2012, 10, 1618–1624 RSC .
- B. Huang, H. Tian, S. Lin, M. Xie, X. Yu and Q. Xu, Tetrahedron Lett., 2013, 54, 2861–2864 CrossRef CAS .
- T. Sonobe, K. Oisaki and M. Kanai, Chem. Sci., 2012, 3, 3249–3255 RSC .
- B. Xu, E. M. Hartigan, G. Feula, Z. Huang, J.-P. Lumb and B. A. Arndtsen, Angew. Chem., Int. Ed., 2016, 55, 15802–15806 CrossRef CAS .
- E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Org. Lett., 2013, 15, 2704–2707 CrossRef CAS .
- K. Gopalaiah and A. Saini, Catal. Lett., 2016, 146, 1648–1654 CrossRef CAS .
- M. Minakawa and T. Sasaki, Synlett, 2019, 30, 1597–1601 CrossRef CAS .
- Y. Zhai, M. Zhang, H. Fang, S. Ru, H. Yu, W. Zhao and Y. Wei, Org. Chem. Front., 2018, 5, 3454–3459 RSC .
- H. Yu, Y. Zhai, G. Dai, S. Ru, S. Han and Y. Wei, Chem. – Eur. J., 2017, 23, 13883–13887 CrossRef CAS .
- J. H. Park, K. C. Ko, E. Kim, N. Park, J. H. Ko, D. H. Ryu, T. K. Ahn, J. Y. Lee and S. U. Son, Org. Lett., 2012, 14, 5502–5505 CrossRef CAS .
- M. Rueping, C. Vila, A. Szadkowska, R. M. Koenigs and J. Fronert, ACS Catal., 2012, 2, 2810–2815 CrossRef CAS .
- A. Berlicka and B. König, Photochem. Photobiol. Sci., 2010, 9, 1359–1366 RSC .
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS .
- J. Jin, H.-W. Shin, J. H. Park, J. H. Park, E. Kim, T. K. Ahn, D. H. Ryu and S. U. Son, Organometallics, 2013, 32, 3954–3959 CrossRef CAS .
- L. Liu, Z. Wang, X. Fu and C.-H. Yan, Org. Lett., 2012, 14, 5692–5695 CrossRef CAS .
- A. Monopoli, P. Cotugno, F. Iannone, F. Ciminale, M. M. Dell'Anna, P. Mastrorilli and A. Nacci, Eur. J. Org. Chem., 2014, 5925–5931 CrossRef CAS .
- J.-L. Gong, X. Qi, D. Wei, J.-B. Feng and X.-F. Wu, Org. Biomol. Chem., 2014, 12, 7486–7488 RSC .
- X.-F. Wu, A. Petrosyan, T. V. Ghochikyan, A. S. Saghyan and P. Langer, Tetrahedron Lett., 2013, 54, 3158–3159 CrossRef CAS .
- L. Liu, S. Zhang, X. Fu and C.-H. Yan, Chem. Commun., 2011, 47, 10148–10150 RSC .
- S. Chandrasekhar, G. Pavan Kumar Reddy, C. Nagesh and C. Raji Reddy, Tetrahedron Lett., 2007, 48, 1269–1271 CrossRef CAS .
- Y. Qin, L. Zhang, J. Lv, S. Luo and J.-P. Cheng, Org. Lett., 2015, 17, 1469–1472 CrossRef CAS .
- X. Cheng, B. Yang, X. Hu, Q. Xu and Z. Lu, Chem. – Eur. J., 2016, 22, 17566–17570 CrossRef CAS .
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2012, 14, 4274–4277 CrossRef CAS .
- K. Singh, A. Kaur, V. S. Mithu and S. Sharma, J. Org. Chem., 2017, 82, 5285–5293 CrossRef CAS .
- S. U. Dighe, S. Kolle and S. Batra, Eur. J. Org. Chem., 2015, 4238–4245 CrossRef CAS .
- C.-p. Dong, A. Uematsu, S. Kumazawa, Y. Yamamoto, S. Kodama, A. Nomoto, M. Ueshima and A. Ogawa, J. Org. Chem., 2019, 84, 11562–11571 CrossRef CAS .
- M. Largeron and K. M. H. Nguyen, Synthesis, 2018, 50, 241–253 CrossRef CAS .
- K. M. H. Nguyen and M. Largeron, Eur. J. Org. Chem., 2016, 1025–1032 CrossRef CAS .
- R. Zhang, Y. Qin, L. Zhang and S. Luo, Org. Lett., 2017, 19, 5629–5632 CrossRef CAS .
- A. D. Hudwekar, P. K. Verma, J. Kour, S. Balgotra and S. D. Sawant, Eur. J. Org. Chem., 2019, 1242–1250 CrossRef CAS .
- Z. Zhou and W. Yang, Synth. Commun., 2014, 44, 3189–3198 CrossRef CAS .
- X.-X. Qi, Z.-Z. Song, J.-L. Gong, Z.-Y. Fang and X.-F. Wu, Chin. Chem. Lett., 2016, 27, 21–24 CrossRef CAS .
- L. Gong, L.-J. Xing, T. Xu, X.-P. Zhu, W. Zhou, N. Kang and B. Wang, Org. Biomol. Chem., 2014, 12, 6557–6560 RSC .
- D. Mao, X. Zhu, G. Hong, S. Wu and L. Wang, Synlett, 2016, 27, 2481–2484 CrossRef CAS .
- S. Ghosh and C. K. Jana, Org. Biomol. Chem., 2019, 17, 10153–10157 RSC .
- B. D. Rupanawar, S. M. Veetil and G. Suryavanshi, Eur. J. Org. Chem., 2019, 6232–6239 CrossRef CAS .
- D. B. Ushakov, K. Gilmore and P. H. Seeberger, Chem. Commun., 2014, 50, 12649–12651 RSC .
- H. Li, H. Feng, J. Zhang, E. V. Van der Eycken and L. Huang, J. Org. Chem., 2019, 84, 10501–10508 CrossRef CAS .
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 6269–6272 CrossRef CAS .
- D. Pingen, C. Altintas, M. Rudolf Schaller and D. Vogt, Dalton Trans., 2016, 45, 11765–11771 RSC .
- S. Kostera, B. Wyrzykiewicz, P. Pawluć and B. Marciniec, Dalton Trans., 2017, 46, 11552–11555 RSC .
- P. T. K. Arachchige, H. Lee and C. S. Yi, J. Org. Chem., 2018, 83, 4932–4947 CrossRef CAS .
- P. T. Kirinde Arachchige and C. S. Yi, Org. Lett., 2019, 21, 3337–3341 CrossRef CAS .
- M.-B. Li, Y. Wang and S.-K. Tian, Angew. Chem., Int. Ed., 2012, 51, 2968–2971 CrossRef CAS .
- X.-S. Wu, Y. Chen, M.-B. Li, M.-G. Zhou and S.-K. Tian, J. Am. Chem. Soc., 2012, 134, 14694–14697 CrossRef CAS .
- Y. Li, Z. Wang and X.-F. Wu, ACS Catal., 2018, 8, 738–741 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2021 |

 and
Mark D.
Levin
and
Mark D.
Levin