
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C(sp2)–H functionalization in non-aromatic azomethine-based heterocycles
Alexey A.
Akulov
 a,
Mikhail V.
Varaksin
ab,
Pieter
Mampuys
c,
Valery N.
Charushin
ab,
Oleg N.
Chupakhin
*ab and
Bert U. W.
Maes
*c
a,
Mikhail V.
Varaksin
ab,
Pieter
Mampuys
c,
Valery N.
Charushin
ab,
Oleg N.
Chupakhin
*ab and
Bert U. W.
Maes
*c
aDepartment of Organic & Biomolecular Chemistry, Ural Federal University, 19 Mira Str., 620002 Ekaterinburg, Russia. E-mail: chupakhin@ios.uran.ru
bInstitute of Organic Synthesis, Ural Branch of the Russian Academy of Sciences, 22 S. Kovalevskaya Str., 620990 Ekaterinburg, Russia
cDivision of Organic Synthesis, Department of Chemistry, University of Antwerp, Groenenborgerlaan 171, B-2020, Antwerp, Belgium. E-mail: bert.maes@uantwerpen.be
First published on 11th September 2020
Abstract
Direct C(sp2)–H functionalization of the endocyclic azomethine and aldonitrone moieties in non-aromatic azaheterocycles has established itself as a promising methodology over the last decade. Transition metal-catalyzed cross-coupling reactions, α-metalation–electrophile quenching protocols, and (metal-free) nucleophilic substitution of hydrogen reactions (SNH) are the major routes applied on cyclic imines and their derivatives. In this overview, we show the tangible progress made in this area during the period from 2008 to 2020.
 Alexey A. Akulov | Alexey A. Akulov obtained his MSc degree in Chemistry at Ural Federal University in 2020. In 2016, he joined the Department of Organic & Biomolecular Chemistry at the same university, where he is currently working as a research engineer. His scientific activities are focused on the C–H functionalization methodology development with regard to azaheterocyclic compounds and, in particular, on the elaboration of both novel transition metal-catalyzed and metal-free cross-dehydrogenative coupling approaches. |
 Mikhail V. Varaksin | Mikhail V. Varaksin graduated from the Ural State Technical University in 2008. In 2010, he earned his PhD in Organic Chemistry. In 2011, he joined the Department of Organic & Biomolecular Chemistry at Ural Federal University as an Associate Professor. His current research interests include the development of novel pot, atom, and step economical approaches based on the methodology of C–H bond functionalization in both (hetero)aromatic and non-aromatic substrates in the design of novel heterocyclic derivatives of nitroxide radicals, calixarenes, carboranes, and also advanced materials for molecular electronics. |
 Pieter Mampuys | Pieter Mampuys obtained his PhD in Organic Chemistry from UAntwerpen and the Vrije Universiteit Amsterdam under the supervision of Profs Bert Maes and Romano Orru in 2017. He also worked at Likat in Germany with Prof. Matthias Beller. In 2018, he was appointed as a post-doctoral researcher at UAntwerpen. His second post-doc is in collaboration with the company Covestro. His current research interests include base metal-catalyzed multicomponent chemistry, organometallics, sulfur chemisty, photoredox catalysis and green metrics analysis. |
 Valery N. Charushin | Valery N. Charushin is a full member of the Russian Academy of Sciences (RAS), Professor at the Department of Organic and Biomolecular Chemistry at Ural Federal University, and Head of Postovsky Institute of Organic Synthesis of the Ural Branch of RAS. Prof. Charushin graduated with honors from Ural Polytechnical Institute in 1973. In 1976, he earned his PhD in Organic Chemistry. In 1981–1982, he joined the research group of Prof. Henk van der Plas (Wageningen University, Wageningen, Netherlands), where he was working on the ring transformations of azines. In 1987, he received a Doctor of Science degree in Organic Chemistry. His current research interests include novel methods in organic synthesis, heterocyclic and medicinal chemistry. |
 Oleg N. Chupakhin | Oleg N. Chupakhin is a full member of the Russian Academy of Sciences, Professor and ex-Head of the Department of Organic & Biomolecular Chemistry at Ural Federal University, and Chief Researcher at Postovsky Institute of Organic Synthesis of Ural Branch of RAS. Prof. Chupakhin graduated with honors from Ural Polytechnical Institute in 1957. He earned his PhD in Organic Chemistry in 1962 and Doctor of Science degree in 1976. Current research interests: new methodologies in organic synthesis, structural analysis of organic compounds, mechanisms of organic reactions, heterocyclic chemistry, green chemistry, industrial chemistry, environmental chemistry, medicinal chemistry, chemosensors, molecular recognition and supramolecular chemistry. |
 Bert U. W. Maes | Bert U. W. Maes obtained his PhD in Organic Chemistry at UAntwerp and subsequently received a Post-Doctoral Fellowship of the Science Foundation (FWO-Flanders) in Belgium. He worked at the École Normale Supérieure in Paris (mechanisms in catalysis) with Prof. Anny Jutand (CNRS). Bert Maes was appointed Assistant Professor (Docent) in the Department of Chemistry at UAntwerp in 2003 and currently holds a Full Professorship (Gewoon Hoogleraar) of Organic Chemistry at UAntwerp; since 2009 he has been a Research Professor. In 2019 he was appointed as Collen-Francqui Research Professor by the Francqui Foundation. In 2015–2016 he acted as chairman of the Department of Chemistry. His research interests cover the fields of heterocyclic chemistry, organometallic chemistry and homogeneous catalysis with a special focus on the development of sustainable synthetic methodology. The research in his group involves base metal catalysis, aerobic oxidation, strong bond activation, renewables, and green metrics analysis. His research group partakes in one of the Excellence Centers of UAntwerp, i.e. CASCH. He is an editor of Topics in Heterocyclic Chemistry and an editorial board member of SynOpen and Advances in Heterocyclic Chemistry. |
1. Introduction
Nitrogen-containing heterocycles certainly belong to one of the most important classes of organic compounds, and their significance for the existence of any living system can hardly be overestimated. The accumulation of knowledge on these structures, which are widely spread in nature, has enhanced the progress in organic chemistry, as well as in other scientific fields, particularly in biomolecular chemistry, biology, medicinal chemistry, and materials science.1 Over the past century, a large number of synthetic methods to modify azaheterocyclic scaffolds have been elaborated, allowing one to obtain novel, often quite complicated molecules, useful for the direct synthesis of fine chemicals (e.g., pharmaceuticals or agrochemicals), dyes, and other industrially important compounds.2 Moreover, since the earliest stage of heterocyclic chemistry, azaaromatic compounds have been considered as very attractive structural motifs, based on their versatile practical applications. The synthesis of these compounds is generally realized through lengthy multi-step approaches.3 With the increasing awareness and importance of green chemistry,4 chemists need to develop alternative efficient synthetic sequences towards azaheterocyclic compounds in accordance with the principles of green chemistry encompassing the pot, atom, and step-economical (PASE) philosophy.5 In particular, the direct functionalization of C(sp2)–H bonds has gained significant interest of the scientific community as a general approach to construct C–C bonds between two pre-designed structural units.6 Such a methodology has the potential to perform better with respect to the principles of green chemistry in comparison to the very powerful classical cross-coupling reactions, which revolutionized the fine chemicals and specialty chemicals synthesis at both discovery and development levels.7 However, regioselectivity problems, directing groups, and requirements of specific [(super)stoichiometric] additives or solvents can hamper its green potential, so a holistic green metrics analysis objectively comparing the different processes, i.e. direct C(sp2)–H bond functionalization versus cross-coupling, will always be required for every specific case.4,8 This does not only involve the C–C bond forming reaction itself, but also how the reactants are synthesized.The progress in exploiting this methodology for the direct C(sp2)–H functionalization of non-aromatic azaheterocycles proved to be not so straightforward and well-studied in comparison with the field of (hetero)aromatic chemistry. However, in the last decade, significant headway in C(sp2)–H functionalization of non-aromatic azaheterocycles has been made. Interestingly, most compounds in those studies contain an endocyclic azomethine (i.e. –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) moiety. Cyclic azomethine derivatives of both natural and synthetic origin are known to exhibit profound biological activities or other practically useful properties.9–13 For instance, the cyclic aldimine nectrisine (Fig. 1a) isolated from the fungus Nectria lucida was found to induce the expression of antigen Ia, thus enabling the restoration of the immune response depressed by immunosuppressive factors of tumors. Nectrisine could also be used for the treatment of type 2 diabetes, since it exhibits α-glucosidase inhibitory activity.9Koranimine (Fig. 1b), another natural product with a cyclic imine moiety, belongs to the family of peptides obtained by non-ribosomal biosynthesis. Compounds of this type are likely to exhibit a wide spectrum of biological activities ranging from antibiotic to antitumor effects.10 Besides that, a number of cycloimines of natural origin are known for their fast-acting toxic effects; the latter include spirocyclic compounds secreted by marine shellfish, (e.g., gymnodimine A, Fig. 1c).11
N–) moiety. Cyclic azomethine derivatives of both natural and synthetic origin are known to exhibit profound biological activities or other practically useful properties.9–13 For instance, the cyclic aldimine nectrisine (Fig. 1a) isolated from the fungus Nectria lucida was found to induce the expression of antigen Ia, thus enabling the restoration of the immune response depressed by immunosuppressive factors of tumors. Nectrisine could also be used for the treatment of type 2 diabetes, since it exhibits α-glucosidase inhibitory activity.9Koranimine (Fig. 1b), another natural product with a cyclic imine moiety, belongs to the family of peptides obtained by non-ribosomal biosynthesis. Compounds of this type are likely to exhibit a wide spectrum of biological activities ranging from antibiotic to antitumor effects.10 Besides that, a number of cycloimines of natural origin are known for their fast-acting toxic effects; the latter include spirocyclic compounds secreted by marine shellfish, (e.g., gymnodimine A, Fig. 1c).11
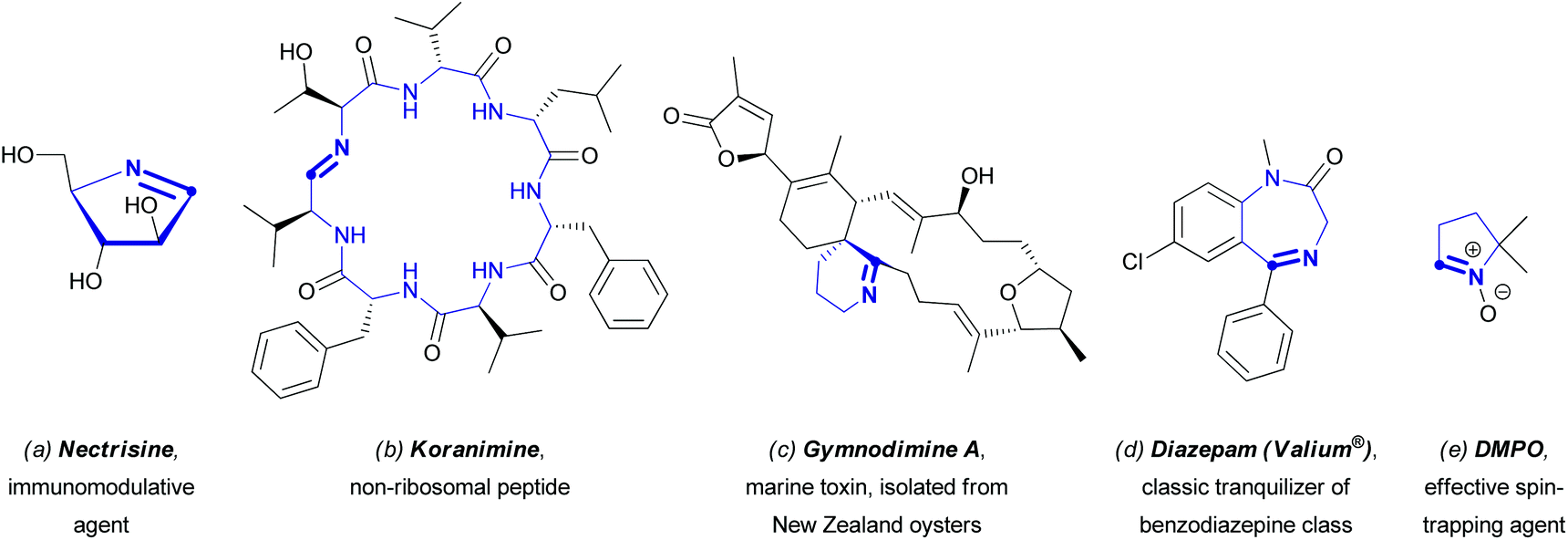 | ||
| Fig. 1 Selected examples of cyclic azomethine derivatives of specific interest for their application. | ||
As for compounds obtained synthetically, one has to highlight the family of psychoactive benzodiazepines used broadly in medicinal practice for the treatment of anxiety disorders, epilepsy, insomnia, and withdrawal syndrome (Fig. 1d shows diazepam, one of the most famous benzodiazepine tranquilizers). Another class of compounds that deserves special attention is the cyclic nitrones, many of which proved to be effective spin traps, used for detection and identification of free radicals,12 and also potential therapeutic agents, particularly nitric oxide donors.13 A classic example of a cyclic nitrone is 5,5-dimethylpyrroline N-oxide (DMPO, Fig. 1e).
Taking into account the versatile applications of cyclic imine derivatives, there is no doubt that the development of efficient synthetic approaches towards such compounds is of importance for modern organic chemistry. In this overview, we discuss in detail the novel approaches based on C(sp2)–H bond functionalization in non-aromatic azomethine-containing azaheterocycles that have been developed over the last decade. The reported methods can be classified into three general categories:
• transition metal-catalyzed C(sp2)–H bond functionalization of azomethine moieties;
• C(sp2)–H metalation of azomethine moieties (followed by treatment with electrophiles);
• nucleophilic substitution of hydrogen (SNH) at the sp2-carbon in azomethine moieties.
We should note that this survey does not embrace the C(sp2)–H activation in planar azomethine-based heterocycles, containing an aromatic resonance structure or aromatic tautomer, which can at least partly be regarded as aromatic ones (e.g., quinoxalin-2(1H)-ones, quinazolin-4(3H)-ones, pyrazin-2(1H)-ones, etc.). The vigorously developing chemistry of some of these valuable compounds has recently been highlighted in other review articles,14 and it keeps updating dynamically with new functionalization examples.15
1.1. Transition metal-catalyzed C(sp2)–H bond functionalization of azomethine moieties
The discovery of transition metal-catalyzed C–C bond forming cross-coupling reactions in the sixties and seventies has had a large impact on the disconnections currently made in organic synthesis. The outstanding achievements of R. Heck, A. Suzuki, E. Negishi, J. Stille, K. Sonogashira, and other distinguished scientists have contributed greatly to the progress in the chemistry of the functionalization of (hetero)aromatics.16 The first results in the ‘conventional’ cross-coupling chemistry involving AG-substituted cyclic azomethines (AG = halogen, SnAlk3, or other auxiliary groups) were reported in the early 1980s,17 whereas the analogous metal-catalyzed transformations exploiting the reactivity of the endocyclic azomethine C(sp2)–H bond remained unexplored until the late 2000s (vide infra).18 Notably, the C–H functionalization methodology can be realized here in two modes.The first approach involves a new carbon–carbon bond formation through the reaction of the imine C(sp2)–H with a (pseudo)halogenated coupling partner (Scheme 1a), while the second one encompasses the formation of a new bond via double C–H bond activation. The latter is also known as cross-dehydrogenative coupling (CDC), requiring an oxidant to obtain a catalytic process (Scheme 1b).19
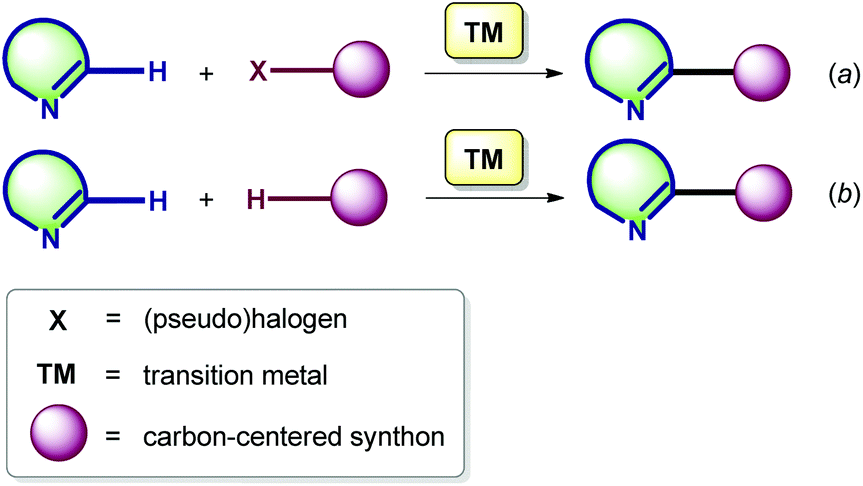 | ||
| Scheme 1 Two possible modes for the TM-catalyzed C(sp2)–H bond functionalization of azomethines. | ||
1.2. C(sp2)–H metalation of azomethine moieties
The inherent insufficient nucleophilicity of the carbon center prevents direct C–H functionalization of azomethine (R–NCH–R′) moieties via electrophilic agents. Therefore, C–C coupling reactions can only be performed by activating the azomethine C(sp2)–H bond first via metalation through deprotonation, e.g., using organolithium or organomagnesium reagents,20 followed by treatment with electrophiles (Scheme 2). The pioneering research studies in the field of electrophilic transformations of cyclic azomethine derivatives were carried out by Voinov & Grigor'ev et al. in the late 1990s.21 They succeeded in developing α-lithiation of cyclic aldonitrones 3 and 7a generating the corresponding lithium salts (e.g., 3-Li), which were reacted with electrophilic reactants, such as aldonitrones 3 as well as ketones/aldehydes 5 (Scheme 3).
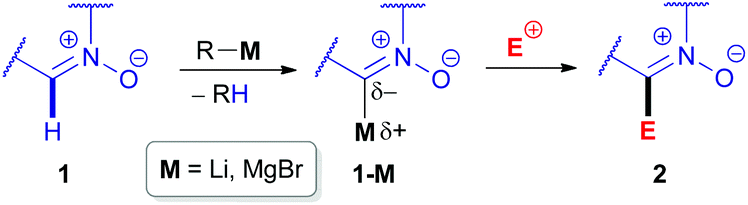 | ||
| Scheme 2 Metalation of the C(sp2)–H bond of a cyclic azomethine derivative (i.e. nitrone 1) followed by electrophilic substitution. | ||
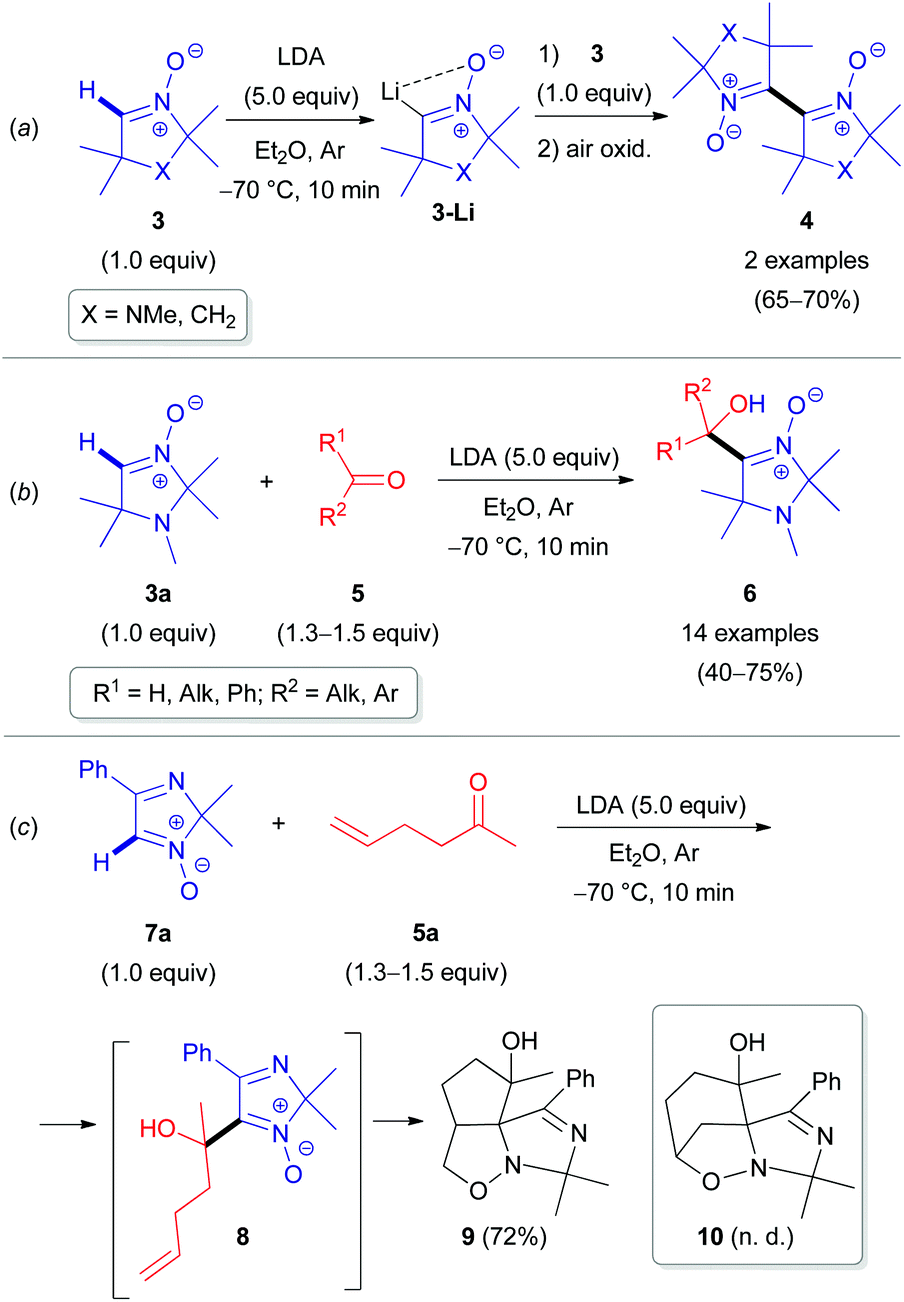 | ||
| Scheme 3 Selected examples of the coupling reaction of α-lithiated aldonitrones with electrophilic reactants. | ||
1.3. Nucleophilic substitution of hydrogen (SNH) in azomethine moieties
Nucleophilic substitution of hydrogen (SNH) has proved to be an efficient synthetic tool to modify (hetero)aromatic compounds with nucleophilic reactants since the seventies, and it was successfully exploited during the next four decades by many researchers.6a,22 Although a few scattered examples employing this strategy for non-aromatic unsaturated heterocycles date back to the early eighties,23 the majority of such SNH reactions have only been reported in the 21st century.A cyclic aldimine derivative is polarized and contains a partial positive charge at the sp2-hybridized carbon, and this center is therefore vulnerable to reaction with nucleophiles (Scheme 4). Nucleophilic substitution of hydrogen proceeds either via an ‘Addition–Elimination’ (SNH(AE), Scheme 4a) or an ‘Addition–Oxidation’ mechanism (SNH(AO), Scheme 4b).22h They both start with the addition of a nucleophilic reactant (Nu−) to the azomethine carbon of 11 to form an anionic σH-adduct 12. According to an SNH(AE) mechanism, subsequent spontaneous or externally-promoted elimination of the leaving group regenerates the azomethine π-bond. Selection of the elimination promoters determines the specific features of the SNH reaction used. On the other hand, via an oxidizing reagent, the equilibrium can also be shifted in favor of the SNH product 14, i.e. the oxidative SNH(AO) mechanism.
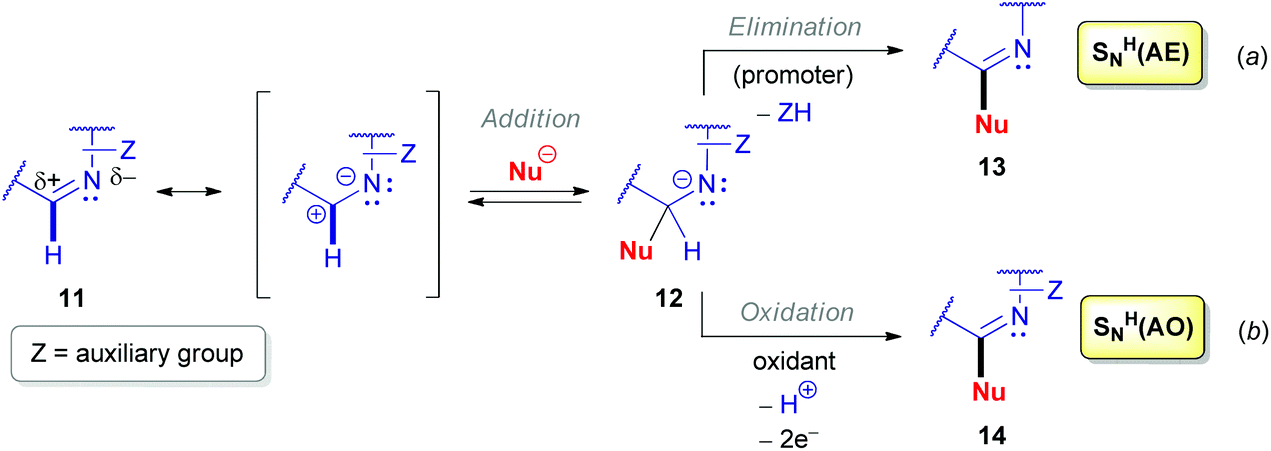 | ||
| Scheme 4 ‘Addition–Elimination’ (a) and ‘Addition–Oxidation’ (b) mechanisms for the nucleophilic substitution of hydrogen (SNH). | ||
Functionalization of azomethine derivatives involving an SNH(AE) pathway was not reported until the last decade.24 Notably, this ‘Addition–Elimination’ strategy has been frequently applied for the modification of (hetero)aromatic substrates, but not for saturated systems.22c In contrast, the alternative oxidative SNH(AO) protocols are well documented here, as illustrated by transformations of aldonitrones 15, 16a–b (Scheme 5).25 Transition metal oxides derived from manganese (Scheme 5b and c), nickel (Scheme 5a), lead,23a and mercury23b are commonly used as oxidizing agents. Oxidations employing air23c or hydrogen peroxide,25a supplemented with catalytic additives, which are more sustainable,26 were also exploited.
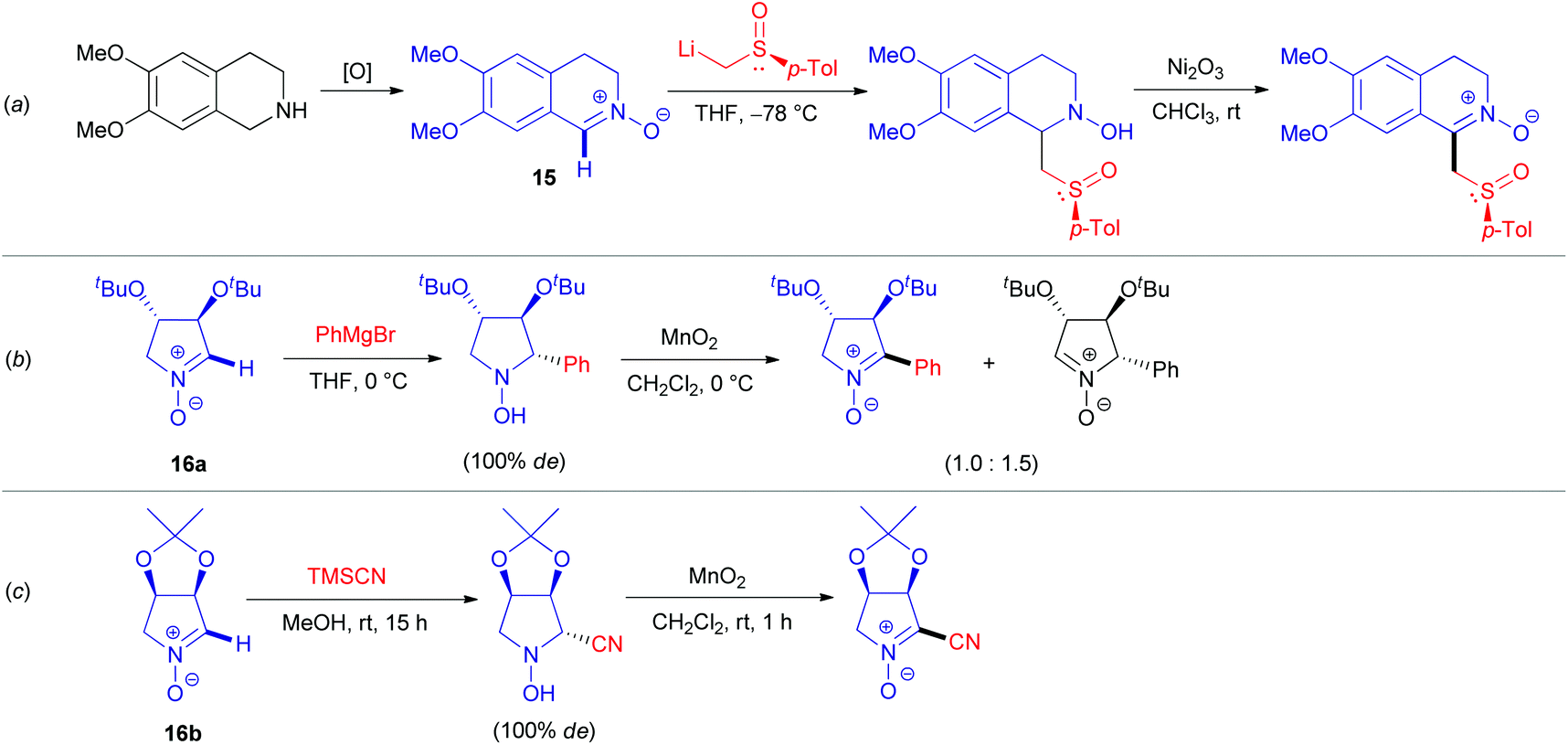 | ||
| Scheme 5 Selected examples of SNH(AO) reactions on cyclic azomethines. | ||
2. C(sp2)–H functionalization in cyclic aldimines and related compounds
The first report describing the direct arylation of a cyclic imine was published in 2008.18 A research team from the University of California (Berkeley) introduced a phenyl moiety at the 2-position of 4,4-dimethyloxazoline (17a), a non-aromatic compound (Scheme 6), in the presence of a rhodium source and (Z)-1-tert-butyl-2,3,6,7-tetrahydrophosphepine (20) as a ligand. The catalytic cycle proposed by the authors (Scheme 7) involves the initial formation of dimeric complex 21 from dimeric [RhCl(coe)2]2 and bidentate phosphepine ligand 20. Dissociation of this complex and coordination with the imidate nitrogen of 4,4-dimethyloxazoline (17a) are likely to promote the formation of intermediate 22 in a similar manner as described for heteroaromatic substrates with rhodium complexes, such as RhCl(PCy3)2.27 A low-valent electron-rich carbene-rhodium complex 23 is then generated via a C–H activation/tautomerization process.27 Subsequent oxidative addition with phenyl bromide 18a delivers a (phenyl)(carbene)rhodium complex 24. Elimination of HBr could then either occur via the amine base or via an intramolecular process, affording intermediate 25. Finally, reductive elimination results in the formation of 19a and regeneration of the rhodium catalyst.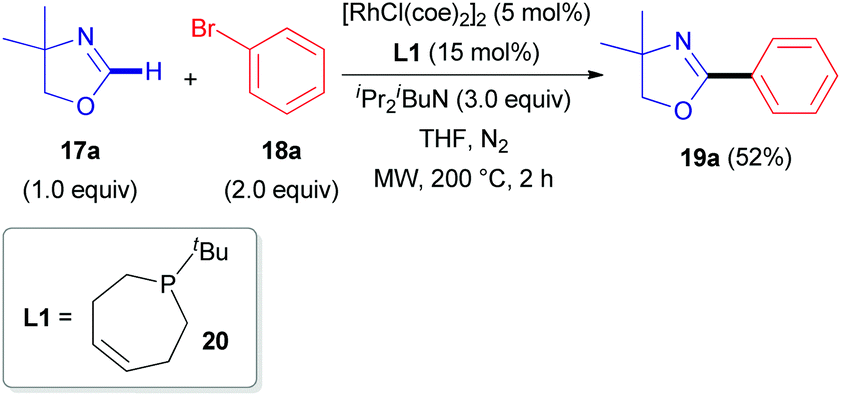 | ||
| Scheme 6 RhI-Catalyzed C(sp2)–H arylation of 4,4-dimethyloxazoline (17a). | ||
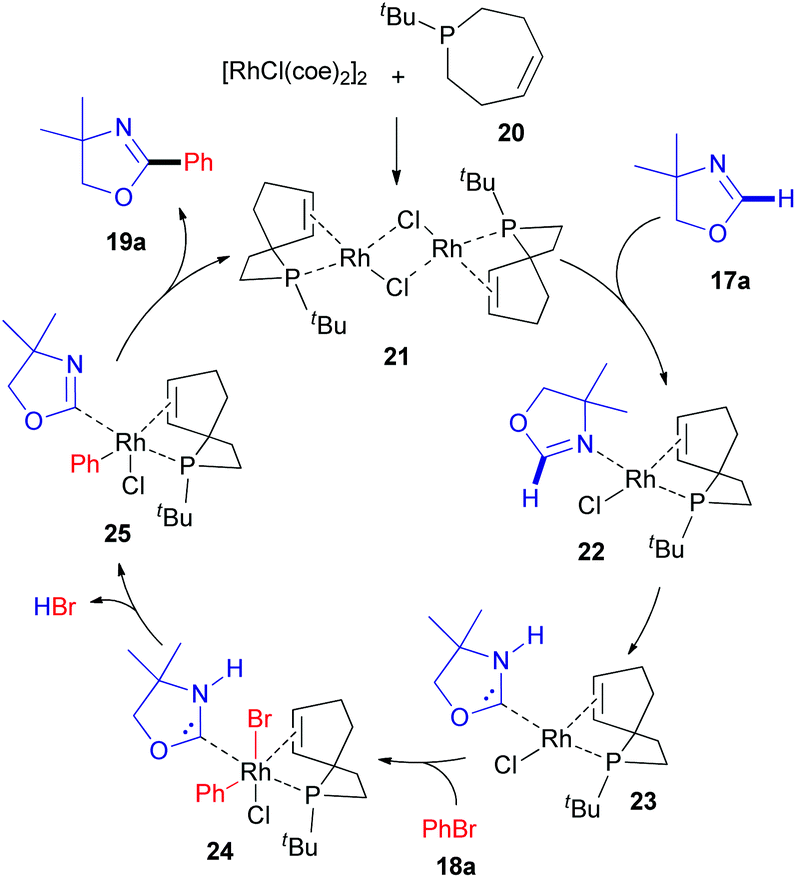 | ||
| Scheme 7 Proposed mechanism for the RhI-catalyzed C(sp2)–H arylation of 4,4-dimethyloxazoline (17a). | ||
In 2011, the Ackermann group reported a similar C(sp2)–H arylation of oxazolines 17 involving Pd rather than Rh catalysis (Scheme 8).28 A palladium complex 26 formed from Pd(OAc)2 and secondary phosphine oxide ‘pre-ligands’ were found to be suitable (Scheme 9). A series of arylated oxazolines (19) were successfully obtained, varying substituents in both the aryl and oxazoline moieties.
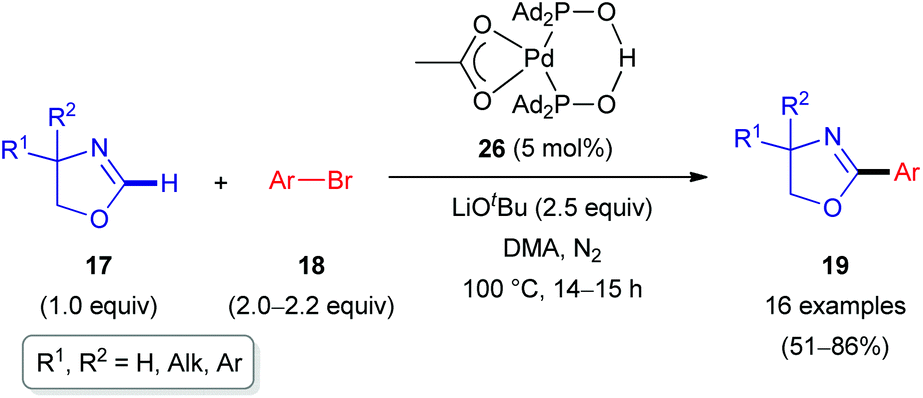 | ||
| Scheme 8 Pd-Catalyzed C(sp2)–H arylation of oxazolines (17). | ||
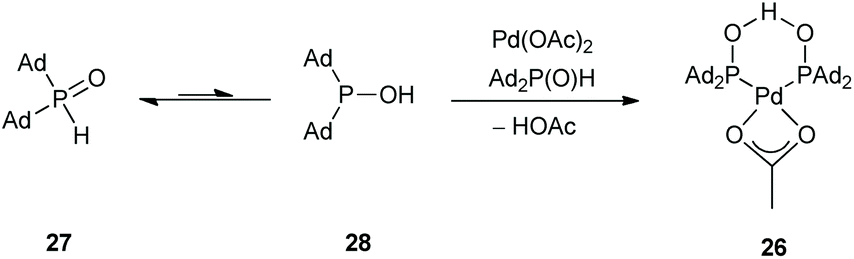 | ||
| Scheme 9 Preparation of Pd catalyst (26) for C(sp2)–H arylation of oxazolines (17). | ||
Another report on the successful C(sp2)–H arylation of non-aromatic heterocycles was published by the Ellman group.29 Herein, benzotriazepinones 29, which can be considered as cyclic hydrazones, were subjected to Cu-catalyzed C–H functionalization with a range of aryl iodides (30, Scheme 10). Being close analogues of pharmaceutically privileged benzodiazepines, these azaheterocyclic derivatives are of interest as biologically active substances. The reaction conditions were initially similar to those proposed by Daugulis for the arylation of aromatic C–H bonds (polyfluoroarene or heteroaromatic) with aryl iodides and bromides.30 However, under those conditions, arylated benzotriazepinone 31c was only obtained in 40% yield. To reach sufficient conversion, a longer reaction time (12 h) was required than that initially reported (10–30 min). Further optimization of the reactant ratio (29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) afforded a range of 5-aryl-substituted benzotriazepinones 31 in rather good yields (Scheme 10).
:30) afforded a range of 5-aryl-substituted benzotriazepinones 31 in rather good yields (Scheme 10).
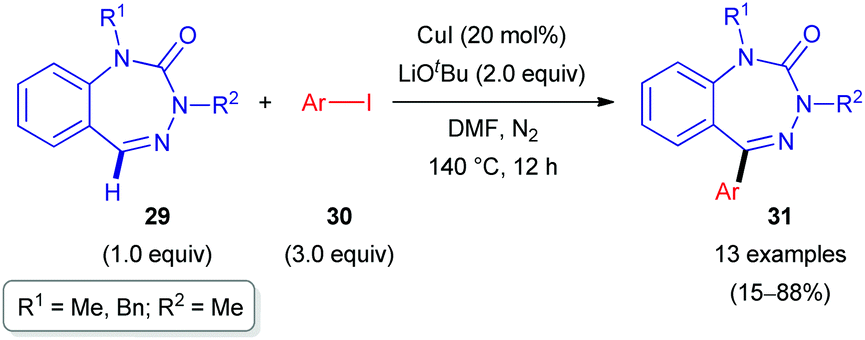 | ||
| Scheme 10 Cu-Catalyzed C(sp2)–H arylation of benzotriazepinones (29). | ||
Palladium in combination with copper catalysis was explored by the scientists from the University of Rouen (France). In 2015, they reported the direct arylation and olefination of imidazolones 32 (Scheme 11).31 Heterocycles of this class are present in nature and exhibit therapeutic and fluorescence properties.31 Aryl or alkenyl halides (33 and 35) were used as coupling partners with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base. It is noteworthy that such arylations and olefinations required different solvents (DMF or toluene) and phosphine ligands (PPh3 or P(o-Tol)3) to obtain appropriate yields of products (34 and 36, see Scheme 11). The potential of this strategy was illustrated for the synthesis of imidazolone-containing fatty acid synthase (FAS) inhibitors.31 This group also extended this approach for the C(sp2)–H functionalization of 4-arylidene imidazolones, enabling a library of GFP and Kaede protein-type fluorophores.32
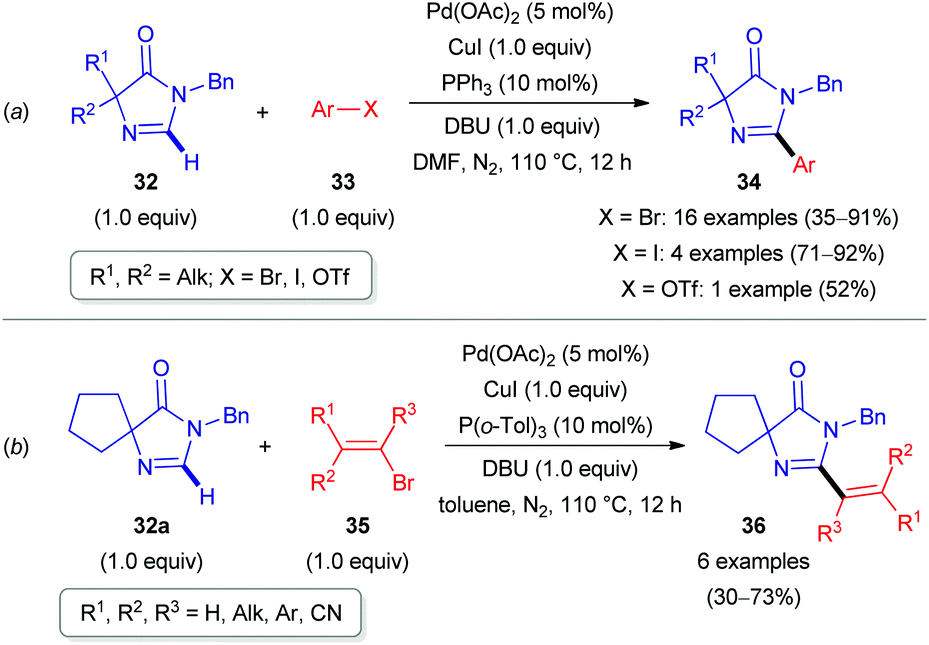 | ||
| Scheme 11 Pd-Catalyzed direct arylation and olefination of 4,4-dialkyl-1H-imidazol-5(4H)-ones (32). | ||
In 2012, researchers from the Zelinsky Institute of Organic Chemistry in Russia reported an atypical C-alkylation of 2-pyrazolines 38 with 2-(hetero)arylcyclopropane-1,1-dicarboxylates 37 in the presence of superstoichiometric gallium trichloride.33 A C,N-bis-alkylation product 39 was obtained as a major compound in refluxing dichloromethane (Scheme 12). However, performing this reaction at room temperature led to a greater proportion of byproducts (40 and 41). Interestingly, other Lewis acids, such as scandium and ytterbium triflates, predominantly resulted in the formation of cycloaddition product 41.34 This cycloadduct 41 is generated via an interaction of the N(2)-atom in 2-pyrazoline 38 with the electrophilic center of cyclopropane 37 after initial activation with a Lewis acid (Scheme 13). In contrast, activation of both reactants (37 and 38) seems to occur at higher temperature, which redistributes the electron density within the pyrazoline ring. This enhances the nucleophilicity at the C(3)-atom and results in the formation of 1-pyrazoline C(3)-adduct 42. Subsequent tautomerization via migration of the hydrogen from C(3)- to N(1)-position, followed by interaction with another equivalent of cyclopropane (37) generates the bis-alkylation product 39.
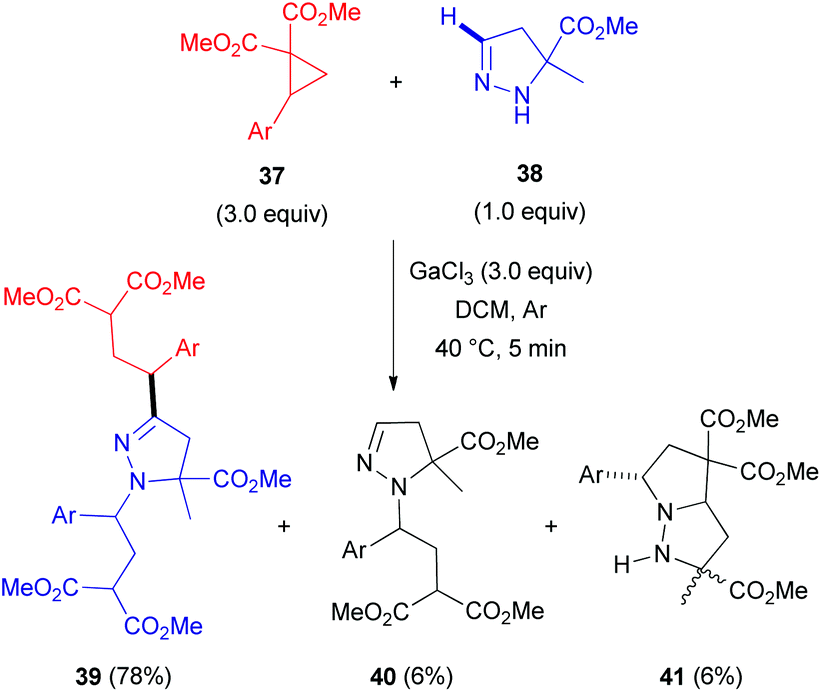 | ||
| Scheme 12 GaCl3-mediated coupling of 5-substituted 2-pyrazolines (38) with 2-arylcyclopropane-1,1-dicarboxylates (37). | ||
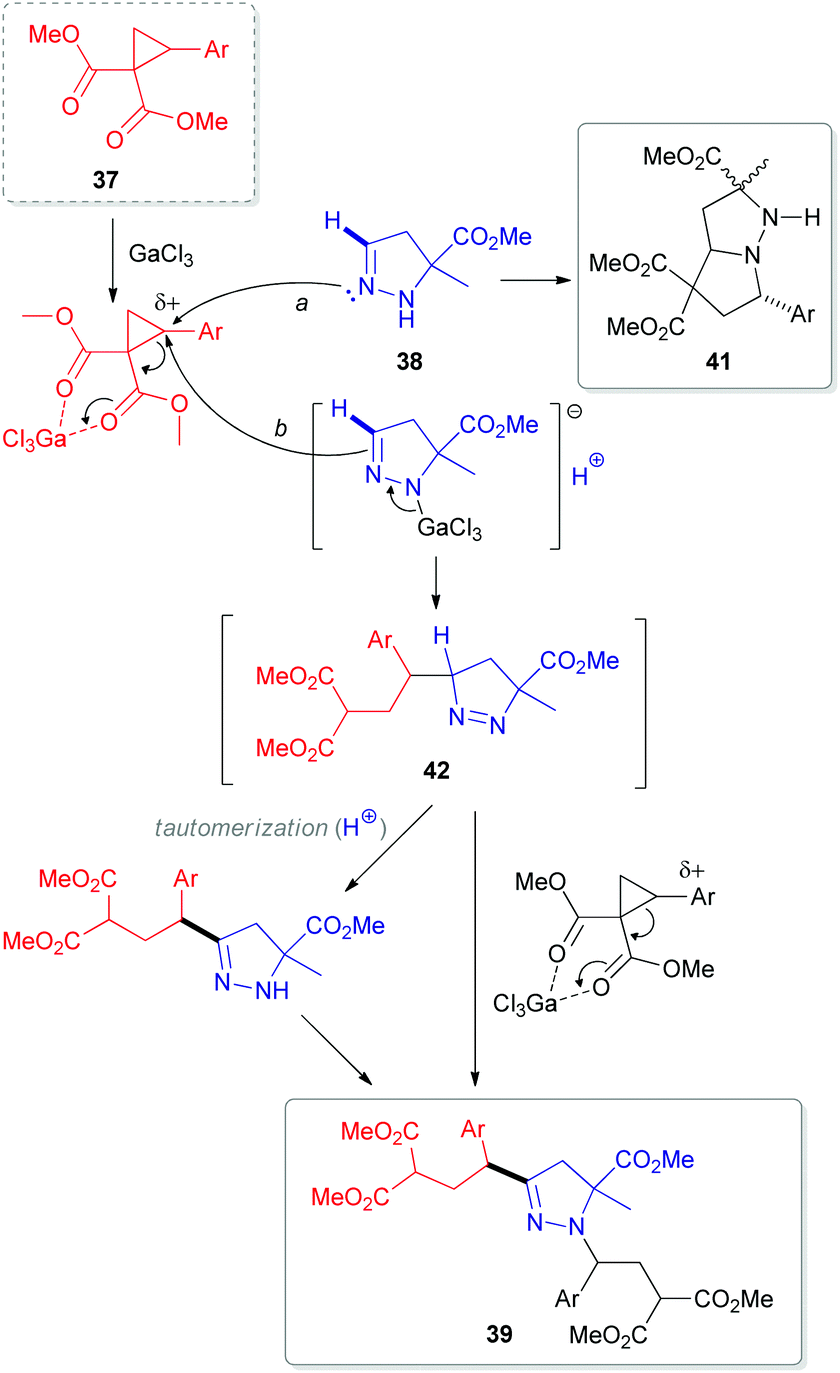 | ||
| Scheme 13 Proposed mechanism for a GaCl3-mediated coupling of 5-substituted 2-pyrazolines (38) with 2-arylcyclopropane-1,1-dicarboxylates (37). | ||
While analyzing the data of various studies focused on the C(sp2)–H functionalization of cyclic azomethines, the tendency to apply compounds of the aldonitrone family (featuring an additional N-oxide on the azomethine) as C–H substrates for these transformations became apparent.35 They are easily accessible via a multitude of procedures (e.g., oxidation reactions (Scheme 14a), condensation reactions of N-monosubstituted hydroxylamines (Scheme 14b), or reactions of oximes with electrophiles (Scheme 14c)).35 The N-oxide group of nitrones has been exploited by various research groups as an elegant directing group for C(sp2)–H functionalization of azomethines.6c In particular, significant attention has been paid to imidazolone N-oxides.
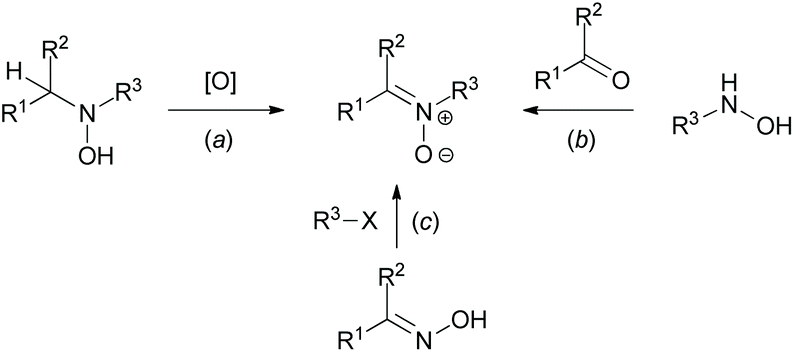 | ||
| Scheme 14 General routes towards nitrones.35 | ||
In 2010, the Merck Research Laboratory in the USA achieved the synthesis of 2-alkenylimidazolones (45) via a 1,3-dipolar cycloaddition (1,3-DC) of alkenes (44) to cyclic nitrones (43, Scheme 15).36 The procedure involves a one-pot three-step process (Scheme 16). Initially, the cycloadduct (46) is formed from imidazolone N-oxide (43) and alkene (44) in refluxing ethanol. After cooling to ambient temperature, subsequent treatment with aqueous NaOH (1 N) (base-induced N–O bond cleavage) and HCl (1 N) (acid-induced water elimination) delivered the target compound 45 in good to excellent yields. Various aromatic and aliphatic alkenes 44 were compatible with the developed reaction conditions.
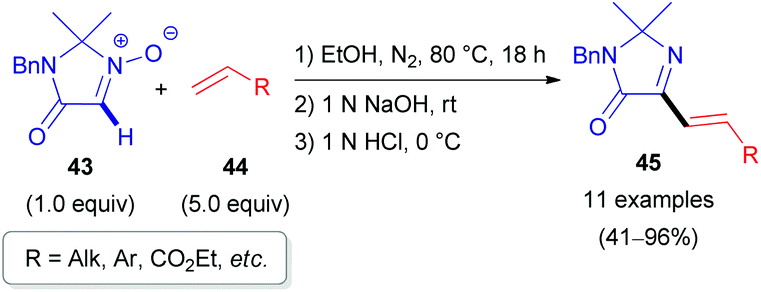 | ||
| Scheme 15 Formal C(sp2)–H alkenylation of imidazolones (43) via the N-oxide. | ||
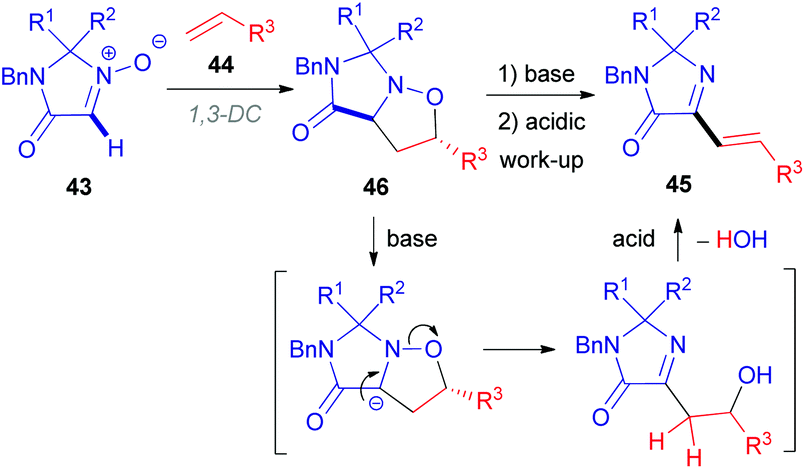 | ||
| Scheme 16 A proposed mechanism for alkenylated imidazolone (45) synthesis from the corresponding imidazolone N-oxide (43). | ||
Two years later, researchers from Albany Molecular Research Inc. reported the first Pd-catalyzed arylation of aldonitrones 47 with aryl bromides (18, Scheme 17).37 The reaction conditions were similar to those previously reported by Fagnou for the arylation of heteroaromatic N-oxides.38 A cross-coupling between 47 and phenyl bromide (18a) successfully delivered 48a; however, only a moderate yield of 49% was obtained (Scheme 17a). A significant improvement in yield was achieved via protection of the imidazolone moiety with either the tert-butoxycarbonyl (Boc) or 2,4-dimethoxybenzyl (DMB) protecting group (48b–c). With the optimal DMB protecting group in hand, the authors were able to arylate aldonitrone 47a with various aryl bromides (18) in good to excellent yields (Scheme 17b). This approach was also successfully applied for the synthesis of GSK2137305 56 (Scheme 18), a known selective inhibitor of glycine transporter type 1 (GlyT1), exhibiting effectiveness for neurological and neuropsychiatric disorders.39 The procedure nicely illustrates the N-oxide directing group, which can be easily introduced via oxidation of cyclic amine 53 with m-CPBA (vide supra) and removed by employing PBr3, yielding cyclic azomethine target compound 56. Other reagents to cleave N-oxides are also available (e.g., CuI/Zn system, PnBu3, Mo(CO)6, InCl3, etc.).35
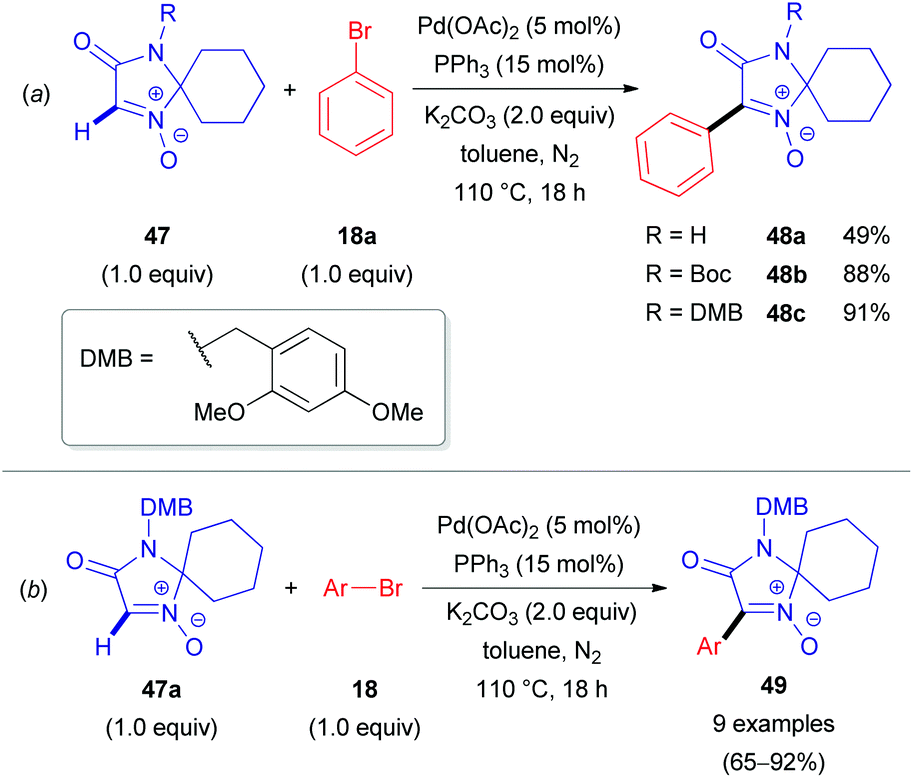 | ||
| Scheme 17 (a) Effects of the N-substituent in lactam 47 on the direct arylation reaction yield; (b) Pd-catalyzed coupling of 4-DMB-protected imidazol-3-one 1-oxide (47a) with aryl bromides (18). | ||
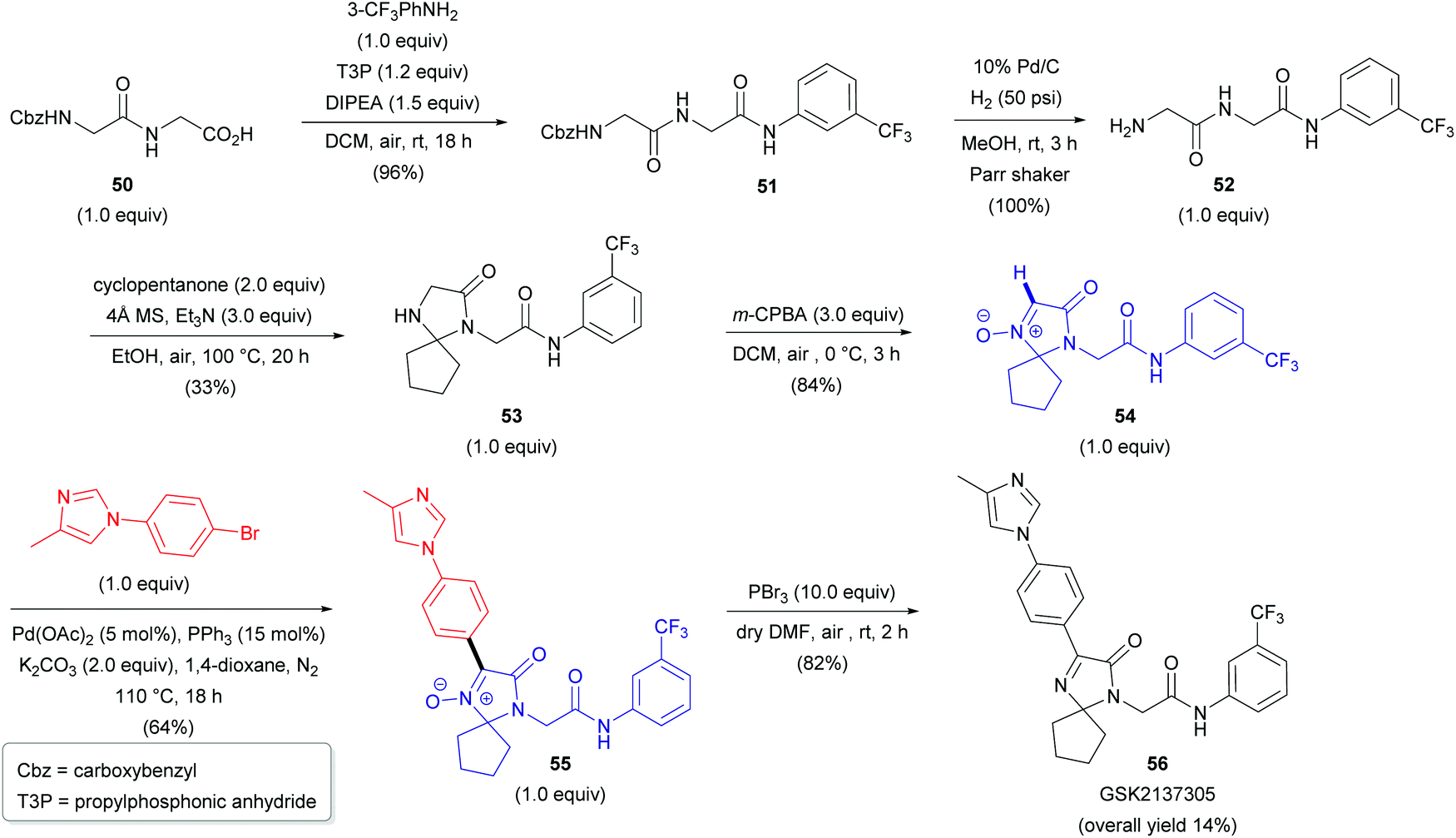 | ||
| Scheme 18 The synthesis of GSK2137305 (56). | ||
In 2011, the synthesis of novel pseudouridine-like compounds (e.g., 62), bearing 1-pyrroline-1-oxide moieties instead of sugar residues, was reported by Koszytkowska-Stawińska (Scheme 19).40 Pseudouridine (ψ-uridine) is a natural C-nucleoside found in various types of RNA. This compound is supposed to play a key role in fine-tuning the functions of RNA, and it contributes likely to the preservation of the RNA structure during translation.41 Moreover, ψ-uridine is also active against ionizing radiation and chemical mutagens.42 These protective effects are associated with the ability of the nucleoside to trap free radicals formed by mutagens. The derivatives of 1-pyrroline 1-oxide are efficient spin-traps for EPR analysis (the most prominent example is 5,5-dimethyl-1-pyrroline N-oxide, DMPO).43 Many azaheterocycles, which are structurally similar to the pyrroline scaffold (pyrrolidines, oxazoles, isoxazoles, etc.), are able to mimic a furanose residue. The replacement of the ribose residue in pseudonucleosides with a 5,5-bis(hydroxymethyl)-1-pyrroline-1-oxide was therefore envisioned to improve the antimutagenic activity. To obtain such modified ψ-uridines, the authors first synthesized O,O′-cyclohexylidene protected 5,5-bis(hydroxymethyl)-1-pyrroline 1-oxide (58) as an aldonitrone precursor. Subsequent addition of either C(5)-lithiated 2,4-dimethoxypyrimidine (57-Li, Scheme 19a) or the analogous Grignard reactant (57-MgBr, Scheme 19b), followed by copper-catalyzed aerobic oxidation of the adduct and acetal deprotection of the resulting nitrone 59 with NaOAc in AcOH (Scheme 19c), afforded the desired pseudouridine aza-analogues 62. The coupling with the lithium derivative (57-Li) was less successful, as nitrone dimerization was observed as the major process, probably due to deprotonation at the C(2)–H of 1-pyrroline 1-oxide (58).
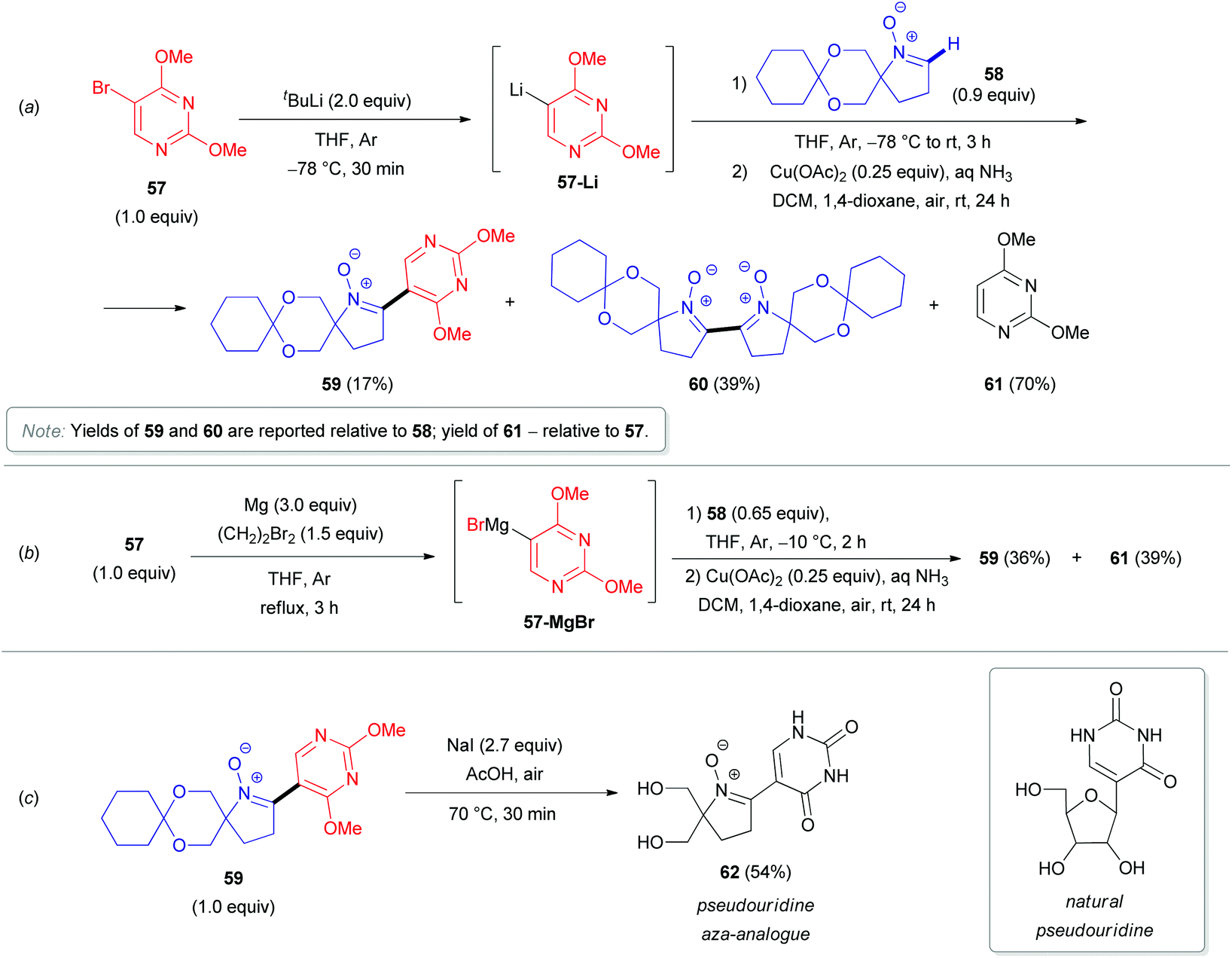 | ||
| Scheme 19 Synthesis of pseudouridine aza-analogue 62. | ||
In 2012, Chavant & Blandin et al. reported an efficient approach for Pd- or Pd/Cu-catalyzed direct arylation of cyclic aldonitrones (63–65),44 which is related to the C–C coupling of imidazolone N-oxides (47) with aryl bromides (18) reported by the AMRI research group (Scheme 17). The cyclic aldonitrone 2-isopropyl-1,2-dimethyl-5-oxo-2,5-dihydro-1H-imidazole 3-oxide (63, MiPNO, Fig. 2) is a versatile precursor for the synthesis of α-amino acids (via 1,3-dipolar cycloaddition reactions with alkenes followed by cleavage of the N–O bond)45 and was one of the key studied substrates. Pivalic acid (20 mol%) was an essential additive for a successful cross-coupling with aryl bromides (18), enhancing the yield dramatically (Scheme 20a).
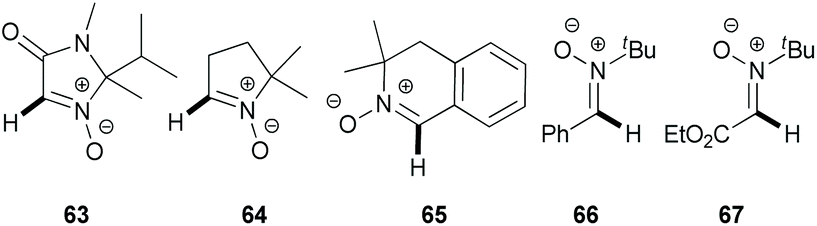 | ||
| Fig. 2 Cyclic and acyclic nitrones studied: MiPNO (63); DMPO (64); 3,3-dimethyl-3,4-dihydroisoquinoline 2-oxide (65); PBN (66); glyoxylic nitrone (67). | ||
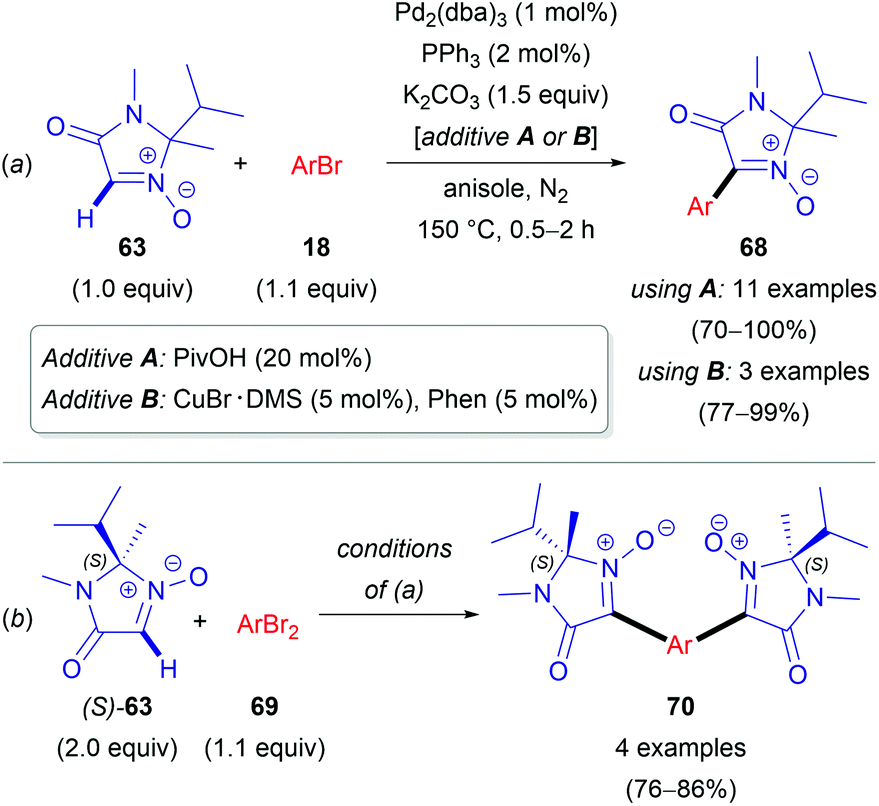 | ||
| Scheme 20 Pd- and Pd-/Cu-Catalyzed direct arylations of MiPNO (63). | ||
Alternatively, CuBr·DMS (5 mol%) with 1,10-phenanthroline (Phen, 5 mol%) as a ligand proved also to be an effective additive to achieve quantitative conversion of 63. It is worth noting that no synergistic effect was observed, when these two additives (PivOH and CuBr·DMS) were used simultaneously. Interestingly, in the absence of the palladium catalyst, a catalytic amount of CuBr·DMS enabled the conversion of aldonitrone 63 into the corresponding homocoupling product (i.e. a nitrone 2,2′-dimer without one of two N-oxide functions). Employing dibromo(hetero)arenes (69) with two equivalents of aldonitrone (S)-63 afforded successfully bis-nitronyl products 70 in good yields (Scheme 20b). The reaction is not specific to MiPNO (63), since a range of other heterocyclic non-aromatic aldonitrones can be employed in this cross-coupling reaction, as illustrated for DMPO (64, Scheme 21a) and 3,3-dimethyl-3,4-dihydroisoquinoline 2-oxide (65, Scheme 21b). The reaction with DMPO is interesting due to the presence of potentially labile allylic H-atoms relative to the imine double bond (Scheme 21a). In contrast, acyclic nitrones, such as N-tert-butyl-α-phenylnitrone (66, PBN) or glyoxylic nitrone (67), were not compatible with this kind of arylation (Fig. 2). It has been suggested that these cross-coupling processes are likely to proceed via the so-called concerted metalation–deprotonation (CMD) mechanism,46 explaining the beneficial effect of PivOH, described previously for Pd-catalyzed direct arylations of (hetero)aromatic substrates (i.e. for azine N-oxides). It is worth noting that this method has been used for the synthesis of chiral imidazolidin-4-one nitroxides starting from enantiopure 63, and they are regarded as potentially active catalysts for aerobic oxidation of alcohols.47 In particular, catalytic application of such compounds in enantiopure form allows one to achieve atroposelective desymmetrization during a diol oxidation process.47
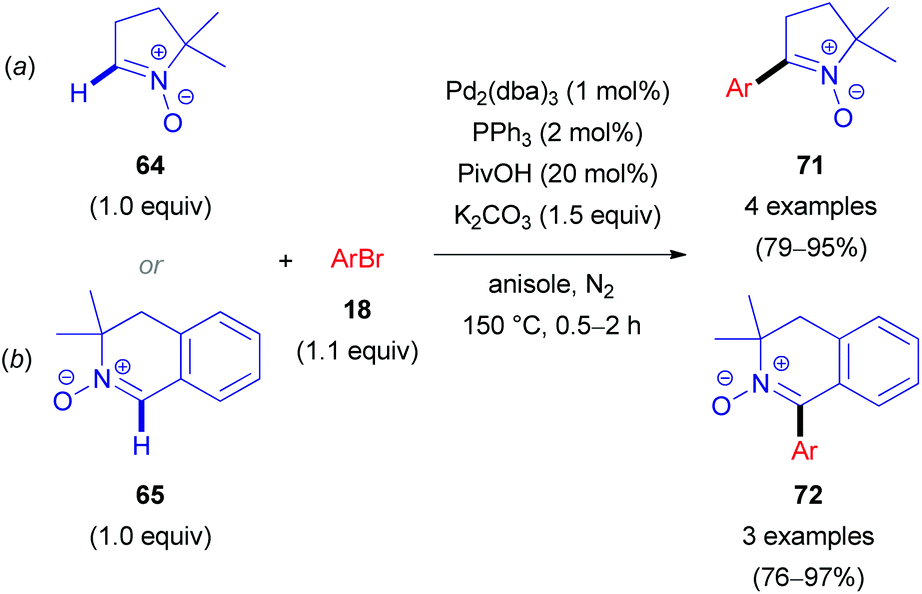 | ||
| Scheme 21 Pd-Catalyzed direct arylation of DMPO (64, a) and 3,3-dimethyl-3,4-dihydroisoquinoline 2-oxide (65, b). | ||
Special attention has recently been paid to C–H functionalization of related non-aromatic 2H-imidazole 1-oxides (7) featuring an extra unsaturation. In 2012, our research group reported the first successful cross-coupling reactions of 2,2-dialkylated 4-phenyl-2H-imidazole 1-oxide (7b) with 1,2,4-triazine 73 (Scheme 22) or its N-oxide 77 (Scheme 23).48 The reaction mechanism includes the initial lithiation of the azomethine C–H bond in aldonitrone 7b, which is assisted by the N-oxide (Directed ‘ortho’ Metalation (DoM)49), followed by reaction with 1,2,4-triazine 73 or its N-oxide 77 to afford NLi- or OLi-adducts (74 or 78). These adducts are subsequently protonated with water into the corresponding NH- or OH-intermediates (75 or 79). Finally, rearomatization of the dihydrotriazine ring in 75/79 occurs either through spontaneous elimination of water, affording the coupling product without the N-oxide function (76/80), or by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give the corresponding N-oxide 81 (Scheme 23). It is worth noting that cleavage of the N-oxide moiety in the triazine fragment of 77 occurs via the ‘Addition–Elimination (AE)’ protocol, while the N-oxide moiety remains untouched via the ‘Addition–Oxidation (AO)’ protocol.
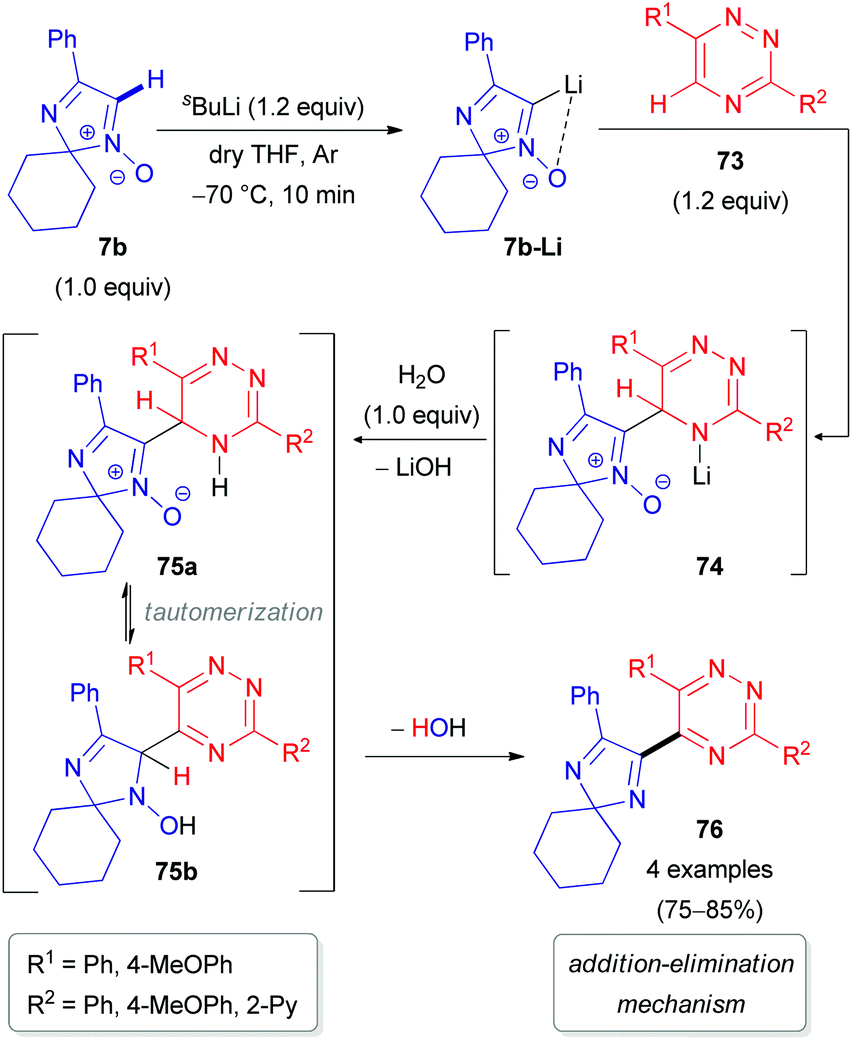 | ||
| Scheme 22 Proposed mechanism for the coupling of 2H-imidazole 1-oxide 7b with 1,2,4-triazines (73) via DoM lithiation. | ||
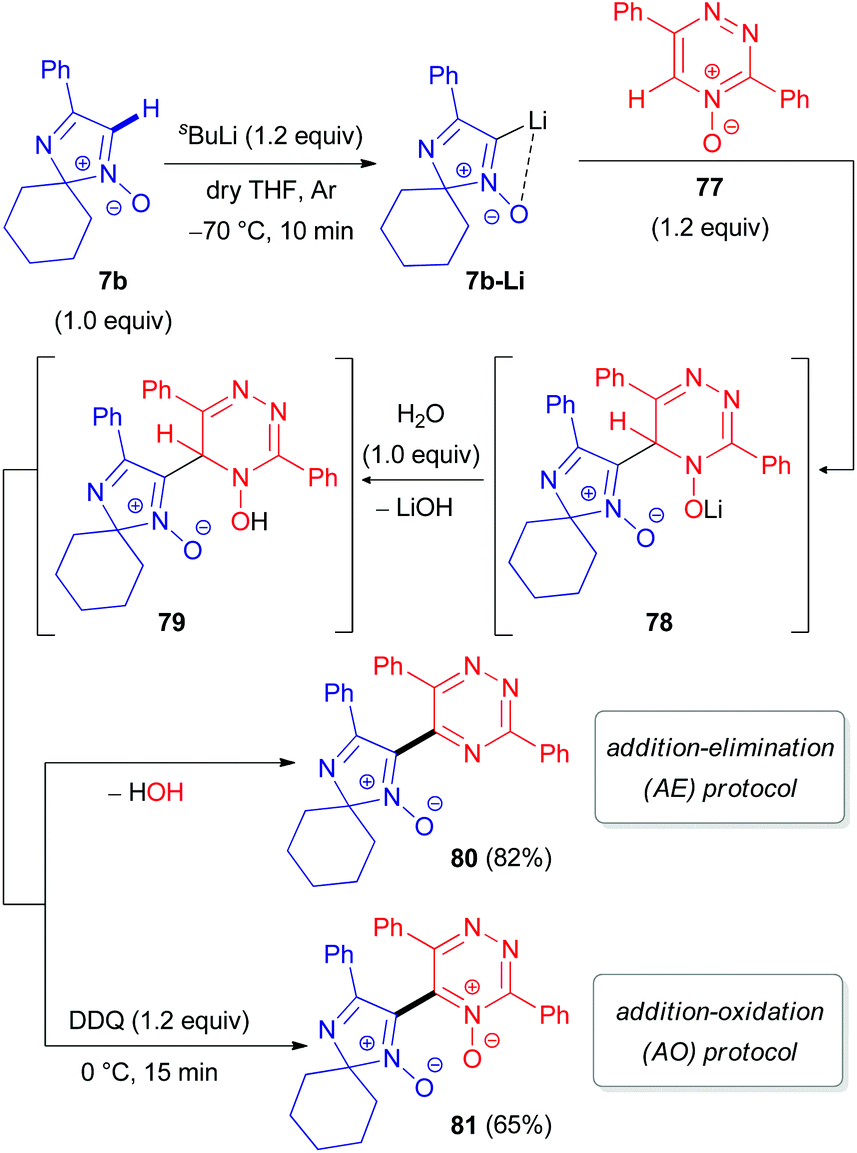 | ||
| Scheme 23 Proposed mechanism for the coupling of 2H-imidazole 1-oxide 7b with 1,2,4-triazine 4-oxide 77via DoM lithiation. | ||
Our group also reported the coupling of 2H-imidazole 1-oxide 7b with 1,10-phenanthroline (82a), quinoxaline (82b), and their N-oxides (85a–b) as electron π-deficient azaheterocyclic substrates (Schemes 24 and 25).50 Interestingly, the N-oxide moiety is eliminated from the imidazole ring in the (AE)-reaction of aldonitrone 7b with quinoline N-oxide 85c, whereas the N-oxide fragment of the pyridine ring remains intact (Scheme 25), in contrast to the reaction of nitrone 7b with triazine N-oxide 77 (Scheme 23). Clearly, retention of the specific N-oxide function in products is controlled by the stability of the tautomeric intermediates (86a and 86b, respectively).
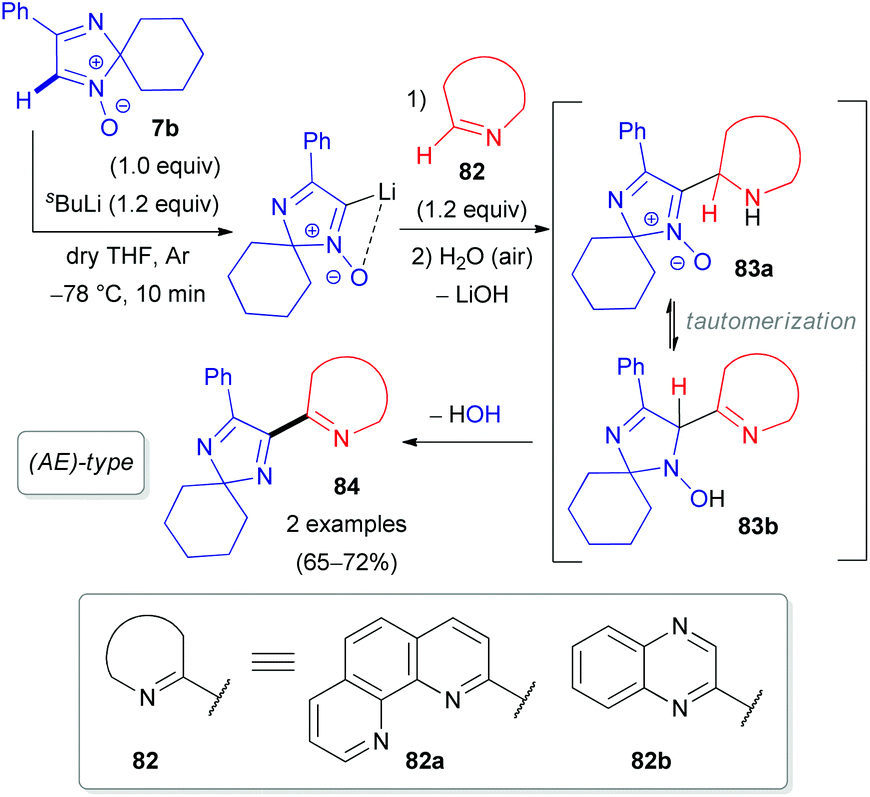 | ||
| Scheme 24 Proposed mechanism for the coupling of 2H-imidazole 1-oxide 7b with azines (82) via DoM lithiation. | ||
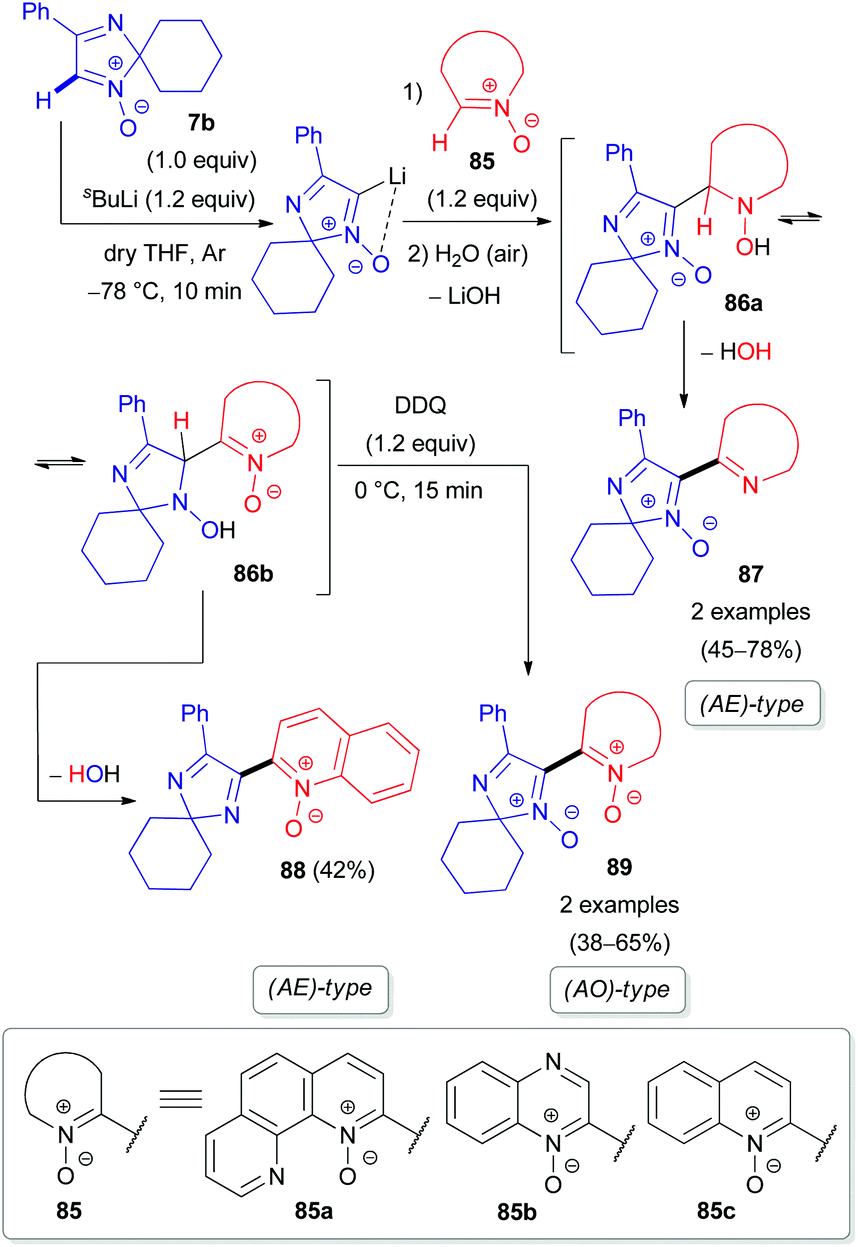 | ||
| Scheme 25 Proposed mechanism for the coupling of 2H-imidazole 1-oxide 7b with azine N-oxides (85) via DoM lithiation. | ||
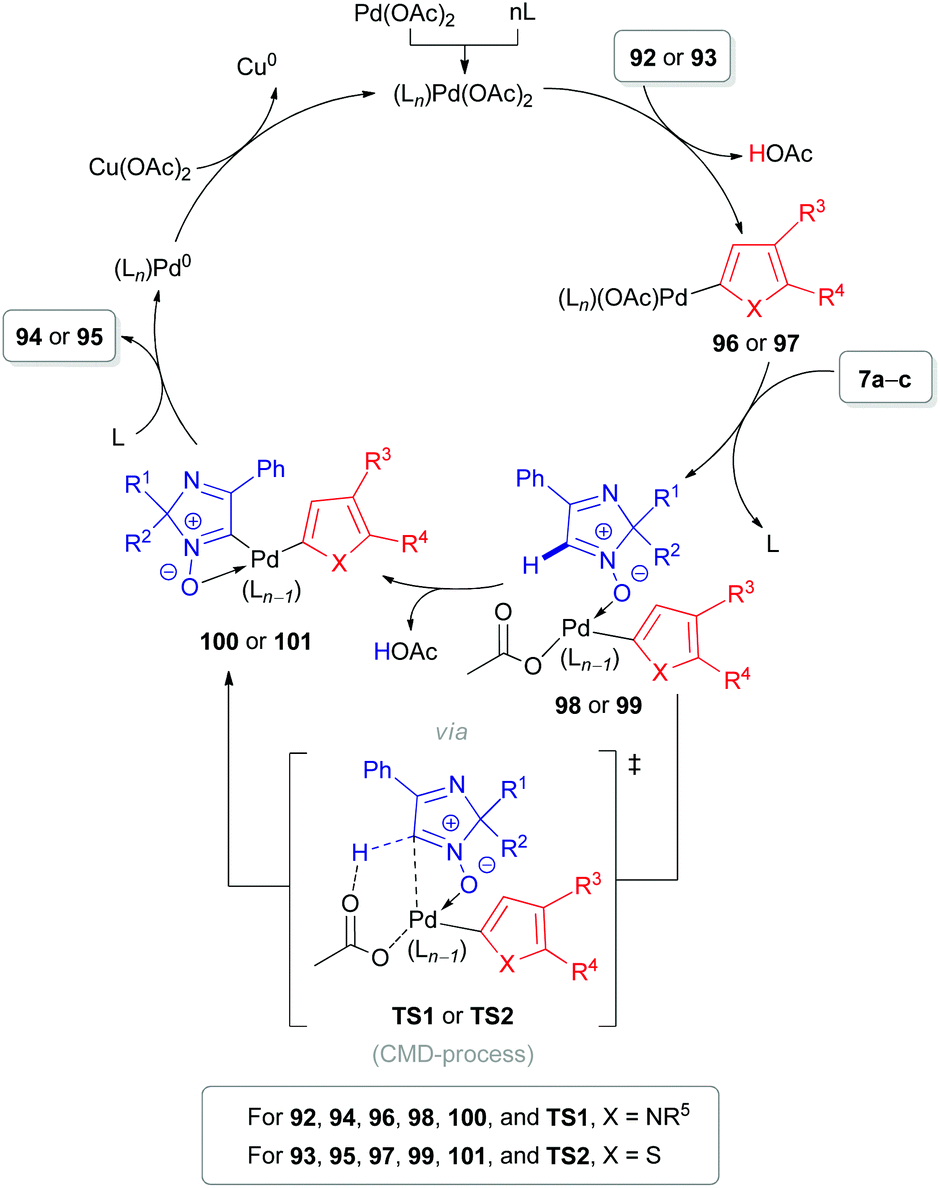
The same substrate 7 can be used as an electrophile rather than as a nucleophile. Our group reported the cross-coupling reaction of 2H-imidazole 1-oxides (7a–c) with electron-rich heteroarenes, such as indoles (90),24 pyrroles (92)51,52 and thiophenes (93) (Schemes 26–28).52 The coupling of these partners can be done in two different ways. The first approach is based on a PdII-catalyzed cross-dehydrogenative coupling (Scheme 26). During the first step of the proposed catalytic cycle (Scheme 27), organopalladium intermediate 96/97 is formed via the reaction of Pd(OAc)2 with a heteroaromatic substrate (92/93). Palladium in the intermediate 96/97 is hypothesized to be coordinated with the oxygen atom of nitrone 7a–cvia a ligand exchange mechanism, leading to the formation of intermediate 98/99. Subsequent carboxylate-assisted functionalization of the nitrone C(1)–H bond takes place (CMD-process, TS1/TS2), generating intermediate 100/101. Finally, reductive elimination affords the target products 94/95 and Pd0. The latter is reoxidized to PdII with the superstoichiometric oxidant Cu(OAc)2. Interestingly, pyridine was found to be an essential additive and presumed to play a role of ligand (Ln) stabilizing organometallic intermediates.
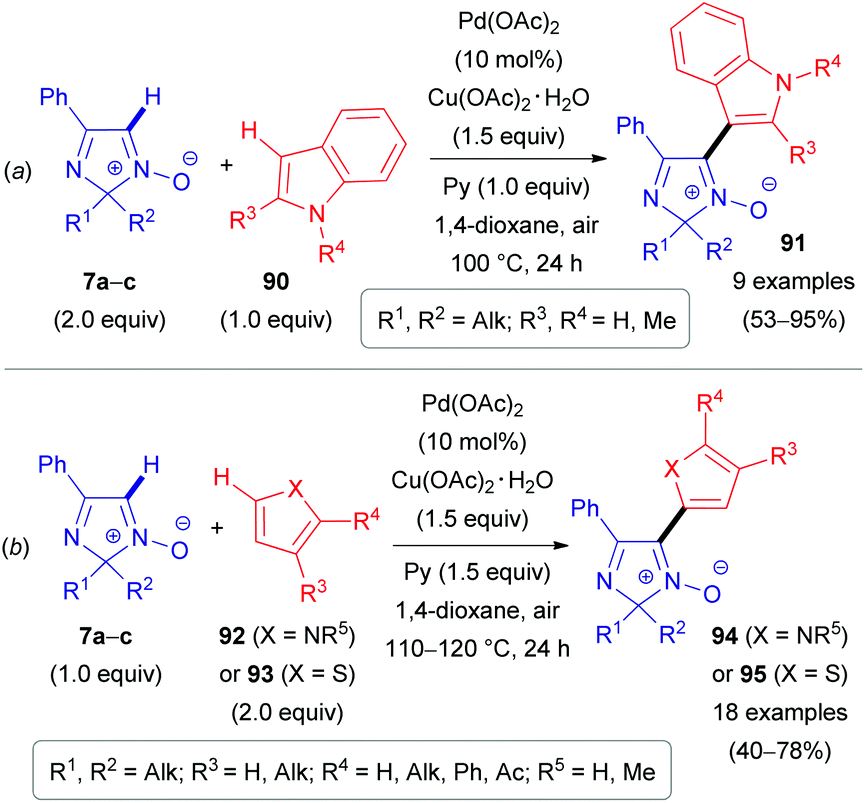 | ||
| Scheme 26 PdII-Catalyzed oxidative coupling of 2H-imidazole 1-oxides (7a–c) with indoles (90, a), pyrroles (92), and thiophenes (93, b). | ||
 | ||
| Scheme 27 Proposed catalytic cycle for the oxidative coupling of 2H-imidazole 1-oxides (7a–c) with pyrroles (92) or thiophenes (93). | ||
 | ||
| Scheme 28 The SNH reaction of 2H-imidazole 1-oxides 7a–c with indoles (90, a) and pyrroles (92, b). | ||
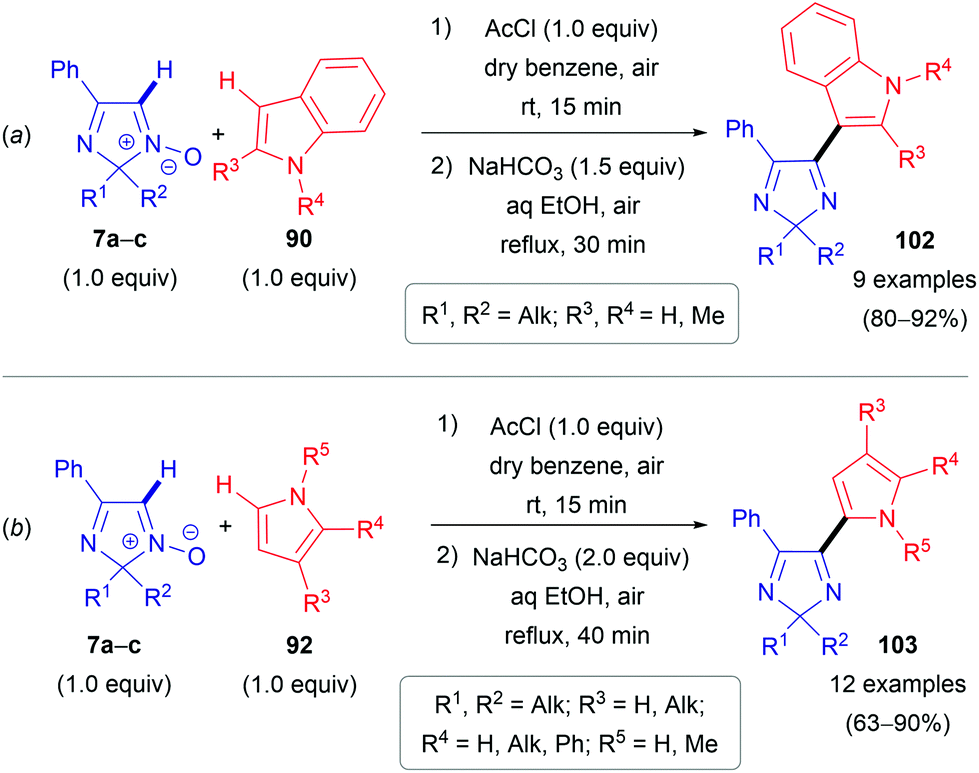
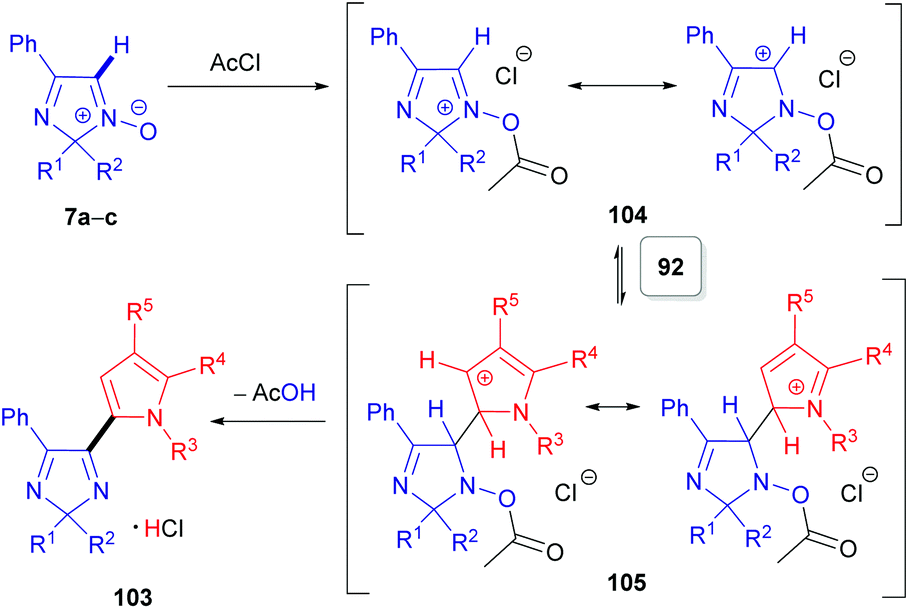
The second approach is a cine-type reaction based on the SNH(AE) process between 2,2-dialkylated 4-phenyl-2H-imidazole 1-oxides (7a–c) and indoles or pyrroles (90 or 92) as coupling partners (Scheme 28). For these transformations, the acylating reagent plays a crucial role, since it reacts with the N-oxide moiety of 7a–c to generate a more electrophilic species 104 and immediately builds in a suitable leaving group (Scheme 29). Simple acetyl chloride can be used. These chloride salts formed then react in electrophilic addition reactions at the C(2)-position of pyrroles (92) or the C(3)-position of indoles (90), generating the corresponding dihydro compounds (e.g., 105). Subsequent spontaneous elimination of acetic acid affords the hydrochloride salts of the desired products 102/103. Notably and in line with the substitution pattern, this method is not compatible with pyrroles (92) and indoles (90) bearing electron-withdrawing groups, i.e. nitro or acetyl groups.
 | ||
| Scheme 29 Proposed reaction mechanism for the SNH reaction of 2H-imidazole 1-oxides (7a–c) with pyrroles (92) (cine reaction). | ||
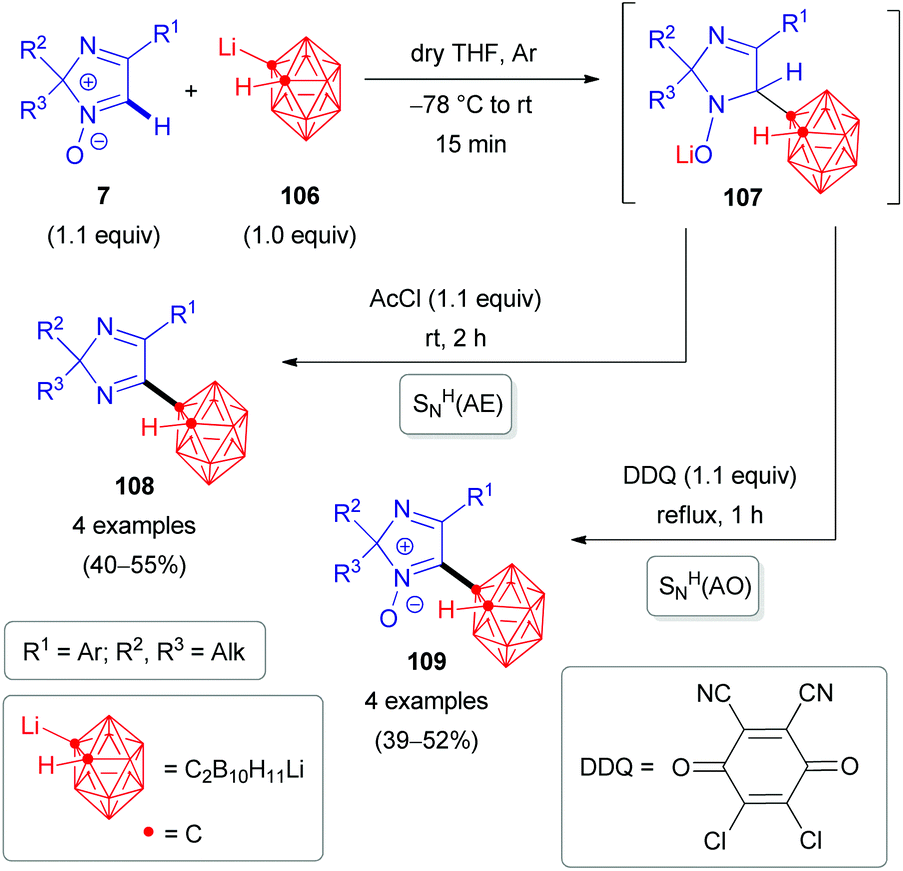
The nucleophile can also be an organometallic reactant (Scheme 30). A challenging example is ortho-carboranyllithium (106). The reaction with 2H-imidazole 1-oxides was reported in 2018.53 The nucleophile is particularly interesting, as organoboron compounds and their heterocyclic derivatives are potential agents for boron neutron capture therapy (BNCT) of cancer.53 Given the nucleophilicity of organolithium species, N-oxide activation is in this case not required; in fact, it could competitively react at the carbonyl of the ester introduced, and a reaction with acetyl chloride is merely used as a quencher to rearomatize. Besides eliminative SNH(AE), an oxidative SNH(AO) strategy keeping the N-oxide in the structure is possible (Scheme 30). Very recently, the analogous protocol has also been applied for the direct C(sp2)–H arylation of 2H-imidazole 1-oxides 7 with another organometallic reactant, pentafluorophenyllithium 110 (Scheme 31).54 Some of the resulting coupling products 112/113 are considered as promising push–pull fluorophores to be useful in the design of novel fluorometric sensors.
 | ||
| Scheme 30 SNH reactions of 2H-imidazole 1-oxides (7) with ortho-carboranyllithium (106) via SNH(AE) and oxidative SNH(AO) strategies. | ||
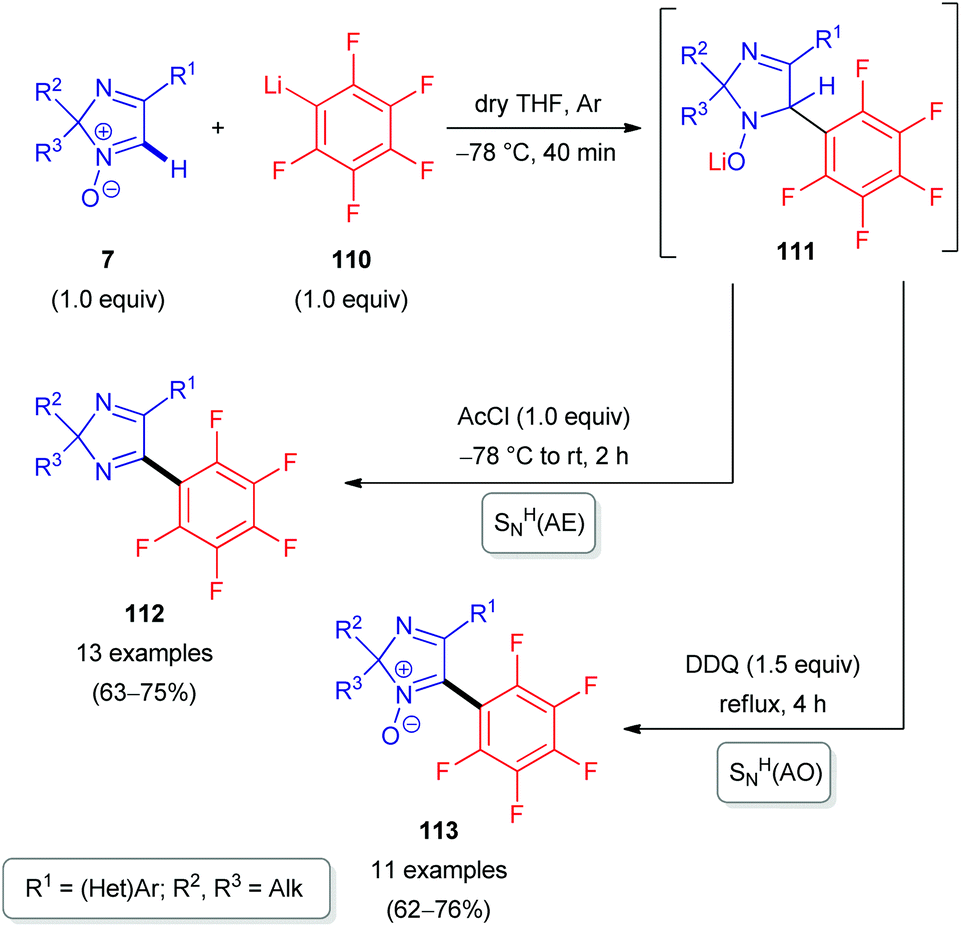 | ||
| Scheme 31 SNH reactions of 2H-imidazole 1-oxides (7) with pentafluorophenyllithium (110) via SNH(AE) and oxidative SNH(AO) strategies. | ||
3. Conclusions and outlook
A systematic overview of C(sp2)–H functionalization reactions for the modification of cyclic azomethine-based compounds has been performed. As functionalizing reactants, both carbon-centered nucleophiles and electrophiles can be used (furthermore, the options for employing heteroatom-centered synthons gradually appear to become accessible as well,55 although this area is still in its infancy). Meanwhile, the use of cyclic aldonitrones as starting azomethine substrates turned out to be prevalent over other related compounds, such as imines or hydrazones. Undoubtedly, this is due to the remarkable properties of the nitrone N-oxide moiety, which can both act as a directing group and as a leaving group.Although aldonitrones are shown to react readily with a number of electrophilic and nucleophilic synthons, it seems to be rather odd that the examples of radical C(sp2)–H functionalization of these compounds are scarcely known at the moment.35 After all, nitrones have become widely applicable exactly due to their radical addition reactions affording relatively stable paramagnetic nitroxides useful for EPR analysis. In this connection, we believe that stepping into the above-mentioned terra incognita can be a milestone for the further development of the discussed topic.
Another certainly considerable objective is the sustainability of the process. Though very effective synthetic methodologies have been developed in the past decade, there is still quite a lot to be improved in terms of greenness and compliance with the PASE principles. Several approaches surveyed in this overview deal with transition metal catalysis. Although catalysis is inherently a green methodology, an aryl bromide or iodide reactant partner, as well as noble metals (sometimes with complex ligands), is typically required. Besides, it often appears to be necessary to use (super)stoichiometric quantities of additives (e.g., bases). Indeed, these reactions are highly valuable for modern organic synthesis and open new horizons compared, for instance, with the conventional ‘electrophile–nucleophile’ interactions, yet there still remains room for further improvement. For example, the use of base metals as catalysts or cross-dehydrogenative procedures, which are inherently more interesting, considering no prefunctionalization of the reactant is required (though this can pose regioselectivity issues in return). However, if no sustainable oxidant is used, nothing can be gained by employing a cross-dehydrogenative approach, as it is still not suitable for scaling, albeit very useful for discovery research purposes. Oxygen,26a hydrogen peroxide,26b and electric current56 are interesting oxidants. Photoredox-catalyzed57 and organocatalyzed58 C–H functionalizations certainly also hold promise with respect to green chemistry. To the best of our knowledge, they have not yet been applied for C(sp2)–H bond functionalization in cyclic azomethines and aldonitrones. Thus, investigations aimed at further improving the sustainability of the current processes are expected to be the focus of research in the next few years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the Russian Science Foundation (Project No. 20-43-01004), a bilateral Russian Science Foundation (RSF) – Fund for Scientific Research Flanders (FWO) project, and the Francqui Foundation.References
- (a) Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley–VCH, Weinheim, 2011 Search PubMed; (b) Green Synthetic Approaches for Biologically Relevant Heterocycles, ed. G. Brahmachari, Elsevier, Oxford, 2014 Search PubMed; (c) J. A. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Publishing, Chichester, 5th edn, 2013 Search PubMed.
- A. Katritzky, C. Ramsden, J. Joule and V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, Amsterdam, 3rd edn, 2010 Search PubMed.
- M. Smith, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, Inc., Hoboken, 7th edn, 2013 Search PubMed.
- (a) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC; (b) A. P. Dicks and A. Hent, Green Chemistry Metrics: A Guide to Determining and Evaluating Process Greenness (SpringerBriefs in Molecular Science), Springer, Cham, 1st edn, 2015 Search PubMed; (c) L. Summerton and A. Constandinou, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, The Royal Society of Chemistry, 2016, pp. 41–53 RSC; (d) J. H. Clark, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, The Royal Society of Chemistry, 2016, pp. 1–11 Search PubMed.
- W. Zhang and W.-B. Yi, Pot, Atom, and Step Economy (PASE) Synthesis, Springer, Cham, 2019 Search PubMed.
- (a) Metal Free C–H Functionalization of Aromatics. Nucleophilic Displacement of Hydrogen, ed. V. Charushin and O. Chupakhin, Springer, Cham, 2014 Search PubMed; (b) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (d) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (e) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2018, 119, 2192–2452 CrossRef; (f) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS.
- M. Beller, A. Varela-Fernandez, J. G. de Vries, S. M. Wong, C. M. So and F. Y. Kwong, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH, Weinheim, 3rd edn, 2017, pp. 411–464 Search PubMed.
- (a) C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC; (b) S. Abou-Shehada, P. Mampuys, B. U. W. Maes, J. H. Clark and L. Summerton, Green Chem., 2017, 19, 249–258 RSC; (c) E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–135 RSC.
- H. Kayakiri, K. Nakamura, S. Takase, H. Setoi, I. Uchida, H. Terano, M. Hashimoto, T. Tada and S. Koda, Chem. Pharm. Bull., 1991, 39, 2807–2812 CrossRef CAS.
- B. S. Evans, I. Ntai, Y. Chen, S. J. Robinson and N. L. Kelleher, J. Am. Chem. Soc., 2011, 133, 7316–7319 CrossRef CAS.
- A. Otero, M.-J. Chapela, M. Atanassova, J. M. Vieites and A. G. Cabado, Chem. Res. Toxicol., 2011, 24, 1817–1829 Search PubMed.
- C. E. Thomas, D. F. Ohlweiler, A. A. Carr, T. R. Nieduzak, D. A. Hay, G. Adams, R. Vaz and R. C. Bernotas, J. Biol. Chem., 1996, 271, 3097–3104 CrossRef CAS.
- (a) M. Rosselin, B. Poeggeler and G. Durand, Curr. Top. Med. Chem., 2017, 17, 2006–2022 CrossRef CAS; (b) C. Oliveira, S. Benfeito, C. Fernandes, F. Cagide, T. Silva and F. Borges, Med. Res. Rev., 2018, 38, 1159–1187 CrossRef.
- (a) Q. Ke, G. Yan, J. Yu and X. Wu, Org. Biomol. Chem., 2019, 17, 5863–5881 RSC; (b) J. Rostoll-Berenguer, G. Blay, J. R. Pedro and C. Vila, Eur. J. Org. Chem., 2020 DOI:10.1002/ejoc.202000746; (c) P. Ghosh, B. Ganguly and S. Das, Org. Biomol. Chem., 2020, 18, 4497–4518 RSC.
- Selected examples of the C(sp2)–H functionalization in planar azomethine-based heterocycles: (a) S. Laclef, M. Harari, J. Godeau, I. Schmitz-Afonso, L. Bischoff, C. Hoarau, V. Levacher, C. Fruit and T. Besson, Org. Lett., 2015, 17, 1700–1703 CrossRef CAS; (b) M. Harari, F. Couly, C. Fruit and T. Besson, Org. Lett., 2016, 18, 3282–3285 CrossRef CAS; (c) S. C. Ruiz, M. Muselli, S. Frippiat, T. M. Diallo, A. Mohamed-Cherif, V. Levacher, C. Baudequin, L. Bischoff and C. Hoarau, Synlett, 2020, 31, 1185–1190 CrossRef; (d) P. Mampuys, T. D. Moseev, M. V. Varaksin, J. De Houwer, C. M. L. Vande Velde, O. N. Chupakhin, V. N. Charushin and B. U. W. Maes, Org. Lett., 2019, 21, 2699–2703 CrossRef CAS; (e) W. Zhang, Y.-L. Pan, C. Yang, L. Chen, X. Li and J.-P. Cheng, J. Org. Chem., 2019, 84, 7786–7795 CrossRef CAS; (f) K. Yin and R. Zhang, Org. Lett., 2017, 19, 1530–1533 CrossRef CAS; (g) W. Wei, L. Wang, H. Yue, P. Bao, W. Liu, C. Hu, D. Yang and H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 17252–17257 CrossRef CAS; (h) W. Wei, L. Wang, P. Bao, Y. Shao, H. Yue, D. Yang, X. Yang, X. Zhao and H. Wang, Org. Lett., 2018, 20, 7125–7130 CrossRef CAS; (i) L.-Y. Xie, J.-L. Hu, Y.-X. Song, G.-K. Jia, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 19993–19999 CrossRef CAS; (j) L. Wang, M. Sun, L. Zhao, Z. Wang and P. Li, ChemCatChem, 2020 DOI:10.1002/cctc.202000459.
- For the Nobel lectures on the development of transition metal-catalyzed cross-coupling reactions, see: (a) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- Selected early examples on the ‘conventional’ cross-coupling reactions of cyclic azomethines: (a) L. N. Pridgen and L. B. Killmer, J. Org. Chem., 1981, 46, 5402–5404 CrossRef CAS; (b) A. Dondoni, G. Fantin, M. Fogagnolo, A. Medici and P. Pedrini, Synthesis, 1987, 693–696 CrossRef CAS; (c) A. I. Meyers and K. A. Novachek, Tetrahedron Lett., 1996, 37, 1747–1748 CrossRef CAS; (d) W. D. Schmitz and D. Romo, Tetrahedron Lett., 1996, 37, 4857–4860 CrossRef CAS; (e) P. A. Jacobi and H. Liu, J. Am. Chem. Soc., 1999, 121, 1958–1959 CrossRef CAS; (f) P. A. Jacobi and H. Liu, J. Org. Chem., 1999, 64, 1778–1779 CrossRef CAS; (g) A. Nadin, J. M. Sánchez López, A. P. Owens, D. M. Howells, A. C. Talbot and T. Harrison, J. Org. Chem., 2003, 68, 2844–2852 CrossRef CAS.
- J. C. Lewis, A. M. Berman, R. J. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2493–2500 CrossRef CAS.
- Relevant reviews on cross-dehydrogenative coupling chemistry: (a) C.-J. Li, Acc. Chem. Res., 2008, 42, 335–344 CrossRef; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (d) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS; (e) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS.
- (a) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS; (b) D. Tilly, J. Magolan and J. Mortier, Chem. – Eur. J., 2012, 18, 3804–3820 CrossRef CAS.
- (a) M. A. Voinov, I. A. Grigor'ev and L. B. Volodarsky, Heterocycl. Commun., 1998, 4, 261–270 CAS; (b) M. A. Voinov, I. A. Grigor'ev and L. B. Volodarsky, Tetrahedron, 2000, 56, 4071–4077 CrossRef CAS; (c) M. A. Voinov and I. A. Grigor'ev, Tetrahedron Lett., 2002, 43, 2445–2447 CrossRef CAS; (d) M. A. Voinov, T. G. Shevelev, T. V. Rybalova, Y. V. Gatilov, N. V. Pervukhina, A. B. Burdukov and I. A. Grigor'ev, Organometallics, 2007, 26, 1607–1615 CrossRef CAS.
- (a) O. N. Chupakhin and I. Y. Postovskii, Russ. Chem. Rev., 1976, 45, 454–468 CrossRef; (b) O. N. Chupakhin, V. N. Charushin and H. C. van der Plas, Tetrahedron, 1988, 44, 1–34 CrossRef CAS; (c) O. N. Chupakhin, V. N. Charushin and H. C. van der Plas, Nucleophilic Aromatic Substitution of Hydrogen, Academic Press, San Diego, 1994 Search PubMed; (d) V. N. Charushin and O. N. Chupakhin, Pure Appl. Chem., 2004, 76, 1621–1631 CAS; (e) V. N. Charushin and O. N. Chupakhin, Mendeleev Commun., 2007, 17, 249–254 CrossRef CAS; (f) A. M. Prokhorov, M. Mąkosza and O. N. Chupakhin, Tetrahedron Lett., 2009, 50, 1444–1446 CrossRef CAS; (g) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef CAS; (h) O. N. Chupakhin and V. N. Charushin, Pure Appl. Chem., 2017, 89, 1195–1208 CAS; (i) V. N. Charushin and O. N. Chupakhin, Russ. Chem. Bull., 2019, 68, 453–471 CrossRef CAS.
- (a) D. Döpp, L. Greci and A. M. Nour-el-Din, Chem. Ber., 1983, 116, 2049–2057 CrossRef; (b) E. Gössinger and B. Witkop, Monatsh. Chem., 1980, 111, 803–811 CrossRef; (c) E. G. Janzen, Y.-K. Zhang and D. L. Haire, Magn. Reson. Chem., 1994, 32, 711–720 CrossRef CAS.
- M. V. Varaksin, I. A. Utepova, O. N. Chupakhin and V. N. Charushin, J. Org. Chem., 2012, 77, 9087–9093 CrossRef CAS.
- (a) S.-I. Murahashi, J. Sun, H. Kurosawa and Y. Imada, Heterocycles, 2000, 52, 557–561 CrossRef CAS; (b) A. Goti, S. Cicchi, V. Mannucci, F. Cardona, F. Guarna, P. Merino and T. Tejero, Org. Lett., 2003, 5, 4235–4238 CrossRef CAS; (c) M. Marradi, S. Cicchi, I. Delso, L. Rosi, T. Tejero, P. Merino and A. Goti, Tetrahedron Lett., 2005, 46, 1287–1290 CrossRef CAS.
- (a) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS; (b) S. P. Teong, X. Li and Y. Zhang, Green Chem., 2019, 21, 5753–5780 RSC.
- (a) K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 3202–3203 CrossRef CAS; (b) S. H. Wiedemann, J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 2452–2462 CrossRef CAS.
- L. Ackermann, S. Barfüsser, C. Kornhaass and A. R. Kapdi, Org. Lett., 2011, 13, 3082–3085 CrossRef CAS.
- S. Yotphan, R. J. Bergman and J. A. Ellman, Org. Lett., 2009, 11, 1511–1514 CrossRef CAS.
- (a) H.-Q. Do, R. M. Kashif Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS; (b) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 1128–1129 CrossRef CAS; (c) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404–12405 CrossRef CAS.
- M. Muselli, C. Baudequin, C. Hoarau and L. Bischoff, Chem. Commun., 2015, 51, 745–748 RSC.
- M. Muselli, C. Baudequin, C. Perrio, C. Hoarau and L. Bischoff, Chem. – Eur. J., 2016, 22, 5520–5524 CrossRef CAS.
- R. A. Novikov, E. V. Shulishov and Y. V. Tomilov, Mendeleev Commun., 2012, 22, 87–89 CrossRef CAS.
- Y. V. Tomilov, R. A. Novikov and O. M. Nefedov, Tetrahedron, 2010, 66, 9151–9158 CrossRef CAS.
- (a) S.-I. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684–4716 CrossRef CAS; (b) I. A. Grigor'ev, in Nitrile Oxides, Nitrones & Nitronates in Organic Synthesis: Novel Strategies in Synthesis, ed. H. Feuer, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2008, pp. 129–435 Search PubMed.
- X. Dai, M. W. Miller and A. W. Stamford, Org. Lett., 2010, 12, 2718–2721 CrossRef CAS.
- H. Zhao, R. Wang, P. Chen, B. T. Gregg, M. M. Hsia and W. Zhang, Org. Lett., 2012, 14, 1872–1875 CrossRef CAS.
- L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291–3306 CrossRef CAS.
- (a) J. P. Graham, N. Langlade, J. M. Northall, A. J. Roberts and A. J. Whitehead, Org. Process Res. Dev., 2011, 15, 44–48 CrossRef CAS; (b) S. Ueda, M. Su and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 700–706 CrossRef CAS.
- M. Koszytkowska-Stawińska, E. Mironiuk-Puchalska and W. Sas, Tetrahedron Lett., 2011, 52, 1866–1870 CrossRef.
- M. Charette and M. W. Gray, IUBMB Life, 2000, 49, 341–351 CrossRef CAS.
- (a) T. Yoshikawa, S. Kimura, T. Hatano, K. Okamoto, H. Hayatsu and S. Arimoto-Kobayashi, Food Chem. Toxicol., 2002, 40, 1165–1170 CrossRef CAS; (b) M. Monobe, S. Arimoto-Kobayashi and K. Ando, Mutat. Res., 2003, 538, 93–99 CrossRef CAS.
- Electron Paramagnetic Resonance, ed. B. C. Gilbert, M. J. Davies and D. M. Murphy, Royal Society of Chemistry, Cambridge, 2002, vol. 18 Search PubMed.
- E. Demory, D. Farran, B. Baptiste, P. Y. Chavant and V. Blandin, J. Org. Chem., 2012, 77, 7901–7912 CrossRef CAS.
- (a) M. Thiverny, E. Demory, B. Baptiste, C. Philouze, P. Y. Chavant and V. Blandin, Tetrahedron: Asymmetry, 2011, 22, 1266–1273 CrossRef CAS; (b) M. Thiverny, D. Farran, C. Philouze, V. Blandin and P. Y. Chavant, Tetrahedron: Asymmetry, 2011, 22, 1274–1281 CrossRef CAS; (c) M. Thiverny, C. Philouze, P. Y. Chavant and V. Blandin, Org. Biomol. Chem., 2010, 8, 864–872 RSC.
- (a) H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180–8189 CrossRef CAS; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS; (c) D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880–6886 CrossRef CAS; (d) D. Garcia-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef CAS; (e) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS.
- M. Carbó-López, G. Royal, C. Philouze, P. Y. Chavant and V. Blandin, Eur. J. Org. Chem., 2014, 4884–4896 CrossRef.
- M. V. Varaksin, I. A. Utepova and O. N. Chupakhin, Chem. Heterocycl. Compd., 2012, 48, 1213–1219 CrossRef CAS.
- G. A. El-Hiti, K. Smith, A. S. Hegazy, M. B. Alshammari and A. M. Masmali, ARKIVOC, 2015, 2015, 19–47 Search PubMed.
- M. V. Varaksin, I. A. Utepova, O. N. Chupakhin and V. N. Charushin, Tetrahedron, 2015, 71, 7077–7082 CrossRef CAS.
- M. Varaksin, T. Moseev, O. Chupakhin, V. Charushin and B. Trofimov, Org. Biomol. Chem., 2017, 15, 8280–8284 RSC.
- A. A. Akulov, M. V. Varaksin, V. N. Charushin and O. N. Chupakhin, ACS Omega, 2019, 4, 825–834 CrossRef CAS.
- L. A. Smyshliaeva, M. V. Varaksin, P. A. Slepukhin, O. N. Chupakhin and V. N. Charushin, Beilstein J. Org. Chem., 2018, 14, 2618–2626 CrossRef CAS.
- T. D. Moseev, M. V. Varaksin, D. A. Gorlov, V. N. Charushin and O. N. Chupakhin, J. Org. Chem., 2020, 85, 11124–11133 CrossRef CAS.
- For a recent example of cyclic azomethine C(sp2)–H bond activation towards the novel C–N bond formation, see: A. A. Akulov, M. V. Varaksin, V. N. Charushin and O. N. Chupakhin, Chem. Heterocycl. Compd., 2019, 55, 783–787 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (b) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS.
- (a) G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |