
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntraband dynamics of mid-infrared HgTe quantum dots†
Matthias
Ruppert
 a,
Hanh
Bui‡
bc,
Laxmi Kishore
Sagar§
de,
Pieter
Geiregat
de,
Zeger
Hens
de,
Gabriel
Bester
bc and
Nils
Huse
*ac
a,
Hanh
Bui‡
bc,
Laxmi Kishore
Sagar§
de,
Pieter
Geiregat
de,
Zeger
Hens
de,
Gabriel
Bester
bc and
Nils
Huse
*ac
aInstitute for Nanostructure and Solid-State Physics, Department of Physics, University of Hamburg and Center for Free-Electron Laser Science, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: nils.huse@uni-hamburg.de
bPhysical Chemistry and Physics departments, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
cThe Hamburg Centre for Ultrafast Imaging, University of Hamburg, Luruper Chaussee, 149, 22761 Hamburg, Germany
dPhysics and Chemistry of Nanostructures, Department of Chemistry, Ghent University, Krijgslaan 281 - S3, B-9000 Gent, Belgium
eCenter for Nano and Biophotonics, Ghent University, Technologiepark Zwijnaarde 15, B-9052 Gent, Belgium
First published on 7th December 2021
Abstract
Femtosecond pump–probe spectroscopy reveals ultrafast carrier dynamics in mid-infrared (MIR) colloidal HgTe nanoparticles with a bandgap of 2.5 μm. We observe intraband relaxation processes after photoexcitation ranging from resonant excitation up to the multi-exciton generation (MEG) regime by identifying initially excited states from atomic effective pseudopotential calculations. Our study elucidates the earliest dynamics below 10 ps in this technologically relevant material. With increasing photon energy, we find carrier relaxation times as long as 2.1 ps in the MEG regime close to the ionization threshold of the particles. For all photon energies, we extract a constant mean carrier energy dissipation rate of 0.36 eV ps−1 from which we infer negligible impact of the density of states on carrier cooling.
Introduction
Intraband cooling is of fundamental interest for technological applications of semiconductors, a process that is most important for light harvesting as well as light-amplification and sensing applications. For instance, slow intraband relaxation may allow the realization of highly efficient solar cells through harnessing of hot carriers1,2 or the utilization of carrier multiplication.3–5 The opposite limit is desirable for lasing applications, i.e. for optimal pumping efficiencies. Compared to bulk materials, the dominant carrier relaxation channels and relaxation rates are heavily altered in colloidal quantum dots for several reasons. First, a discrete level structure with spacings of several LO phonon energies makes phonon-mediated relaxation processes significantly less efficient.6 At the same time, the wave packet nature and close vicinity of carriers enhance Auger scattering, a good overview of which can be found in a recent review.7 A larger surface to volume ratio also enhances surface effects and the influence of ligands.8–11 Yet, the resulting relaxation pathways are by no means universal for all quantum dot systems. This observation becomes very clear when comparing the well described quantum dot systems CdSe and PbSe. In CdSe quantum dots, electrons in the conduction band transfer their energy almost instantaneously (sub 100 fs) to hole states in the valence band. This is known as Auger cooling (of the electron) or Auger heating (of the hole).12,13 Hole states then relax through phonon and/or ligand mediated processes. In PbSe quantum dots on the other hand, both, electrons and holes, seem to independently relax in a ladder hopping manner through ligand or phonon assisted processes.14–16In contrast to these well described quantum dot systems, the dynamics during the first 10 picoseconds after photo-excitation are largely unknown in MIR HgTe quantum dots, while the slower band edge dynamics are well understood.17,18 The possibility of tuning their band-gap through the entire infrared down to the Terahertz spectral range by varying the size of the crystallites19–21 makes these particles very promising for technological applications such as infrared sensing22–27 and emission.27–29 MEG has been demonstrated in these particles as well,18 meaning that a photon of sufficient energy may efficiently excite several electron hole pairs upon being absorbed, as also observed in other quantum dot systems.3,5,30
Here, we report the early dynamics in MIR HgTe quantum dots with a band-gap of 2.5 μm using ultrafast visible/infrared pump–probe spectroscopy. We observe the early state filling dynamics of the energetically lowest exciton state upon non-resonant excitation, which allow assigning timescales for intraband carrier relaxation mechanisms in different excitation regimes. By varying pump wavelengths from 2.4 μm up to 400 nm, we gain insight into the exciton cooling process from the lowest optical excitation up to the highly non-resonant MEG regime, where a single photon may excite up to four electron hole pairs in HgTe quantum dots.18
Experimental
Our infrared pump–probe setup consists of two independently tunable optical parametric amplifiers31 (OPAs) for pump and probe pulses. Additionally, the 800 nm radiation of the Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sa regenerative amplifier (Spitfire Ace, Spectra Physics, 800 nm, 90 fs, 1.7 mJ, 3 kHz) and its second harmonic (400 nm) were used to excite the sample at these wavelengths. For all measurements, the probe at 2.5 μm was generated using the idler of one OPA, which is in resonance with the optical bandgap of the nanoparticles. Depending on the excitation wavelength the idler (2 μm, 2.4 μm) or signal (1.2 μm) of the second OPA were used to excite the sample at infrared wavelengths. The power of all beams was controlled by a combination of polarizers and waveplates. To keep the fluence low, while still achieving reasonable photon counts on the detector, the sample was mounted outside the focal plane, where the spot size is approximately 1 mm in diameter (FWHM). A peristaltic pump (Cole Parmer Masterflex) and a flow-cell (Harrick Scientific, 1 mm thick CaF2 windows, 2 mm sample thickness) was used to provide fresh sample for each consecutive shot. Spectra were resolved using an imaging monochromator (Triax 180, Horiba) in combination with a nitrogen cooled 2× 32 MCT pixel array (Infrared Associates).
:Sa regenerative amplifier (Spitfire Ace, Spectra Physics, 800 nm, 90 fs, 1.7 mJ, 3 kHz) and its second harmonic (400 nm) were used to excite the sample at these wavelengths. For all measurements, the probe at 2.5 μm was generated using the idler of one OPA, which is in resonance with the optical bandgap of the nanoparticles. Depending on the excitation wavelength the idler (2 μm, 2.4 μm) or signal (1.2 μm) of the second OPA were used to excite the sample at infrared wavelengths. The power of all beams was controlled by a combination of polarizers and waveplates. To keep the fluence low, while still achieving reasonable photon counts on the detector, the sample was mounted outside the focal plane, where the spot size is approximately 1 mm in diameter (FWHM). A peristaltic pump (Cole Parmer Masterflex) and a flow-cell (Harrick Scientific, 1 mm thick CaF2 windows, 2 mm sample thickness) was used to provide fresh sample for each consecutive shot. Spectra were resolved using an imaging monochromator (Triax 180, Horiba) in combination with a nitrogen cooled 2× 32 MCT pixel array (Infrared Associates).
Synthesis
The dodecanethiol-capped particles were synthesized following Keuleyan et al.20 and dispersed in toluene. Additional details can be found in a more recent publication29 with a deviating temperature of 90° instead of 60° and a growth time of 3 minutes.Theoretical description
The single-particle Schrödinger equation is solved using the atomistic effective pseudopotential approach32–34 which allows us to study structures with several thousand atoms, so that we can model experimentally relevant system sizes. The approach is based on density functional theory in the local density approximation (LDA) but includes an empirical correction applied to the non-local part of the pseudopotential,35 and which corrects the failure of the LDA to obtain accurate quasiparticle bandgap states including the effective masses. In the next step, the excitonic states are obtained using the screened configuration interaction (CI) approach35,36 where a microscopic screening model has been used in the calculation of the Coulomb and exchange matrix elements. The structure used in our calculation is spherical with a diameter of 5.25 nm. It includes 1253 Hg atoms and 1328 Te atoms using a so-called aspherical pseudo-hydrogen passivation33 which accurately represent the case of a fully passivated and defect free surface.Results and discussion
Fig. 1 (panel A) shows an exemplary time trace of the band edge bleach signal for excitation at 800 nm. The band-edge dynamics show an initial build-up during the first few picoseconds, followed by a fast decay component on the order of several tens of picoseconds and a slower component decaying on the order of 100 s of picoseconds. The long-lived components of HgTe quantum dots are well known and can be attributed to exciton recombination in the 100 s of picoseconds to nanoseconds and Auger decay auf bi-excitons on the order of tens of picoseconds.17 The signal grows in much more slowly than our temporal resolution of ≲150 fs and can therefore attributed to a delayed arrival of carriers at the bandgap as a consequence of the non-resonant excitation. To obtain a quantitative description of this data, we employ a rate-equation model as depicted in Fig. 1 (panel B, see ESI† for details). Only a single cooling time for the carrier relaxation to the bandgap is considered here, as the bandgap dynamics do not allow for distinction between electron and hole intraband cooling. The bandgap relaxation is modelled using the commonly employed bi-exponential approach37,38 to account for the above mentioned Auger decay of multi-excitons. The number of electron hole-pairs at the band-gap, N0(t), can then be described by: | (1) |
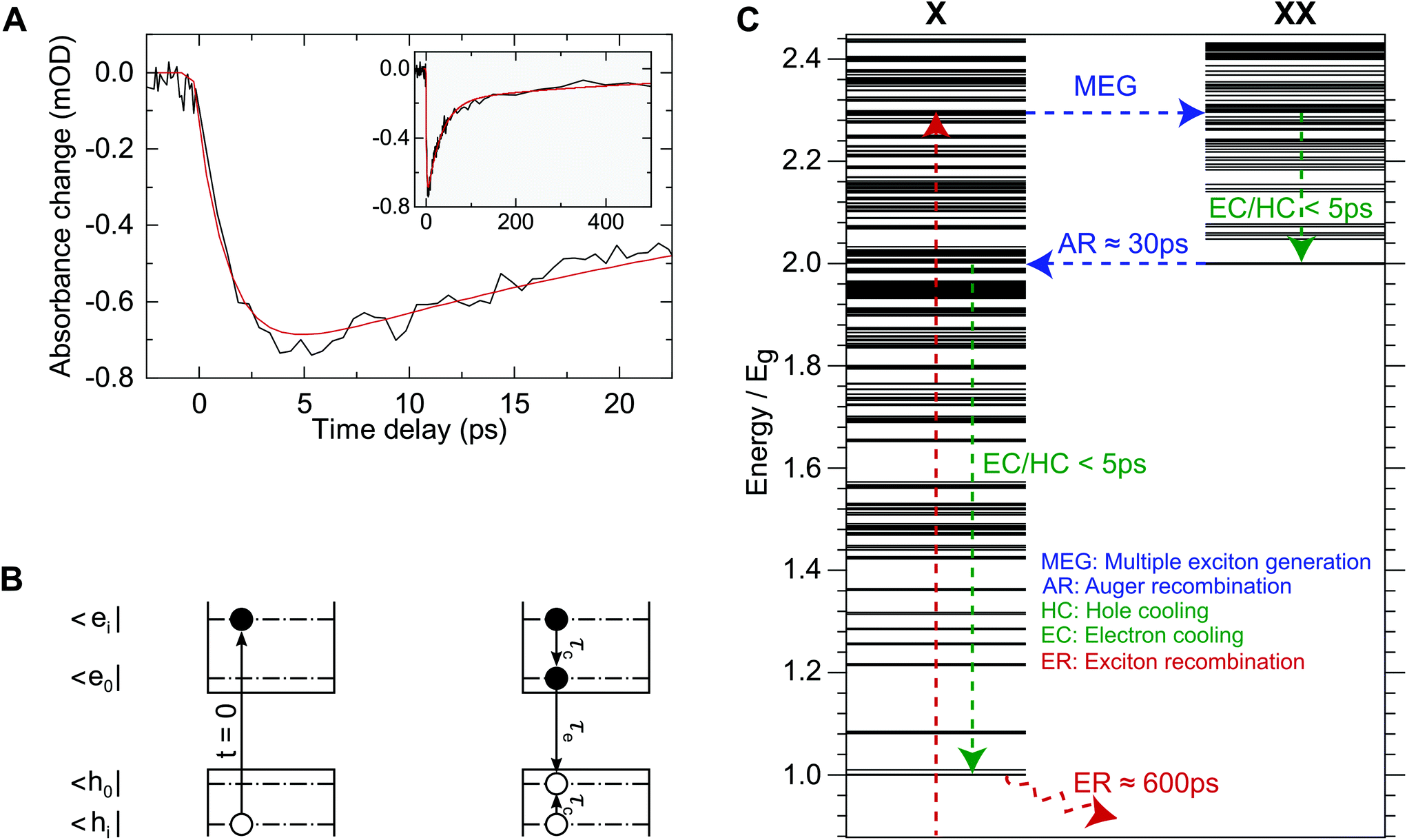 | ||
| Fig. 1 (A) Exemplary band-gap bleach signal at a probe wavelength of 2.5 μm for excitation at 800 nm with 50 nJ pulse energy (black) along with a fit of the two-step model (red) as depicted in (B). Early, sub-10 picosecond dynamics can be mainly attributed to filling of the lowest excitonic state. (B) Illustration of the two-step decay model used for data analysis. At t = 0 an electron–hole-pair, |ei,hi> is created by laser excitation. This state then relaxes to the optical bandgap |e0,h0> with the exciton cooling time, τc. The exciton recombination time is denoted by τe. This model does not treat electron and hole cooling times individually, as only the population of |e0,h0> is accessible using our experimental method. (C) Calculated exciton (X) and bi-exciton (XX) spectra and schematic carrier dynamics in the MEG regime and associated experimentally obtained time-scales. Energy is displayed in units of the band-gap, Eg. After photo-excitation a bi-exciton is created through MEG. This hot bi-exciton then relaxes to the energetically lowest bi-exciton state through intraband cooling, which can be observed as the rising edge in (A). This bi-excitonic state then undergoes inverse Auger-recombination on a 30 picosecond time-scale. The lifetime of the remaining exciton is roughly 600 ps. | ||
Here, Ni,e(0) and Ni,b(0) denote the number of initially excited electron hole pairs in particles with a single exciton only and particles with two excitons, respectively. τe, τb and τc, denote the exciton recombination time, the bi-exciton recombination time and the intraband cooling time. The first term in Eqn (1) accounts for the finite width of the instrument response where erf denotes the Gaussian error function and IRF denotes the full width at half maximum (FWHM) of the instrument response function. All pump and probe pulses are close to transform limited with a pulse duration of 100 fs (FWHM), which results in a signal rise time (10%-to-90%) of ≤150 fs. IRF was therefore fixed to 150 fs so as not to overparameterize the fit.
Eqn (1) was used to simultaneously fit data for different excitation densities and wavelengths. For this global fit, the exciton and bi-exciton lifetime parameters, τe and τb, were shared across this large data set and only the amplitudes, Ni,e(0) and Ni,b(0), and the intraband cooling time, τc, vary between different time traces. An exemplary fitted time trace is also shown in Fig. 1 (panel A, red curve), demonstrating that the data can be adequately modelled using only 3 time-constants covering both, the initial rise and the long-lived interband recombination. We obtain τE = 623 ± 150 ps and τB = 31 ± 10 ps for the exciton recombination and biexciton Auger relaxation times. These lifetimes have already been reported for HgTe NCs with a similar bandgap.17 Our findings for the bi-exciton lifetime are identical within the margins of error. However, we find 5 times shorter exciton lifetimes, which may be caused by differences in synthesis and ligands used.
We now discuss the influence of the excitation wavelength on the band edge dynamics. Fig. 2 shows time traces for excitations at 2.4 μm, 2.0 μm, 1.2 μm, 800 nm and 400 nm. The excitation densities for the data shown correspond to less than 0.2 photons absorbed per quantum dot on average, meaning these measurements are not distorted by multi-photon processes (details of the excitation density analysis can be found in the ESI†). All time traces have been normalized to their peak signal for easier comparability.
 | ||
| Fig. 2 Exemplary bleach signal at the band-gap of 0.5 eV (2.5 μm) for low excitation density (solid lines) and fits according to eqn (1) (dotted lines) for different pump wavelengths: 2.4 μm (red), 2.0 μm (orange), 1.2 μm (yellow), 800 nm (green) and 400 nm (blue). For 400 nm and 800 nm excitation, the long-lived dynamics are governed by multi-excitons generated through MEG. | ||
For 2.4 μm, 2.0 μm and 1.2 μm the long-lived dynamics are well described by a single exponential decay with a lifetime of 623 ± 150 ps, shown as dotted lines. For 800 nm and 400 nm excitation, the photon energy exceeds the MEG threshold.18 Consequently, the long-lived dynamics are governed by multi-excitons. These are no longer well described by a single exponential decay due to Auger recombination of multi-excitons as can be clearly seen in the inset. Apart from carrier multiplication, the excitation wavelength does not affect carrier dynamics past 5 ps after excitation in our data. This means that all intraband relaxation occurs within this time window. By comparing the early dynamics, a trend of longer rise times with higher photon energy can be observed. This trend can be expected because the carriers need to bridge increasingly larger energy differences between photon energy and particle band-gap. We also note that the nearly instantaneous rise of the bleach signal for resonant excitation at 2.4 μm within 111 ± 43 fs is faster than the instrument response function and therefore confirms a temporal resolution of ≲150 fs.
Fig. 3 shows the cooling time as a function of excitation density for all excitation wavelengths. For better comparison we normalize the fluence with respect to signal saturation (see ESI† for details). Below the MEG onset (for 2.4 μm, 2.0 μm and 1.2 μm excitation wavelength), Poissionian statistics can be employed to calculate the mean exciton number per quantum dot, 〈N〉, for which a direct correspondence to the normalized fluence, F/F0, is found in the linear regime as detailed in the ESI.† We do not observe a dependence of intraband cooling time on excitation density. I.e. a ratio of F/F0 = 0.5 corresponds to 25% of excited particles being doubly excited. Yet, no significant change in the cooling time is apparent within our experimental precision. Consequently, multi-particle scattering appears to play only a minor role in intraband relaxation in these quantum dots as also reported for much larger THz-gap HgTe crystallites.39
 | ||
| Fig. 3 Exciton cooling time as a function of excitation fluence for different wavelengths: 2.4 μm (red), 2.0 μm (orange), 1.2 μm (yellow), 800 nm (green) and 400 nm (blue). The dotted lines correspond to the weighted average for each excitation wavelength (also see Table 1). | ||
In the following discussion, the mean value of the cooling times for each excitation wavelength in Fig. 3 is used as a more precise value. Here, the inverse uncertainty of the fit results (error bars in Fig. 3) has been used to calculate the weighted average. The uncertainties correspond to the standard deviation of the fit results for the cooling time. These values are summarized in Table 1. The top left panel of Fig. 4 shows the relation between photon energy and cooling time. This representation also reflects the increase of cooling time with higher photon energy as already apparent in the time traces in Fig. 2. Moreover, the increment of cooling time compared to photon energy appears to stagnate past 1200 nm excitation wavelength, suggesting much higher energy dissipation rates for 800 nm and 400 nm excitation. However, these wavelengths lie in the MEG regime, resulting in the generation of more than a single exciton from a single photon. The initial intraband cooling process is complete within less than 5 ps regardless of excitation wavelength (cf. Fig. 2) while Auger recombination of multi-excitons requires several tens of picoseconds. This means that the electronic configuration after intraband cooling consists of several excitons in the MEG regime.
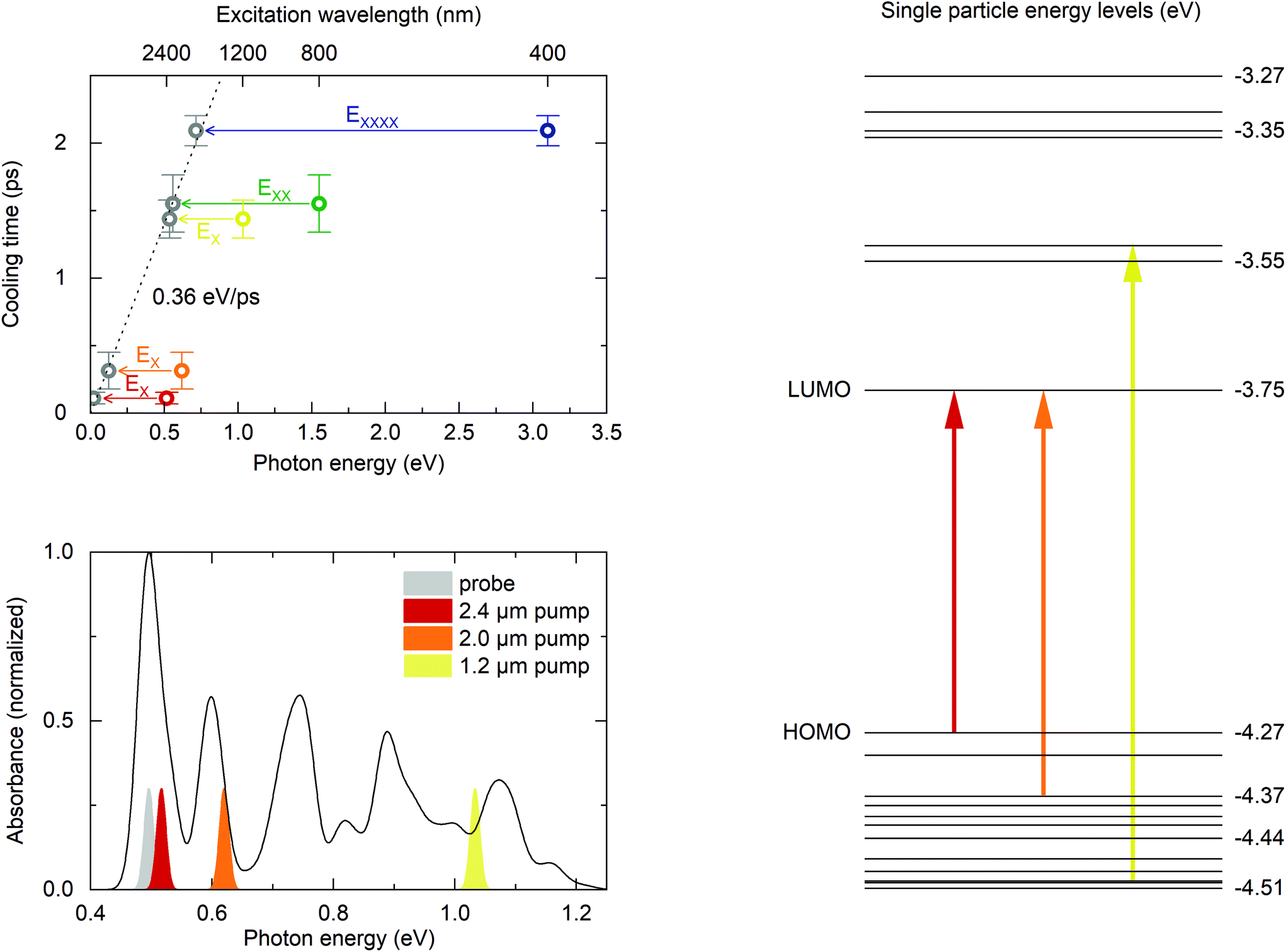 | ||
| Fig. 4 Top left. Exciton cooling times as a function of photon energy. Colored data points represent mean values for the respective excitation wavelength, error bars correspond to the standard deviation of all measured cooling times in the linear regime. Arrows indicate the energy corresponding to the energetically lowest exciton (EX), bi-exciton (EXX) and quad-exciton (EXXXX). This assignment of exciton multiplicity to photon energies is based recent results on MEG in HgTe NCs.18 Subtracting the (multi-) exciton energies from the photon energies accordingly (grey symbols), reveals a linear relation between cooling time and energy to be dissipated from the electronic system at a rate of 0.36 eV ps−1, independent of excitation wavelength or density of states. Bottom left. Calculated absorption spectrum and experimental spectra for pump and probe pulses. The calculated spectrum has been shifted by +33 meV to match the experimental bandgap. Right. Calculated level scheme and optical transitions. | ||
| λ exc (nm) | t cool (fs) | k diss (eV ps−1) |
|---|---|---|
| For 800 nm and 400 nm carrier multiplication was considered in the calculation of the excess energy. The rates for the resonant excitation at 2.4 μm are governed by the instrument response function. | ||
| 2400 | (111) (±43) | — |
| 2000 | 314 ± 136 | 0.39 ± 0.17 |
| 1200 | 1438 ± 141 | 0.37 ± 0.04 |
| 800 | 1552 ± 212 | 0.36 ± 0.05 |
| 400 | 2092 ± 152 | 0.34 ± 0.02 |
Given these different band-gap states for the different excitation regimes, we subtract the energy of the corresponding lowest (multi-) exciton energy from the photon energy in order to obtain a meaningful comparison for the energy dissipation during intraband cooling. Following results from photoconductivity measurements of HgTe quantum dots,18 we subtract the lowest bi-exciton energy, EXX, for 800 nm excitation and the energy of the lowest quadruple exciton, EXXXX, for 400 nm excitation. Our approach is illustrated in the top left panel of Fig. 4 by arrows that correspond to the energy of the respective (multi-)excitons. The grey data points then correspond to the excess of absorbed photon energy that is dissipated during intraband cooling. This representation of our data reveals an essentially linear relationship between dissipated energy and cooling time with a mean energy dissipation rate 0.36 eV ps−1. Individual cooling times and energy dissipation rates for all excitation wavelengths are summarized in Table 1.
Also shown in Fig. 4 is the single particle energy spectrum obtained from the pseudopotential calculations and the corresponding absorption spectrum. These calculations allow assigning the most probable electron and hole states after photo-excitation with 2.4 μm, 2.0 μm and 1.2 μm, shown as arrows in the level scheme. Most importantly, excitation at 2.0 μm creates only hot holes, allowing quantification of the 2Sh-to-1Sh relaxation time of 314 ± 136 fs, which is comparable to the rates found in CdSe NCs with similar 2sh–1sh energy spacings40 and roughly one order of magnitude faster than in PbSe quantum dots with comparable hole energy spacing.41 Excitation at 1.2 μm excites both, hot electrons and holes. Yet, the energy dissipation rates are identical within the margin of error for both excitations. We would like to note that our measurements are blind to the cooling rate associated to any states outside the band-gap, such as long-lived (surface)trap states as we do not measure depopulation of the hot state but population of the cold state. This means that these energy dissipation rates represent lower limits.
Several conclusions can be drawn from these observations. Most striking is the almost universal energy dissipation rate independent of the initially excited state and hence, the density of states. By comparing the theoretically assigned initial states for excitation at 2.0 μm and 1.2 μm (as indicated in the right-hand side of Fig. 4), the presence of an excitation in the conduction band does not seem to influence the cooling rate of the system. This suggests mainly two options for intraband carrier relaxation. Firstly, the cooling rate in the conduction band could be identical to the cooling rate in the valence band. This seems unlikely given the large difference of level spacings in the conduction and valence bands, and much slower electron intraband relaxation times reported for doped MIR HgSe quantum dots in the range of tens to hundreds of picoseconds9 and intraband lifetimes of more than 4 picoseconds reported for self-doped Terahertz-gap HgTe nanoparticles with roughly ten-fold smaller intraband gaps.39 Instead, these findings strongly point to Auger cooling as the dominant decay channel for conduction band relaxation in HgTe quantum dots, similar to CdSe quantum dots12,13 where the process is well understood: electrons rapidly transfer energy to holes in the valence band through Auger coupling. The hole subsequently relaxes back to the band-gap by dissipating energy to phonons and/or ligands. Assuming Auger cooling to be much quicker than the relaxation of the hole, the overall energy dissipation rate would be equal to the dissipation rate of holes alone, which would explain the observation of similar energy dissipation rates for 2.0 μm and 1.2 μm excitation. In this scenario, electron intraband cooling via an Auger mechanism in HgTe quantum dots might be substantially slowed down by spatial separation of electrons and holes in heterostructured core–shell quantum dots.8
This still leaves the question of a universal hole cooling rate unanswered, as the higher density of states for hole excitations larger than 0.1 eV (see Fig. 4, right hand side) should lead to more efficient energy dissipation through coupling to phonons and, consequently, larger cooling rates for higher photon energies. This is clearly not what we observe, not even in the MEG regime close to the ionization threshold of the particles. A possible explanation could be nonadiabatic coupling of states as already discussed in carrier relaxation for CdSe and PbSe quantum dots11,41 where it was argued that the transition rate for two states is scaling with the inverse of their energy spacing.11,40 Such a scaling law would explain the observed constant energy loss rate independent of excitation energy.
Confirmation of this proposed mechanism requires further experiments. These could include similar experiments on MIR HgTe quantum dots of various sizes which will give insight into the influence of intraband energy level spacings and localization effects on carrier cooling. The Auger cooling processes could be addressed by using type II heterostructured quantum dots. Here, spatial separation of electrons and holes should slow down carrier cooling in the picture that we propose.8 Further information on the role of ligands in carrier cooling can be accessed by ligand exchange studies or the use of type I core shell particles.
The cooling time found for 800 nm excitation also imposes an upper limit for the MEG process in MIR HgTe quantum dots which is at most 1.5 ps. Taking intraband-cooling into account as well, the process is likely much quicker than our temporal resolution of ≲150 fs. By estimating a mean carrier cooling time on the order of 1.4 ps after inverse Auger-recombination at an excess energy of one band-gap (see Fig. 4, excitation at 1.2 μm), a time-scale for electron–electron scattering during inverse Auger-recombination of 30 ps is retrieved in these particles. The time-scales obtained for the electron–electron interactions match theoretical calculations for inverse Auger-recombination and MEG in PbSe quantum dots42 where the mismatch between the ultrafast sub-100 fs Auger process during MEG and inverse Auger recombination was attributed to the differences in the density of final states. If this reasoning also holds for HgTe quantum dots will have to be addressed in future theoretical studies.
Conclusion
We investigated the intraband relaxation in thiol-capped mid-infrared HgTe QDs by ultrafast infrared spectroscopy. This relaxation occurs with time constants of less than 2.1 ps independent of excitation wavelength and excitation density (within the linear regime) and without an apparent bottleneck process for carrier cooling. We identify the decay constant for the 2Sh-to-1Sh transition as 314 ± 136 fs. During intraband relaxation, the energy dissipation from the electronic system in MIR HgTe quantum dots is constant up to excitation energies close to the work function of HgTe which includes the MEG regime. A mean energy dissipation rate of 0.36 eV ps−1 indicates only a minor influence of the density of states on intraband cooling. These observations imply either identical cooling rates for electrons and holes or rapid energy transfer from electrons to holes through Auger cooling/heating as found in CdSe quantum dots. The latter cooling mechanism seems more likely given the much slower intraband cooling times reported for electrons in HgSe9 and THz HgTe39 quantum dots. While these findings can be brought to agreement with already existing concepts reminiscent of the carrier relaxation mechanisms found in CdSe quantum dots, further experiments are required to identify the underlying processes. Such mechanistic insight will ultimately allow for tailoring intraband dynamics in MIR quantum dots by either enhancing or suppressing relaxation pathways through e.g. heterostructuring8 and/or choice of ligands and particle size. Furthermore, negligible fluence dependence on the cooling rates suggests that multiparticle scattering processes play only a minor role in carrier cooling which is consistent with a very recent report of intraband dynamics in THz HgTe quantum dots.39 This conclusion is further supported by the unaltered cooling rate found for excitations in the MEG regime.Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 925 – project 170620586. M. R. gratefully acknowledges funding from the International Max Planck Graduate School for Ultrafast Imaging & Structural Dynamics (IMRPS-UFAST). P. G. gratefully acknowledges funding from FWO Vlaanderen. Z. H. acknowledges funding from Ghent University (GOA 01G01019).References
- M. Li, et al., Slow cooling and highly efficient extraction of hot carriers in colloidal perovskite nanocrystals, Nat. Commun., 2017, 8, 3–12 CrossRef.
- M. Li, J. Fu, Q. Xu and T. C. Sum, Slow Hot-Carrier Cooling in Halide Perovskites: Prospects for Hot-Carrier Solar Cells, Adv. Mater., 2019, 31, 1–17 Search PubMed.
- R. D. Schaller and V. I. Klimov, High efficiency carrier multiplication in PbSe nanocrystals: Implications for solar energy conversion, Phys. Rev. Lett., 2004, 92, 1–4 CrossRef PubMed.
- J. T. Stewart, et al., Carrier multiplication in quantum dots within the framework of two competing energy relaxation mechanisms, J. Phys. Chem. Lett., 2013, 4, 2061–2068 CrossRef CAS PubMed.
- M. Li, et al., Low threshold and efficient multiple exciton generation in halide perovskite nanocrystals, Nat. Commun., 2018, 9, 3–9 CrossRef.
- U. Bockelmann and G. Bastard, Phonon scattering and energy relaxation in two-, one-, and zero-dimensional electron gases, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 8947–8951 CrossRef CAS.
- C. Melnychuk and P. Guyot-Sionnest, Multicarrier dynamics in quantum dots, Chem. Rev., 2021, 121, 2325–2372 CrossRef CAS PubMed.
- A. Pandey and P. Guyot-Sionnest, Slow Electron Cooling in Colloidal, Science, 2008, 322, 929–932 CrossRef CAS.
- C. Melnychuk and P. Guyot-Sionnest, Auger Suppression in n-Type HgSe Colloidal Quantum Dots, ACS Nano, 2019, 13, 10512–10519 CrossRef CAS PubMed.
- P. Guyot-Sionnest, B. Wehrenberg and D. Yu, Intraband relaxation in CdSe nanocrystals and the strong influence of the surface ligands, J. Chem. Phys., 2005, 123, 074709 CrossRef PubMed.
- R. R. Cooney, S. L. Sewall, K. E. H. Anderson, E. A. Dias and P. Kambhampati, Breaking the phonon bottleneck for holes in semiconductor Quantum dots, Phys. Rev. Lett., 2007, 98, 1–4 CrossRef.
- P. Kambhampati, Hot exciton relaxation dynamics in semiconductor quantum dots: Radiationless transitions on the nanoscale, J. Phys. Chem. C, 2011, 115, 22089–22109 CrossRef CAS.
- E. Hendry, et al., Direct observation of electron-to-hole energy transfer in CdSe quantum dots, Phys. Rev. Lett., 2006, 96, 1–4 CrossRef PubMed.
- F. C. M. Spoor, et al., Hole cooling is much faster than electron cooling in pbse quantum dots, ACS Nano, 2016, 10, 695–703 CrossRef CAS PubMed.
- F. C. M. Spoor, S. Tomić, A. J. Houtepen and L. D. A. Siebbeles, Broadband Cooling Spectra of Hot Electrons and Holes in PbSe Quantum Dots, ACS Nano, 2017, 11, 6286–6294 CrossRef CAS PubMed.
- P. Geiregat, et al., A phonon scattering bottleneck for carrier cooling in lead-chalcogenide nanocrystals, Mater. Res. Soc. Symp. Proc., 2015, 1787, 1–5 CrossRef CAS.
- C. Melnychuk and P. Guyot-Sionnest, Slow Auger Relaxation in HgTe Colloidal Quantum Dots, J. Phys. Chem. Lett., 2018, 9, 2208–2211 CrossRef CAS.
- C. Livache, et al., Band Edge Dynamics and Multiexciton Generation in Narrow Band Gap HgTe Nanocrystals, ACS Appl. Mater. Interfaces, 2018, 10, 11880–11887 CrossRef CAS.
- E. Lhuillier, S. Keuleyan, H. Liu and P. Guyot-Sionnest, Mid-IR colloidal nanocrystals, Chem. Mater., 2013, 25, 1272–1282 CrossRef CAS.
- S. E. Keuleyan, P. Guyot-Sionnest, C. Delerue and G. Allan, Mercury telluride colloidal quantum dots: Electronic structure, size-dependent spectra, and photocurrent detection up to 12 μm, ACS Nano, 2014, 8, 8676–8682 CrossRef CAS PubMed.
- N. Goubet, et al., Terahertz HgTe Nanocrystals: Beyond Confinement, J. Am. Chem. Soc., 2018, 140, 5033–5036 CrossRef CAS.
- A. Chu, et al., HgTe Nanocrystals for SWIR Detection and Their Integration up to the Focal Plane Array, ACS Appl. Mater. Interfaces, 2019, 11, 33116–33123 CrossRef CAS PubMed.
- X. Tang, M. M. Ackerman, M. Chen and P. Guyot-Sionnest, Dual-band infrared imaging using stacked colloidal quantum dot photodiodes, Nat. Photonics, 2019, 13, 277–282 CrossRef CAS.
- P. Guyot-Sionnest, M. M. Ackerman and X. Tang, Colloidal quantum dots for infrared detection beyond silicon, J. Chem. Phys., 2019, 151, 06090 CrossRef.
- M. M. Ackerman, M. Chen and P. Guyot-Sionnest, HgTe colloidal quantum dot photodiodes for extended short-wave infrared detection, Appl. Phys. Lett., 2020, 116, 083502 CrossRef CAS.
- S. S. Chee, et al., Correlating Structure and Detection Properties in HgTe Nanocrystal Films, Nano Lett., 2021, 21, 4145–4151 CrossRef CAS.
- C. Gréboval, et al., Mercury Chalcogenide Quantum Dots: Material Perspective for Device Integration, Chem. Rev., 2021, 121, 3627–3700 CrossRef.
- J. Qu, et al., Electroluminescence from HgTe Nanocrystals and Its Use for Active Imaging, Nano Lett., 2020, 20, 6185–6190 CrossRef CAS PubMed.
- P. Geiregat, et al., Continuous-wave infrared optical gain and amplified spontaneous emission at ultralow threshold by colloidal HgTe quantum dots, Nat. Mater., 2018, 17, 35–41 CrossRef CAS.
- H. Goodwin, T. C. Jellicoe, N. J. L. K. Davis and M. L. Böhm, Multiple exciton generation in quantum dot-based solar cells, Nanophotonics, 2018, 7, 111–126 Search PubMed.
- R. A. Kaindl, et al., Generation, shaping, and characterization of intense femtosecond pulses tunable from 3 to 20 μm, J. Opt. Soc. Am. B, 2000, 17, 2086 CrossRef CAS.
- J. R. Cárdenas and G. Bester, Atomic effective pseudopotentials for semiconductors, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 30–37 CrossRef.
- A. Karpulevich, H. Bui, D. Antonov, P. Han and G. Bester, Nonspherical atomic effective pseudopotentials for surface passivation, Phys. Rev. B, 2016, 94, 1–10 CrossRef.
- F. Zirkelbach, P. Y. Prodhomme, P. Han, R. Cherian and G. Bester, Large-scale atomic effective pseudopotential program including an efficient spin-orbit coupling treatment in real space, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 1–16 CrossRef.
- H. Bui, A. Karpulevich and G. Bester, Excitonic fine structure of zinc-blende and wurtzite colloidal CdSe nanocrystals and comparison to effective mass results, Phys. Rev. B, 2020, 101, 1–9 CrossRef.
- G. Bester, Electronic excitations in nanostructures: An empirical pseudopotential based approach, J. Phys.: Condens. Matter, 2009, 21, 023202 CrossRef.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Quantization of multiparticle Auger rates in semiconductor quantum dots, Science, 2000, 287, 1011–1014 CrossRef CAS PubMed.
- V. I. Klimov, J. A. McGuire, R. D. Schaller and V. I. Rupasov, Scaling of multiexciton lifetimes in semiconductor nanocrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–12 CrossRef.
- T. Apretna, et al., Few picosecond dynamics of intraband transitions in THz HgTe nanocrystals, Nanophotonics, 2021, 10, 2753–2763 CrossRef.
- R. R. Cooney, et al., Unified picture of electron and hole relaxation pathways in semiconductor quantum dots, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 1–14 CrossRef.
- R. D. Schaller, et al., Breaking the phonon bottleneck in semiconductor nanocrystals via multiphonon emission induced by intrinsic nonadiabatic interactions, Phys. Rev. Lett., 2005, 95, 1–4 CrossRef.
- A. Franceschetti, J. M. An and A. Zunger, Impact ionization can explain carrier multiplication in PbSe quantum dots, Nano Lett., 2006, 6, 2191–2195 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07007j |
| ‡ Current address: Faculty of Fundamental Science, Phenikaa University, Yen Nghia, Ha-Dong District, Hanoi, 10000, Viet Nam. |
| § Current address: Department of Electrical and Computer Engineering, University of Toronto, 10 King's College Road, Toronto, Ontario M5S 3G4, Canada. |
| This journal is © The Royal Society of Chemistry 2022 |