
Construction of a hetero-epitaxial nanostructure at the interface of Li-rich cathode materials to boost their rate capability and cycling performances†
Jun
Cao
a,
Haijian
Huang
 *ab,
Yifan
Qu
a,
Weijian
Tang
b,
Zeheng
Yang
*a and
Weixin
Zhang
*ab
*ab,
Yifan
Qu
a,
Weijian
Tang
b,
Zeheng
Yang
*a and
Weixin
Zhang
*ab
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology and Anhui Key Laboratory of Controllable Chemical Reaction & Material Chemical Engineering, 230009 Hefei, Anhui, PR China. E-mail: wxzhang@hfut.edu.cn; haijian.huang@hfut.edu.cn; zehengyang@hfut.edu.cn
bInstitute of Energy, Hefei Comprehensive National Science Center, Hefei, Anhui 230071, P. R. China
First published on 17th November 2021
Abstract
Lithium-rich cathode materials are considered to be promising candidate cathode materials for next-generation Li-ion batteries owing to their high specific capacities and low cost. Nevertheless, they still suffer from undesirable capacity loss and voltage decay during cycling. In this work, we propose a facile strategy to coat lithiated transition metal phosphates on the surface of Li-rich cathode materials. Strikingly, the coated material shows a hetero-epitaxial nanostructure at the interface between the coating layer and the cathode material. Such a coating layer with a unique interfacial structure could effectively boost the Li+ solid-state diffusion kinetics, protect the cathode material from the corrosion of the electrolyte, and suppress the oxygen loss during the charge–discharge processes. Moreover, the lithiated phosphate coating layer can inhibit the formation of residual Li compounds upon long-term storage under an ambient atmosphere. Based on the above favorable properties, the lithiated phosphate coated Li-rich cathode material shows a high rate capability with a discharge capacity of 156 mA h g−1 obtained at 5 C and decent cyclic stability with a capacity retention of 93.4% achieved at 0.5 C after 140 cycles. This study investigates the interfacial engineering of Li-rich cathode materials via the construction of a Li+-conductive lithiated phosphate coating layer with a hetero-epitaxial interfacial nanostructure, which may offer an effective way to further improve the electrochemical performances of Li-rich cathode materials.
 Haijian Huang | Haijian Huang was born in Hefei, China in 1991. He received his bachelor's degree in science at Northwestern Polytechnical University in 2012. From 2012 to 2015, he continued at the same university to earn his master's degree in engineering under the supervision of Prof. Ying Huang. He then moved to Switzerland and concluded his Ph.D study at Eidgenössische Technische Hochschule Zürich (ETH Zürich) with a diploma in 2019 under the supervision of Prof. Markus Niederberger. From July to October in 2019, he worked as a researcher at ETH Zürich. In 2020, he joined the School of Chemistry and Chemical Engineering at Hefei University of Technology as Associate Professor. The focus of his current research is directed towards the micro-structural engineering of electrode materials for applications in advanced energy storage systems. |
1. Introduction
Over the past 30 decades, the rapid development of the Li-ion battery (LIB) technology has created exuberance in the market of portable electronics. However, with the reach of the batteries’ applications expanding to large-scale energy storage such as electric vehicles and grid energy storage, the energy density of the current commercial LIBs is still far from satisfactory for customers.1–3 In this context, the exploration of high-capacity LIB cathode materials is a timely and highly relevant endeavor to strengthen the position of LIB technology in the energy storage market.4 Compared with commercial LIB cathode materials, Li-rich Mn-based oxide (LRMO) cathode materials [xLi2MnO3·(1 − x)LiTMO2 (0 < x < 1), TM = Ni, Co, Mn, etc.] feature the advantages of prominent specific capacity, low cost, and environmental friendliness, which make LRMO promising alternative cathode materials in next-generation LIBs.5,6 For instance, the theoretical capacity of Li1.2Mn0.54Co0.13Ni0.13O2 (when x = 0.5 in LRMO) can reach almost 380 mA h g−1 for 1.2 Li+ extraction.7 The high charge storage capability of such LRMO cathode materials may presumably originate from both cation redox reactions and anion redox reactions.8 On the one hand, due to the high O/TM (transition metal) ratio in LRMO, one of the O 2p orbitals will be loosely bonded with TM. As a result, such an O-based anion will behave as peroxo-like On− and is thus liable to be engaged in reversible redox reactions.9 As evidence, the transition between divalent O2− and higher-valency O (e.g., O−) has been reported in previous studies.9–13 On the other hand, at high voltages, Mn4+ ions occupying the octahedral site will be oxidized and transformed into Mn7+ ions and migrated to the tetrahedral sites, further contributing to the high specific capacity of LRMO.14Nonetheless, despite the above appealing properties, LMR cathode materials still suffer from poor cyclic performance and insufficient rate capability. Specifically, during the first charge, Li2O will be irreversibly removed from the monoclinic Li2MnO3 phase (one of the two primary phases of LRMO)8,15 based on the reaction Li2MnO3 → MnO2 + Li2O, which will result in the structural instability of LRMO during cycling. Besides, though the involvement of oxygen in the redox reactions can bring about decent charge storage capability for LRMO, the extraction of oxygen based on the above reaction will, however, trigger the dissolution of TM. Such a process will be followed by the phase transformations of LRMO, thereby speeding up its structural degradation during operation.15,16 Moreover, when charging at high voltages, the extensive side reactions at the interface of the LRMO cathode and the electrolyte will lead to the formation of a thick, non-conductive, and inactive cathode/electrolyte interface (CEI), which will significantly hinder Li-ion diffusion, thus lowering the rate capability of the cathode.8,17 Accordingly, considering the above critical issues, the realization of high performance for LRMO remains a great challenge.
Note that the structural degradation of LRMO always starts at the surface of the cathode prior to its expansion to the inner structure.8 Meanwhile, the CEI formation is highly related to the surface properties of the cathode material.8 Hence, surface and/or interface engineering of LRMO plays a vital role in tackling the above-mentioned problems of LRMO. Reflecting its importance, massive efforts have been devoted to coating LRMO with various types of materials, including metal oxides, metal fluorides, metal phosphates, etc.18 The coating layers can effectively reduce the side reactions between the cathode and electrolyte, and in the meantime suppress the oxygen release and structural degradation during cycling. Among the many investigated coating materials for optimizing the Li-ion storage performances of LIB cathode materials, metal phosphates have been considered to be feasible candidate coating species because of their stable structure and high Li-ion conducting nature.19–21 However, the coating strategy does have its drawbacks. In particular, due to the blocking effects of the coating layer on Li-ion diffusion across the interface, the coating species would introduce additional resistance for Li-ion transport, which remains to be appropriately addressed.6,22,23
In this study, a facile etching-induced coating strategy is combined with a subsequent co-lithiation step to construct the LRMO@LiMPO4 (M = Ni, Co, Mn) composite material. The coating layer of the lithiated phosphate with high Li-ion conductivity significantly improves the electrochemical performances of Li-rich cathode materials via restraining the corrosion of the electrode surface by the electrolyte. More intriguingly, a hetero-epitaxial nanostructure is observed at the interface between LRMO and LiMPO4, which significantly facilitates fast Li-ion diffusion across the interface. Furthermore, the coating of LiMPO4 on LRMO can efficiently suppress the oxygen release during charge/discharge, favoring the structural stability during cycling. On the basis of the above favorable properties, the LRMO@LiMPO4 composite achieves an outstanding Li-ion storage performance with a high capacity of 238.8 mA h g−1 and a decent capacity retention of 93.4% delivered at 0.5 C after 140 cycles, implying the great potential of such a coating strategy for boosting the Li-ion storage performances of Li-rich cathode materials.
2. Results and discussion
The coating process of LiMPO4 on LRMO is illustrated in Fig. 1. In the first step, NaH2PO4 was employed to etch the MCO3 (M = Ni, Co, and Mn) precursors, which were prepared based on the method previously reported by us.24 The etching step results in the interfacial phase conversion, resulting in the oriented coating of M3(PO4)2 on MCO3 (denoted as MCO3@M3(PO4)2). Next, the MCO3@M3(PO4)2 composite was mixed with Li2CO3 and calcined at 800 °C under an ambient atmosphere. During this step, co-lithiation of M3(PO4)2 and MCO3 is triggered, which finally leads to the formation of LRMO coated with LiMPO4 (denoted as LRMO@LiMPO4). Besides, to study how the content of the coating species affects the electrochemical performance of LRMO, different feeding ratios of NaH2PO4 were applied during synthesis. The obtained LRMO samples coated with varied contents of the coating LiMPO4 layer are labelled as LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3, respectively (see details in the Experimental section). | ||
| Fig. 1 Schematic diagram of the fabrication of LRMO@ LiMPO4 with a hetero-epitaxial structure at the interface. | ||
To achieve uniform coating, NaH2PO4 etching was performed in a mixed solvent comprising ethylene glycol (EG) and water (volume ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, see the Experimental section) first. At this stage, NaH2PO4 was ionized in the solvent, during which a certain amount of H+ ions and PO43− would be generated. The H+ ions would then etch the surface of the MCO3 precursor, resulting in the release of TM ions (M2+). The reaction of the surface-adsorbed M2+ with PO43− dissolved in the solution would eventually produce the M3(PO4)2 coating layer on the surface of MCO3. The reaction mechanism of the above etching and coating steps can be described as below:
:1, see the Experimental section) first. At this stage, NaH2PO4 was ionized in the solvent, during which a certain amount of H+ ions and PO43− would be generated. The H+ ions would then etch the surface of the MCO3 precursor, resulting in the release of TM ions (M2+). The reaction of the surface-adsorbed M2+ with PO43− dissolved in the solution would eventually produce the M3(PO4)2 coating layer on the surface of MCO3. The reaction mechanism of the above etching and coating steps can be described as below:
| H2PO4− → H+ + HPO42− | (1) |
| HPO42− → H+ + PO43− | (2) |
| 2H+ + MCO3 → M2+ + H2O + CO2↑ | (3) |
| 3M2+ + 2PO43− → M3(PO4)2↓ | (4) |
In the following annealing step, M3(PO4)2 and MCO3 both underwent a lithiation step in the presence of Li2CO3, which resulted in the generation of the final LiMPO4@LRMO. The reactions can be expressed as:
| 5MCO3 + 3Li2CO3 + O2 → 5Li1.2MO2 + 8CO2↑ | (5) |
| M3(PO4)2 + Li2CO3 → LiMPO4 + CO2↑ | (6) |
The mixed solvent (H2O–EG), acting as the reaction medium, plays a crucial role in realizing the uniform coating of LiMPO4. To demonstrate this, 1 mmol NaH2PO4 was gradually added to 50 mL of the mixed H2O–EG solvent (solution A) and pure water (solution B), respectively, and the ionic conductivities of the two solutions were measured. As shown in Fig. S1,† the ionic conductivity of solution A is found to scale up much slower than that of the control solution B. The results suggest that the mixed solvent can retard the ionization of NaH2PO4 to a large extent. In addition, a certain amount of the carbonate precursors was added into the above two solutions, while the pH evolution of the two dispersions with time was recorded (see the Experimental section). Due to the generation of H+ during the ionization of NaH2PO4, the pH values of both dispersions are observed to decrease during the initial step (step I). However, for the carbonate precursors dissolved in solution B, the pH value drops much more sharply from 7.781 to 5.612 within a very short time (Fig. S2a†). For comparison, the pH of the carbonate precursor dispersion in solution A decreases from 7.723 to 6.659 at a relatively much more moderate rate (Fig. S2b†). According to the above results, NaH2PO4 is more liable to be ionized and to release H+ in water than in the H2O–EG system. Consequently, the more acidic solution in the pure water system will provoke fast etching of the carbonate precursor MCO3, as reflected by the fast increase of the pH value in the following procedure (step II, see Fig. S2a†). In such a case, the nucleation speed of the transition metal phosphates is not easy to control, based on which the homogeneous coating of NaH2PO4 on LRMO becomes difficult. As evidence, separate coating particles, instead of a uniform coating layer, were observed when using pure water as the solvent (Fig. S3†). In contrast, for the H2O–EG system, the more moderate pH evolution offers a mild environment for the etching reaction to take place, which leads to the homogeneous growth of the metal phosphate on LRMO at a slow rate during step II (Fig. S2b†).
The main XRD reflections of the pristine sample (LRMO) and the coated samples (LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3) can be well indexed to LiMO2 (M = Ni, Co, and Mn) that possesses the layered α-NaFeO2-type structure with a R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (Fig. 2a).25 Meanwhile, the weak diffraction peaks between 20° and 25° found for the four samples (Fig. 2b) correspond to the LiMn6 superstructures and are induced by the ordered arrangement of Li ions and TM ions in the TM layer of the monoclinic Li2MnO3 phase (C2/m space group).26 For the Li-rich layered structure, the TM ions are located at the 3b octahedral sites, while the Li+ ions occupy the 3a tetrahedron sites. Meanwhile, there is a small portion of Li+ ions that are located at the 3b octahedral sites in the TM layer, as shown in Fig. S4.† The missing of the XRD peaks corresponding to the coating layer of LiMPO4 can likely be attributed to its low content. To verify that the intermediate product M3(PO4)2 in the coating layer is also transferred into lithiated LiMPO4 during the Li-implanting process, we designed a control experiment by mixing M3(PO4)2 with Li2CO3 and conducted the same lithiation procedure as that for the synthesis of LRMO@LiMPO4. Here, M3(PO4)2 was synthesized based on the reaction of NaH2PO4 with MSO4 in the mixed solvent of H2O–EG (see the Experimental section). As shown in Fig. S5,† the XRD patterns of the annealed samples obtained via the control experiment are well in line with the characteristics of LiMnPO4, LiCoPO4 and LiNiPO4, respectively, which indicates the lithiation of transition metal phosphates during the Li-implanting procedure.
m space group (Fig. 2a).25 Meanwhile, the weak diffraction peaks between 20° and 25° found for the four samples (Fig. 2b) correspond to the LiMn6 superstructures and are induced by the ordered arrangement of Li ions and TM ions in the TM layer of the monoclinic Li2MnO3 phase (C2/m space group).26 For the Li-rich layered structure, the TM ions are located at the 3b octahedral sites, while the Li+ ions occupy the 3a tetrahedron sites. Meanwhile, there is a small portion of Li+ ions that are located at the 3b octahedral sites in the TM layer, as shown in Fig. S4.† The missing of the XRD peaks corresponding to the coating layer of LiMPO4 can likely be attributed to its low content. To verify that the intermediate product M3(PO4)2 in the coating layer is also transferred into lithiated LiMPO4 during the Li-implanting process, we designed a control experiment by mixing M3(PO4)2 with Li2CO3 and conducted the same lithiation procedure as that for the synthesis of LRMO@LiMPO4. Here, M3(PO4)2 was synthesized based on the reaction of NaH2PO4 with MSO4 in the mixed solvent of H2O–EG (see the Experimental section). As shown in Fig. S5,† the XRD patterns of the annealed samples obtained via the control experiment are well in line with the characteristics of LiMnPO4, LiCoPO4 and LiNiPO4, respectively, which indicates the lithiation of transition metal phosphates during the Li-implanting procedure.
 | ||
| Fig. 2 (a–c) XRD patterns and (d) Raman spectra of the pristine LRMO and the three coated samples (LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3). (e) Peak-differentiated Raman spectra of LRMO and LRMO@LiMPO4-2 in the range of 300–900 cm−1. | ||
The analysis of the lattice parameters for the four samples (Table S1†) uncovers that the crystal structure of the as-obtained LRMO samples is influenced, to some extent, by the feeding ratio of NaH2PO4 during synthesis. Specifically, with more NaH2PO4 being added during synthesis, a larger c-lattice parameter is found, which is also reflected by the left shift of the (003) reflections as illustrated in Fig. 2c. The results implicate the slight increase of the Li-layer distance in the structure, which can likely be ascribed to the possible occurrence of PO43− doping in LRMO when a larger amount of NaH2PO4 is introduced during synthesis. Such doping of PO43− in LRMO was also previously reported in the literature.27,28 Note that the doping process involves the replacement of O2− in the structure by tetrahedral PO43− that possesses a much larger ionic radius than the O2− anions, which leads to the expansion of the Li layer. The expanded Li layer will benefit the fast Li-ion extraction/intercalation processes, thus boosting the rate capability of the cathode material as discussed later. The peak intensity ratios of (003)/(104) for the four samples are investigated based on the XRD results, which offer information on the ion mixing degree of Li/Ni in the structures.29 As summarized in Table S1,† the values of LRMO, LRMO@ LiMPO4-1, LRMO@ LiMPO4-2 and LRMO@ LiMPO4-3 samples are all larger than the criterion value of 1.2.29 The results clarify the small degree of Li/Ni mixing in the four samples, which has been proved to be beneficial for the structural stability of the cathode materials during cycling.29
Fig. 2d presents the Raman spectra of the original and the coated Li-rich cathode materials. For the pristine sample, the two main Raman bands at 485 and 598 cm−1 are indexed to the Eg bending vibration and A1g stretching vibration of LiMO2 with the layered Rm structure, respectively.30 Relative to the original sample, the blue shifts of the Raman peaks for the coated samples are observed. The phenomenon can be likely related to the shortened bond in the structure, which may originate from the internal stress imposed by the coating layer.22 Except for the above two strong signals, the deconvolution of these two peaks results in additional fitted peaks located at around 415 and 560 cm−1 (Fig. 2e), which are assigned to the phonon vibrations of Li2MnO3.22 Moreover, the shoulder peak at around 650 cm−1 can be ascribed to the Mn–O stretching vibrations in cubic spinel phases.31 Intriguingly, a stronger intensity of this shoulder peak is observed for the coated sample of LRMO@LiMPO4-2 when compared with the primary LRMO, which indicates that the coating process gives rise to more substantial spinel phases on the surface of LRMO. Considering that the spinel phase can effectively prevent the release of oxygen upon deep charging,22 the larger content of the spinel phase coating layer in the LRMO@LiMPO4-2 composite can facilitate the structural stability of the sample during cycling, as discussed later.
The SEM images of the pristine LRMO sample and the coated samples demonstrate that all the four samples exhibit a porous spherical morphology with a diameter of around 2 μm (Fig. S6†). The porous structures can be due to the CO2 emission during the decomposition of the carbonate precursors. The high-resolution transmission electron microscopy (HRTEM) images of LRMO@LiMPO4-2 (Fig. 3a and b) show its core–shell structure, with the thickness of the coating layer determined to be about 7 nm. The selected area within the core of the sample (area A, marked in Fig. 3b) shows well-defined lattice fringes with a d-spacing of 0.47 nm (Fig. 3c), which corresponds to the (003) plane of the typical layered LRMO phase. The HRTEM image of the selected area B (Fig. 3d, marked in Fig. 3b) displays the interface between the core and the shell of the composite, which evidences the well compatible combination of the core and shell sample. Besides, Fig. S7a† shows the existence of the LiMn2O4 spinel phase in the core area, which is in accord with the Raman results as discussed above. Moreover, FFT analysis of the selected area in the HRTEM image of Fig. S7b† points to the (400) reflection of spinel-type LiMn2O4 (PDF #88-1030) and the (003) reflection of LRMO (PDF #85-1980) (Fig. S7c†), further confirming the existence of the spinel phase in the structure. Within the shell of the particle, the lattice fringes with a d-spacing of 0.25 nm correspond to the (311) planes of LiMnPO4, verifying the successful coating of the lithiated manganese phosphate on the surface of LRMO. Interestingly, the crystallographic orientation of the coating species is found to be parallel to that of the LRMO core, which can be due to the epitaxial growth during preparation. Such a hetero-epitaxial nanostructure effectively avoids the blocking effects of the coating layer on Li+ diffusion across the interface, thus significantly facilitating fast Li+ storage kinetics. The selected area electron diffraction (SAED) patterns of area A show well-resolved diffraction spots, which are ascribed to the (003) and (101) planes of LRMO (Fig. 3e). In the SAED patterns of area B, two sets of electron diffractions are observed, which can be indexed to the (009) plane for LRMO and the (200) and (111) planes for the LiMPO4 coating layer, respectively (Fig. 3f). Similar morphological features are also found for the LRMO@LiMPO4-1 sample, except that its coating layer is much thinner than that of the LRMO@LiMPO4-2 sample (Fig. S8†). However, for the LRMO@LiMPO4-3 sample, an obvious transition area is observed at the interface, with the hetero-epitaxial structure nearly disappearing (Fig. S8†). The absence of the hetero-epitaxial structure in LRMO@LiMPO4-3 can be likely due to the larger content of the coating species in LRMO@LiMPO4-3, which gives rise to a higher degree of PO43− doping in LRMO that damages the interfacial structure. To further clarify the structure of the coating area (area C in Fig. 3b), the SAED pattern of area C is given in Fig. 3g. The indexing of the diffraction spots points to the (022) and (041) planes of LiMnPO4 (PDF 77-0178), further evidencing the successful coating of LiMnPO4 on the surface of LRMO. The energy dispersive spectroscopy (EDS) results reveal the uniform distribution of O, Mn, Ni, Co, and P within the composite particles, attesting to the homogeneous coating of LiMPO4 on LRMO (Fig. 3h, i and Fig. S9†).
 | ||
| Fig. 3 (a–d) HRTEM images of LRMO@LiMPO4-2. SAED patterns of (e) area A, (f) area B, and (g) area C in (b). (h) EDS spectrum of LRMO@LiMPO4-2. (i) SEM image of a typical LRMO@LiMPO4-2 particle and the corresponding element distribution maps of O, Mn, Ni, Co, and P. | ||
Based on the above analysis, the construction of the LRMO@LiMPO4 composite with a hetero-epitaxial interfacial structure is proposed as below: upon annealing during preparation, the carbonate precursor is decomposed into metal oxides, which are subsequently lithiated. Due to the different lithiation temperatures for the carbonate precursor in the core and M3(PO4)2 in the coating layer, the Li+ ion will migrate from the lithiated metal oxides to the coating layer, leading to the formation of Li vacancies in the core structure. The TM ions will then migrate to the lithium layer and occupy the Li vacancies to form a structure with a lower energy. As a structure with sufficient free space typically tends to rotate during the crystal growth until a lower energy of the structure is achieved, the epitaxial heterostructure at the interface can be eventually formed.
The envelope of the Mn 2p2/3 XPS spectra for both LRMO and LRMO@LiMPO4-2 can be deconvoluted into two components that are assigned to Mn3+ (641.8 eV) and Mn4+ (642.9 eV), respectively. Moreover, the quantitative analysis of Mn 2p reveals the Mn3+/Mn4+ ratios in the samples. For the pristine LRMO, Mn3+/Mn4+ is determined to be 0.257, which is lower than those of the coated LRMO@LiMPO4-1 (0.290), LRMO@LiMPO4-2 (0.328), and LRMO@LiMPO4-3 (0.344) samples (Fig. 4b). Hence, the average valence state of Mn in the coated samples is relatively lower than that in LRMO, which can be due to the doping of PO43− into the surface of LRMO. Such a doping process will induce the decomposition of Li2MnO3 (Mn4+) into Li2O and LiMnO2 (Mn3+) accompanied by the release of oxygen upon annealing, thus reducing the average valence state of Mn and meanwhile, giving rise to the formation of oxygen vacancies. The O 1s spectra provide further proof. The fitted peaks of O 1s at 529.2, 531.3, and 533.4 eV can be indexed to the lattice oxygen, the oxygen vacancies (OV) and the chemisorbed oxygen, respectively. Compared with 28.6% of OV in the primary LRMO, the coated LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3 samples demonstrate higher OV contents of 33.9%, 36.5%, and 38.0%, respectively, which is consistent with the above analysis.32 Note that the oxygen vacancies on the surface of LRMO can efficiently inhibit the oxygen loss from the structure upon deep charging, thus contributing to enhancing the structural stability during cycling.31 The Ni 2p spectra of LRMO and LRMO@LiMPO4-2 reveal that the intensity ratios of Ni3+/Ni2+ are higher in the coated samples relative to those of the pristine one (Fig. 4d and Fig. S8†). The results can be ascribed to the doping of PO43−, which inevitably increases the average valence state of Ni on the basis of charge balance. For Co, the absence of the satellite peaks in both samples, together with the same peak position difference between the split spin–orbit components of Co 2p3/2 and Co 2p1/2, points to the fact that the chemical state of Co in LRMO is not significantly influenced by the coating process (Fig. 4e). Moreover, the appearance of the P XPS signal in the coated samples further corroborates the successful coating of LiMPO4 on the surface of LRMO (Fig. 4f and Fig. S10†).
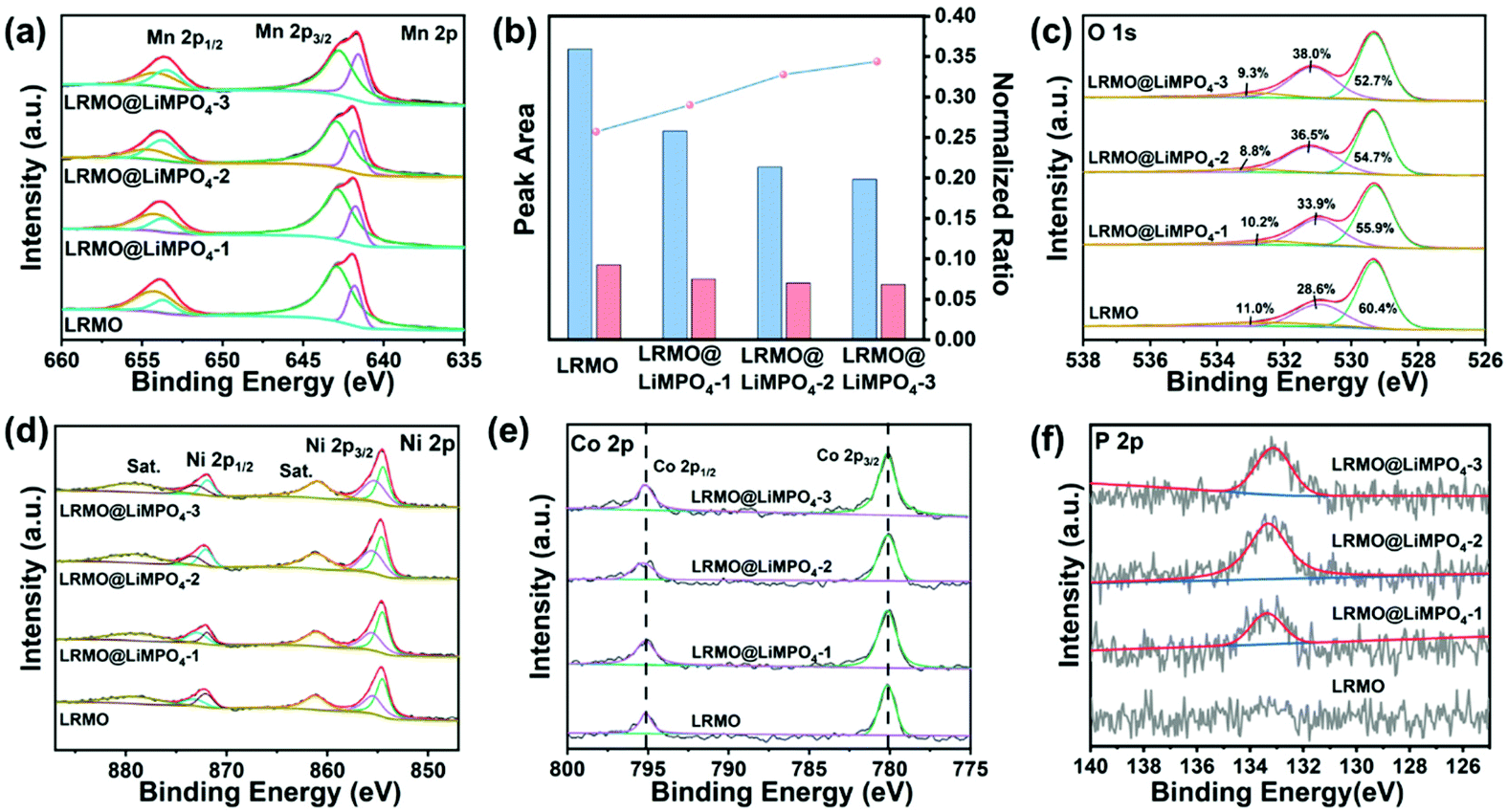 | ||
| Fig. 4 (a) High-resolution XPS spectra of Mn 2p3/2 and (b) peak areas of Mn3+ and Mn4+ for the pristine LRMO and the three coated samples (LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3). High-resolution XPS spectra of (c) O 1s, (d) Ni 2p, (e) Co 2p, and (f) P 2p for LRMO and the coated samples. | ||
To investigate the effectiveness of the LiMPO4 coating, the electrochemical performances of the pristine and coated cathode materials were evaluated and compared as shown in Fig. 5 and Table S2.†Fig. 5a, b and Fig. S11† present the charge–discharge profiles of LRMO and the coated samples. According to the results, the initial coulombic efficiencies of LRMO@LiMPO4-1, LRMO@LiMPO4-2, and LRMO@LiMPO4-3 are determined to be about 82.1%, 82.6%, and 78.8%, respectively, which are all higher than 76.8% achieved for the primary LRMO. Besides, a small discharge plateau at around 2.7 V (as marked by the small circle in Fig. 5b and Fig. S11†) is observed for the coated samples, and it is nearly absent in the case of the pristine LRMO sample. Such a discharge plateau can be related to the redox reaction of the spinel phase, of which the coated sample possesses a higher content as discussed above. The cycling performances of the original and the coated samples were evaluated at 0.5 C within the voltage range of 2.0–4.8 V (Fig. 5c). The results demonstrate that the coated LRMO@LiMPO4-2 sample shows the best cycling performance among the investigated samples. Specifically, after 140 cycles, the LRMO@LiMPO4-2 sample achieves a high discharge capacity of 238.8 mA h g−1 with a capacity retention of 93.4%, while the pristine LRMO cathode delivers a discharge capacity of only 181.0 mA h g−1 with a capacity retention of 78.0%. During cycling, severe voltage decay is found for the pristine LRMO sample. To be specific, the average discharge voltages are shown in Fig. 5d. After 200 cycles, the voltage decay reaches almost 0.64 V. In comparison, after the same number of cycles, the coated LRMO@LiMPO4-2 shows the smallest voltage decay of 0.46 V, confirming its decent cyclic stability. When evaluated at a higher rate of 1 C after 100 cycles, LRMO could deliver a discharge capacity of 142.8 mA h g−1, corresponding to 76.8% of the initial capacity (Fig. S12†). For comparison, LRMO@LiMPO4-2 shows a much higher discharge capacity of 210.8 mA h g−1 that represents 94.2% of the initial capacity, further evidencing the outstanding cycling performance of the coated LRMO@LiMPO4-2 sample. The rate performances of the samples are compared in Fig. 5e. When tested at 0.1 C, 0.2 C, 0.5 C, 1 C, 2 C, and 5 C, LRMO@LiMPO4-2 can show high specific capacities of 277.2, 271.0, 252.4, 235.6, 210.7, and 159.3 mA h g−1, respectively, which are superior to those of the other three samples. Even at a current density of 10 C, LRMO@LiMPO4-2 can still achieve a high discharge capacity of 91.1 mA h g−1, shedding light on its outstanding rate performance. The improved rate performances of the coated LRMO@LiMPO4-2 could be attributed to the improvement of the Li-ion solid-state diffusion kinetics resulting from the high Li+ conductivity of LiMPO4-2 and the hetero-epitaxial nanostructure. Besides, it should be noted that the LRMO@LiMPO4-3 sample, with a higher content of the coating layer, shows an inferior rate performance relative to that of the LRMO@LiMPO4-2 sample. The results can be ascribed to the damaged interfacial hetero-epitaxial structure in LRMO@LiMPO4-3 as discussed above, which impedes the fast Li+ diffusion across the interface. Fig. 5f and g display the charge–discharge profiles of LRMO and LRMO@LiMPO4-2 at different rates, respectively. Interestingly, at high current densities, more serious polarization is observed for LRMO relative to LRMO@LiMPO4-2, further evidencing the enhanced Li-ion diffusion kinetics in the LRMO@LiMPO4-2 electrode.
 | ||
| Fig. 5 Charge–discharge profiles of (a) LRMO and (b) LRMO@LiMPO4-2 at different cycles. (c) Cycling performances at 0.5 C, (d) changes of the average potential during cycling, and (e) rate performances for the pristine LRMO and the three coated samples (LRMO@LiMPO4-1, LRMO@LiMPO4-2 and LRMO@LiMPO4-3). Charge–discharge profiles of (f) LRMO and (g) LRMO@LiMPO4-2 at different current rates. (h) pH evolution of the aqueous dispersions of LRMO and the coated samples upon long-term storage under an ambient atmosphere. | ||
For Li-rich cathode materials, there are usually residual Li species left on the surface during synthesis. The residual Li components could form LiOH and Li2CO3 upon exposure to the atmospheric moisture and CO2 during storage. LiOH would react with the electrolyte and generate HF,33 which could further lead to the dissolution of the TM ions. Meanwhile, the Li2CO3 layer possesses a rather low Li+ conductivity, which contributes to a high interfacial impedance.34,35 The above factors could cause severe capacity attenuation and voltage decay, thus impeding the practical applications of Li-rich cathode materials.36 To probe into the influence of the LiMPO4 coating layer on reducing the residual alkali, the pristine sample and the coated sample were both stored under an ambient atmosphere for a week prior to the performance evaluation. As shown in Fig. S13,† LRMO@LiMPO4-2 shows a similar cycling performance to the fresh sample, with a capacity retention of 88.5% achieved after 100 cycles. However, for the pristine sample, the capacity retention drops significantly to 37.9% after 100 cycles at 0.5 C. In addition, the investigations of the pH values of the aqueous suspensions that contain 1 g of the samples and 50 mL of deionized water (kept for 30 min) were performed (Fig. 5h). With the storage time lasting for 3 weeks, a significant increase of the pH value of the LRMO aqueous suspension is observed, which could be due to the formation of LiOH and Li2CO3 on the surface. However, the pH change in the LRMO@LiMPO4-1 aqueous suspension is much smaller. For the LRMO@LiMPO4-2 and LRMO@LiMPO4-3 suspensions, the pH evolution is even more negligible, indicating that the coating layer can effectively reduce the content of the residual Li compounds on the surface of the material.
The cyclic voltammetry (CV) curves of LRMO and LRMO@LiMPO4-2 are displayed in Fig. 6a and b. The CV curve during the initial cycle for LRMO shows two distinct oxidation peaks at about 4.0 and 4.6 V, which can be attributed to the oxidation reaction of Ni2+ and Co3+ and the loss of lattice oxygen, respectively.37 Because of the irreversibility of the oxygen loss process, the oxidation peak assigned to such a process emerges only in the initial charging process. For LRMO@LiMPO4-2, the intensity of this peak is much weaker, pointing to the suppressed oxygen loss in the coated sample. The two reduction peaks appearing at about 3.75 and 3.3 V correspond to the reduction of Ni4+ and Co4+ and the reduction of Mn4+, respectively.16 Compared with the LRMO sample, the degree of coincidence of the CV curves of LRMO@LiMPO4-2 is much higher, indicating that the coating layer could effectively suppress the phase transitions, thus leading to a better structural stability during the cycling process. However, relatively larger polarization is observed for the coated sample as revealed by the higher voltage difference between the cathodic and anodic peaks, which could be possibly attributed to the undesirable electronic conductivity of the LiMPO4 coating layer.38–40 Nevertheless, as the Li-ion storage performance of the coated sample has been verified to be superior to that of the pristine sample, the positive role of the LiMPO4 coating layer, including the inhibition of the electrolyte corrosion and oxygen release as well as facilitation of Li-ion diffusion kinetics, should still dominate in affecting the electrochemical performance of LRMO. Fig. 6c and e show the EIS spectra of the pristine LRMO and the coated samples obtained after 1 cycle and 100 cycles, respectively. All the spectra are composed of a semicircle and an uplifted straight line, which reflect the charge transfer resistance (Rct) and the Warburg impedance (Zw) of the Li+ ion diffusion, respectively. Through simulation, an equivalent circuit is obtained as displayed in Fig. S14.† The results suggest that the LRMO@LiMPO4-2 sample possesses the smallest Rct value among the four samples. Moreover, the Li+ diffusion coefficient (DLi+) of the samples could be calculated based on the equation DLi+ = R2T2/(2A2F4n4C2σω2). Accordingly, the DLi+ value of the LRMO@LiMPO4-2 material is determined to be 1.21 × 10−15 cm2 s−1, which is much higher than 3.99 × 10−16 cm2 s−1 obtained for the pristine LRMO. Even after 100 cycles, LRMO@LiMPO4-2 still shows the smallest Rct and the highest DLi+, as shown in Fig. 6f, corroborating that the high Li+ conductivity of the coating LiMPO4 layer and the hetero-epitaxial structure at the interface together lead to a much reduced Li+ diffusion barrier.
 | ||
| Fig. 6 CV curves of (a) LRMO (a) and (b) LRMO@LiMPO4-2 at 0.1 mV s−1. Electrochemical impedance spectra (EIS) of the samples collected after (c) 1 cycle and (e) 100 cycles. The relationship between Z′ and ω−1/2 derived from the electrochemical impedance spectra collected after (d) 1 cycle and (f) 100 cycles, respectively. | ||
3. Conclusions
In summary, a facile coating strategy is proposed to construct the LRMO@LiMPO4 composite with a hetero-epitaxial nanostructure at the interface. The LiMPO4 coating layer remarkably improves the cycling performance and rate capability of Li-rich cathode materials. Collectively, the performance enhancement of the coated sample can be ascribed to the following reasons: firstly, the coating layer prevents the corrosion of the electrolyte towards the cathode material, thus promoting the cycling stability. Secondly, the coating of LiMPO4 leads to the formation of more oxygen vacancies and the LiMn2O4 spinel phase, both of which could effectively reduce the irreversible capacity loss via inhibiting the oxygen release during cycling. Thirdly, the high Li+ conductivity nature of the LiMPO4 coating layer reduces the charge-transfer resistance between the electrode and the electrolyte, thus boosting the Li+ diffusion kinetics. More importantly, the hetero-epitaxial nanostructure effectively facilitates fast Li+ transportation across the interface between the coating layer and the cathode material. Furthermore, the coating process can reduce the residual Li species on the surface of the cathode material upon long-term storage, which further underscores the practical application value of this LiMPO4 coating strategy. Accordingly, this facile approach for constructing the LiMPO4 coating layer with a hetero-epitaxial interface provides a promising avenue to further improve the electrochemical performances of Li-rich cathode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (NSFC 91834301, 91534102, 2210080709, and 22108053) and the Anhui Provincial Science and Technology Department Foundation (201903a05020021 and 202003a05020046).References
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed
.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed
- H. Cheng, J. Shapter, Y. Li and G. Gao, J. Energy Chem., 2021, 57, 451–468 CrossRef
- G. Ma, S. Li, W. Zhang, Z. Yang, S. Liu, X. Fan, F. Chen, Y. Tian, W. Zhang, S. Yang and M. Li, Angew. Chem., Int. Ed., 2016, 55, 3667–3671 CrossRef CAS PubMed
- L. de Biasi, B. Schwarz, T. Brezesinski, P. Hartmann, J. Janek and H. Ehrenberg, Adv. Mater., 2019, 31, 1900985 CrossRef PubMed
- B. Wu, X. Yang, X. Jiang, Y. Zhang, H. Shu, P. Gao, L. Liu and X. Wang, Adv. Funct. Mater., 2018, 28, 1803392 CrossRef
- M. Freire, N. V. Kosova, C. Jordy, D. Chateigner, O. I. Lebedev, A. Maignan and V. Pralong, Nat. Mater., 2016, 15, 173–177 CrossRef CAS PubMed
- W. He, W. Guo, H. Wu, L. Lin, Q. Liu, X. Han, Q. Xie, P. Liu, H. Zheng, L. Wang, X. Yu and D.-L. Peng, Adv. Mater., 2021, 2005937 CrossRef PubMed
- S. Zhao, Z. Guo, K. Yan, S. Wan, F. He, B. Sun and G. Wang, Energy Storage Mater., 2021, 34, 716–734 CrossRef
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák and G. Van Tendeloo, Science, 2015, 350, 1516–1521 CrossRef CAS PubMed
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvère and J.-M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed
- X. Li, Y. Qiao, S. Guo, Z. Xu, H. Zhu, X. Zhang, Y. Yuan, P. He, M. Ishida and H. Zhou, Adv. Mater., 2018, 30, 1705197 CrossRef PubMed
- M. D. Radin, J. Vinckeviciute, R. Seshadri and A. Van der Ven, Nat. Energy, 2019, 4, 639–646 CrossRef CAS
- S. Zhao, K. Yan, J. Zhang, B. Sun and G. Wang, Angew. Chem., Int. Ed., 2021, 60, 2208–2220 CrossRef CAS PubMed
- Y. Liu, Z. Yang, J. Zhong, J. Li, R. Li, Y. Yu and F. Kang, ACS Nano, 2019, 13, 11891–11900 CrossRef CAS PubMed
- X. Ding, D. Luo, J. Cui, H. Xie, Q. Ren and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 7778–7782 CrossRef CAS PubMed
- C. Zhang, Y. Feng, B. Wei, C. Liang, L. Zhou, D. G. Ivey, P. Wang and W. Wei, Nano Energy, 2020, 75, 104995 CrossRef CAS
- D. Zhang, L.-L. Hu, Y.-G. Sun, J.-Y. Piao, X.-S. Tao, Y.-S. Xu, A.-M. Cao and L.-J. Wan, J. Mater. Chem. A, 2018, 6, 8992–8999 RSC
- J. Duan, C. Wu, Y. Cao, K. Du, Z. Peng and G. Hu, Electrochim. Acta, 2016, 221, 14–22 CrossRef CAS
- A. Mauger, C. M. Julien, M. Armand, J. B. Goodenough and K. Zaghib, Curr. Opin. Electrochem., 2017, 6, 63–69 CrossRef CAS
- X.-D. Zhang, J.-L. Shi, J.-Y. Liang, Y.-X. Yin, J.-N. Zhang, X.-Q. Yu and Y.-G. Guo, Adv. Mater., 2018, 30, 1801751 CrossRef PubMed
- S. Ramakrishnan, B. Park, J. Wu, W. Yang and B. D. McCloskey, J. Am. Chem. Soc., 2020, 142, 8522–8531 CrossRef CAS PubMed
- B. Guo, J. Zhao, X. Fan, W. Zhang, S. Li, Z. Yang, Z. Chen and W. Zhang, Electrochim. Acta, 2017, 236, 171–179 CrossRef CAS
- D. Gao, Z. Zeng, H. Mi, L. Sun, X. Ren, P. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 23964–23972 RSC
- K. Shi, K. Liang, J. Zheng and Y. Qiu, Appl. Surf. Sci., 2019, 483, 1–9 CrossRef CAS
- H. Z. Zhang, Q. Q. Qiao, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 7454–7460 RSC
- Y. Zhao, J. Liu, S. Wang, R. Ji, Q. Xia, Z. Ding, W. Wei, Y. Liu, P. Wang and D. G. Ivey, Adv. Funct. Mater., 2016, 26, 4760–4767 CrossRef CAS
- K. Kleiner, B. Strehle, A. R. Baker, S. J. Day, C. C. Tang, I. Buchberger, F.-F. Chesneau, H. A. Gasteiger and M. Piana, Chem. Mater., 2018, 30, 3656–3667 CrossRef CAS
- G. Singh, W. C. West, J. Soler and R. S. Katiyar, J. Power Sources, 2012, 218, 34–38 CrossRef CAS
- Y. Liu, Z. Yang, J. Li, B. Niu, K. Yang and F. Kang, J. Mater. Chem. A, 2018, 6, 13883–13893 RSC
- Z. Ding, C. Zhang, S. Xu, J. Liu, C. Liang, L. Chen, P. Wang, D. G. Ivey, Y. Deng and W. Wei, Energy Storage Mater., 2019, 21, 69–76 CrossRef
- S.-Q. Yang, P.-B. Wang, H.-X. Wei, L.-B. Tang, X.-H. Zhang, Z.-J. He, Y.-J. Li, H. Tong and J.-C. Zheng, Nano Energy, 2019, 63, 103889 CrossRef CAS
- A. Sharafi, S. Yu, M. Naguib, M. Lee, C. Ma, H. M. Meyer, J. Nanda, M. Chi, D. J. Siegel and J. Sakamoto, J. Mater. Chem. A, 2017, 5, 13475–13487 RSC
- L. Cheng, E. J. Crumlin, W. Chen, R. Qiao, H. Hou, S. F. Lux, V. Zorba, R. Russo, R. Kostecki, Z. Liu, K. Persson, W. Yang, J. Cabana, T. Richardson, G. Chen and M. Doeff, Phys. Chem. Chem. Phys., 2014, 16, 18294–18300 RSC
- S. Hy, H. Liu, M. Zhang, D. Qian, B.-J. Hwang and Y. S. Meng, Energy Environ. Sci., 2016, 9, 1931–1954 RSC
- Q. Li, D. Zhou, L. Zhang, D. Ning, Z. Chen, Z. Xu, R. Gao, X. Liu, D. Xie, G. Schumacher and X. Liu, Adv. Funct. Mater., 2019, 29, 1806706 CrossRef
- F. Zhou, K. Kang, T. Maxisch, G. Ceder and D. Morgan, Solid State Commun., 2004, 132, 181–186 CrossRef CAS
- J. Wolfenstine and J. Allen, J. Power Sources, 2004, 136, 150–153 CrossRef CAS
- M. Johannes, K. Hoang, J. Allen and K. Gaskell, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115106 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06746j |
| This journal is © The Royal Society of Chemistry 2021 |