
Heterogeneous multi-compartmental DNA hydrogel particles prepared via microfluidic assembly for lymphocyte-inspired precision medicine†
Sophie Wan Mei
Lian‡
ab,
Song
Guo‡
 ab,
Kewei
Ren
a,
Ying
Xu
c,
John S.
Ho
ab and
Chia-Hung
Chen
*c
ab,
Kewei
Ren
a,
Ying
Xu
c,
John S.
Ho
ab and
Chia-Hung
Chen
*c
aDepartment of Biomedical Engineering, National University of Singapore, 4 Engineering Drive 3, 04-08, 117583, Singapore
bInstitute for Health Innovation and Technology (iHealthtech), MD6, 14 Medical Drive #14-01, Singapore 117599
cDepartment of Biomedical Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: chiachen@cityu.edu.hk
First published on 19th November 2021
Abstract
Lymphocytes play a vital role in immunosurveillance through sensing biomolecules and eliminating targeted invaders. Compared with conventional therapies that depend on drug loading, lymphocytes are advantageous as they are able to ensure self-regulated therapeutics. Here, novel multi-compartmental DNA hydrogel particles were synthesized using a microfluidic assembly for intelligent cancer treatment via the logic-based control of siRNA release without external stimulation. The sensing sequence (D1) was compartmentalized from the treatment sequence (D2) with the use of core–shell DNA hydrogel particles. When D1 detects a cancer-associated biomarker, miRNA-21, a sequence cascade is triggered to release siRNA from D2, effectively eliminating the targeted cancer cells via lymphocyte-inspired precision medicine.
Introduction
In the human system, lymphocytes are essential because of their ability to recognize and eliminate invaders such as pathogens and cancer cells. For example, during cancer immunosurveillance, lymphocytes sense biomarkers associated with cancer pathogenesis and infiltrate tumors to trigger an anti-cancer immune response.1 Lymphocytes have at least three features that would be advantageous for therapeutics: (1) the inherent drug-free approach, (2) long-term biostability and biosafety, and (3) a smart system to quickly sense and eliminate the target invaders to manage diseases in their early stages.2 Recently, engineered lymphocytes have been developed for immunotherapy, such as adoptive cell therapy, to offer curative responses in patients with cancer.3 However, this therapy usually relies on T-cell receptor modification.4 Therefore, the challenges of the limited production yield, quality controls and unpredictable bio-fabrication in cell culture remain.5 In fact, engineered lymphocytes may cause a serious cytokine release syndrome, such as fever-like symptoms and potential neurological difficulties, giving rise to biomedical concerns.6,7Hydrogels are a three-dimensional chemically or physically crosslinked polymeric network with the ability to absorb a large volume of reagents for sustainable delivery.8 Hydrogels can be formed with synthetic polymers, such as poly(ethylene glycol) (PEG), and these are promising biocompatible/degradable materials for physiological applications.9 By tailoring the monomers and crosslinkers used, functional hydrogel systems can be developed to be responsive to various external stimuli, such as pH, temperature, enzymes, magnetic fields or light, and can be used for sustainable treatment in a programmable manner.10 For example, poly(N-isopropylacrylamide) (pNIPAM) composite hydrogels incorporating polypyrrole (PPy) nanoparticles were fabricated for on-demand treatment utilizing near-infrared light stimulation.11 Notably, unlike lymphocytes with biological variation, synthetic hydrogels are highly uniform, ensuring biomedical safety and reliability.11 However, most hydrogel systems still rely on the manual application of external stimuli, which limits their biomedical applications in long-term healthcare monitoring, in on time treatment and in intelligent therapeutics.12 Moreover, due to the porous nature of the hydrogel network, there is often the issue of drug cargo diffusion out of the network, which could also present a risk for drug toxicity.10–12 To overcome this challenge, functional nanoparticles were developed for target delivery by antigen-cell receptor binding, but the challenges for long-term treatment still remain.13,14
RNA interference (RNAi) is a biological process of post-transcriptional gene translation and expression suppression and has been widely investigated for its potential in drug-free targeting therapeutics.15 The process utilizes double-stranded RNA (dsRNA), such as microRNA (miRNA) or short interfering RNA (siRNA), to release target-specific, gene-silencing agents via degradation or translational repression.16 The efficiency of siRNA has been demonstrated in the ability to treat a range of diseases, such as metabolic and genetic disorders due to aberrant gene expression, viral infections, chronic infections and cancers.17–19
Despite all the advantages that siRNA therapeutics have to offer, the instability of siRNA in the bloodstream during both delivery and long-term treatment needs to be addressed. In the bloodstream, unmodified siRNA can be degraded by endogenous enzymes, and have non-specific interactions with serum proteins or non-target cells, causing a decrease in treatment efficiency.15 Therefore, functional hydrogels have been investigated to improve the stability and delivery efficiency of siRNA. Utilizing PEG or poly(caprolactone) (PCL) to encapsulate siRNA has been shown to minimize nonspecific interactions for sustainable and effective delivery.20,21 Additionally, issues of low local bioavailability and off target silencing can be addressed by the use of hydrogel systems.22 However, these siRNA hydrogel systems either still rely on external stimulation or have a degradation mechanism that is sensitive, but not specific to the target.23 Recently, the compartmentalized systems have been close to being used to create organelle mimics, which are able to detect biomolecules and process spatially segregated reaction pathways.24–29 They can also be used for biomedical treatments.30,31 For example, heterogeneous multi-compartmental hydrogel particles were constructed for use in tandem reactions against cancers, which were triggered by the presence of glucose.32 However, the challenge of developing a lymphocyte-like intelligent hydrogel system to automatically eliminate cancers without external stimulation for healthcare applications, remains.
In this study, multicompartmental core–shell DNA hydrogel particles were fabricated using PEG and a microfluidic assembly and incorporated with a tandem DNA sequence logical circuit for intelligent treatment against cancers. The DNA sequence circuit contains two parts: the sensing sequence (D1) and the treatment sequence (D2). D1 could interact with a cancer biomarker, miRNA-21, to release a specific intermediate sequence for cascade transduction. The D2 would then react with the intermediate from D1 to release siRNA for treatment. The D1 and D2 are independently loaded into the compartments of the core–shell PEG hydrogel particles via a microfluidic assembly.25 The D1 is loaded into the core, and the D2 into the shell to allow multistep tandem sequence interactions, while reducing crosstalk (Fig. 1). Notably, the DNA hydrogel particles made using PEG were highly porous for reagent transportation33 and biocompatible/biodegradable.34,35 The PEG hydrogels could be dissolved in vivo within ∼40 d.36 To demonstrate its capability, D1 is used to detect the cancer biomarker, miRNA-21, and the D2 releases siRNA for targeted cancer elimination. When miRNA-21 diffuses into the hydrogels to interact with D1 in the core, part of the D1 sequence is replaced to release the intermediate sequence (complex 2–3′), triggering a cascade of interactions. The intermediate sequence reacts with D2 in the shell to release the siRNA for cancer elimination. Notably, the synthesized DNA hydrogel particles are biocompatible and drug-free and ideal for long-term biomedical applications. They can sense the presence of cancer cells around them and administer siRNA for automatic elimination.
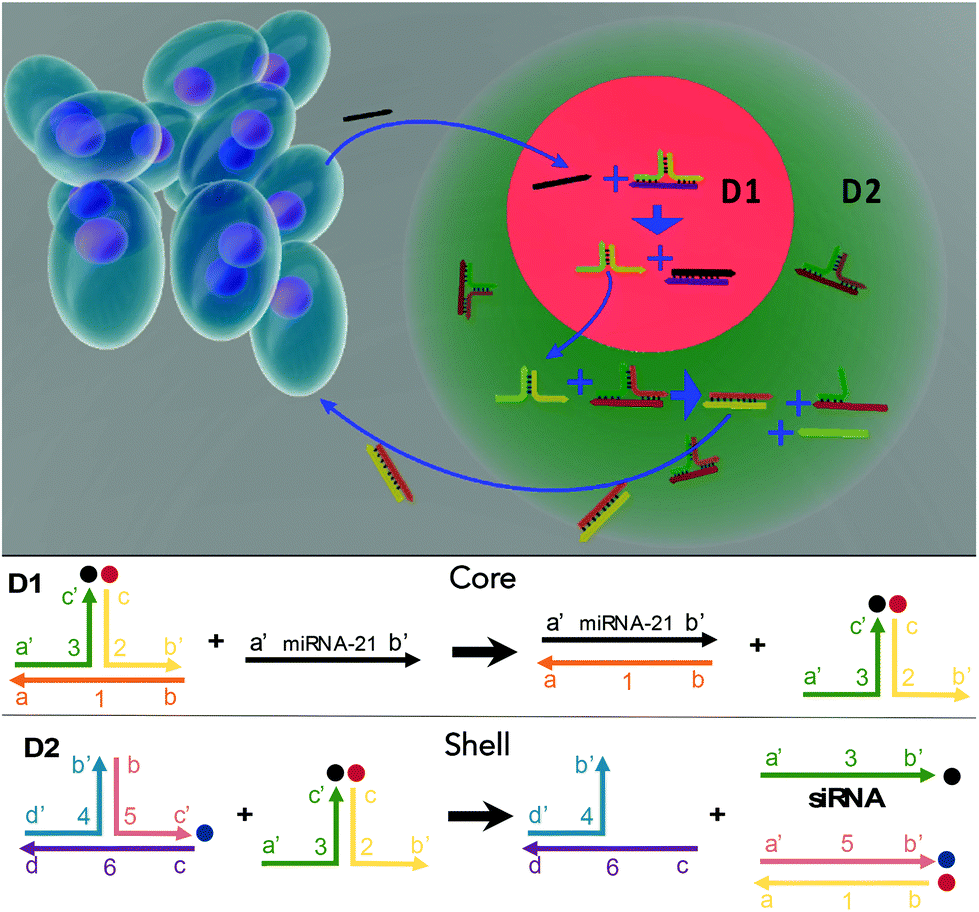 | ||
| Fig. 1 Multicompartmental hydrogel particles with integrative DNA circuits were fabricated via a microfluidic assembly. For intelligent treatment, the sensing sequence (D1) and treatment sequence (D2) were loaded into the core and the shell of a hydrogel particle, respectively. Upon detecting the miRNA-21 target associated with cancer, a degradation cascade is triggered to generate anticancer siRNA. The D1 sequence consists of a three-arm RNA conjugation construction that responds to the targeted miRNAs with matched sequences, releasing an intermediate sequence. The D2 sequence then responds to the intermediate sequence and releases siRNA to kill the cancer cells. | ||
Materials and methods
Oligonucleotide reagent and sample preparation
To fabricate the treatment sequences with logical controls (to detect miRNA-21 and release siRNA), HPLC-purified custom oligonucleotides (Table S1, ESI†) were purchased from Integrated DNA Technologies, (USA). The deionized water was filtered with a Minisart 0.22 μm pore-sized filter (Sartorius) to prepare the stock solution. The concentration was then determined using an NanoDrop 2000c ultraviolet-visible (UV-Vis) spectrophotometer (ThermoScientific). The oligonucleotides were prepared by diluting the stock solution with Tris-HCl buffer (40 mM Tris-HCl, 1 mM EDTA, 12.5 mM MgCl2, deionized water) in DNAse- and RNAse-free tubes (AITbiotech). The oligonucleotides in the stock solutions were stored at −20 °C. To form the D1 and D2 complexes, the sequences were heated to 95 °C for 5 min, followed by snap cooling on ice for at least 1 h. To verify the preparation of the oligonucleotides, polyacrylamide gel electrophoresis (PAGE) was carried out. The samples were loaded into a 15% polyacrylamide gel in Tris base, acetic acid and EDTA (TAE) buffer and run at 90 V for 100 min.Sequence replacement process
The D1/D2 sequence replacement by miRNA-21 was characterized using fluorescence measurements. The three-armed junction oligonucleotides D1 and D2 were designed, and by logically combining D1 and D2, a cancer biomarker, miRNA-21 can be detected causing the release of siRNA for treatment. To demonstrate the feasibility of sequence replacement for hybridization via miRNA-21, 300 nM D1/D2 was mixed with miRNA-21 at various concentrations (0, 0.05, 0.2, 1, 5 and 20 nM). The miRNA-21 binds to D1 to replace and release the intermediate sequence. This intermediate sequence then replaces the siRNA in D2, which was labeled with a fluorescent probe (excitation: 588 nm, emission: 608 nm). Release of the siRNA then causes an increase in fluorescence intensity. The fluorescence intensity was measured with a Spark microplate reader (Tecan, Männedorf, Switzerland) every 12 s for 90 min.Microfluidic device
The microfluidic device consisted of poly(dimethylsiloxane) (PDMS) chips connected by poly(tetrafluoroethylene) tubes. The device was made from a PDMS elastomer using soft lithography methods. The PDMS with microchannels was bonded to glass slides with a thin PDMS layer coating to ensure that the channel walls were hydrophobic. To prevent aggregation and adhesion of the hydrogel particles, the microchannels were treated with 1% trichloro(1H,1H,2H,2H-perfluorooctyl) silane in a desiccator overnight. To fabricate the core hydrogel particles (diameter ∼ 23 μm), an aqueous stream-containing sequence, poly(ethylene glycol) diacrylate (PEGDA) monomer (Mn = 575), PEG (Mn = 4000), 2 wt% of the photoinitiator, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, and an oil stream of HFE 7500 with 5 wt% Pico-Surf, were co-injected into a microfluidic droplet generator (D: 30 μm W: 25 μm) (Fig. S1, ESI†) using PHD 2000 syringe pumps (Harvard Apparatus, MA, USA). The aqueous solution flow rate was ∼2 μL min−1, and the oil flow rate was ∼4 μL min−1. The core droplets were then treated with an OmniCure S1500 spot UV curing system (40–1151 mW cm−2, Profiber, Mississauga, Ontario, Canada) for 10 min to polymerize and form the core hydrogels. These core hydrogels were then washed with isopropanol and Tris-HCl buffer.To form the multicompartmental core–shell hydrogel particles, the core hydrogel particles, DNA sequences, PEGDA (Mn = 575), PEG (Mn = 4000), photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, and oil were coflowed into a microfluidic droplet generator (D: 30 μm W: 30 μm) for droplet encapsulation (Fig. S2, ESI†). The encapsulated droplet particles were then polymerized by UV irradiation, forming core–shell hydrogel particles. Next, the particles were washed with isopropanol and Tris-HCl to remove the oils and they were then ready for redispersion in an aqueous solution. Notably, this microfluidic strategy was suitable for fabricating multicompartmental hydrogel particles, in which different components are spatially confined to distinct domains.
For the formation of the D1 core/D2 shell-structured hydrogel particles, pregel solutions containing 10% (v/v) PEGDA (Mn = 575), 5% (v/v) PEG (Mn = 4000), 2 wt% photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, and three-armed junction oligonucleotides (D1 for the core, D2 for the shell) were used. By exchanging the positions of the three-armed junction oligonucleotides, particles with D2 cores and D1 shells were obtained.
miRNA detection
The first step in miRNA-21 detection consists of the combination of sequence 1 with miRNA-21, to remove BHQ-2. The release of complex 2–3′ (the intermediate sequence) can be monitored using fluorescence signals (excitation: 588 nm, emission: 608 nm). To characterize the first step in the DNA reaction, 100 μL of hydrogel particles were loaded with D1 (1 μM) and resuspended in 100 μL of the miRNA-21 buffer solution (0, 1, 5, 10, 20, 50 and 100 nM). The fluorescence intensity was measured using a Spark plate reader (Tecan) every 5 min for 70 min. Notably, the half lifetime of miRNA-21 in a bio-solution was longer than 10.5 h which allowed the interactions with the D1 sequence to trigger the cascade reactions.37siRNA release process
The second step of the hybridization consisted of replacing CholTEG on sequence 5 with TAMRA. To characterize this reaction, 100 μL of hydrogel particles loaded with D2 (1 μM) were resuspended in 100 μL of buffer solution containing various concentrations of oligonucleotide complex 2–3′ (0, 1, 5, 10, 20, 50 and 100 nM). Similarly, the fluorescence intensity was measured using a Spark plate reader (Tecan) every 5 min for 70 min (excitation: 546 nm, emission: 575 nm). To test the siRNA release from the core–shell particles, a solution loaded with particles (100 μL) was mixed with miRNA-21 buffer solution (100 μL). The core–shell particles contained both D1 (core, 1 μM) and D2 (shell, 1 μM). The remaining steps are the same as those described previously.Long-term siRNA release
To test for long-term siRNA stability, identical procedures with siRNA release from the core–shell particles were carried out. However, fluorescent measurements were taken after longer times for the long-term study (0, 1, 2, 4, 18, 16, 24, 48, 72 d). Additionally, at the end of the 72 d, the hydrogel particles were resuspended in pure buffer and annealed by heating the particles to 95 °C. The fluorescence intensity was once again measured using a plate reader.Cell cultures
The human colon cancer cell line HCT116 and the neonatal human dermal fibroblasts (NHDFs) were obtained from the American Type Culture Collection (ATCC). The HCT116 cells were cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Logan, UT, USA), penicillin (100 μg ml−1; Life Technologies, Grand Island, NY, USA), streptomycin (100 μg ml−1; Life Technologies) and 2 mM L-glutamine (Life Technologies). The NHDFs were cultured in Dulbecco's modified Eagle's medium (DMEM) with the same supplements as those used for the HCT116 cells. All the cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.Confocal imaging
To characterize the siRNA delivery, the cells were observed using confocal laser scanning microscopy (CLSM) at a wavelength of 588 nm. The cells were seeded at a density of 1 × 105 cells per well in Nunc Lab-Tek 8-well chambered slides (ThermoFisher, USA). The cells were incubated for 4 h, and then 100 μL of hydrogel particles were loaded into each well followed by culturing for 8 h. The particles were removed, and the cells were washed with phosphate-buffered saline (PBS) at pH 7.4, to remove free siRNA from the culture medium.Calcein-AM live/dead assay
A live/dead stain-based assay was carried out to evaluate cell viability. The cells were seeded at a density of 1 × 104 per well in a Costar 96-well plate (Corning) and left to adhere for 18 h. Then, the prepared particles were loaded into each well and incubated with the cells for 48 h. An equal volume of the live/dead stain was then added to each well followed by incubation at room temperature for 20 min. The fluorescence images were obtained with an ECLIPSE Ti2 fluorescence microscope (Nikon). Calcein-AM showed a green fluorescence (excitation: 494 nm, emission: 517 nm) in the presence of live cells, whereas ethidium homodimer-1 showed a bright red fluorescence (excitation: 528 nm, emission: 617 nm) in the presence of dead cells. The images were processed using ImageJ software.MTT assay
A colorimetric assay (MTT) was conducted to estimate metabolic activity and indicate cell viability. Identical procedures were carried out for both the calcein-AM stain and the MTT assays, and the cells and particles were incubated in a 96-well plate for 48 h. The MTT labeling solution (0.5 mg ml−1, Roche) was added to each well and incubated with the cells for 4 h. Then, 100 μL of solubilization solution was added, and the cells were incubated overnight. The absorbance at 570 nm was measured with a Multiskan SkyHigh plate reader (ThermoScientific). Cell viability was determined by comparing the absorbance of the sample group against that of a control group.Statistical analysis
The graphs were plotted using Prism (Graphpad, San Diego, CA, USA). A one-way ANOVA with Bartlett's test was carried out, with p < 0.05 being statistically significant.Results and discussion
Oligonucleotide sequence circuit
The sequences for the two, three-arm oligonucleotide conjugated assemblies (D1: 1,2,3 combination and D2: 4,5,6 combination) were commercially produced by IDT. The sequences used are summarized in Table S1 (ESI).† In the presence of miRNA-21, sequence 1 in D1 conjugates with the target miRNA-21, releasing the intermediate complex sequence 2–3′, of which sequence 3 codes for a siRNA. When the intermediate sequence 2–3′ approaches D2, sequence 5 interacted with sequence 2 because of its better binding efficiency and this degraded the intermediate sequence 2–3′ and to release sequence 3 (siRNA). To determine the efficacy of this process, D1 and D2, with and without the addition of miRNA-21, were characterized by using PAGE. The results showed distinct bands that were compared to a well-known prepared DNA ladder (control) to indicate the sequence size. A successful hybridization reaction would result in the release of siRNA, as indicated by the presence of a low bp (<50 bp) product in the PAGE gel (Fig. 2A). For the lanes in which only D1 was loaded, a single clear band of approximately 100 bp was observed, whereas the bands representing siRNA were not observed. The same result was observed when only D2 was loaded, signifying that no siRNAs were released by adding miRNA-21. However, in the lane loaded with a mixture of D1, D2 and miRNA-21, two additional bands corresponding to the pure siRNA were present. This indicated that siRNA can be released due to sequence-cascade-based replacement based on the relevant binding affinities. Notably, in the lane with only D1 and D2, the siRNA bands were present but only faintly. This showed that a cross-interaction between D1 and D2 in a one-pot system is inevitable. Therefore, to ensure the siRNA release by miRNA-21, it is necessary to confine D1 and D2 to separate hydrogel compartments. The release of siRNA from D1/D2 (D1: 1 μM, D2 1 μM) in buffer solutions with different miRNA-21 concentrations was characterized. The release profiles were recorded by the separation of the fluorophore ROXN from the quencher BHQ2, resulting in fluorescence emission (excitation: 546 nm, emission: 575 nm) (Fig. 2B). When the concentration of miRNA-21 was increased, the release rate of siRNA also increased. For example, 5 nM miRNA-21 resulted in a steep increase in siRNA release (∼80%) within 90 min and 20 nM miRNA-21 showed a ∼100% siRNA release. Notably, due to the spontaneous cross-interaction between D1 and D2, without miRNA-21, a release of ∼20% siRNA was observed.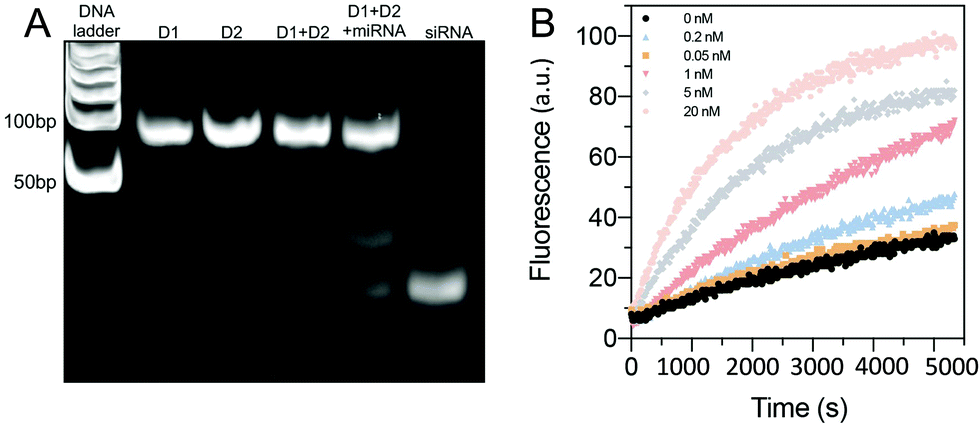 | ||
| Fig. 2 (A) The DNA hybridization cascade was determined based on polyacrylamide gel electrophoresis (PAGE). D1 and D2 are larger complexes with similar distinct bands at 100 bp, whereas siRNA is a smaller 24 bp DNA fragment. The addition of miRNA-21 to a D1/D2 mixture resulted in both D1/D2 bands at 100 bp and a fainter siRNA band. However, in the D1/D2 mixture alone, bands corresponding to siRNA were present as well, indicating that a cross-interaction between D1 and D2 occurs despite the absence of miRNA-21. (B) The siRNA release profiles of D1/D2 in buffer solutions with different miRNA-21 concentrations, determined using fluorescence measurements. As the miRNA-21 concentration increased, the amount of siRNA released also increased. | ||
Multicompartmental particle fabrication via microfluidics
A two-step sequential gelation process together with droplet assembly in a microfluidic device was conducted to fabricate the core–shell hydrogel particles (Fig. 3A). In step 1, two immiscible fluids were both added to a droplet generator microfluidic device to generate monodispersed droplets. D1 (1 μM), PEGDA (Mn = 575), PEG (Mn = 4000) and a photoinitiator (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone) were encapsulated by oil with a surfactant (HFE-7000 + 5% Pico-Surf), which wetted the hydrophobic walls and formed monodispersed water-in-oil droplets. The volume flow rates were optimized to 2 μL min−1 for the aqueous solution and 4 μL min−1 for the oil. The D1 hydrogel particles (diameter ∼23 μm) were then fabricated via UV irradiation-based polymerization of the droplet templates.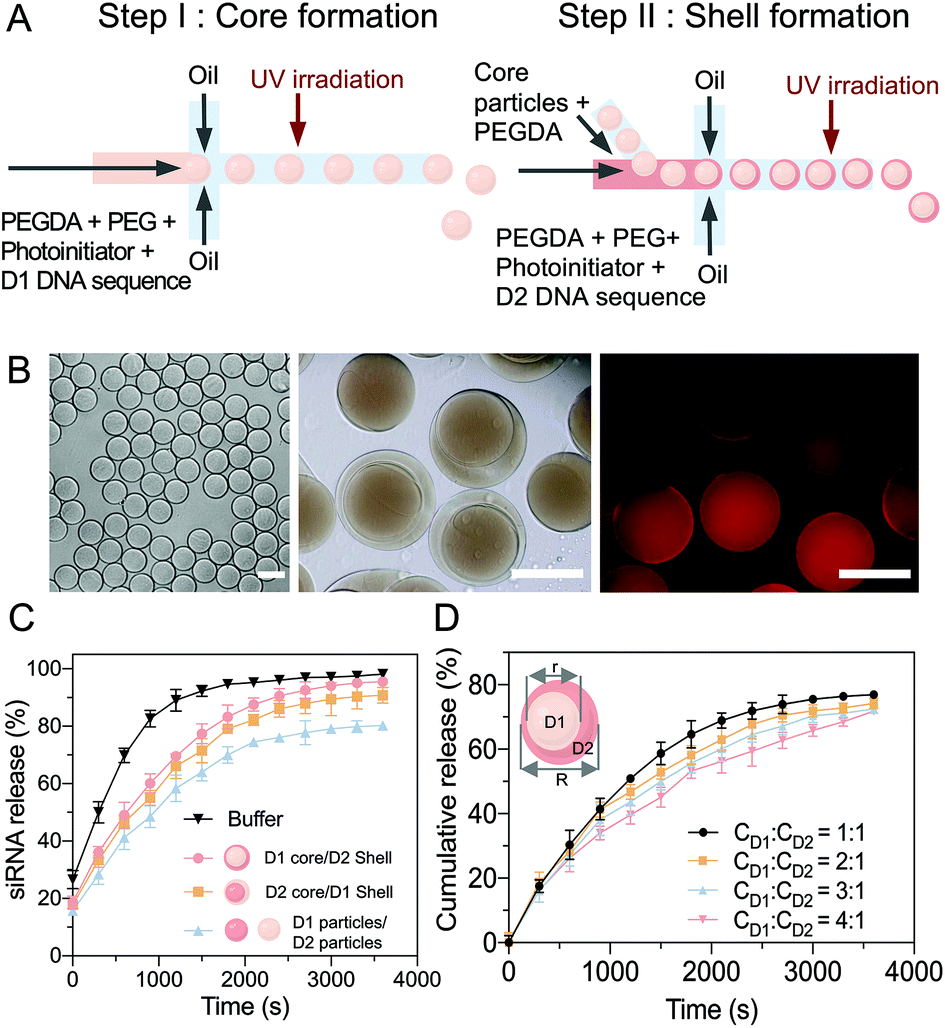 | ||
Fig. 3 (A) A two-step microfluidic gelation process was developed to fabricate core–shell-structured PEGDA hydrogel particles. Firstly, the core particles encapsulating D1 were fabricated via droplet templates and crosslinked using UV irradiation. Secondly, the prepared core (D1) particles were injected into a second device for droplet encapsulation, forming core (D1)–shell (D2) structured particles. (B) Images of core particles (20×), core/shell particles (40×), and D1 distribution in the core particle (40×); scale bar: 30 μm. (C) The siRNA release efficiencies of different permutations of the hydrogel particles. (D) The core–shell volume ratio was altered from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:4 via controlling the flow rates. The siRNA release rates via miRNA-21 input were investigated, showing that the optimized core/shell ratio was 1:1. :1 to 1:4 via controlling the flow rates. The siRNA release rates via miRNA-21 input were investigated, showing that the optimized core/shell ratio was 1:1. | ||
In step 2, microfluidics were used to fabricate the core–shell multicompartmental hydrogel particles, which allowed for the different components to be spatially confined into distinct domains. The prepared D1 hydrogel particles were mixed with D2 (1 μM), PEGDA and the photoinitiator and injected into the second microfluidic droplet generator. By optimizing the flow rates of 1 μL min−1 for the aqueous solution and 8 μL min−1 for the oil solution, single D1 hydrogel particles were encapsulated into single droplets (diameter ∼30–40 μm) (Video S1, ESI†). After UV irradiation-based polymerization, the hydrogel particles were monodispersed with well-defined D1 cores (diameter ∼ 23 μm) and D2 shells (diameter ∼30–40 μm) (Fig. 3B). As sequences 1 and 4 were modified with acrydite, once the hydrogel particles were polymerized, the oligonucleotide molecules were trapped within the hydrogel network, to minimize leakage. Sequence 2 in the D1 complex was modified with a fluorophore ROXN (excitation: 588 nm, emission: 608 nm), and the homogeneous molecular distribution in the hydrogel particles could be observed. Moreover, due to the multicomponent aspect of these core–shell hydrogel particles, D1 and D2 were separated, limiting their cross-reactivity.
The siRNA release efficiency of the core (D1)–shell (D2) hydrogel particles was evaluated (Fig. 3C). Upon the addition of miRNA-21, the D1/D2 cascade was triggered, and the miRNA-21 reacted with D1 in the core to release an intermediate sequence (complex 2–3′), which then diffused into the shell to react with D2 for the siRNA release. The induction of the siRNA release based on the sequence cascade replacement was shown to be time-dependent after the miRNA-21 addition. Similar siRNA release trends were observed with different component combinations, core (D2)–shell (D1), and particle (D1)–particle (D2).
Notably, as long as D1 and D2 interacted with each other, siRNA was released. However, together with the diffusion path, the most efficient combination was the core (D1)–shell (D2) hydrogel particles, which achieved faster and overall higher siRNA release. Additionally, by changing the flow rates, the size ratio between the core (D1) and the shell (D2) was tuned with a range of core/shell volumes from 1:1 to 1:4 (Fig. 3D and Table S2, ESI†). The siRNA release profiles of the hydrogels with various core–shell ratios in the presence of miRNA-21 (20 nM) were examined. To estimate the cumulative release, the siRNA release was measured, giving an indication of the release rate (released siRNA/total siRNA). A core/shell ratio of 1:1 was suggested to have the most effective siRNA release rate, with ∼95% of the siRNA released under miRNA stimulation within 50 min. However, when the core/shell ratio was 1:4, the siRNA release rate decreased to ∼75% over 50 min of miRNA stimulation. Therefore, to maximize the treatment efficiency, the core shell size ratio used for the particle fabrication was kept to a maximum of 1:1.
Sequence transportation between the hydrogel compartments
To ensure the efficiency of the oligonucleotide hybridization cascade via both intermediate sequence transportation from the core to the shell and siRNA transportation from the shell to the surroundings, the effects of the hydrogel pore size were investigated. As the hydrogel pore size (PEG gels) increased with an increase in the length of constructed polymeric chains, the diffusion profiles with different PEG gels of different molecular weights were systematically examined. The intermediate sequence (complex 2–3′) was labeled with a fluorescence dye (excitation: 588 nm, emission: 608 nm). After 2 h, ∼72% of the intermediate sequence (complex 2–3′ in the supernatant) had diffused from the hydrogel when the PEG200 gel (30 wt%) was added. For the PEG600 gel (20 wt%), ∼80% diffusion was observed. For the PEG 4 K gel (5 wt%), ∼80% diffusion was recorded (Fig. 4A). To ensure efficient intermediate sequence diffusion for the reaction with D2, the PEG 4 K hydrogel was used to fabricate the core hydrogel.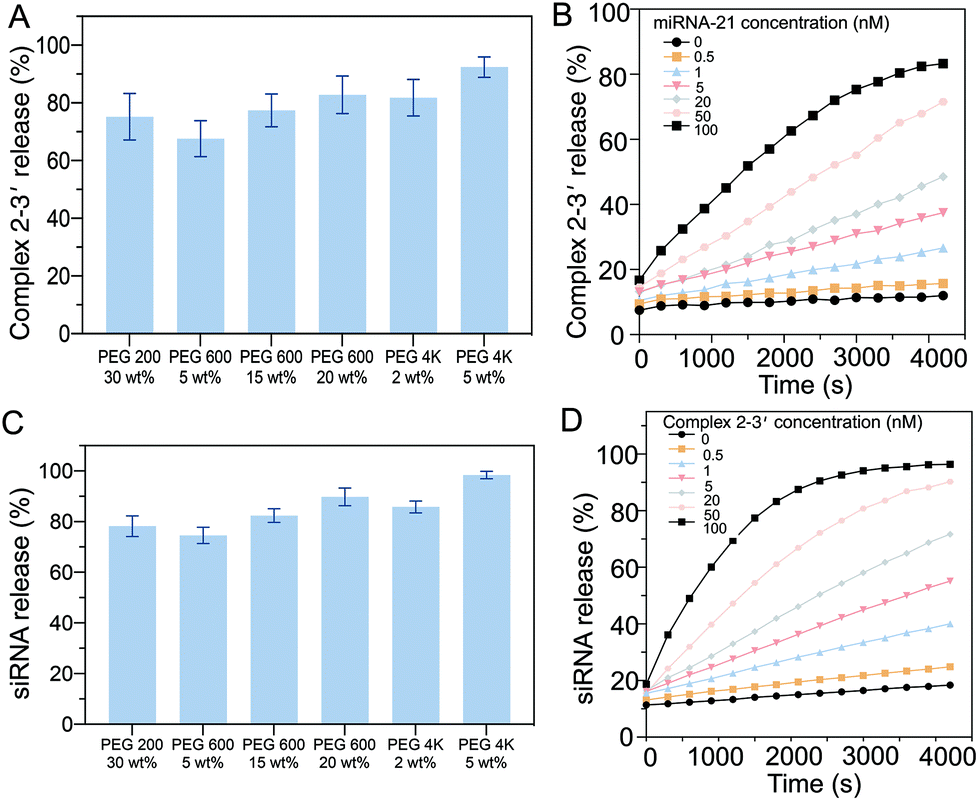 | ||
| Fig. 4 (A) Diffusion of the intermediate sequences from hydrogel particles with different PEG molecular weights were investigated and PEG 4 K (5 wt%) gave the optimum gel with 95% diffusion in ∼70 min. (B) The release profiles of the intermediate sequences from PEG 4 K gels with various miRNA-21 concentrations, showing a positive trend with the miRNA-21 concentration. (C) Characterization of the diffusion of siRNA from hydrogel particles with different molecular weight PEG gels. The optimum release rate was observed from PEG 4 K (5 wt%). (D) Release profiles of siRNA from PEG 4 K gels at various intermediate sequence concentrations, showing the positive trend of the intermediate sequence. | ||
The kinetics of the diffusion of the intermediate sequence through the PEG 4 K hydrogels (5 wt%) under various miRNA-21 concentrations (0–100 nM) were characterized (Fig. 4B). At the lowest concentration of 0.05 nM, less than 7% of the intermediate sequence was released within 1 h, showing that there was limited leakage. It is worth noting that a small amount of the intermediate sequence was detached from D1 without miRNA-21 interaction (4% at 0 nM miRNA-21), however, this would not trigger the cascade reaction to release sufficient siRNA. By increasing the concentration of miRNA-21 in the buffer, the amount of the intermediate sequence released, increased. For example, with 100 nM miRNA-21, 66% of the siRNA was released in 1 h. Notably, because of the nonspecific molecular trapping in a polymeric network, the maximum release rate was ∼80%. The release profiles of the intermediate sequence with PEGs of different molecular weights were investigated. The presence of siRNA was correlated with the replacement of CholTEG with TAMRA to show the fluorescence signal (excitation: 546 nm, emission: 588 nm). Approximately 75% of the siRNA was released from PEG200 (30 wt%) within 1 h. When using PEG 4 K (5 wt%), the release rate increased to ∼93% over 1 h (Fig. 4C). The kinetics of the siRNA release from the PEG 4 K hydrogel (5 wt%) with different intermediate sequence (complex 2–3′) concentrations (0–100 nM) were also characterized (Fig. 4D). The results showed that a higher concentration of siRNA was released when the gels were placed in a higher concentration of complex 2–3′. For example, in the presence of 0.5 nM of complex 2–3′, minimal siRNA was released (only ∼11% after 1 h). However, when 100 nM complex 2–3′ was added, ∼77% of the siRNA was released. Moreover, a small concentration of siRNA (∼7%) was released in the absence of the intermediate sequence (complex 2–3′). Accordingly, when 5 wt% of PEG 4 K was used as a porogen to fabricate the shell compartment of the core–shell hydrogel particles.
siRNA release
The long-term siRNA release profiles of the D1 core/D2 shell particles with different concentrations of miRNA-21 were investigated (Fig. 5A). The hydrogel particles were placed in buffer containing different concentrations of miRNA-21 for 70 d to assess, using fluorescence measurements (excitation: 546 nm, emission: 588 nm), their long-term stability and release. The fluorescence profiles indicated that saturated release occurred for ∼30 d. Without miRNA-21 (negative control), a very low concentration of siRNA was released (∼6%) from the hydrogels. Comparatively, in a solution with 20 nM of miRNA-21, a substantial amount of siRNA (∼81%) was released over 60 d. Moreover, decreasing the miRNA-21 concentration resulted in a decrease in the release rate. For example, 0.05 nM of miRNA-21 showed only ∼17% siRNA release over 60 d.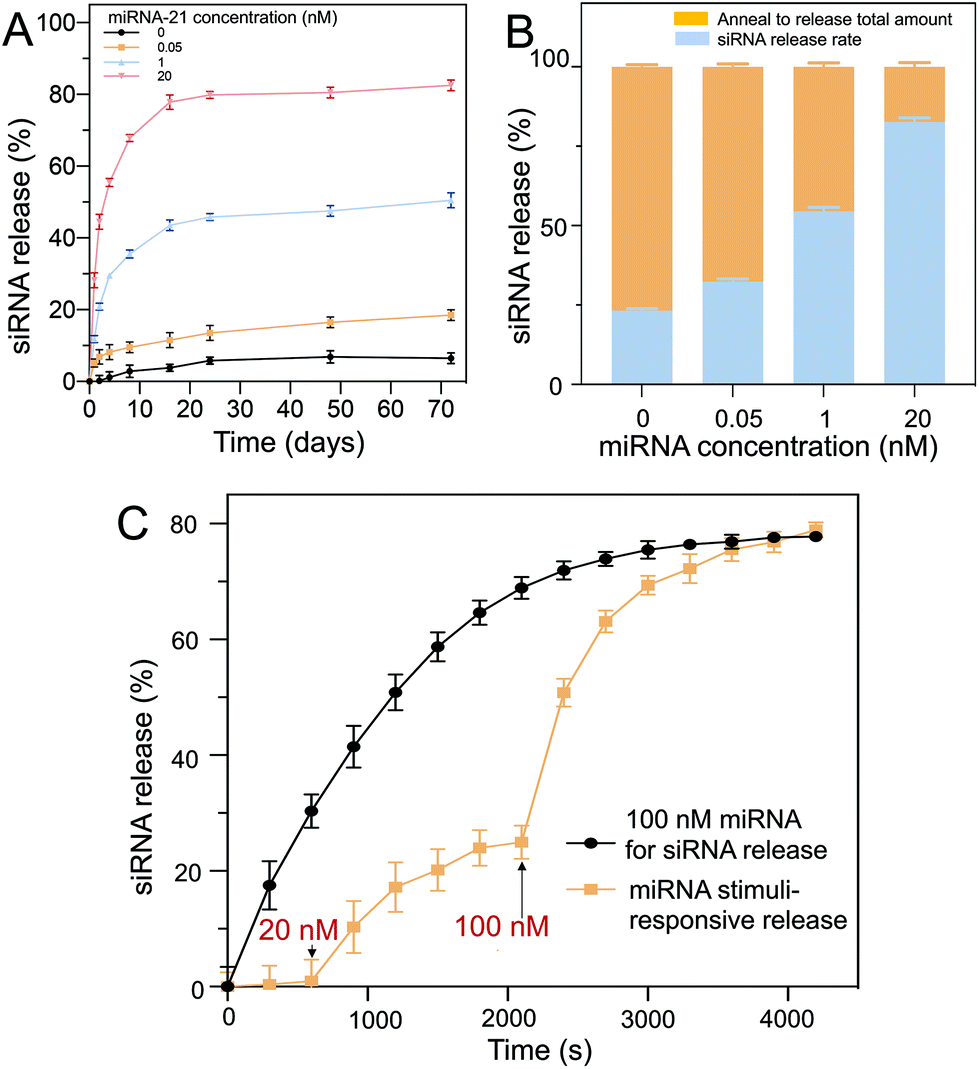 | ||
| Fig. 5 (A) The siRNA release profiles at different miRNA-21 concentrations. Release reached saturation in ∼20 days. The release increased ∼2.5-fold as the miRNA-21 concentration was increased from 0.05 nM to 20 nM. (B) When increasing the miRNA-21 concentration, the total amount of siRNA released also increased. In 20 nM miRNA-21, ∼80% of the total siRNA was released. (C) The siRNA release based on miRNA-21 pulse input, showing dynamic control. | ||
To further coordinate the long-term release profile, the hydrogel particles were resuspended in pure buffer and annealed to release the remaining siRNA trapped in the polymeric networks (Fig. 5B). By heating the hydrogel particles to 95 °C, the oligonucleotide complexes separated and diffused out from the network into the surrounding buffer. It was observed that increasing the miRNA-21 concentration allowed more siRNA to be released from the hydrogel. In contrast, at low miRNA-21 concentrations, the siRNA release was limited. Moreover, D1 and D2 were stable in the PEG hydrogels for up to 70 d, which was suitable for long-term treatment.
The miRNA-21 stimuli-responsive siRNA release from the core (D1)–shell (D2) hydrogel particles was demonstrated, and it showed an intelligent delivery based on the miRNA-21 concentration. With 100 nM miRNA-21 as a stimulus, a large amount of siRNA was immediately released and approached saturation (∼80% release) over 50 min (Fig. 5C, black line). A two-step release via miRNA-21 input was also demonstrated (Fig. 5C, yellow line). In the first step (15 min), a low concentration of miRNA-21 (20 nM) was added to trigger a portion of the siRNA to be released (∼25% release). In the second step (40 min), when a higher concentration of miRNA-21 (100 nM) was added, more siRNA was released (∼78% release) within ∼65 min, showing intelligent release occurred as a result of the miRNA-21 input. The level of miRNA-21 concentrations in cancer cells has previously been proven to be upregulated in tumor growth.38 Liu et al. quantified miRNA-21 concentration in gastric tumors and surrounding tissues, and found the concentration to be ∼20 nM.39 The core/shell DNA hydrogel particles fabricated can be triggered by low levels of miRNA-21 (20 nM) to release the siRNA for cancer cell elimination.
Biocompatibility
To ensure hydrogel particle biocompatibility for future clinical applications, the cell survival rates of the NHDFs mixed with the D1 core/D2 shell hydrogel particles (∼7520 particles per 10 μL) were evaluated after incubating the cells (∼104 cells per 100 μL) for 2 d. Both calcein-AM live/dead staining and MTT assays were performed. The calcein-AM assay was used for qualitative analysis, and the MTT assay was used for quantitative analysis. Calcein-AM was used as an indicator of live cells. In the presence of live cells, calcein-AM shows a green fluorescence (excitation: 494 nm, emission: 517 nm). However, in the presence of dead cells, the ethidium homodimer-1 shows a red fluorescence (excitation: 528 nm, emission: 617 nm) (Fig. 6A). It was observed that without the hydrogel particles (control), the survival rate was ∼99%, as indicated by the green fluorescence within the cells. Similar high cell survival rates were observed after incubating the cells with the core hydrogel particles only, shell hydrogel particles only and core–shell hydrogel particles without loading of D1 and D2. Cell viability was then quantified against the control group (Fig. 6B). High cell viabilities were observed in all cases. Both the calcein-AM live/dead staining and MTT assay results indicated that the hydrogel particles were biocompatible for potential physiological treatment. | ||
| Fig. 6 (A) The biocompatibility of PEG hydrogels determined using live/dead stains. High biocompatibility (>96%) is observed when mixing the hydrogels with core particles, shell particles, and core–shell particles. The biocompatibility of the D1 (core)/D2 (shell) hydrogel particles in NHDFs was examined based on calcein/AM live/dead staining. The live cells are represented by green fluorescence signals (excitation: 492 nm, emission: 517 nm), whereas the dead cells are represented by red fluorescence. High cell viability was observed in all treatment groups (control, core only, shell only and core–shell hydrogel particles). Scale bar = 100 μm. (B) MTT assays were also carried out. After 2 days of incubation, cell viability was not affected in any of the treatment groups. | ||
Target cancer cell elimination
The treatment efficiency of the core (D1)–shell (D2) hydrogel particles was evaluated by using HTC116 human colon cancer cells. The HTC116 cells have been shown to produce miRNA-21 for cell proliferation.40 The HTC116 cells were incubated in 96-well plates with different components of the core–shell hydrogel particles for 2 d. The PEG gels were used to fabricate the hydrogel particles. The pore size of the PEG gels was ∼1–10 μm for extracellular vesicle transportation of miRNA-21 to trigger the reactions. To verify the treatment efficiency of the target cells, the core (D1)/shell (D2) hydrogel particles were incubated with HTC116 and NHDF cells for 8 h. When the HTC116 cells were incubated with the hydrogel particles, the miRNA-21 released from the HTC116 cells triggered a DNA degradation cascade for the siRNA delivery (Fig. 7A). Consequently, the siRNAs released were taken up by the HTC116 cells, showing red fluorescence signals (excitation: 528 nm, emission: 617 nm). It is worth noting that although the local concentration of miRNA-21 in a solution was not easy to measure, the experimental results suggested that the concentration of miRNA-21 produced by the HTC-116 cells needed to trigger the DNA cascade in the hydrogel particles to release sufficient siRNA for cancer cell elimination could be higher than ∼20 nM. However, due to the lack of miRNA-21 secretion by NHDFs, a DNA cascade did not occur and siRNA delivery to the cells was not observed by CLSM (Fig. 7B). Notably, a small amount of cross talk between D1 and D2 might occur in the absence of miRNA-21, releasing a low level of siRNA. However, in this case, the siRNA concentration was too low to cause cell death.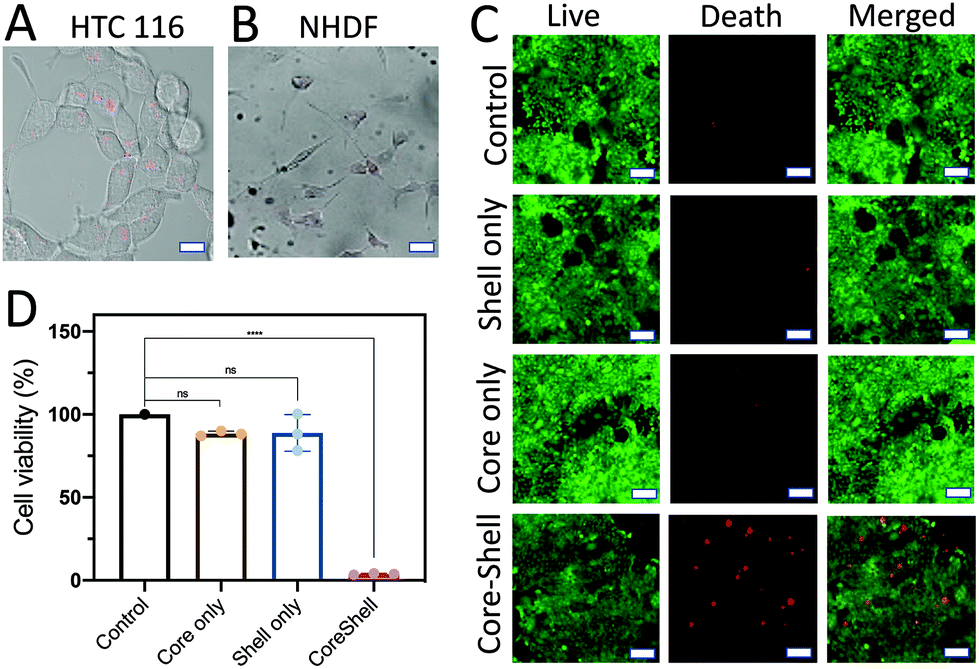 | ||
| Fig. 7 (A) Confocal images showing that siRNA delivery to the target cells had occurred. The HTC 116 cells secreted miRNA-21 to trigger the DNA cascade in the D1 core/D2 shell DNA hydrogel particles to release siRNA into the cells for killing. Scale bar is 300 μm. (B) Because there was no miRNA-21 in the NHDF cells, siRNA was not released from the D1 core/D2 shell DNA hydrogel particles to kill the cells. Scale bar is 300 μm. (C) Different types of DNA hydrogel particles (control, shell only, core only, and D1 core/D2 shell) were combined with HTC116 cancer cells to determine their killing efficiencies using the MTT assay. Scale bar is 100 μm. (D) HTC116 cells were only killed upon loading them with the core–shell DNA hydrogel particles. In other cases (control, core only, shell only), the killing efficiencies were limited, due to the lack of a complete DNA circuit for the miRNA-21-triggered cascade to release siRNAs. | ||
The siRNA could silence a proto-oncogene, Kristen rat sarcoma (K-ras), a member of the RAS family, thus killing the cancers.41,42 The cell viability was assessed by both a calcein-AM live/dead assay (Fig. 7C) and an MTT assay (Fig. 7D). The absorbance was measured using a microplate reader (ThermoFisher). The fluorescence signals of cells without the addition of any hydrogel particles were measured as the background signals. The cell viability was estimated using the fluorescence signals from the samples and dividing the background fluorescence signals with the cell fluorescence to give a percentage.
As expected, without loading of the hydrogels, the cells showed a high viability. This was also seen in cells treated with D1 particles only and D2 particles only. This result was further corroborated by the MTT assay results, in which there was no significant difference between the previously mentioned two treatment groups and the control group. In both cases, the DNA cascade was incomplete, obstructing the production of siRNA, and even in the presence of the colon cancer cells that secrete miRNA-21, the siRNA release was not triggered to cause cell death. However, when the HTC116 cells were treated with the D1 (core)/D2 (shell) hydrogel particles, a strong red fluorescence signal was observed, which indicated cell death. The MTT assay also showed a dramatic decrease in cell viability after incubation with the D1 (core)/D2 (shell) hydrogel particles compared to the control (p < 0.05).
Conclusions
In summary, multicompartmental DNA hydrogel particles with an integrated DNA circuit were synthesized via microfluidics for the logic-controlled release of siRNAs via detecting miRNA-21 to spontaneously eliminate target cancer cells in the vicinity. The positional assembly of the sensing sequence (D1) in the core and treatment sequence (D2) in the shell was used to intelligently sense the biomarker miRNA-21 and deliver siRNA for target cancer cell elimination. The presence of these hydrogels resulted in the death of ∼95% of the targeted colon cancer cells, and the survival rate of ∼100% of the other cells. The segregation of D1 and D2 into separate hydrogel compartments allowed for a synergistic effect – there was no cross-reaction between the two sequences, and the structural integrity of the sequences was preserved for biomarker monitoring and biomedical treatment. Notably, the DNA cascade reaction was triggered only when the DNA sequence matches the specific cancer-associated biomarker, such as miRNA-21. The DNA degradation cascade was then initiated for siRNA release to eliminate the target cancer. Automatic cancer elimination treatment can therefore be obtained via introducing the multi-compartmental DNA hydrogel particles, and these may emerge as a new drug-free therapeutic option for cancer intervention.Notably, the hydrogel particles were micro-sized (∼30 μm in diameter) and, hence, could be implanted around the tumor site. For example, after surgically removing a tumor mass, these hydrogel particles can be inserted around the area for postoperative treatment to prevent cancer reoccurrence. Nevertheless, other DNA circuits could be investigated in the future by a similar concept, for example, by constructing a range of intelligent DNA hydrogel particles for drug-free automatic pathogen elimination. The multicompartmental DNA hydrogel particles are therefore likely to be widely beneficial to researchers in the fields of intelligent soft micro/nanosystems, functional hydrogels, smart delivery, medical devices, and precision medicine.
Author contributions
S. L., G. S. and C. H. C. designed the research and wrote the paper. S. L. and G. S. performed the experiments and data analysis. J. H. and C. H. C. contributed to the design of the hydrogel particles. G. S. and K. W. R. designed the DNA circuits. S. L. performed the biological experiments. S. L. and X. Y. contributed to the discussions about the drug-free cancer treatment. C. H. C. supervised the research. All the authors discussed the results and commented on, proofread, and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding provided by the City University of Hong Kong (9610467, 7005436), the National Natural Science Foundation of China (NSFC, 22074129), the National Research Foundation Singapore, Synthetic Biology Research Program (NRF, SBP, R-397-000-323-592), the National Medical Research Council Singapore, Open Fund – Individual Research Grant (NMRC, OFIRG, R-397-000-289-213), and the Ministry of Education (MOE) Singapore, Academic Research Fund Tier-2 (R-397-000-271-112, R-397-000-253-112).References
- G. J. Yuen, E. Demissie and S. Pillai, Trends Cancer, 2016, 2, 747–757 CrossRef.
- A. Hamaï, H. Benlalam, F. Meslin, M. Hasmim, T. Carré, I. Akalay, B. Janji, G. Berchem, M. Z. Noman and S. Chouaib, Tissue Antigens, 2010, 75, 1–8 CrossRef PubMed.
- M. W. Rohaan, S. Wilgenhof and J. B. A. G. Haanen, Virchows Arch., 2019, 474, 449–461 CrossRef PubMed.
- C. H. June, J. T. Warshauer and J. A. Bluestone, Nat. Med., 2017, 23, 540–547 CrossRef.
- M. L. Davila, I. Riviere, X. Wang, S. Bartido, J. Park, K. Curran, S. S. Chung, J. Stefanski, O. Borquez-Ojeda, M. Olszewska, J. Qu, T. Wasielewska, Q. He, M. Fink, H. Shinglot, M. Youssif, M. Satter, Y. Wang, J. Hosey, H. Quintanilla, E. Halton, Y. Bernal, D. C. G. Bouhassira, M. E. Arcila, M. Gonen, G. J. Roboz, P. Maslak, D. Douer, M. G. Frattini, S. Giralt, M. Sadelain and R. Brentjens, Sci. Transl. Med., 2014, 6, 224ra25–224ra25 Search PubMed.
- R. C. Sterner and R. M. Sterner, Blood Cancer J., 2021, 11, 1–11 CrossRef.
- T. R. Abreu, N. A. Fonseca, N. Gonçalves and J. N. Moreira, J. Controlled Release, 2020, 319, 246–261 CrossRef.
- F. P. Pons-Faudoa, A. Ballerini, J. Sakamoto and A. Grattoni, Biomed. Microdevices, 2019, 21, 47 CrossRef PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- H. Zhang, H. Zeng, A. Priimagi and O. Ikkala, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- R. C. Luo, S. Ranjan, Y. Zhang and C. H. Chen, Chem. Commun., 2013, 49, 7887–7889 RSC.
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 CrossRef CAS.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62(2), 90–99 CrossRef CAS.
- J. Y. Yhee, S. Lee and K. Kim, Nanoscale, 2014, 6, 13383–13390 RSC.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- G. J. Hannon, Nature, 2002, 418, 244–251 CrossRef CAS PubMed.
- M. L. Bobbin and J. J. Rossi, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 103–122 CrossRef CAS.
- A. Levanova and M. M. Poranen, Front. Microbiol., 2018, 9, 2151 CrossRef PubMed.
- F. Petrocca and J. Lieberman, J. Clin. Oncol., 2011, 29, 747–754 CrossRef CAS PubMed.
- H. Shen, T. Sun and M. Ferrari, Cancer Gene Ther., 2012, 19, 367–373 CrossRef CAS.
- M. S. Martina, V. Nicolas, C. Wilhelm, C. Ménager, G. Barratt and S. Lesieur, Biomaterials, 2007, 28, 4143–4153 CrossRef CAS PubMed.
- L. L. Wang, J. J. Chung, E. C. Li, S. Uman, P. Atluri and J. A. Burdick, J. Controlled Release, 2018, 285, 152–161 CrossRef CAS PubMed.
- H. Wang, S. Zhang, J. Lv and Y. Cheng, VIEW, 2021, 2, e146 Search PubMed.
- X. Hu, Z. Tong and L. A. Lyon, J. Am. Chem. Soc., 2010, 132, 11470–11472 CrossRef CAS.
- L. Schoonen and J. C. M. Van Hest, Adv. Mater., 2016, 28, 1109–1128 CrossRef CAS.
- H. Yang, L. Fu, L. Wei, J. Liang and B. P. Binks, J. Am. Chem. Soc., 2015, 137, 1362–1371 CrossRef CAS PubMed.
- G. C. Le Goff, R. L. Srinivas, W. A. Hill and P. S. Doyle, Eur. Polym. J., 2015, 72, 386–412 CrossRef CAS PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 1–5 Search PubMed.
- A. Guiseppi-Elie, S. I. Brahim and D. Narinesingh, Adv. Mater., 2002, 14, 743–746 CrossRef CAS.
- F. He, W. Wang, X. H. He, X. L. Yang, M. Li, R. Xie, X. J. Ju, Z. Liu and L. Y. Chu, ACS Appl. Mater. Interfaces, 2016, 8, 8743–8754 CrossRef CAS PubMed.
- H. Tan, S. Guo, N. D. Dinh, R. Luo, L. Jin and C. H. Chen, Nat. Commun., 2017, 8, 663 CrossRef PubMed.
- Z. Xu, J. Li, H. Zhou, X. Jiang, C. Yang, F. Wang, Y. Pan, N. Li, X. Li, L. Shi and X. Shi, RSC Adv., 2016, 6, 43626–43633 RSC.
- B. Reid, M. Gibson, A. Singh, J. Taube, C. Furlong, M. Murcia and J. Elisseeff, J. Tissue Eng. Regener. Med., 2015, 9(3), 315–318 CrossRef CAS.
- K. Lei, Y. Chen, J. Wang, X. Peng, L. Yu and J. Ding, Acta Biomaterialia, 2017, 55, 396–409 CrossRef CAS PubMed.
- R. Kelmansky, A. Shagan, B. Rozenblit, R. Omar, M. Lufton and B. Mizrahi, Mol. Pharmaceutics, 2017, 14(10)), 3609–3616 CrossRef CAS.
- O. A. Patutina, S. K. Miroshnichenko, N. L. Mironova, A. V. Sen'kova, E. V. Bichenkova, D. J. Clarke, V. V. Vlassov and M. A. Zenkova, Front. Pharmacol., 2019, 10, 879 CrossRef CAS.
- D. Bautista-Sánchez, C. Arriaga-Canon, A. Pedroza-Torres, I. A. D. La Rosa-Velázquez, R. González-Barrios, L. Contreras-Espinosa, R. Montiel-Manríquez, C. Castro-Hernández, V. Fragoso-Ontiveros, R. M. Álvarez-Gómez and L. A. Herrera, Mol. Ther.–Nucleic Acids, 2020, 20, 409 CrossRef.
- Q. Liu, J. Fan, C. Zhou, L. Wang, B. Zhao, H. Zhang, B. Liu and C. Tong, Int. J. Anal. Chem., 2018, 3625823 Search PubMed.
- Y. Wu, Y. Song, Y. Xiong, X. Wang, K. Xu, B. Han, Y. Bai, L. Li, Y. Zhang and L. Zhou, Cell. Physiol. Biochem., 2017, 43, 945–958 CrossRef CAS PubMed.
- J. Liu, R. Kang and D. Tang, Cancer Gene Ther., 2021, 2021, 1–4 CrossRef CAS PubMed.
- B. Papke, S. H. Azam, A. Y. Feng, C. Gutierrez-Ford, H. Huggins, P. S. Pallan, A. E. D. Van Swearingen, M. Egli, A. D. Cox, C. J. Der and C. V. Pecot, ACS Pharmacol. Transl. Sci., 2021, 4, 703–712 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06594g |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2021 |