
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-equilibrium diffusion of dark excitons in atomically thin semiconductors†
Roberto
Rosati
 *a,
Koloman
Wagner
bf,
Samuel
Brem
a,
Raül
Perea-Causín
c,
Jonas D.
Ziegler
bf,
Jonas
Zipfel
bg,
Takashi
Taniguchi
d,
Kenji
Watanabe
e,
Alexey
Chernikov
bf and
Ermin
Malic
ac
*a,
Koloman
Wagner
bf,
Samuel
Brem
a,
Raül
Perea-Causín
c,
Jonas D.
Ziegler
bf,
Jonas
Zipfel
bg,
Takashi
Taniguchi
d,
Kenji
Watanabe
e,
Alexey
Chernikov
bf and
Ermin
Malic
ac
aDepartment of Physics, Philipps-Universität Marburg, Renthof 7, D-35032 Marburg, Germany. E-mail: roberto.rosati@physik.uni-marburg.de
bDepartment of Physics, University of Regensburg, Regensburg D-93053, Germany
cChalmers University of Technology, Department of Physics, 412 96 Gothenburg, Sweden
dInternational Center for Materials Nanoarchitectonics, National Institute for Materials Science, Tsukuba, Ibaraki 305-004, Japan
eResearch Center for Functional Materials, National Institute for Materials Science, Tsukuba, Ibaraki 305-004, Japan
fDresden Integrated Center for Applied Physics and Photonic Materials (IAPP) and Würzburg-Dresden Cluster of Excellence ct.qmat, Technische Universität Dresden, 01062 Dresden, Germany
gMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 16th November 2021
Abstract
Atomically thin semiconductors provide an excellent platform to study intriguing many-particle physics of tightly-bound excitons. In particular, the properties of tungsten-based transition metal dichalcogenides are determined by a complex manifold of bright and dark exciton states. While dark excitons are known to dominate the relaxation dynamics and low-temperature photoluminescence, their impact on the spatial propagation of excitons has remained elusive. In our joint theory-experiment study, we address this intriguing regime of dark state transport by resolving the spatio-temporal exciton dynamics in hBN-encapsulated WSe2 monolayers after resonant excitation. We find clear evidence of an unconventional, time-dependent diffusion during the first tens of picoseconds, exhibiting strong deviation from the steady-state propagation. Dark exciton states are initially populated by phonon emission from the bright states, resulting in creation of hot (unequilibrated) excitons whose rapid expansion leads to a transient increase of the diffusion coefficient by more than one order of magnitude. These findings are relevant for both fundamental understanding of the spatio-temporal exciton dynamics in atomically thin materials as well as their technological application by enabling rapid diffusion.
Excitonic phenomena are known to determine the properties of atomically thin transition metal dichalcogenides (TMDs). A plethora of Coulomb-bound states including bright, spin- and momentum-dark excitons1–9 as well as spatially-separated interlayer excitons in van der Waals heterostructures10–12 dominate optical response and ultrafast dynamics of these technologically promising materials. Particularly rich excitonic manifolds are observed in tungsten-based TMDs7,8,13–20 that exhibit several sharp resonances in their photoluminescence (PL) spectra. Most of these resonances stem from dark exciton states, which can not directly interact with in-plane polarized light due to either spin- or momentum-conservation. However, they are sufficiently long-lived and can recombine either through out-of-plane polarized or phonon-assisted emission resulting in pronounced PL signatures located energetically below the optically bright exciton.21–28
Following optical excitation resonant with bright X0 peak, energetically favorable dark states are populated on a sub-picosecond timescale via emission of phonons from the energetically higher bright states (Fig. 1(a)).61 Due to the mismatch between exciton valley separation and phonon energy, this results in hot dark excitons exhibiting a considerable excess energy, as recently evidenced in spectrally and temporally resolved PL spectra.29 Overheated excitons in an otherwise cold lattice represent a particularly interesting scenario that should directly lead to a rapid expansion of excitons, cf.Fig. 1(a). These non-equilibrated excitons appear independently from the excitation density – in contrast to longer-lived non-linear effects previously observed at room temperature such as non-conventional diffusion30 and halo formation31,32 or also phonon wind and drag phenomena.33 Interestingly, while spatio-temporal dynamics of excitons in TMD mono- and few-layer materials has attracted broad attention,30–42 only little is known regarding transient propagation of dark excitons, before they reach the regime of conventional, time-independent diffusion. A direct demonstration of a transient dark exciton diffusion observing hot exciton populations on a picosecond time scale has yet remained unexplored in monolayer materials.
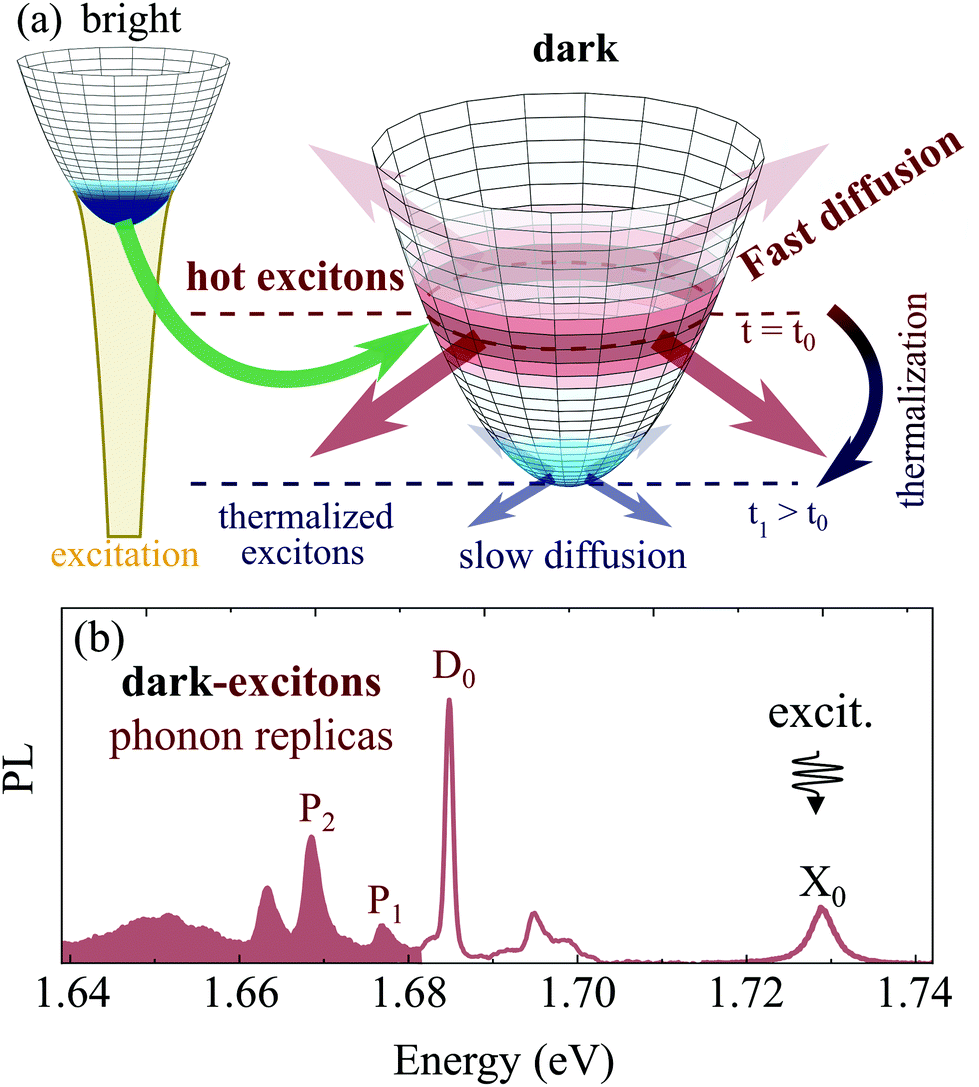 | ||
| Fig. 1 Hot exciton formation. (a) Optically excited bright excitons scatter via emission of phonons into energetically lower momentum-dark exciton states. The resulting excess energy of dark excitons leads to a fast, transient diffusion of the non-equilibrium population prior to thermal equilibrium. (b) Experimentally measured PL spectrum of hBN-encapsulated WSe2 monolayer at T = 5 K. Indicated are resonances stemming from the direct emission of bright excitons (X0) and out-of-plane polarized, spin-dark excitons (D0). Shaded area denotes the spectral range of phonon replicas, including those attributed to momentum-dark states (P1 and P2). | ||
In this work, we address this intriguing exciton diffusion regime in a joint theory-experiment study, where we combine microscopic many-particle modeling with spatially and temporally resolved luminescence microscopy. While the diffusion after excitation in the continuum has been investigated in different materials,30,43–46 we focus here on optical excitation resonant to the deeply-bound bright exciton X0, cf.Fig. 1(b). We explicitly avoid high-excitation effects and focus on the linear regime.31–33,37 To suppress environmental disorder we employ hBN-encapsulated WSe2 monolayers, cooled down to cryogenic temperatures. These conditions allow us to take advantage of prolonged thermalization times29 but also gain direct optical access to dark excitons which recombine regardless of their center-of-mass momenta via characteristic, spectrally-narrow phonon sidebands,21–28cf.Fig. 1(b). This allows access to non-equilibrium regime of exciton diffusion via optical observation of dark excitons beyond commonly-studied bright states. In both theory and experiment we demonstrate an unconventional, time-dependent diffusion during thermalization and relaxation of dark excitons. We show a pronounced increase of the effective diffusion coefficient by more than one order of magnitude few picoseconds after the excitation, in contrast to the stationary case of locally-thermalized exciton population. The agreement between microscopic theory and spatio-temporal PL measurements provides a clear evidence of accelerated dark-exciton propagation in tungsten-based TMD monolayers.
1 Microscopic modeling
First, we present theoretical calculations with the goal to microscopically describe spatially and temporally resolved dynamics of the phonon-assisted emission from dark excitons. The energies and wavefunctions of bright and dark exciton states7,13,16,17,22 are accessed via numerical solution of the Wannier equation8,47,48,62 including material-specific parameters for the electronic bandstructure.48 Due to the resonant optical excitation and considerable binding energies, we focus our study on the three energetically lowest 1s states, which in our calculations are KK′ excitons followed by KΛ and bright KK ones. Here, K, K′, Λ denote the high-symmetry points in the reciprocal space that host electrons and holes forming an exciton. We restrict our analysis to spin-allowed states emitting through low-energy phonon replicas, even as the emission from spin-forbidden states is also observed in experiments25,26,49,50 (e.g. D0 resonance in Fig. 1(b)). While spin-dark states are found to exhibit similar diffusion coefficients at longer timescales after relaxation,35,51 their larger formation times compete with the non-equilibrium processes studied here, in particular with a minor presence of phonon sidebands from spin-dark states in the first few tens of picoseconds.29To obtain a microscopic access to the spatio-temporal exciton dynamics, we introduce the excitonic Wigner function NvQ(r,t), which provides the quasi-probability of finding excitons with momentum Q at time t in position r and exciton valley v.39 We derive its equation of motion using the Heisenberg equation and transforming into the Wigner representation.52 In the low-density regime it reads39
 | (1) |
The first term describes free propagation of excitons depending on the total mass Mv and the spatial gradient in the exciton occupation, resulting in faster spatial variations for more confined exciton distributions (cf. ESI†). The second term takes into account losses due to the radiative recombination γ within the light cone (δQ,0δv,KK). The third and fourth contributions represent phonon-assisted formation and dynamics of excitons, respectively.
For our study, we consider a short and confined optical pulse in resonance with bright exciton states: this initially (t = 0) creates an excitonic polarization PQ≈0(r,t) in the light cone (i.e. “coherent excitons”53,54), which can then be converted into population of “incoherent excitons” via scattering. In particular exciton–phonon scattering with the rates Γ\,vv′QQ′ drives the formation of incoherent excitons also in other valleys via intervalley mechanisms.55,56 Finally, the exciton–phonon interaction leading e.g. to thermalization is described via the Boltzmann scattering term Ṅ\,vQ(r,t)|th = Γ\,in,vQ(r,t) − Γ\,out,vQNQ(r,t) with phonon-driven in- and out-scattering rates Γ\,in/out,vQ(r,t).57 Here, we neither consider non-thermal phonons nor Auger-like recombination processes, which can be described via additional non-linear terms in eqn (1),32 that become important only for strong excitations and can lead to phenomena, such as halo formation on comparatively long timescales.31–33 Thus, exciton–phonon scattering in the linear density regime provides the leading contribution to the interplay between energy relaxation and spatio-temporal dynamics, determining transient diffusion studied in this work.
The first contribution in eqn (1) alone leads to a ballistic exciton propagation, where each state moves in space along the direction of Q with a velocity proportional to |Q|. Exciton–phonon scattering (second line in eqn (1)), however, redistributes exciton occupation primarily toward states with smaller energies (energy-relaxation) and those of different orientation in the reciprocal space (momentum-relaxation). In contrast to the ballistic regime, these scattering processes lead to the typically slower, conventional exciton diffusion.
Different propagation regimes can be quantified by studying the evolution of the spatial width w(t) of a given exciton distribution N(r,t). This is evaluated via , with the area w2 corresponding for Gaussian distributions to the denominator in the exponent (N(r,t) ∝ exp[−x2/w2(t)]). In the ballistic regime, w2 increases quadratically in time, while in the conventional diffusive regime the dependence is strictly linear.58 Deviations from this conventional linear law can be described by defining an effective time-dependent diffusion coefficient
, with the area w2 corresponding for Gaussian distributions to the denominator in the exponent (N(r,t) ∝ exp[−x2/w2(t)]). In the ballistic regime, w2 increases quadratically in time, while in the conventional diffusive regime the dependence is strictly linear.58 Deviations from this conventional linear law can be described by defining an effective time-dependent diffusion coefficient  .39 This coefficient typically converges to a constant D = kBTτs/MX, when the conventional diffusion is reached, with τs being a state-independent scattering time. As we demonstrate in the following, the regime of unconventional, time-dependent diffusion can last for tens of picoseconds in WSe2 monolayers at cryogenic temperatures, where equilibration mechanisms are sufficiently slow.
.39 This coefficient typically converges to a constant D = kBTτs/MX, when the conventional diffusion is reached, with τs being a state-independent scattering time. As we demonstrate in the following, the regime of unconventional, time-dependent diffusion can last for tens of picoseconds in WSe2 monolayers at cryogenic temperatures, where equilibration mechanisms are sufficiently slow.
2 Results
2.1 Spatio-temporal exciton dynamics
To investigate the spatially and temporally dependent optical response of hBN-encapsulated WSe2 monolayers, we solve the equation of motion for the Wigner function [eqn (1)] and the generalized Elliot formula for the PL (cf. ESI†). In the calculations, we set the lattice temperature to 20 K and consider pulsed excitations confined to an initial spatial width of w ≈ 0.5 μm. In Fig. 2 we present an overview of the resulting energy-resolved spatial profiles of excitonic distributions (Fig. 2(a–c)) and phonon sidebands (Fig. 2(d–f)) at given times. This gives access to the relaxation of excitons in time and energy and associated evolution of PL spectra, while the resulting spatial propagation behavior is analyzed and discussed in detail in Fig. 3 and 4. Fig. 2(a–c) illustrate the resulting occupation of the energetically lowest momentum-dark KK′ excitons, NKK′Q(r,t) at different times after the optical excitation. We focus on only one spatial direction due to rotational symmetry of the system.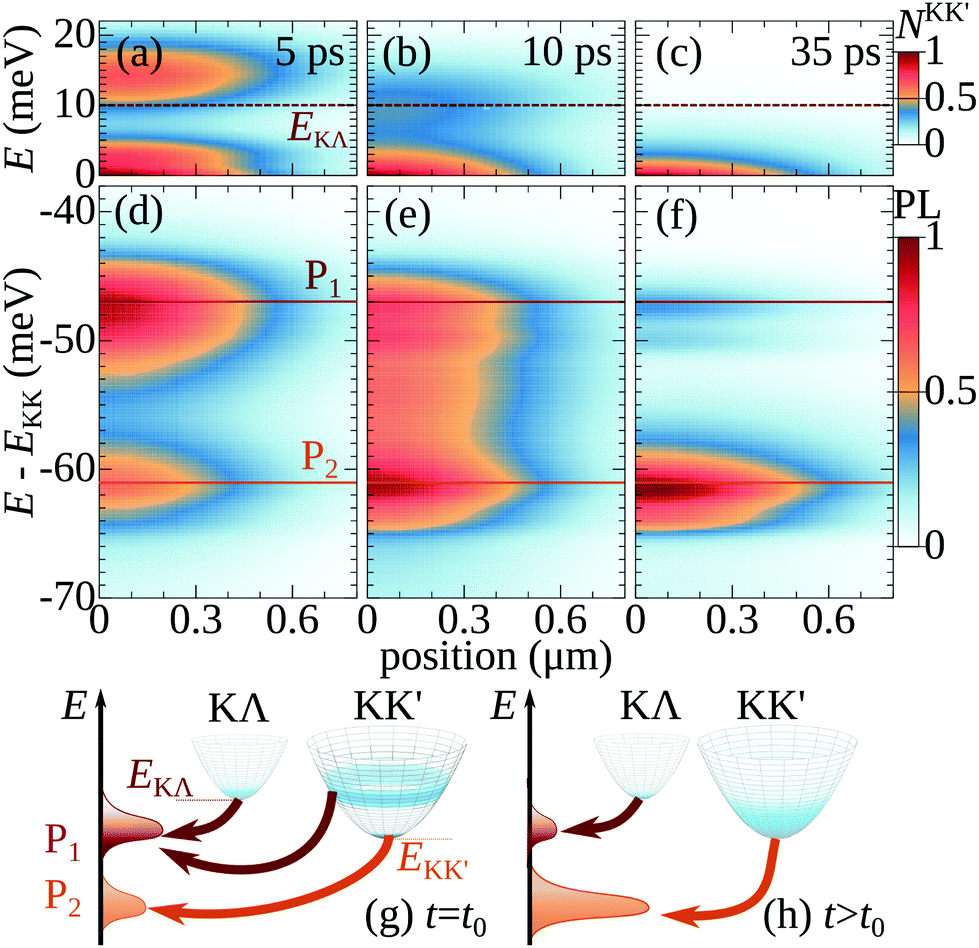 | ||
| Fig. 2 Non-equilibrium excitons dynamics. (a)–(c) Spatio-temporal dynamics of momentum-dark KK′ excitons at 20 K illustrating the interplay between exciton propagation away from the excitation spot and their thermalization towards thermal equilibrium. (d)–(f) Corresponding spatio-temporal evolution of the PL from momentum-dark excitons emitting through phonon-assisted recombination. The two phonon sidebands P1 and P2 can be traced back mainly to KK′ excitons with some contribution from KΛ states at early times. Schematic illustration of the origin of phonon sidebands (g) directly after optical excitation and (h) after thermalization into an equilibrium distribution. | ||
 | ||
| Fig. 3 Spatio-temporal dynamics of phonon sidebands. Exciton emission profiles at representative times (upper panel) and spatially and temporally resolved surface plots (lower panel) evaluated at the spectral position of the (a) P1 and (b) P2 sideband. It demonstrates both an initially accelerated (a) and slower (b) exciton propagation due to non-equilibrium KK′ excitons emitting predominantly at P1 first and subsequently at P2 spectral positions, following relaxation. (c) and (d) Direct comparison between the spatial intensity of P1 and P2 phonon sidebands at different times (normalized w.r.t. P2). | ||
 | ||
| Fig. 4 Theory-experiment comparison of hot exciton diffusion. Calculated and measured representative spatial profiles of the PL at fixed times (upper panel) and two-dimensional spatially and temporally resolved PL (lower panel). The data is normalized to the maximum signal intensity at each time step. The solid lines in the upper experimental panel show Gaussian fits, while in the lower panel the white dashed lines show the extracted profile width w(t). (b) Selected differential PL profiles at fixed times after the excitation IPL(x,t) − IPL(x,t = 0) obtained by subtracting the normalized, fitted lineshape of the initial profile at t = 0 from the measured, normalized profiles at later times. The data is averaged over 0.08 μm on the spatial axis and integrated over 4 ps in time. Differential fits by Gaussian peak functions are shown by solid lines. (c) Temporal evolution of the extracted squared spatial width w2 and associated transient diffusion coefficient D (upper and lower panel, respectively). In experiments, each data point corresponds to the automatically fitted width w of measured PL profiles extracted from (a). In the calculations, the shaded area between full evolution (solid thin line) and assuming initial thermal equilibrium (dashed line) illustrates the impact of non-equilibrated, hot excitons. | ||
Interestingly, the distributions of both excitons and PL in energy are notably different between the three considered times of 5, 10, and 35 ps after the excitation. In particular, after 5 ps (Fig. 2(a)) excitons are still far from the thermal equilibrium. We find a pronounced hot-exciton region, where excitons carry a considerable excess energy on the order of 15 meV. This energy is acquired after phonon-assisted relaxation from the optically-excited bright to the dark state, (Fig. 1) and stems from the difference between the bright-dark energy separation and the energy of the inter-valley phonons involved. These overheated excitons subsequently thermalize and cool down, losing their kinetic energy mainly via scattering with intravalley acoustic modes, so that the distribution starts spreading approximately 10 ps after the excitation. After 20 to 30 ps, excitons finally form a Boltzmann distribution with a temperature corresponding to that of the lattice (Fig. 2(c)). The presented relaxation dynamics in momentum space is consistent with the case of a spatially-homogeneous excitation.29 For spatially-localized excitation, however, it has major implications for the exciton propagation, as discussed below.
To obtain a key observable accessible in experiments, we present the corresponding spatially and spectrally dependent PL in Fig. 2(d–f). In the studied regime, it is dominated by phonon sidebands of momentum-dark excitons, located approximately 50 to 60 meV below the energy of the bright KK exciton.28 The majority of the PL signal is traced back to the KK′ excitons that recombine under emission of the zone-edge acoustic phonons with an energy around 15 meV.55,56 Notably, phonon-assisted processes allow for all exciton states to emit, regardless of their center-of-mass momenta. As a consequence, the profiles in Fig. 2(d–f) largely follow the exciton distribution presented in Fig. 2(a–c). In addition to KK′ sidebands, we also find contributions from KΛ excitons at early times (Fig. 2(g) and (h)) that partially overlap the emission from hot KK′ excitons with an excess energy of about 15 meV (Fig. 2(a and d)). This overlap results in an initially more pronounced P1 sideband, cf.Fig. 2(d).
2.2 Transient exciton diffusion
In the following, we consider the consequences of hot dark excitons with high excess energies for the exciton diffusion. Spatio-temporal PL signals, evaluated at the energies of the two pronounced P1 and P2 phonon sidebands are presented in Fig. 3(a) and (b). We find a fast spatial broadening of the P1 signal in the first 10 ps (reaching a transient diffusion coefficient D of many 10's of cm2 s−1, cf. ESI†). The driving force for the initial increased spatial broadening are hot KK′ excitons, which emit light approximately at the same energy as the KΛ states (Fig. 2(g)). This initial spatial broadening of KK′ excitons is much faster than that of KΛ excitons due to their higher excess energy, cf.Fig. 1(a), as further confirmed by calculating energy- and exciton-valley-specific diffusion coefficients DE,v (cf. ESI†). Over time, the spatial broadening slows down, and eventually results in a small narrowing, i.e. effectively negative diffusion: At 35 ps the profile is narrower than at 10 ps, since former hot KK′ excitons have cooled down. Hence, their signal does not contribute anymore to the emission in the spectral range of P1, cf.Fig. 2(h) and ESI.†The situation in P2 is qualitatively different, exhibiting a much slower spatial broadening in the first 10 ps (Fig. 3(b)). Here, the main contribution of the PL signal stems from nearly thermalized KK′ excitons with a vanishing excess energy (Fig. 2(g)). As a consequence, the initial spatial broadening is considerably slower compared to P1. However, it lasts longer due to the cooling of hot KK′ excitons, which initially diffuse faster but emit spectrally away from P2. When they relax, their emission increasingly occurs in the spectral range of P2 that becomes spatially broader over time (cf. ESI†). This leads to a broader P2 profile compared to that of P1 at 35 ps. Finally, after a few tens of ps the stationary situation of conventional diffusion is recovered. Besides the qualitatively different diffusion behavior, P1 and P2 phonon sidebands also differ in their intensity with the signal in P1 being initially stronger (Fig. 3(c)) then weaker than P2 after approx. 10 ps (Fig. 3(d)). This reflects the cooling of hot KK′ excitons, whose PL consequently spectrally shift from P1 to P2 (Fig. 2(g and h)).
2.3 Measurement of transient exciton diffusion
To study the theoretically predicted impact of hot dark excitons on the transient diffusion, we perform measurements of spatially and temporally resolved PL on hBN-encapsulated WSe2 monolayers, cooled to liquid helium temperature. The samples are obtained by mechanical exfoliation and stamping of bulk crystals onto SiO2/Si substrates60 and allow for an effective suppression of the environmental disorder, thus offering clean access to the phonon-assisted emission from dark states. For excitation, we use a 80 MHz, 140 fs-pulsed Ti:sapphire source tuned into resonance conditions with the bright exciton X0 at 1.726 eV and focused to a spot with a sub-micron diameter. The resulting emission is collected from a lateral cross-section and guided through an imaging spectrometer equipped with a mirror and a grating to provide spatial and spectral resolutions, respectively. For time-resolved detection we use a streak camera operated in the single-photon-counting mode.31Spatially-resolved PL is acquired in the spectral region of the phonon sidebands including P1 and P2, i.e. approximately 50 meV below the bright exciton resonance, cf.Fig. 1(b). A direct comparison between theoretically predicted and experimentally measured spatio-temporal PL is presented in Fig. 4. Representative PL profiles at 0, 10, and 35 ps after the excitation are presented in the upper panel of Fig. 4(a) for calculated and measured distributions. The lower panel shows corresponding spatially and temporally resolved surface plots of the emission. In addition, we present differential PL profiles in Fig. 4(b), corresponding to the relative change of the emission as compared to the initial one at t = 0. Both theory and experiment show an initially fast spatial broadening in the first 10 ps followed by smaller variations. Finally, in Fig. 4(c) we show the extracted, time-dependent variance w(t)2, obtained by automated procedures. The width w(t) is further indicated in Fig. 4(a) by dashed white lines. Time-dependent effective diffusion coefficient is then extracted from  and presented in Fig. 4(c).
and presented in Fig. 4(c).
During the first 10's of ps we observe deviations from the standard diffusion law w(t)2 ∝ t. This initially fast increase stems from rapidly diffusing hot excitons with high excess energies that later converges towards steady-state diffusion after exciton thermalization and cooling. The corresponding effective diffusion coefficients are found experimentally to be in the range of 30 to 50 cm2 s−1 immediately after optical excitation, more than an order of magnitude higher than the equilibrated value of about 3 cm2 s−1. The experimental findings agree overall very well with theoretical predictions. Theoretically predicted timescales are slightly longer and could be related to a slight underestimation of exciton–phonon matrix elements, eventual residual strain and weak unintentional free carrier concentrations in the studied materials. We note that the initial increase of the diffusion coefficient predicted by theory and reflecting an initial quasi-ballistic propagation39,59 could not be observed in the experiment due to the limited time-resolution. The initially rapid diffusion from hot excitons and a substantially slower propagation in the steady-state, inherently limited by the exciton scattering with linear acoustic phonons, are captured both in experiment and theory.
In addition, it is further instructive to consider a direct comparison of spectrally and spatially resolved data. In particular, we measure the spectral shifts ΔE(t) of the phonon sideband emission from the time-dependent PL spectrum in the region of P1 and P2 resonances (cf. ESI†). This can be interpreted as an excess energy affecting the diffusion coefficient via D(t) = (ΔE(t) + kBT) × τ/MX, setting T = 5 K to the lattice temperature, the scattering time τ = 2.8 ps from the steady-state linewidth and MX = 0.75m0.48 As shown by the circles in Fig. 4(c), this results in a good agreement with the direct measurement of the time-dependent diffusion, further supporting the interpretation of hot exciton propagation.
The diffusion behavior is found to be drastically different when neglecting the impact of hot excitons, i.e. considering an initial exciton distribution at thermal equilibrium in our calculations, cf. the dashed lines in Fig. 4(c). Similarly to the case of bright MoSe2 monolayers (see ESI†), without hot excitons there is no fast transient diffusion: we find a transient diffusion coefficient D(t) < 4 cm2 s−1, i.e. one order of magnitude smaller compared to the case with hot excitons. The timescale for the small maximum of D(t) corresponds to τ and indicates the momentum relaxation after around one exciton–phonon scattering event. In contrast the energy relaxation in the real scenario requires emission of various (quasi-elastic) phonons: this results in the 10 ps timescale with an intermediate regime between ballistic and diffusive exciton propagation. Furthermore, at approximately 40 ps after the optical excitation, we also predict small negative diffusion values (Fig. 4(c)) induced by the interstate thermalization and back-scattering processes.39 This weak feature could not be resolved in the experiment at the current stage, motivating future studies. At later times approaching 100 ps, the regime of conventional diffusion is reached, so that dashed and solid lines converge to the same value of almost 2 cm2 s−1.51
3 Conclusions
In summary, we have demonstrated non-equilibrium propagation of hot dark excitons in atomically thin semiconductors by combining microscopic many-particle theory with low-temperature transient PL microscopy. Focusing on the linear regime of low-excitation densities, we find a fast and unconventional, time-dependent exciton diffusion with an initial increase of the diffusion coefficient of up to 50 cm2 s−1 stemming from hot dark excitons with substantial excess energies. This rapid expansion is followed by thermalization and cooling of the exciton distribution on a timescale of a few tens of picoseconds, converging towards a steady-state diffusivity of about 2 to 3 cm2 s−1. Our findings provide fundamental insights into the non-equilibrium transport of excitons in atomically thin semiconductors with potential implications towards their technological application in optoelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mikhail M. Glazov (Ioffe Institute) for valuable discussions on exciton diffusion. This project has received funding from the European Union's Horizon 2020 Research and Innovation programme under grant agreement no. 881603 (Graphene Flagship) and from the 2D Tech VINNOVA competence Center (Ref. 2019-00068). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC). Financial support by the DFG via SFB 1083 (project B9) and SFB 1244 (project B05), SPP 2196 Priority Program (CH 1672/3-1) and Emmy Noether Initiative (CH 1672/1, Project-ID: 287022282), and the Würzburg-Dresden Cluster of Excellence on Complexity and Topology in Quantum Matter ct.qmat (EXC 2147, Project-ID: 390858490) is gratefully acknowledged. K. W. and T. T. acknowledge support from the Elemental Strategy Initiative conducted by the MEXT, Japan, grant number JPMXP0112101001, JSPS KAKENHI grant numbers JP20H00354 and the CREST (JPMJCR15F3), JST.Notes and references
- G. Wang, A. Chernikov, M. M. Glazov, T. F. Heinz, X. Marie, T. Amand and B. Urbaszek, Rev. Mod. Phys., 2018, 90, 021001 CrossRef CAS.
- T. Mueller and E. Malic, npj 2D Mater. Appl., 2018, 2, 29 CrossRef.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- D. Xiao, G.-B. Liu, W. Feng, X. Xu and W. Yao, Phys. Rev. Lett., 2012, 108, 196802 CrossRef PubMed.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- A. Chernikov, T. C. Berkelbach, H. M. Hill, A. Rigosi, Y. Li, O. B. Aslan, D. R. Reichman, M. S. Hybertsen and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 076802 CrossRef CAS PubMed.
- X.-X. Zhang, Y. You, S. Y. F. Zhao and T. F. Heinz, Phys. Rev. Lett., 2015, 115, 257403 CrossRef PubMed.
- M. Selig, G. Berghäuser, A. Raja, P. Nagler, C. Schüller, T. F. Heinz, T. Korn, A. Chernikov, E. Malic and A. Knorr, Nat. Commun., 2016, 7, 13279 CrossRef CAS PubMed.
- T. Chervy, S. Azzini, E. Lorchat, S. Wang, Y. Gorodetski, J. A. Hutchison, S. Berciaud, T. W. Ebbesen and C. Genet, ACS Photonics, 2018, 5, 1281–1287 CrossRef CAS.
- M. M. Fogler, L. V. Butov and K. S. Novoselov, Nat. Commun., 2014, 5, 4555 CrossRef CAS PubMed.
- P. Rivera, J. R. Schaibley, A. M. Jones, J. S. Ross, S. Wu, G. Aivazian, P. Klement, K. Seyler, G. Clark, N. J. Ghimire, J. Yan, D. G. Mandrus, W. Yao and X. Xu, Nat. Commun., 2015, 6, 6242 CrossRef CAS PubMed.
- P. Merkl, F. Mooshammer, P. Steinleitner, A. Girnghuber, K.-Q. Lin, P. Nagler, J. Holler, C. Schüller, J. M. Lupton, T. Korn, S. Ovesen, S. Brem, E. Malic and R. Huber, Nat. Mater., 2019, 18, 691–696 CrossRef CAS PubMed.
- W.-T. Hsu, L.-S. Lu, D. Wang, J.-K. Huang, M.-Y. Li, T.-R. Chang, Y.-C. Chou, Z.-Y. Juang, H.-T. Jeng, L.-J. Li and W.-H. Chang, Nat. Commun., 2017, 8, 929 CrossRef PubMed.
- A. Steinhoff, M. Florian, M. Rösner, G. Schönhoff, T. O. Wehling and F. Jahnke, Nat. Commun., 2017, 8, 1166 CrossRef CAS PubMed.
- O. B. Aslan, M. Deng and T. F. Heinz, Phys. Rev. B, 2018, 98, 115308 CrossRef CAS.
- T. Deilmann and K. S. Thygesen, 2D Mater., 2019, 6, 035003 CrossRef CAS.
- G.-H. Peng, P.-Y. Lo, W.-H. Li, Y.-C. Huang, Y.-H. Chen, C.-H. Lee, C.-K. Yang and S.-J. Cheng, Nano Lett., 2019, 19, 2299–2312 CrossRef CAS PubMed.
- J. Madéo, M. K. L. Man, C. Sahoo, M. Campbell, V. Pareek, E. L. Wong, A. Al-Mahboob, N. S. Chan, A. Karmakar, B. M. K. Mariserla, X. Li, T. F. Heinz, T. Cao and K. M. Dani, Science, 2020, 370, 1199–1204 CrossRef PubMed.
- S. Dong, M. Puppin, T. Pincelli, S. Beaulieu, D. Christiansen, H. Hübener, C. W. Nicholson, R. P. Xian, M. Dendzik, Y. Deng, Y. W. Windsor, M. Selig, E. Malic, A. Rubio, A. Knorr, M. Wolf, L. Rettig and R. Ernstorfer, Nat. Sci., 2021, 1, e10010 Search PubMed.
- R. Wallauer, R. Perea-Causin, L. Münster, S. Zajusch, S. Brem, J. Güdde, K. Tanimura, K.-Q. Lin, R. Huber, E. Malic and U. Höfer, Nano Lett., 2021, 21, 5867–5873 CrossRef CAS PubMed.
- E. Courtade, M. Semina, M. Manca, M. M. Glazov, C. Robert, F. Cadiz, G. Wang, T. Taniguchi, K. Watanabe, M. Pierre, W. Escoffier, E. L. Ivchenko, P. Renucci, X. Marie, T. Amand and B. Urbaszek, Phys. Rev. B, 2017, 96, 085302 CrossRef.
- J. Lindlau, C. Robert, V. Funk, J. Förste, M. Förg, L. Colombier, A. Neumann, E. Courtade, S. Shree, T. Taniguchi, et al., 2017, arXiv:1710.00988, arXiv preprint.
- Z. Ye, L. Waldecker, E. Y. Ma, D. Rhodes, A. Antony, B. Kim, X.-X. Zhang, M. Deng, Y. Jiang, Z. Lu, D. Smirnov, K. Watanabe, T. Taniguchi, J. Hone and T. F. Heinz, Nat. Commun., 2018, 9, 3718 CrossRef PubMed.
- M. Barbone, A. R.-P. Montblanch, D. M. Kara, C. Palacios-Berraquero, A. R. Cadore, D. De Fazio, B. Pingault, E. Mostaani, H. Li, B. Chen, K. Watanabe, T. Taniguchi, S. Tongay, G. Wang, A. C. Ferrari and M. Atatüre, Nat. Commun., 2018, 9, 3721 CrossRef PubMed.
- Z. Li, T. Wang, C. Jin, Z. Lu, Z. Lian, Y. Meng, M. Blei, S. Gao, T. Taniguchi, K. Watanabe, T. Ren, S. Tongay, L. Yang, D. Smirnov, T. Cao and S.-F. Shi, Nat. Commun., 2019, 10, 2469 CrossRef PubMed.
- E. Liu, J. van Baren, T. Taniguchi, K. Watanabe, Y.-C. Chang and C. H. Lui, Phys. Rev. Res., 2019, 1, 032007 CrossRef CAS.
- M. He, P. Rivera, D. Van Tuan, N. P. Wilson, M. Yang, T. Taniguchi, K. Watanabe, J. Yan, D. G. Mandrus, H. Yu, H. Dery, W. Yao and X. Xu, Nat. Commun., 2020, 11, 618 CrossRef CAS PubMed.
- S. Brem, A. Ekman, D. Christiansen, F. Katsch, M. Selig, C. Robert, X. Marie, B. Urbaszek, A. Knorr and E. Malic, Nano Lett., 2020, 20, 2849–2856 CrossRef CAS PubMed.
- R. Rosati, K. Wagner, S. Brem, R. Perea-Causín, E. Wietek, J. Zipfel, J. D. Ziegler, M. Selig, T. Taniguchi, K. Watanabe, A. Knorr, A. Chernikov and E. Malic, ACS Photonics, 2020, 7, 2756–2764 CrossRef CAS.
- D. F. Cordovilla Leon, Z. Li, S. W. Jang and P. B. Deotare, Phys. Rev. B, 2019, 100, 241401 CrossRef CAS.
- M. Kulig, J. Zipfel, P. Nagler, S. Blanter, C. Schüller, T. Korn, N. Paradiso, M. M. Glazov and A. Chernikov, Phys. Rev. Lett., 2018, 120, 207401 CrossRef CAS PubMed.
- R. Perea-Causín, S. Brem, R. Rosati, R. Jago, M. Kulig, J. D. Ziegler, J. Zipfel, A. Chernikov and E. Malic, Nano Lett., 2019, 19, 7317–7323 CrossRef PubMed.
- M. M. Glazov, Phys. Rev. B, 2019, 100, 045426 CrossRef CAS.
- T. Kato and T. Kaneko, ACS Nano, 2016, 10, 9687–9694 CrossRef CAS PubMed.
- F. Cadiz, C. Robert, E. Courtade, M. Manca, L. Martinelli, T. Taniguchi, K. Watanabe, T. Amand, A. C. H. Rowe, D. Paget, B. Urbaszek and X. Marie, Appl. Phys. Lett., 2018, 112, 152106 CrossRef.
- S. Z. Uddin, H. Kim, M. Lorenzon, M. Yeh, D.-H. Lien, E. S. Barnard, H. Htoon, A. Weber-Bargioni and A. Javey, ACS Nano, 2020, 14, 13433–13440 CrossRef CAS PubMed.
- D. F. Cordovilla Leon, Z. Li, S. W. Jang, C.-H. Cheng and P. B. Deotare, Appl. Phys. Lett., 2018, 113, 252101 CrossRef.
- J. Wang, Y. Guo, Y. Huang, H. Luo, X. Zhou, C. Gu and B. Liu, Appl. Phys. Lett., 2019, 115, 131902 CrossRef.
- R. Rosati, R. Perea-Causín, S. Brem and E. Malic, Nanoscale, 2020, 12, 356–363 RSC.
- M. G. Harats, J. N. Kirchhof, M. Qiao, K. Greben and K. I. Bolotin, Nat. Photonics, 2020, 14, 324–329 CrossRef CAS.
- H. Moon, G. Grosso, C. Chakraborty, C. Peng, T. Taniguchi, K. Watanabe and D. Englund, Nano Lett., 2020, 20, 6791–6797 CrossRef CAS PubMed.
- R. Rosati, S. Brem, R. Perea-Causín, R. Schmidt, I. Niehues, S. M. de Vasconcellos, R. Bratschitsch and E. Malic, 2D Mater., 2021, 8, 015030 CrossRef CAS.
- H. Zhao, B. Dal Don, S. Moehl, H. Kalt, K. Ohkawa and D. Hommel, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035306 CrossRef.
- H. Kalt, H. Zhao, B. D. Don, G. Schwartz, C. Bradford and K. Prior, J. Lumin., 2005, 112, 136–141 CrossRef CAS.
- J. Sung, C. Schnedermann, L. Ni, A. Sadhanala, R. Y. S. Chen, C. Cho, L. Priest, J. M. Lim, H.-K. Kim, B. Monserrat, P. Kukura and A. Rao, Nat. Phys., 2020, 16, 171–176 Search PubMed.
- T. Hotta, S. Higuchi, A. Ueda, K. Shinokita, Y. Miyauchi, K. Matsuda, K. Ueno, T. Taniguchi, K. Watanabe and R. Kitaura, Phys. Rev. B, 2020, 102, 115424 CrossRef CAS.
- H. Haug and S. W. Koch, Quantum Theory of the Optical and Electronic Properties of Semiconductors, World Scientific Publishing Company, 5th edn, 2009 Search PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 085413 CrossRef.
- A. Kormányos, G. Burkard, M. Gmitra, J. Fabian, V. Zólyomi, N. D. Drummond and V. Fal'ko, 2D Mater., 2015, 2, 022001 CrossRef.
- C. Robert, T. Amand, F. Cadiz, D. Lagarde, E. Courtade, M. Manca, T. Taniguchi, K. Watanabe, B. Urbaszek and X. Marie, Phys. Rev. B, 2017, 96, 155423 CrossRef.
- G. Wang, C. Robert, M. M. Glazov, F. Cadiz, E. Courtade, T. Amand, D. Lagarde, T. Taniguchi, K. Watanabe, B. Urbaszek and X. Marie, Phys. Rev. Lett., 2017, 119, 047401 CrossRef CAS PubMed.
- K. Wagner, J. Zipfel, R. Rosati, E. Wietek, J. D. Ziegler, S. Brem, R. Perea-Causín, T. Taniguchi, K. Watanabe, M. M. Glazov, E. Malic and A. Chernikov, Phys. Rev. Lett., 2021, 127, 076801 CrossRef CAS PubMed.
- O. Hess and T. Kuhn, Phys. Rev. A, 1996, 54, 3347–3359 CrossRef CAS PubMed.
- M. Kira and S. Koch, Prog. Quantum Electron., 2006, 30, 155–296 CrossRef.
- F. Lengers, T. Kuhn and D. E. Reiter, Phys. Rev. B, 2020, 101, 155304 CrossRef CAS.
- Z. Jin, X. Li, J. T. Mullen and K. W. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 045422 CrossRef CAS.
- I. Paradisanos, G. Wang, E. M. Alexeev, A. R. Cadore, X. Marie, A. C. Ferrari, M. M. Glazov and B. Urbaszek, Nat. Commun., 2021, 12, 538 CrossRef CAS PubMed.
- S. Brem, M. Selig, G. Berghaeuser and E. Malic, Sci. Rep., 2018, 8, 8238 CrossRef PubMed.
- F. Steininger, A. Knorr, P. Thomas and S. W. Koch, . Phys. B: Condens. Matter, 1997, 103, 45–52 CAS.
- A. Castellanos-Gomez, M. Buscema, R. Molenaar, V. Singh, L. Janssen, H. S. J. van der Zant and G. A. Steele, 2D Mater., 2014, 1, 011002 CrossRef CAS.
- G. Berghäuser, P. Steinleitner, P. Merkl, R. Huber, A. Knorr and E. Malic, Phys. Rev. B, 2018, 98, 020301(R) CrossRef.
- G. Berghäuser, I. Bernal-Villamil, R. Schmidt, R. Schneider, I. Niehues, P. Erhart, S. Michaelis de Vasconcellos, R. Bratschitsch, A. Knorr and E. Malic, Nat. Commun., 2018, 9, 971 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06230a |
| This journal is © The Royal Society of Chemistry 2021 |