
Molecular design of heterogeneous electrocatalysts using tannic acid-derived metal–phenolic networks
Nayeong
Kim
 ab,
Inhui
Lee
ab,
Yuri
Choi
*ab and
Jungki
Ryu
*ab
ab,
Inhui
Lee
ab,
Yuri
Choi
*ab and
Jungki
Ryu
*ab
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jryu@unist.ac.kr; choiyuri@unist.ac.kr
bEmergent Hydrogen Technology R&D Center, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 25th October 2021
Abstract
Electrochemistry could play a critical role in the transition to a more sustainable society by enabling the carbon-neutral production and use of various chemicals as well as efficient use of renewable energy resources. A prerequisite for the practical application of various electrochemical energy conversion and storage technologies is the development of efficient and robust electrocatalysts. Recently, molecularly designed heterogeneous catalysts have drawn great attention because they combine the advantages of both heterogeneous solid and homogeneous molecular catalysts. In particular, recently emerged metal–phenolic networks (MPNs) show promise as electrocatalysts for various electrochemical reactions owing to their unique features. They can be easily synthesized under mild conditions, making them eco-friendly, form uniform and conformal thin films on various kinds of substrates, accommodate various metal ions in a single-atom manner, and have excellent charge-transfer ability. In this minireview, we summarize the development of various MPN-based electrocatalysts for diverse electrochemical reactions, such as the hydrogen evolution reaction, the oxygen evolution reaction, the CO2 reduction reaction, and the N2 reduction reaction. We believe that this article provides insight into molecularly designable heterogeneous electrocatalysts based on MPNs and guidelines for broadening the applications of MPNs as electrocatalysts.
 Nayeong Kim | Nayeong Kim obtained her bachelor's degree in Chemical Engineering from Ulsan National Institute of Science and Technology (UNIST) in 2019. She is currently a PhD student at the School of Energy and Chemical Engineering at UNIST and studying under the supervision of Prof. Jungki Ryu. She is interested in the design of photo(electro)chemical CO2 conversion systems using organic/inorganic hybrid materials. |
 Inhui Lee | Inhui Lee obtained her bachelor's degree in Energy Engineering from Kyungpook National University in 2021. She is currently a PhD student at the School of Energy and Chemical Engineering at UNIST and studying under the supervision of Prof. Jungki Ryu. She is interested in the design of biomass utilization systems using polyoxometalate catalysts. |
 Yuri Choi | Dr Yuri Choi is a research assistant professor at the School of Energy and Chemical Engineering at UNIST. She received her PhD from the Department of Energy Engineering, UNIST. Her current research interest is focused on the synthesis and functionalization of carbon and inorganic nanomaterials for energy conversion and storage. |
 Jungki Ryu | Jungki Ryu is an associate professor at the UNIST School of Energy and Chemical Engineering. He received his BS and PhD in Materials Science and Engineering from Yonsei University in 2006 and Korea Advanced Institute of Science and Technology (KAIST) in 2011, respectively. Before joining UNIST in 2014, he had studied bioinspired functional materials as a postdoctoral associate at the Massachusetts Institute of Technology for 3 years. His current research interests include electrocatalysis, solar fuel production, biomass utilization, and electrochemical waste refinery. |
1. Introduction
Electrochemistry can provide an efficient and sustainable route for the production and use of localized and intermittent renewable energy sources.1 Electricity generated from renewable energy resources can be (1) stored in electrochemical energy storage devices for smart grid applications and electrification of various systems2–5 or (2) converted to chemical compounds for long-term energy storage and use in the sustainable chemical industry.6–11 Examples include next-generation rechargeable batteries, such as Li–air2,3 and Zn–air,4 fuel cells,12 and electrolyzers, such as water,6 CO2,7–10 and N2 electrolyzers.11 The first step towards realizing these technologies is to develop efficient, inexpensive, and robust electrocatalysts that enable energy- and cost-efficient conversion of renewable energy sources to various forms.13–16Electrocatalysts can be categorized into homogeneous or heterogeneous according to whether the catalysts and reactants are in the same phase (Table 1).17 In general, homogeneous electrocatalysts are molecular in nature and have the advantage of uniform active sites (beneficial for a better understanding and rational design of electrocatalysts) and strong interactions between catalysts and reactants in the same phase (beneficial for high catalytic activity).17 However, their molecular nature can also pose many problems for practical application, such as low stability under harsh redox conditions, difficulty separating products, and low recyclability of catalysts.18 In contrast, heterogeneous electrocatalysts, mostly inorganic, are generally inexpensive, robust, readily separable from the reaction mixture, and highly recyclable.17,18 As a result, they are preferred over their homogenous counterparts for industrial applications. However, heterogeneous electrocatalysts have non-uniform active sites and low catalytic activity in terms of turnover frequency and selectivity.19
| Homogeneous | Heterogeneous | |
|---|---|---|
| Active site | Uniform, well-defined | Non-uniform, mainly undefined |
| Catalyst modification | Easy | Difficult |
| Activity | High | Low |
| Selectivity | High | Low |
| Stability | Low | High |
| Separation | Difficult | Easy |
| Recyclability | Low | High |
The complementary nature of homogeneous and heterogeneous electrocatalysts has spurred researchers to develop molecularly designed heterogeneous electrocatalysts that combine only the advantages of both. Notable examples include (1) heterogeneous catalysts assembled from molecular building blocks, such as metal–organic frameworks (MOFs)20–22 and covalent organic frameworks (COFs),22,23 and (2) single-atom catalysts based on the alloying of metallic elements24 or carbonization of metal-incorporated macrocycles.13–16 Although their promising electrocatalytic activity has been demonstrated, they also have some limitations for practical applications. For example, they often require harsh and environmentally unfriendly processing conditions. MOFs and COFs are generally synthesized in the form of microparticles in organic solvents by solvothermal methods,25,26 and alloying and carbonization necessitate calcination at high temperatures.27 Such harsh conditions can also pose a limitation on the selection of underlying substrates and the deposition of these catalysts onto the substrates.
Recently, metal–phenolic networks (MPNs) have emerged as molecularly designable heterogeneous electrocatalysts. Unlike the aforementioned, MPNs can be produced using natural polyphenol molecules, such as tannic acid (TA), under mild conditions (e.g., in an aqueous solution at room temperature) and are thus more sustainable for the synthesis of electrocatalysts. In addition, they can readily form a conformal thin film on various substrates and thus allow more versatile applications. In this review article, we highlight recent advances in the development of molecularly designable heterogeneous electrocatalysts based on MPNs that can be prepared in a simple and facile manner. We believe that this article provides a comprehensive review of, and insights into, MPN-based electrocatalysts, as well as guidelines for broadening the application of MPNs to heterogeneous electrocatalysts.
2. Synthesis and properties of metal–phenolic networks
MPNs have supramolecular network structures formed by coordinate bonds between metal ions and polyphenols (Fig. 1a). In particular, among various plant-derived polyphenols, TA has been widely used in catalysis because it can strongly chelate various metal ions due to its numerous phenol groups (Fig. 1a).28 In addition, as a catechol- and pyrogallol-containing molecule,29,30 TA has universal adhesion properties,31,32 allowing the deposition of a stable MPN-based catalytic thin film on any substrate for various target reactions (Fig. 1b). MPN films can be deposited on the desired substrate by various simple and quick methods, such as self-assembly,33 spray assembly,34,35 dipping,36 layer-by-layer assembly,37 and electrophoretic deposition38 (Table 2).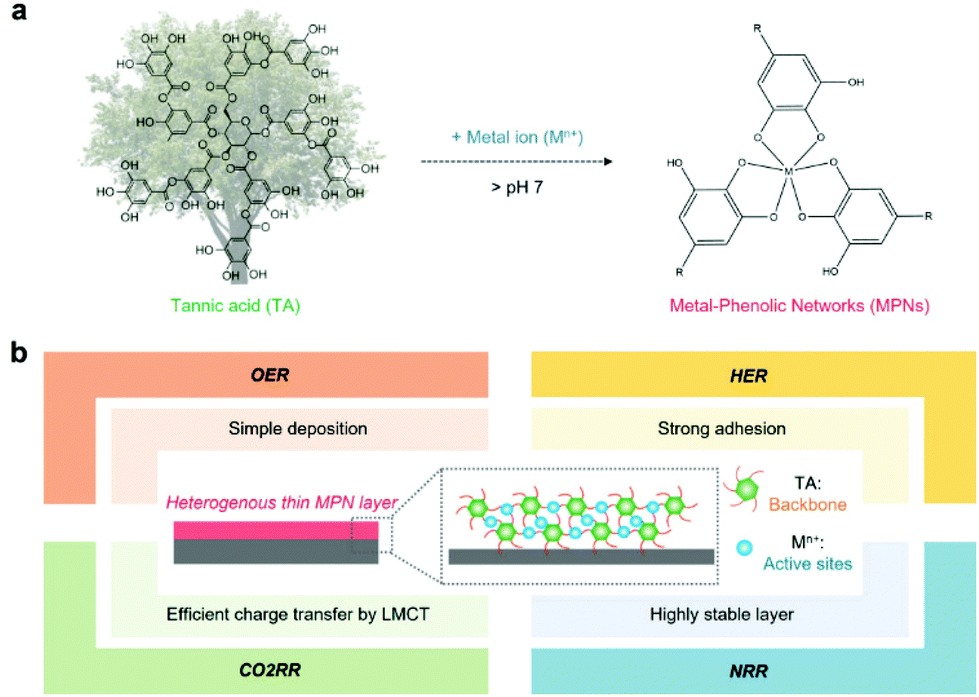 | ||
| Fig. 1 (a) Schematic illustration of the formation of metal–phenolic networks (MPNs) in the presence of tannic acid and metal ions. (b) Properties of heterogeneous thin MPN films and their application in various electrochemical reactions. | ||
| Self-assembly | Spray assembly | Layer-by-layer assembly | Electrophoretic deposition | |
|---|---|---|---|---|
| Pros | -Simple and inexpensive | -Rapid and scalable | -Precise control of film thickness on nm to micron scale | -Rapid and uniform |
| -Independent of substrate morphology | -Independent of substrate morphology | |||
| Cons | -Often time-consuming | -Poor control over molecular and nanoscale structure | -Multiple steps necessary | -External bias is necessary |
| -Poorly scalable | -Material-inefficient | -Often time-consuming | -Substrate should be conductive | |
| -Difficult to control |
The formation of MPNs is highly affected by pH because the coordinating mode between metal ions and polyphenol groups depends highly on their pH values. Caruso et al. reported the effect of pH on the formation and growth of MPN films by the spray method (Fig. 2a).34 They found that Fe3+ ions and TA can form mono-, bis-, and tris-type complexes at pH 1, pH 3–7, and pH 11, respectively, and that TA can undergo oxidative polymerization at high pHs. Thus, at low pHs, molecularly thin MPN layers (∼1.5 nm) were formed due to insufficient intermolecular crosslinking between metal–TA complexes because mono-complexes were favorably formed. Meanwhile, at high pHs, thicker but still relatively thin MPN films (∼5.1 nm) were obtained due to the slow diffusion of larger metal–TA complexes—formed by tris-complex formation and oxidative polymerization of TA—to the substrate surface. As a result, neutral pH was found to be the most efficient for the growth of heterogeneous MPN thin films (11.9–13.8 nm). Furthermore, Caruso et al. reported that diverse MPN films can be formed on various types of substrates, from flat to even hierarchical substrates.28 It has been found that TA can form MPN thin films with metal ions of various sizes, groups, and periods, such as main group metal ions (e.g., Al), transition metal ions (e.g., V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Nr, Mo, Ru, Cd, etc.), and lanthanide ions (e.g., Ce, Eu, Gd, Tb, etc.) (Fig. 2b).
 | ||
| Fig. 2 Characteristics of MPN films. (a) pH-dependent coordination mode of metal ions and TA in MPN films. Reproduced from ref. 34. Copyright 2018, American Chemical Society. (b) Wide selection of metals for synthesizing MPNs. Reproduced with permission from ref. 28. Copyright 2014, Wiley-VCH. (c) Tunable properties of MPN films such as thickness, mechanical modulus, permeability, and stability according to the counteranions of the metal ions. Reproduced from ref. 40. Copyright 2020, American Chemical Society. (d) LMCT charge transfer ability of MPNs. Reproduced from ref. 34. Copyright 2018, American Chemical Society. | ||
In addition to pH, the ionic strength and type of ion significantly influence the growth and properties of MPN films. Caruso et al. reported that a high concentration of NaCl can promote the formation of thicker and rougher MPN films because Na+ can prevent the formation of intermolecular hydrogen bonding between metal–TA monomers and thus free phenolic groups can further react with metal ions or other metal–TA monomers.39 Choi et al. found that type of anion also affects the thickness, porosity, and mechanical properties of MPN films (Fig. 2c).40 When depositing Fe3+–TA MPN films with different anions, such as SO42−, Cl−, and Br−, they found that the thickness, mechanical modulus, permeability, and stability of the film varied in the order of Br− > Cl− > SO42− (i.e., reversed Hofmeister series) due to the structure of the MPN film being extended more by more chaotropic anions.
Interestingly, it has been reported that MPNs can facilitate the charge separation and/or transfer process for enhanced catalytic activity. It is well-known that ligand-to-metal charge transfer (LMCT) can take place between phenolic groups and metal ions in MPNs.41,42 This suggests that the catalytic efficiency of MPNs and MPN-based hybrid systems can be further improved by enhancing the charge separation and reducing the charge recombination via the LMCT process from the LUMO of phenolic groups to metal ions in MPNs (Fig. 2d).34 Furthermore, the LMCT properties of MPNs can improve the kinetics of various chemical reactions by improving electron mobility as well as having intramolecular charge transport between coordinated metal ions and organic linkers.
3. Application of MPNs
In this part, we review recent reports about the development of MPN-based (photo)electrocatalysts for various target reactions. In addition to unique properties, such as simple and universal applicability, high tunability of structure and properties, and LMCT properties, in principle, MPNs can also be considered as single-atom electrocatalysts that can exhibit high mass-specific activity. In MPNs, metal ions can act as active sites for various electrochemical reactions and polyphenolic groups can act as both the backbone of MPNs and the host for metal ions. As a result, MPNs have recently emerged as promising molecularly designable heterogeneous catalysts for various (photo)electrochemical reactions, such as the oxygen evolution reaction (OER), hydrogen evolution reaction (HER), CO2 reduction reaction (CO2RR), and nitrogen reduction reaction (NRR) (Table 3).| Target reaction | Metal | Electrode | Electrolyte | pH | Light intensity | Performance | Ref. |
|---|---|---|---|---|---|---|---|
| OER | Co | BiVO4 | 0.15 M borate buffer | 8.5 | 100 mW cm−2 (AM 1.5 G filter) | 4.8 mA cm−2 (@ 1.23 V) | 72 |
| Ni, Fe | Mo:BiVO4 | 0.5 M borate buffer | 8.5 | 100 mW cm−2 (AM 1.5 G filter) | 5.10 mA cm−2 (@ 1.23 V) | 70 | |
| Ni, Fe | WO3/BiVO4 | 0.5 M borate buffer | 8.5 | 100 mW cm−2 (AM 1.5 G filter) | 3.7 mA cm−2 (@ 1.23 V) | 71 | |
| Ni, Fe | Carbon fiber paper | 1 M KOH | ∼14 | No light | 290 mV (@ 10 mA cm−2) | 53 | |
| Ni, Fe | CNT | 1 M KOH | ∼14 | No light | 287 mV (@ 10 mA cm−2) | 54 | |
| HER | Ru | Carbon cloth | 1 M KOH | ∼14 | No light | 29 mV (@ 10 mA cm−2) | 77 |
| Water splitting | Fe | Ni(OH)2/NF | 1 M KOH | ∼14 | No light | 160 mV (@ 10 mA cm−2) | 78 |
| NRR | Pd | Carbon fiber paper | 0.1 M Na2SO4 | Neutral | No light | 24.12 μg h−1 mg−1 | 88 |
| Au NWs | Carbon fiber paper | 0.1 M Na2SO4 | Neutral | No light | 15.71 μg h−1 mg−1 | 89 | |
| CO2RR | Pb | Carbon fiber paper | 0.5 M NaHCO3 | Neutral | No light | Formate FE of 90.1% (@ −0.92 V) | 90 |
3.1 Electrochemical OER
The electrochemical OER is an important half reaction for various energy conversion systems, such as water, CO2, and N2 electrolyzers, because it can act as a clean source of electrons.43–46 However, it also acts as an obstacle for practical application of these systems due to its sluggish kinetics and high overpotential.44–46 In order to improve OER efficiency, tremendous efforts have been devoted to the development of heterogeneous OER catalysts via control of the size, crystal structure, morphology, defects, and dopants/impurities of catalysts.45,46 Among various OER catalysts, ruthenium-47,48 and iridium-based electrocatalysts49 show high efficiency for the electrochemical OER. However, they have some limitations, such as high cost, low stability, and difficulty in scaling up. In particular, these OER catalysts can be deposited, frequently non-uniformly, on the electrode by complex methods that require specific equipment, such as electrodeposition,50 atomic-layer-deposition,51 and spin coating.52In this regard, MPN was first utilized as a simple and versatile platform for the development of efficient OER catalysts with Earth-abundant transition metals. For example, Zhang et al. reported the development of MPN-based heterogeneous OER electrocatalysts by coating carbon fiber paper (CP) with TA-based MPNs with various metal ions of Fe, Co, and Ni (Fig. 3a).53 These MPN films with a thickness of ∼7.6 nm were readily deposited on CP by simple dipping methods. TA-based MPNs with Co or Fe ions (TA–Co and TA–Fe, respectively) showed improved OER efficiency in 1 M KOH (Fig. 3b). Interestingly, TA-based MPNs containing a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Ni and Fe (TANF) showed much better performance with the lowest overpotential of 290 mV at 10 mA cm−2 and a Tafel slop of 28 mV dec−1. Furthermore, it exhibited a faradaic efficiency close to 100% and excellent stability for 24 h. However, they found that partial dissociation of the TA ligand can occur under anodic potential, leading to the conversion of TANF to NixFe1−xOyHz. Based on similar approaches, Liu et al. reported the fabrication of composite electrodes by coating carbon nanotubes (CNTs) with TA-based MPNs for OER (Fig. 3c).54 They found that the composite electrode with both Ni and Fe ions (Ni3Fe/TA@CNT) exhibited higher OER activity than those with Ni (Ni/TA@CNT) or Fe ions alone (Fe/TA@CNT) and a representative OER catalyst IrO2. Ni3Fe/TA@CNT showed a low overpotential of 287 mV at 10 mA cm−2 and a Tafel slop of 70.24 mV dec−1 in 1 M KOH (Fig. 3d). Through a combination of experiments and density functional theory calculations, they suggested that the hybridization of MPNs and CNTs synergically improves the OER catalytic activity by the electron transfer from CNTs to MPNs, which lowers the energy barrier for the formation of the key intermediate O*. Interestingly, unlike in the study by Zhang et al., the MPN layer remained stable for 10 h at 10 and 50 mA cm−2 without conversion to metal oxides or metal hydroxides.
:1 ratio of Ni and Fe (TANF) showed much better performance with the lowest overpotential of 290 mV at 10 mA cm−2 and a Tafel slop of 28 mV dec−1. Furthermore, it exhibited a faradaic efficiency close to 100% and excellent stability for 24 h. However, they found that partial dissociation of the TA ligand can occur under anodic potential, leading to the conversion of TANF to NixFe1−xOyHz. Based on similar approaches, Liu et al. reported the fabrication of composite electrodes by coating carbon nanotubes (CNTs) with TA-based MPNs for OER (Fig. 3c).54 They found that the composite electrode with both Ni and Fe ions (Ni3Fe/TA@CNT) exhibited higher OER activity than those with Ni (Ni/TA@CNT) or Fe ions alone (Fe/TA@CNT) and a representative OER catalyst IrO2. Ni3Fe/TA@CNT showed a low overpotential of 287 mV at 10 mA cm−2 and a Tafel slop of 70.24 mV dec−1 in 1 M KOH (Fig. 3d). Through a combination of experiments and density functional theory calculations, they suggested that the hybridization of MPNs and CNTs synergically improves the OER catalytic activity by the electron transfer from CNTs to MPNs, which lowers the energy barrier for the formation of the key intermediate O*. Interestingly, unlike in the study by Zhang et al., the MPN layer remained stable for 10 h at 10 and 50 mA cm−2 without conversion to metal oxides or metal hydroxides.
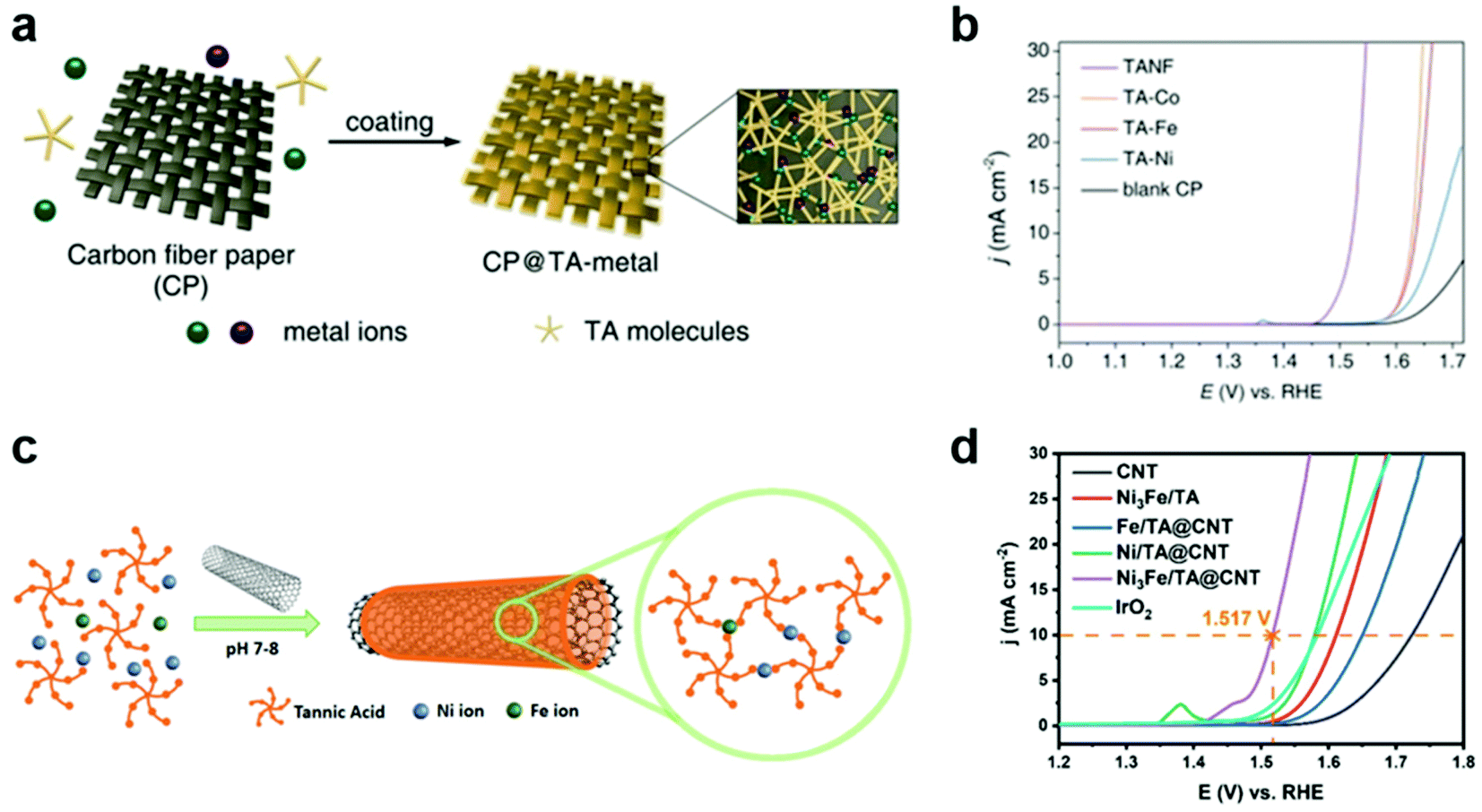 | ||
| Fig. 3 MPNs as OER electrocatalysts. (a) Deposition of CP@TA-metal on CP by simple dipping method. (b) OER performance of various TA–metal complexes in 1 M KOH. TA–metal complexes of TA–Co, TA–Fe, TA–Ni, and TANF were prepared using Co, Fe, Ni and Ni3Fe, respectively. Reproduced with permission from ref. 53. Copyright 2019, Wiley-VCH. (c) Schematic illustration for the synthesis for Ni3Fe/TA@CNT by coating CNTs with MPNs incorporating Ni and Fe in a 3:1 ratio. (d) OER performance of Ni3Fe/TA@CNT superior to those of IrO2 and other MPN-coated CNT electrodes for the OER. Adapted with permission from ref. 54. Copyright 2020, Elsevier. | ||
These results suggest that the factors influencing the stability of MPNs under OER conditions, such as deposition methods/conditions, types of substrates, and testing conditions, need to be further investigated. For example, Zhu et al. demonstrated that the stability of MPNs can vary depending on the pH and types of buffer solutions using TANF-coated BiVO4 as a model system.55 They found that TANF was disassembled and agglomerated during the OER without buffer because the decrease of the local pH near the electrode surface destabilizes the metal–TA complexes. In addition, it was found that in neutral buffer solutions, the type of electrolyte also has a significant influence on the performance of TANF catalysts; both the activity and stability of TANF decreased in the order of borate, carbonate, and phosphate. As studies on MPN electrocatalysts are still in their infancy, further studies on the stability of MPN-based OER catalysts are required.
3.2 Photoelectrochemical OER
OER catalysts also play a crucial role in photoelectrochemical (PEC) cells using semiconductor photoelectrodes to produce solar fuels, such as hydrogen and hydrocarbons. In PEC cells, representative photoanodes for PEC water oxidation include BiVO4,32,56–58 Fe2O3,59–61 WO3,62,63 and others. Unfortunately, these semiconductors have intrinsic limitations, such as fast charge recombination, low electrical conductivity, poor catalytic activity, and photocorrosion.64,65 Thus, the modification of photoanodes with OER cocatalysts is inevitably required to address these issues. However, among the numerous OER catalysts developed to date, only a few of them, such as cobalt phosphate (Co–Pi),66 ferrihydrite,67 NiOOH,68 and FeOOH,57 have been successfully utilized as OER cocatalysts due to the more stringent requirements for PEC applications, such as good electrical contact with the underlying semiconductors, high transparency of cocatalysts, formation of conformal and uniform films with high coverage, and mild deposition conditions that do not degrade the performance of underlying semiconductors.69In this regard, MPNs can be utilized as ideal OER cocatalysts for PEC applications. For example, Zhang et al. synthesized a uniform thin layer of TANF (5.3 nm) on highly porous Mo-doped BiVO4 (Mo:BiVO4@TANF) for PEC water oxidation by simple dip-coating method (Fig. 4a).70 For comparison, they also deposited conventional OER cocatalysts, such as ferrihydrite (Fh) and Co–Pi—that form non-uniform islands and thus result in incomplete protection of the underlying photoanodes—on Mo:BiVO4 (Mo:BiVO4@Fh and Mo:BiVO4@Co-Pi, respectively). Among various Mo:BiVO4 photoanodes tested, Mo:BiVO4@TANF showed the highest photocurrent density of 5.1 mA cm−2 at 1.23 V vs. a reversible hydrogen electrode (RHE) and remained stable for 3 h in 0.5 M borate buffer (pH 8.5) (Fig. 4b). In contrast, Mo:BiVO4@Co-Pi and Mo:BiVO4@Fh exhibited low stability due to photocorrosion of partially exposed (i.e., unprotected) Mo:BiVO4. It was also found that TANF can be successfully utilized as an OER cocatalyst for undoped BiVO4 and TiO2 photoanodes. Later, Wang et al. also showed that the PEC performance of WO3/BiVO4 heterojunction photoanodes can be significantly improved through modification with TA-based MPNs (Fig. 4c).71 WO3/BiVO4 modified with TA-based MPNs containing both Ni and Fe ions (WO3/BiVO4@TANiFe) showed significantly improved PEC performance with a photocurrent density of 3.7 mA cm−2 at 1.23 V vs. RHE and excellent stability for 5 h in 0.5 M borate buffer (pH 8.5) (Fig. 4d). These results again confirm the dual roles of TANiFe as an efficient OER cocatalyst and a protective coating. In addition to Ni- and Fe-containing ones, Co-containing MPNs were also tested as OER cocatalysts for PEC water oxidation. For instance, Ding et al. deposited TA-coordinated Co-based MPNs (6–12 nm thick) on BiVO4 (BiVO4/TACo) by simple dip-coating method for PEC water oxidation in 0.15 M borate buffer (Fig. 4e).72 After modification with TACo, BiVO4 photoanodes showed an increase in the photocurrent density from 1.45 to 4.8 mA cm−2 at 1.23 V vs. RHE and a cathodic shift of the onset potential by 200 mV (Fig. 4f). Mott–Schottky and open circuit potential analyses showed that the TACo cocatalyst film results in a negative shift of the flat band potential and an increase of photovoltage. These results imply that the TACo film not only improves the catalytic charge-transfer efficiency but also facilitates charge separation.
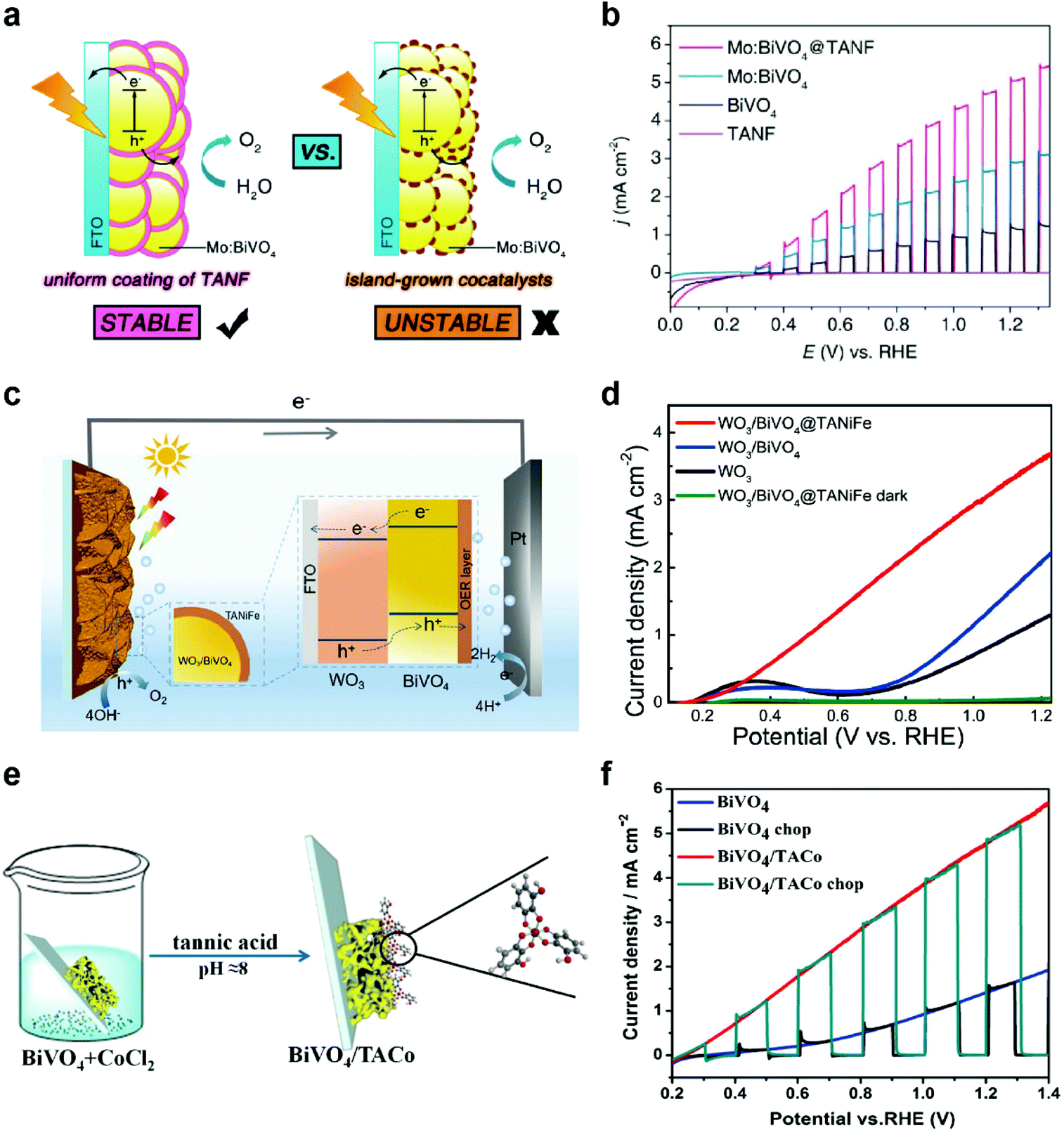 | ||
| Fig. 4 MPNs as OER cocatalysts for PEC water. (a) Schematic illustration showing the advantage of the TANF layer on Mo:BiVO4 compared to general island-grown cocatalyst layers and (b) linear sweep voltammetry (LSV) curves of bare undoped BiVO4, Mo-doped BiVO4 (Mo:BiVO4), TANF-coated Mo:BiVO4 (Mo:BiVO4@TANF), and TANF during PEC water oxidation under chopped AM 1.5 G illumination. Reproduced from ref. 70. Copyright 2018, American Chemical Society. (c) Schematic illustration and (d) LSV curves of WO3/BiVO4 heterojunction photoanodes with and without MPN layers composed of TA, Ni ions, and Fe ions (TANiFe). Reproduced from ref. 71. Copyright 2020, American Chemical Society. (e) Schematic illustration and (f) PEC water oxidation performance of BiVO4 photoanodes modified with MPNs of TA and Co ions. Adapted with permission from ref. 72. Copyright 2019, Elsevier. | ||
Interestingly, most studies on the application of MPN-based OER cocatalysts have been limited to BiVO4 photoanodes. The narrow choice of photoanode may be partly attributed to the photoanodes and MPNs possibly having different pH windows for stable operation. For example, WO3, BiVO4, and Fe2O3 are stable at acidic, neutral, and alkaline pHs, respectively, while MPNs are expected to be unstable at lower pHs. Considering that there are numerous photoanode candidates and diverse MPNs can form a uniform and conformal multifunctional coating on virtually any substrate, it is highly worth utilizing MPNs as OER cocatalysts on various photoanodes for PEC water oxidation.
3.3 HER and overall water splitting
Overall water splitting has been considered a promising method to produce clean hydrogen fuels, and enormous effort has been made to develop efficient, durable, and cost-effective HER and bifunctional HER/OER catalysts over the past five decades.73 There are two different approaches for the development of these catalysts: development of highly active catalysts (1) with minimal use of expensive noble metal elements such as Pt,74 Ru,5,75 and Ir;76 or (2) with cheap and abundant elements.MPN-based HER and HER/OER bifunctional catalysts have also been investigated in two different approaches. For example, Wang et al. first reported the development of MPN-based HER catalysts. An MPN-based single-atom catalyst composed of Ru3+ ions and TA was deposited on porous activated carbon cloth (TARu/ACC) with a one-step dip-coating method (Fig. 5a).77 The as-prepared TARu/ACC showed very low overpotentials of 29, 60, and 112 mV at 10 mA cm−2 in 1 M KOH, 1 M phosphate buffered saline, and 1 M H2SO4, respectively, which are comparable to the state-of-the-art HER electrocatalysts reported to date. Interestingly, the overpotential of TARu/ACC at 50 mA cm−2 (95 mV) was lower than that of commercial Pt/C electrocatalysts (146 mV) (Fig. 5b). This catalyst was stable for more than 130 h in 1 M KOH because Ru3+ strongly binds to the phenolic oxygen of TA, and the cross-linked Ru–TA network is strongly coated on the ACC due to the binding affinity of TA. On the other hand, Zhang et al. utilized MPNs containing various metal ions, such as Fe3+, Co2+, and Ni2+, as bifunctional HER/OER electrocatalysts. They used a highly porous Ni(OH)2 nanosheet grown on nickel foam (Ni(OH)2/NF) as a substrate to grow MPNs with Fe and Ni ions (TAFe and TANi, respectively) by the interfacial coordination assembly of TA–metal complexes (Fig. 5c).78 TAFe@Ni(OH)2/NF showed the lowest overpotential of 280 mV for the OER in 1 M KOH, while the overpotentials of TANi@Ni(OH)2/NF and Ni(OH)2/NF were 373 and 380 mV, respectively. TAFe@Ni(OH)2/NF electrode also showed superior HER performance with an overpotential of 160 mV at 10 mA cm−2. Based on these results, they found that TAFe@Ni(OH)2/NF is an excellent bifunctional HER/OER electrocatalyst with a low cell voltage of 1.7 V at 10 mA cm−2 and high stability for more than 100 h (Fig. 5d). Of note, although there have been only a few reports about the synthesis of MPN-based HER and HER/OER bifunctional catalysts, there have been no reports about the disassembly of MPNs and conversion to other species during HER. This suggests that MPNs may exhibit higher stability under the HER than under the OER.
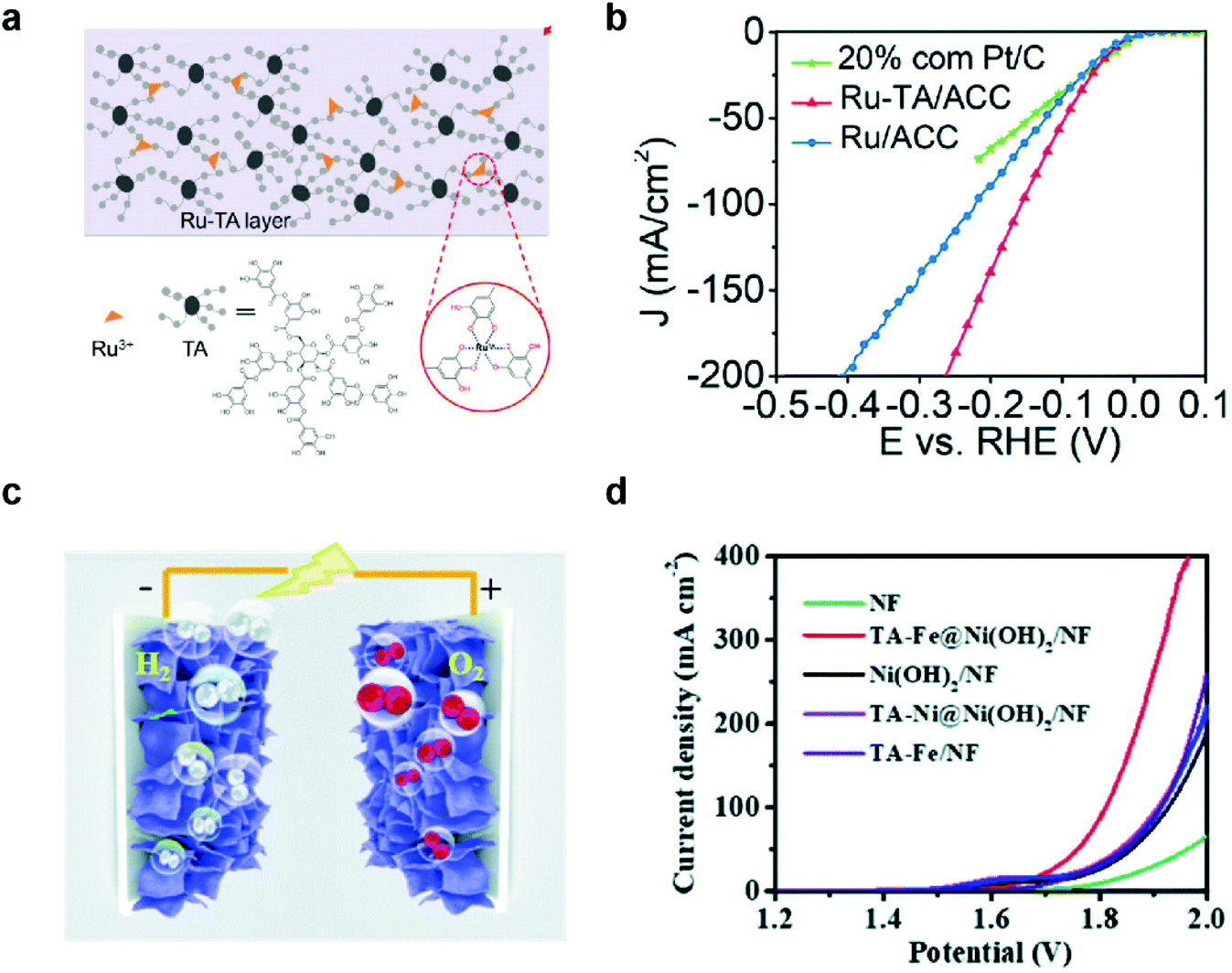 | ||
| Fig. 5 MPN electrocatalysts for HER and overall water splitting. (a) Schematic illustration and (b) HER performance of Ru–TA MPNs. Ru–TA was coated on activated carbon cloth (Ru–TA/ACC) and compared with a commercial Pt/C catalysts for HER in 1 M KOH. Reproduced from ref. 77 with permission from The Royal Society of Chemistry. (c) Illustration of the 3D structure of bifunctional electrodes (TA-metal@Ni(OH)2/NF) prepared by depositing TA–metal complexes on Ni(OH)2-grown nickel foam. (d) Outstanding performance of TA-metal@Ni(OH)2/NF as HER/OER bifunctional electrocatalysts for overall water splitting in 1 M KOH. Reproduced from ref. 78 with permission from The Royal Society of Chemistry. | ||
3.4 Electrocatalytic NRR and CO2RR
Ammonia and hydrocarbons are important raw chemicals for chemical industries but have been produced in energy-intensive and unsustainable ways, such as the Haber–Bosch process and the cracking of oils.79–83 These traditional methods inevitably emit huge amounts of CO2, and thus there have been recent, extensive studies on the synthesis of ammonia and hydrocarbons by electrochemical methods combined with renewable energy resources.79,84 However, both the electrochemical NRR and CO2RR are highly challenging due to the presence of competing reactions such as the HER,85 the high stability of N2 and CO2 requiring high overpotentials,86 and complex reaction pathways87 leading to the formation of diverse end products.In this context, MPNs have also been investigated as NRR electrocatalysts for ammonia synthesis. For example, Sun et al. proposed oxygen-rich TA-modified Pd nanoparticles (Pd–TA) for the synthesis of NH3 through the electrochemical NRR (Fig. 6a).88 Of note, Pd is more Earth-abundant than Ru and Rh and cheaper than Au, which are conventionally utilized as the main catalytic elements of NRR catalysts. They found that TA modification of Pd nanoparticles significantly improved the NRR activity. Pd–TA nanoparticles deposited on carbon paper (Pd–TA/CP) showed a NH3 yield of 24.12 μg h−1 mgcat−1 and a high faradaic efficiency of 9.49% at −0.45 V vs. RHE in N2-saturated 0.1 M Na2SO4 solution (Fig. 6b). On the other hand, Wang et al. recently reported that modification of Au nanowires with oxygen-rich TA considerably improved the NRR activity by suppressing the competing HER, enhancing N2 adsorption, and activating N2 (Fig. 6c and d).89 Despite such encouraging results, no detailed mechanism for the observed performance improvement was provided and only a limited number of metals, such as Pd and Au, have been employed in combination with TA.
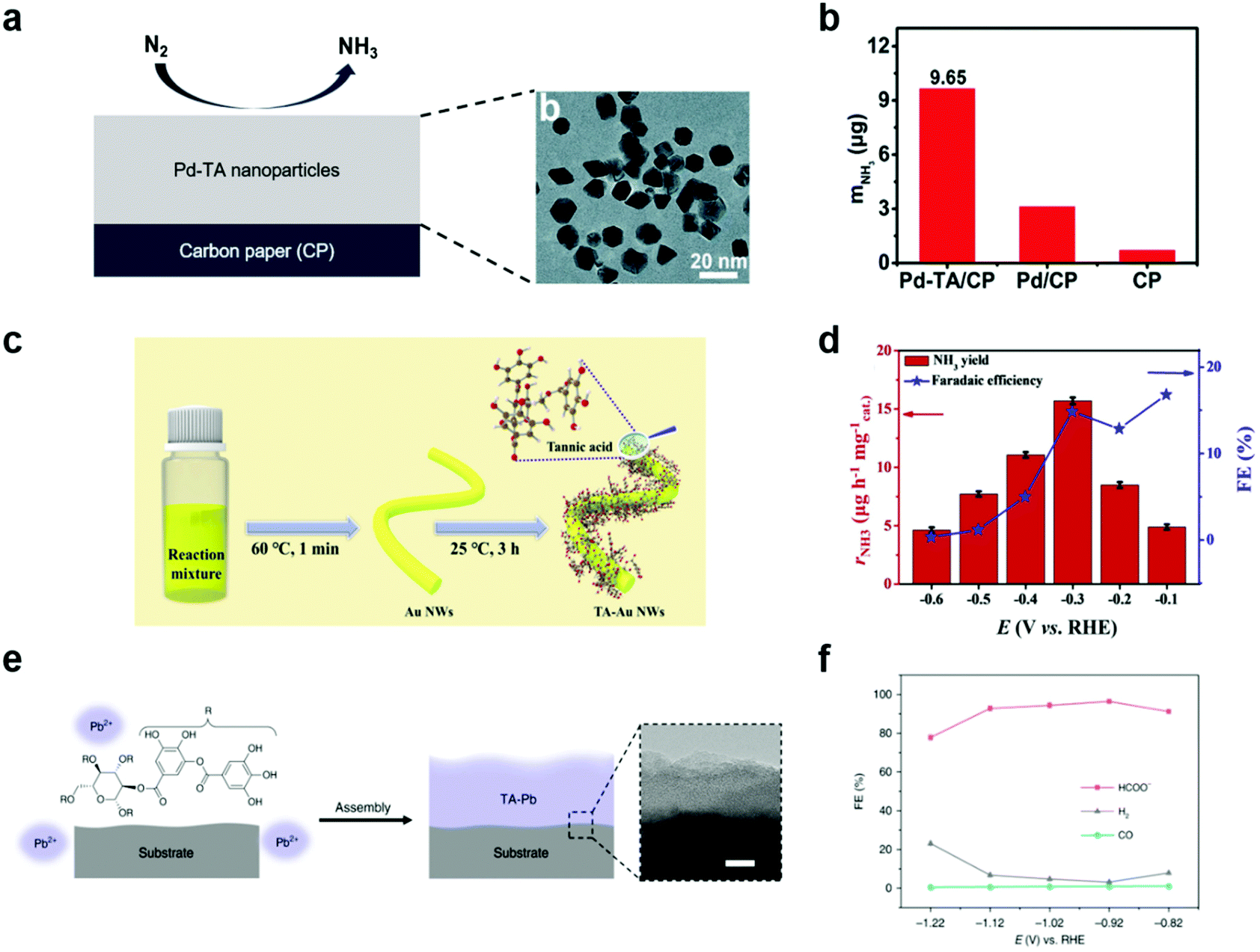 | ||
| Fig. 6 Electrochemical NRR and CO2RR using MPN-based electrocatalysts. (a) Schematic illustration (left) and high-resolution transmission electron micrographs (right) of Pd–TA nanoparticles on CP. (b) Production yield of NH3 with different electrodes after 2 h reaction at −0.45 V vs. RHE in N2-saturated 0.1 M Na2SO4. Reproduced from ref. 88 with permission from The Royal Society of Chemistry. (c) Two-step synthesis of Au nanowires coated with TA (TA–Au NWs) and (d) their application in NRR. FE stands for faradaic efficiency. Adapted with permission from ref. 89. Copyright 2021, Elsevier. (e) Schematic illustration of TA–Pb MPNs on CP and (f) their application in CO2RR in 0.5 M NaHCO3. Reproduced with permission from ref. 90. Copyright 2020, Springer Nature. | ||
MPNs have also been studied to develop CO2RR electrocatalysts. For example, Zhang et al. studied hydrocerussite as a stable electrocatalyst through in situ conversion of the tannin–lead(II) (TA–Pb) nanoparticles (Fig. 6e).90 TA–Pb is converted to PbCO3 (t-PCO) and Pb3(CO3)2(OH)2 (t-PCOH) in a step-wise manner, which acts as an active site for formate (HCOOH) production during the electrochemical CO2RR. t-PCOH showed a high faradaic efficiency of 90.1% at −0.92 V vs. RHE and stability for 10 h, while Pb foil showed low faradaic efficiency of HCOOH of 40% (Fig. 6f). Through DFT calculation, t-PCOH showed stronger adsorption of *HCOO than *H and *COOH, confirming that HCOOH was mainly produced. In this paper, TA–Pb was not directly used as an electrocatalyst for the CO2RR, but they suggested that MPNs can be electrochemically converted to new metal complexes with high catalytic activity for the CO2RR.
3.5 MPN-derived hybrid materials for electrocatalysis
In addition to applying MPNs themselves as electrocatalysts for various reactions, polyphenol-derived hybrid materials can also be used as efficient electrocatalysts by utilizing the strong binding properties of polyphenol groups toward both metal ions and various substrates. Wang et al. first synthesized metal/carbon composites through hydrothermal treatment of Co (or Fe)–polyphenol coordination crystals (Co–TA-x, where x = carbonization temperature) (Fig. 7a).91 They found that Co–TA-x showed high performance for the ORR under both alkaline and acid conditions. The onset potential of Co–TA-800 in O2-saturated 0.1 M KOH solution was 0.85 V, which was slightly higher than that of commercial Pt/C (Fig. 7b). Moreover, Co–TA-800 showed the best ORR efficiency among reported non-precious metal catalysts in acidic conditions. As the tannic acid is carbonized, the catalytic efficiency is improved due to the strong electronic interaction between the metal and the carbon layers. Next, several efforts have been made to develop multifunctional electrocatalysts that can simplify electrode design as well as reduce side reactions. Cho et al. succeeded in synthesizing tannic acid-derived, N,P-codoped carbon-supported iron-based nanocomposites (FePx/Fe–N–C/NPC) for trifunctional electrocatalysts.92 Based on the strong metal-binding properties of tannic acid, FePx/Fe–N–C/NPC catalysts were synthesized by one-pot pyrolysis of a mixture of tannic acid, ferrous chloride, and sodium hydrogen phosphate (Fig. 7c). While FePx, Fe–N–C, and NPC showed excellent efficiency for the HER, ORR, and OER-ORR, respectively, the FePx/Fe–N–C/NPC nanocomposite catalyst showed excellent catalytic activity for all of the HER, OER, and ORR. Moreover, it showed high performance for overall water splitting and rechargeable zinc–air batteries. Surprisingly, FePx/Fe–N–C/NPC||FePx/Fe–N–C/NPC cells showed a low overpotential of 1.58 V at 10 mA cm−2, which was more efficient than the IrO2||Pt/C cell (Fig. 7d). On the other hand, TA-derived carbon materials can also be utilized as cocatalysts for photoelectrochemical cells. Recently, Choi et al. reported that N-doped graphene quantum dots (N-TAGQDs) can be produced from TA and that they can be uniformly and conformally coated on the underlying BiVO4 photoanodes together with Co2+ ions for photoelectrochemical water oxidation (Fig. 7e).32 By taking advantage of TA's strong adhesion properties, N-TAGQDs formed an ultrathin and uniform layer on the substrate. They found that BiVO4/Co/N-TAGQD showed a low onset potential of 0.21 V vs. RHE because N-TAGQDs and Co2+ ions played a role in improving the charge separation, hole storage, and catalytic activity (Fig. 7f). This study showed the potential to improve the PEC performance and stability of photoelectrodes via surface modification using Earth-abundant, inexpensive materials by simple synthetic methods.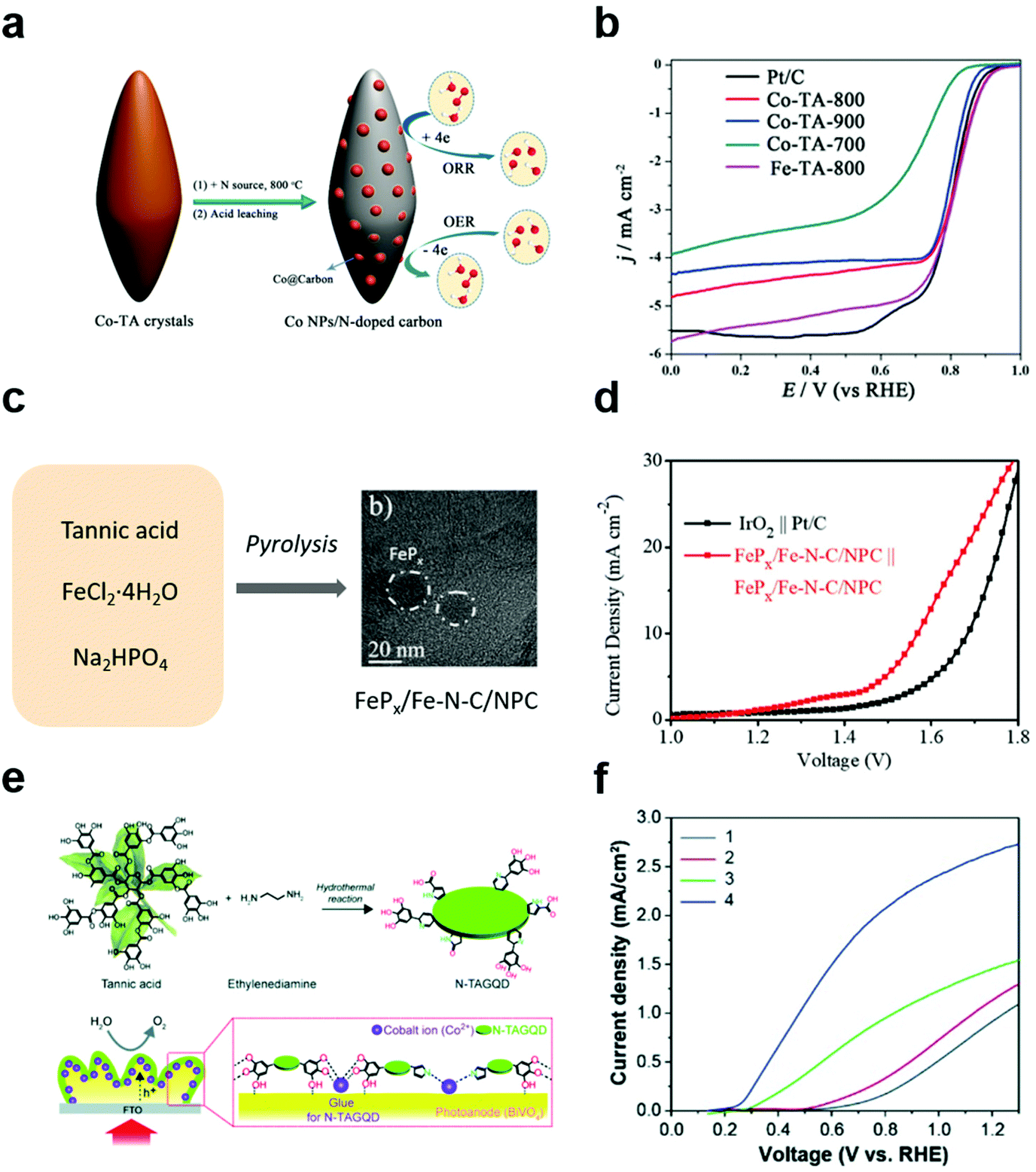 | ||
| Fig. 7 Polyphenol-derived metal–carbon composites for electrocatalysis and photoelectrocatalysis. (a) Schematic illustration of synthesis method of Co nanoparticles embedded in the N-doped carbon and (b) linear sweep voltammetry (LSV) curves of commercial Pt/C, Co–TA-x (x = carbonization temperature) for ORR in O2-saturated 0.1 M KOH solution. Reproduced with permission from ref. 91. Copyright 2016, Wiley-VCH. (c) Schematic illustration and (d) LSV curves of IrO2||Pt/C and FePx/Fe–N–C/NPC||FePx/Fe–N–C/NPC cells in 1 M KOH for overall water splitting with a two-electrode system. Reproduced with permission from ref. 92. Copyright 2019, Wiley-VCH. (e) Schematic illustration and (f) LSV curve of (1) BiVO4, (2) BiVO4/N-TAGQD, (3) BiVO4/Co, and (4) BiVO4/Co/N-TAGQD for PEC water oxidation. Reproduced with permission from ref. 32. Copyright 2021, Wiley-VCH. | ||
4. Future perspectives and conclusions
In order to utilize MPNs for a variety of applications, we believe that the following issues should be addressed in the future. First, more diverse metal ions should be utilized for the development of MPN-based electrocatalysts. To date, only a few metal ions, including Fe, Ni, Co, and Ru, have been investigated for MPN-based electrocatalysts. In addition, a relatively small number of studies with MPNs have been reported for the HER, NRR, and CO2RR compared to for the OER. However, as discussed above, there is a high possibility that MPNs undergo degradation and structural transformation under the same harsh oxidative conditions as the OER. Nevertheless, by rational selection of metal ions appropriate for a specific target reaction, the activity and applicability of MPN-based electrocatalysts can be extended further. It is well-known that ions of certain metals have higher activity for a specific target reaction in conventional heterogeneous electrocatalysis (Fig. 8a). For example, Fe, Mo, and Ru can also be incorporated or hybridized with TA to develop efficient NRR electrocatalysts while Pd and Au with TA have been investigated to date. | ||
| Fig. 8 (a) The periodic table summarizing the experimentally observed relationship between the main elements of catalysts and the preferred target electrochemical reactions. (b) Theoretical redox potential for various electrochemical reactions. (c) Various plant-derived polyphenolic molecules. | ||
Second, MPNs can be utilized as electrocatalysts for more diverse electrochemical reactions, such as the oxygen reduction reaction (ORR) and the chlorine evolution reaction (CER) (Fig. 8b). The ORR is an important reaction in energy conversion systems such as fuel cells, Li–air, and Zn–air batteries. On the other hand, chlorine is considered an important building block in the material and chemical compound synthesis industry. However, expensive noble metals, such as Pt and Ru, have been conventionally used for the ORR and CER, respectively. Thus, these reactions are promising target reactions for MPN-based electrocatalysts because a relatively small amount of the noble metal elements can be used; MPNs can be considered as single-atom catalysts. Of course, MPNs can still be utilized to develop cheap and efficient electrocatalysts for these reactions by using abundant metallic elements.
Third, to date, only TA has been used as a phenolic ligand for the development of MPN-based electrocatalysts. The popular use of TA can be attributed to its abundant phenolic groups that allow both strong coordination with metal ions and strong adhesion to a substrate. In principle, various small phenolic molecules, such as gallic acid, caffeic acid, and pyrogallol, can also be utilized (Fig. 8c). One can expect that these small molecules might have poor activity and stability due to a limited number of phenolic groups. However, it is well-known that these molecules undergo oxidative polymerization at high pHs, have an affinity toward various metal ions, and are conformally coated on the desired substrate, as is TA.29,30 Thus, in principle, MPNs with more diverse polyphenol molecules can be utilized in the development of efficient and stable electrocatalysts. Indeed, there was a recent report that polydopamine formed by oxidative polymerization of dopamine can be used as a CO2RR catalyst, although no metallic element was employed.93
In addition, most MPN films reported to date are amorphous, rather than crystalline. While highly crystalline electrocatalysts can have higher stability and electrical conductivity in general, amorphous counterparts can have more exposure of active sites and allow diverse bond lengths between constituting atoms (and thus flexible tuning of the electronic structure of electrocatalysts).94,95 In this regard, controlling the crystallinity of MPN films to tailor their electrocatalytic activity should be studied in the future.
Lastly, although MPNs show excellent electrocatalytic activity for various electrochemical reactions, there is a lack of studies about the detailed mechanisms for their enhanced catalytic activity. For example, phenolic groups can have unexpected roles, such as enhancing proton-coupled electron transfer and acting as a reservoir of protons. Molecular understanding of MPNs will open a new avenue for the development of unprecedented electrocatalysts.
In this article, we have summarized recent efforts in the development of molecularly designable heterogeneous electrocatalysts based on MPNs. MPNs have many advantages for the development of heterogeneous electrocatalysts: they (i) can be quickly and simply synthesized in aqueous solution, (ii) form uniform and conformal thin films on various substrates, (iii) have excellent charge-transfer ability through LMCT, and (iv) can effectively use catalytic elements by providing a platform for single-atom catalysts. Based on the unique structural and chemical properties of MPNs, promising MPN-based electrocatalysts for various target reactions have been reported. However, the development of MPN-based electrocatalysts is still in its infancy, and thus more diverse and versatile MPN-based electrocatalysts are expected to be developed in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Technology Development Program to Solve Climate Changes (2019M1A2A2065616), the Basic Science Research Program (2021R1A2C2013684) and the Nano-Material Technology Development Program (2021M3H4A1A03051390) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT of Korea. This work was also supported by the Basic Science Research Program (2020R1I1A1A01057924 and 2021R1A6A3A13042213) through the NRF funded by the Ministry of Education of Korea.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J.-S. Lee, C. Lee, J.-Y. Lee, J. Ryu and W.-H. Ryu, ACS Catal., 2018, 8, 7213–7221 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- Y. Li, M. Gong, Y. Liang, J. Feng, J. E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- C. Lee, D. Jeon, J. Park, W. Lee, J. Park, S. J. Kang, Y. Kim and J. Ryu, ACS Appl. Mater. Interfaces, 2020, 12, 32689–32697 CrossRef CAS PubMed.
- X. Yan, J. Biemolt, K. Zhao, Y. Zhao, X. Cao, Y. Yang, X. Wu, G. Rothenberg and N. Yan, Nat. Commun., 2021, 12, 4143 CrossRef CAS PubMed.
- F. A.-P. Andrew, A. Pterson, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- N. Kim, J. S. Nam, J. Jo, J. Seong, H. Kim, Y. Kwon, M. S. Lah, J. H. Lee, T.-H. Kwon and J. Ryu, Nanoscale Horiz., 2021, 6, 379–385 RSC.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550–1606555 CrossRef PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- S. Farid, S. Ren and C. Hao, Inorg. Chem. Commun., 2018, 94, 57–74 CrossRef CAS.
- C. Wang, J. Kim, J. Tang, M. Kim, H. Lim, V. Malgras, J. You, Q. Xu, J. Li and Y. Yamauchi, Chem, 2020, 6, 19–40 CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 2014, 645–659 CrossRef CAS.
- M. García-Melchor, L. Vilella, N. López and A. Vojvodic, ChemCatChem, 2016, 8, 1792–1798 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- H. Kim, N. Kim and J. Ryu, Inorg. Chem. Front., 2021, 8, 4107–4148 RSC.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- A. I. B. Adrien, P. Côté, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 RSC.
- S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660–12668 CrossRef CAS PubMed.
- J. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- Q. Li, W. Xiao, F. Zhang, Q. Liu, J. Ye, H. Dong and X. Cao, J. Mater. Chem. B, 2018, 6, 2734–2738 RSC.
- Y. Choi, S. Bae, B.-S. Kim and J. Ryu, J. Mater. Chem. A, 2021, 9, 13874–13882 RSC.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- Q. Z. Zhong, S. Pan, M. A. Rahim, G. Yun, J. Li, Y. Ju, Z. Lin, Y. Han, Y. Ma, J. J. Richardson and F. Caruso, ACS Appl. Mater. Interfaces, 2018, 10, 33721–33729 CrossRef CAS PubMed.
- Y. Liu, J. Jia, Z. Liu, N. Pu, G. Ye, W. Wang, T. Hu, T. Qi and J. Chen, Chem. Mater., 2021, 33, 4733–4744 CrossRef CAS.
- H. T. Zheng, H. L. Bui, S. Chakroborty, Y. Wang and C. J. Huang, Langmuir, 2019, 35, 8829–8839 CrossRef CAS PubMed.
- H. Wu, Q. He, L. Li, L. Li, Z. Zhou, N. Chen, M. Yang, Q. Luo, B. Zhang, R. Luo, L. Yang and Y. Wang, Chem. Eng. Sci., 2022, 427, 130932–130946 CrossRef CAS.
- G. Yun, W. Youn, H. Lee, S. Y. Han, M. B. Oliveira, H. Cho, F. Caruso, J. F. Mano and I. S. Choi, Chem. Mater., 2020, 32, 7746–7753 CrossRef CAS.
- J. Guo, J. J. Richardson, Q. A. Besford, A. J. Christofferson, Y. Dai, C. W. Ong, B. L. Tardy, K. Liang, G. H. Choi, J. Cui, P. J. Yoo, I. Yarovsky and F. Caruso, Langmuir, 2017, 33, 10616–10622 CrossRef CAS PubMed.
- G. Yun, D. G. Kang, H. B. Rheem, H. Lee, S. Y. Han, J. Park, W. K. Cho, S. M. Han and I. S. Choi, Langmuir, 2020, 36, 15552–15557 CrossRef CAS PubMed.
- J. Chen, S. Pan, J. Zhou, R. Seidel, S. Beyer, Z. Lin, J. J. Richardson and F. Caruso, Chem. Mater., 2021, 33, 2557–2566 CrossRef CAS.
- S. Pan, E. Goudeli, J. Chen, Z. Lin, Q. Z. Zhong, W. Zhang, H. Yu, R. Guo, J. J. Richardson and F. Caruso, Angew. Chem., 2021, 60, 14586–14594 CrossRef CAS PubMed.
- C. F. D. Linsey, C. Seitz, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, J. K. N. Harold, Y. Hwang and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef PubMed.
- K. Zhang and R. Zou, Small, 2021, 2100129–2100168, DOI:10.1002/smll.202100129.
- A. Badruzzaman, A. Yuda, A. Ashok and A. Kumar, Inorg. Chim. Acta, 2020, 511, 119854–119882 CrossRef CAS.
- Y. Pan, H. Ren, H. Du, F. Cao, Y. Jiang, H. Du and D. Chu, J. Mater. Chem. A, 2018, 6, 22497–22502 RSC.
- Z. Li, S. Wang, Y. Tian, B. Li, H. J. Yan, S. Zhang, Z. Liu, Q. Zhang, Y. Lin and L. Chen, Chem. Commun., 2020, 56, 1749–1752 RSC.
- J. Yi, W. H. Lee, C. H. Choi, Y. Lee, K. S. Park, B. K. Min, Y. J. Hwang and H.-S. Oh, Electrochem. Commun., 2019, 104, 106469–106473 CrossRef CAS.
- L.-W. Chen and H.-W. Liang, Catal. Sci. Technol., 2021, 11, 4673–4689 RSC.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587–4597 CrossRef CAS PubMed.
- S. Schlicht, P. Büttner and J. Bachmann, ACS Appl. Energy Mater., 2019, 2, 2344–2349 CrossRef CAS.
- M. E. C. Pascuzzi, J. P. Hofmann and E. J. M. Hensen, Electrochim. Acta, 2021, 366, 137448–137447 CrossRef.
- Y. Shi, Y. Yu, Y. Liang, Y. Du and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 3769–3773 CrossRef CAS PubMed.
- H. Huang, J. Zhao and R. Liu, J. Colloid Interface Sci., 2021, 582, 396–404 CrossRef CAS PubMed.
- M. Huang, Z. Huang and H. Zhu, Nano Energy, 2020, 70, 104487–104493 CrossRef CAS.
- H. S. Han, S. Shin, D. H. Kim, I. J. Park, J. S. Kim, P.-S. Huang, J.-K. Lee, I. S. Cho and X. Zheng, Energy Environ. Sci., 2018, 11, 1299–1306 RSC.
- M. Wang, Z. Wang, B. Zhang, W. Jiang, X. Bao, H. Cheng, Z. Zheng, P. Wang, Y. Liu, M.-H. Whangbo, Y. Li, Y. Dai and B. Huang, ACS Catal., 2020, 10, 13031–13039 CrossRef CAS.
- H. Kim, S. Bae, D. Jeon and J. Ryu, Green Chem., 2018, 20, 3732–3742 RSC.
- L. Jia, K. Harbauer, P. Bogdanoff, I. Herrmann-Geppert, A. Ramírez, R. van de Krol and S. Fiechter, J. Mater. Chem. A, 2014, 2, 20196–20202 RSC.
- R. Zhang, Y. Fang, T. Chen, F. Qu, Z. Liu, G. Du, A. M. Asiri, T. Gao and X. Sun, ACS Sustainable Chem. Eng., 2017, 5, 7502–7506 CrossRef CAS.
- Y. Choi, D. Jeon, Y. Choi, D. Kim, N. Kim, M. Gu, S. Bae, T. Lee, H. W. Lee, B. S. Kim and J. Ryu, ACS Nano, 2019, 13, 467–475 CrossRef CAS PubMed.
- J. Zhang, I. Salles, S. Pering, P. J. Cameron, D. Mattia and S. Eslava, RSC Adv., 2017, 7, 35221–35227 RSC.
- A. Jelinska, K. Bienkowski, M. Jadwiszczak, M. Pisarek, M. Strawski, D. Kurzydlowski, R. Solarska and J. Augustynski, ACS Catal., 2018, 8, 10573–10580 CrossRef CAS.
- X. Liu, F. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7894–7911 RSC.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2017, 3, 53–60 CrossRef.
- G. M. Carroll and D. R. Gamelin, J. Mater. Chem. A, 2016, 4, 2986–2994 RSC.
- H. Yin, D. Li, X. Wang and C. Li, J. Phys. Chem. C, 2021, 125, 8369–8375 CrossRef CAS.
- F. Malara, A. Minguzzi, M. Marelli, S. Morandi, R. Psaro, V. Dal Santo and A. Naldoni, ACS Catal., 2015, 5, 5292–5300 CrossRef CAS.
- P. Varadhan, H. C. Fu, Y. C. Kao, R. H. Horng and J. H. He, Nat. Commun., 2019, 10, 5282–5290 CrossRef PubMed.
- Y. Shi, Y. Yu, Y. Yu, Y. Huang, B. Zhao and B. Zhang, ACS Energy Lett., 2018, 3, 1648–1654 CrossRef CAS.
- H. Sun, W. Hua, Y. Li and J.-G. Wang, ACS Sustainable Chem. Eng., 2020, 8, 12637–12645 CrossRef CAS.
- T. Tian, C. Dong, X. Liang, M. Yue and Y. Ding, J. Catal., 2019, 377, 684–691 CrossRef CAS.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- J. Klein, A. K. Engstfeld, S. Brimaud and R. J. Behm, Phys. Chem. Chem. Phys., 2020, 22, 19059–19068 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, Nano Energy, 2020, 78, 105270–105297 CrossRef CAS.
- J. Chen, H. Wang, Y. Gong and Y. Wang, J. Mater. Chem. A, 2019, 7, 11038–11043 RSC.
- Y. Wang, S. Chen, S. Zhao, Q. Chen and J. Zhang, J. Mater. Chem. A, 2020, 8, 15845–15852 RSC.
- S. K. Sahoo, J. Heske, M. Antonietti, Q. Qin, M. Oschatz and T. D. Kuhne, ACS Appl. Energy Mater., 2020, 3, 10061–10069 CrossRef CAS PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- M. H. Plunkett, C. M. Knutson and B. M. Barney, Microb. Cell Fact., 2020, 19, 107–118 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Mater. Today Adv., 2020, 7, 100074–100097 CrossRef.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- W. da Silva Freitas, A. D'Epifanio and B. Mecheri, J. CO2 Util., 2021, 50, 101579–101596 CrossRef CAS.
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- S. Liu, S. Yin, S. Jiao, H. Zhang, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Mater. Today Energy, 2021, 21, 100828–100834 CrossRef CAS.
- Y. Shi, Y. Ji, J. Long, Y. Liang, Y. Liu, Y. Yu, J. Xiao and B. Zhang, Nat. Commun., 2020, 11, 3415–3424 CrossRef CAS PubMed.
- J. Wei, Y. Liang, Y. Hu, B. Kong, J. Zhang, Q. Gu, Y. Tong, X. Wang, S. P. Jiang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 12470–12474 CrossRef CAS PubMed.
- Q. Qin, H. Jang, P. Li, B. Yuan, X. Liu and J. Cho, Adv. Energy Mater., 2019, 9, 1803312–1803324 CrossRef.
- H. Coskun, A. Aljabour, P. De Luna, D. Farka, T. Greunz, D. Stifter, M. Kus, X. L. Zheng, M. Liu, A. W. Hassel, W. Schofberger, E. H. Sargent, N. S. Sariciftci and P. Stadler, Sci. Adv., 2017, 3, e1700686 CrossRef PubMed.
- S. Liu, L. Dai, Y. Qu, Y. Qiu, J. Fan, X. Li, Q. Zhang and X. Guo, Mater. Chem. Front., 2021, 5, 6648–6658 RSC.
- Y. Han, K. Choi, H. Oh, C. Kim, D. Jeon, C. Lee, J. H. Lee and J. Ryu, J. Catal., 2018, 367, 212–220 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |