
Virus-inspired nanosystems for drug delivery
Zhihuan
Liao
a,
Li
Tu
a,
Xuejian
Li
a,
Xing-Jie
Liang
 *abc and
Shuaidong
Huo
*a
*abc and
Shuaidong
Huo
*a
aFujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, Xiamen 361102, China. E-mail: huosd@xmu.edu.cn
bCAS Center for Excellence in Nanoscience, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: liangxj@nanoctr.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 15th October 2021
Abstract
With over millions of years of evolution, viruses can infect cells efficiently by utilizing their unique structures. Similarly, the drug delivery process is designed to imitate the viral infection stages for maximizing the therapeutic effect. From drug administration to therapeutic effect, nanocarriers must evade the host's immune system, break through multiple barriers, enter the cell, and release their payload by endosomal escape or nuclear targeting. Inspired by the virus infection process, a number of virus-like nanosystems have been designed and constructed for drug delivery. This review aims to present a comprehensive summary of the current understanding of the drug delivery process inspired by the viral infection stages. The most recent construction of virus-inspired nanosystems (VINs) for drug delivery is sorted, emphasizing their novelty and design principles, as well as highlighting the mechanism of these nanosystems for overcoming each biological barrier during drug delivery. A perspective on the VINs for therapeutic applications is provided in the end.
 Zhihuan Liao | Zhihuan Liao obtained his B.S. degree from the Guangdong Medical University in 2020. Subsequently, he joined the group of Prof. Shuaidong Huo at Xiamen University as a postgraduate student. His research interests include bioinspired and biofunctionalized nanomaterials for drug delivery. |
 Xing-Jie Liang | Xing-Jie Liang obtained his Ph.D. degree at the National Key Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences (CAS). He finished his post-doctorate under the guidance of Dr Michael M. Gottesman, a member of NAS and Deputy Director of NIH. Then, he worked as a research fellow at Surgical Neurology Branch, NINDS, NIH. Dr Liang is currently a principal investigator at the National Center for Nanoscience and Technology of China and a chair-professor at Xiamen University. His research interests are elucidating mechanisms to improve nanomedicinal drugability and developing novel strategies to increase therapeutic efficiency. |
 Shuaidong Huo | Shuaidong Huo obtained his Ph.D. degree from the National Center for Nanoscience and Technology of China in 2016 under the supervision of Prof. Xing-Jie Liang. After that, he worked as a post-doctoral researcher in Prof. Andreas Herrmann's team at the DWI-Leibniz Institute for Interactive Materials, Germany. In January 2020, he joined the School of Pharmaceutical Sciences, Xiamen University as a Professor. His research topic focuses on taking advantage of nanotechnology to understand the biological effect of nanomaterials on living systems and develop activity-regulatable drug delivery systems for disease treatment. |
1. Introduction
Viruses are core–shell entities co-assembled from nucleic acids and proteins, with sizes ranging from 20–900 nm.1 As a non-cellular life, a virus is inert outside, but it can replicate itself inside the host cell.2 With over millions of years of evolution, viruses have been able to bypass the immune system's surveillance effectively.3 By taking advantage of the amphoteric charges, they can prolong their blood circulation time to obtain enough time for cell infection.4–6 Attachment is the first step of viral infection. The polyvalent ligand–receptor interactions or spiky surface morphology can transform the viral envelope proteins, allowing viruses to enter and penetrate the cell.7 For some viruses, the replication step mainly takes place in the cell nucleus. The presence of nuclear localization signals (NLS) enables viruses to specifically target the nucleus, thus entering and releasing their genomes for replication.8 In addition, the interaction between the viral surface with cellular components and their microenvironment is likewise conducive to the viral infection steps.9 It is noteworthy that the coat protein and structural morphology of viruses play irreplaceable roles in crossing various physiological barriers and attaching to host cells during the whole infection process.Inspired by the efficient viral infection process, various bioengineered vectors have been developed for cargo delivery in biomedical applications.10,11 For example, adenoviral and lentiviral vectors with high transfection efficiency have been used to treat multiple diseases,12 such as hemophilia,13 cystic fibrosis,14 tumors,15etc. However, the direct use of viruses as a conveyance still has many safety risks, including broad viral tropism, high immunogenicity, and pathogenicity, significantly limiting their further applications.16–19 In recent decades, the prosperous development of nanotechnology has resulted in unprecedented strategies for biomedicine, especially for drug delivery.20–22 A number of conventional nanocarriers have been developed for drug delivery, including polymeric nanoparticles (NPs),23 solid lipid NPs,24 inorganic NPs,25etc. Despite numerous efforts, limitations are still multifaceted. First, the carrier must effectively escape the clearance of immune systems, then cross the biological barriers and cell membrane while avoiding endosomal entrapment. Moreover, the nanocarrier requires excellent stability outside the cell, and must effectively target and release the payload inside the nucleus. To overcome these challenges, fortunately, virus-inspired nanosystems (VINs) have been constructed and shown unique superiority for drug delivery.26–29 By simulating the original composition and topological structure of viruses, VINs with high biocompatibility have been successfully exploited. In general, VINs have editable physicochemical properties, and can flexibly cross the barriers and deal with the changes of the surrounding physiological environment for delivery.30,31 In addition to delivering genes, VINs can also be used to deliver a range of therapeutic drugs.32,33 Thanks to the large space of the hollow structure, encapsulation of therapeutics in VINs can improve their water solubility, loading efficiency, and biocompatibility.34,35 Furthermore, the virus-like design of VINs can promote a better biological distribution and higher cellular uptake efficiency.36,37 More importantly, the characteristic of easy surface functionalization of VINs might further push them to cross the boundary of clinical translation for disease treatment.38
In this review, we summarized the state of the art of strategy for constructing VINs for drug delivery, with an emphasis on their novelty and design principles. Meanwhile, the mechanism of these nanosystems for overcoming each biological barrier during drug delivery is highlighted, including prolonged blood half-life, endothelial barrier breaking, enhanced cellular uptake, endosomal escape and nuclear targeting (Fig. 1). Finally, the challenges and an outlook on the VINs for drug delivery are discussed.
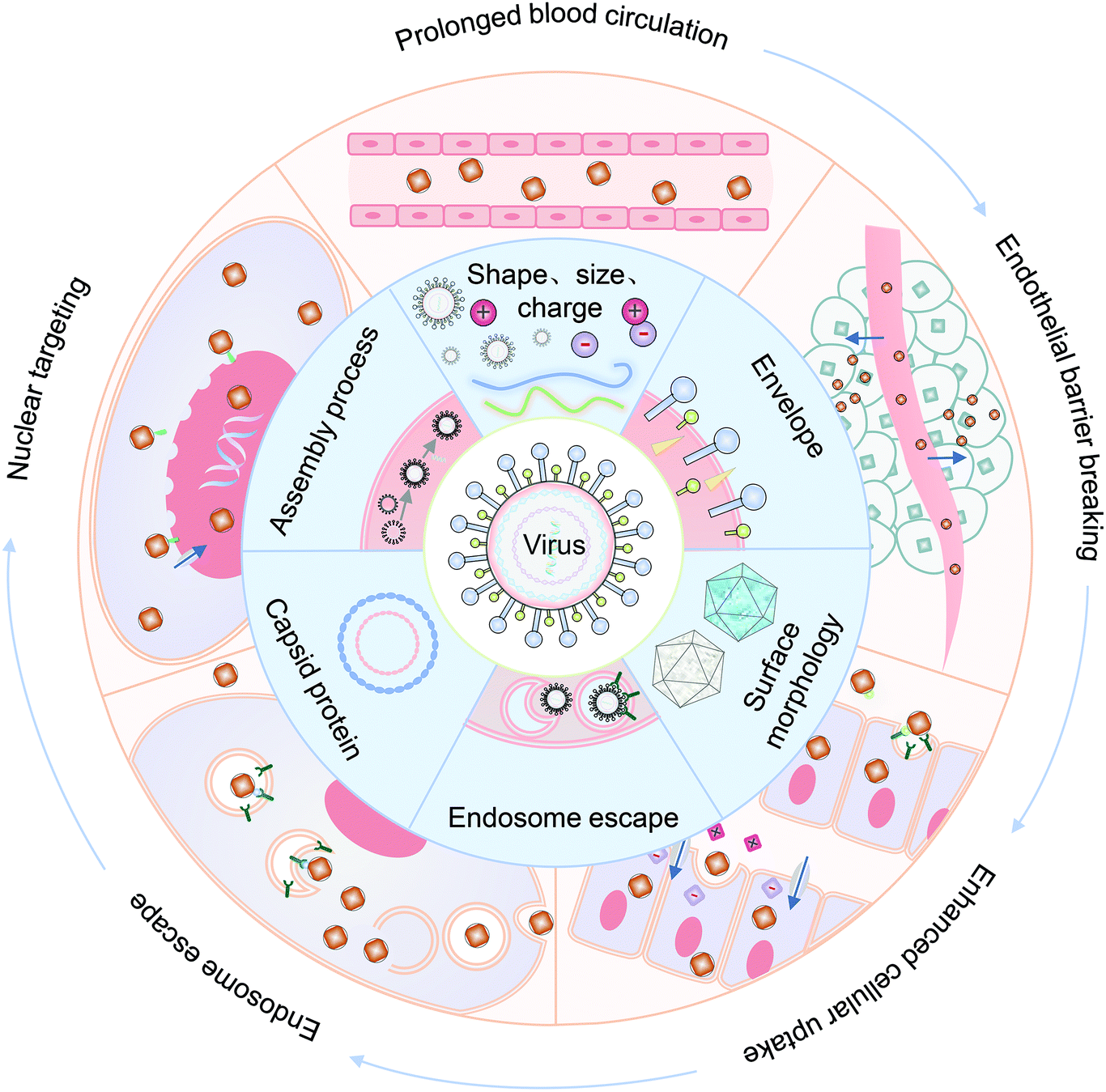 | ||
| Fig. 1 Schematic illustration of VINs used to overcome physiological barriers for drug delivery. Common physiological barriers that prevent drug delivery in the body (the outer ring). Structure and function that affect the drug delivery process of VINs (the inner ring). | ||
2. Prolonged blood circulation
The blood half-life of drug vectors refers to their retention time in the bloodstream, which directly reflects the pharmacokinetic properties of the carriers. Generally, foreign nanocarriers tend to bind with proteins or biomolecules non-specifically in the blood, forming the so-called corona that promotes their clearance by the immune system. Remarkably, some viruses have the ability to evade the capture of the immune system, which inspires us to design similar structures for drug delivery.39,40 This section describes virus-inspired strategies to prolong blood circulation by simulating the inherent viral traits.The blood half-life of NPs is dependent on their shape, size, and surface charge.41–43 Compared with spherical NPs, the velocity gradient of the fluid environment in the blood circulation leads to the uneven distribution of forces, which more likely affects the trajectory and drift of the rod-shaped NPs in the blood flow and promotes their adhesion to the target position.44 Filoviruses, single-stranded RNA viruses, have a filamentous or branched polymorphic structure with a diameter of about 80 nm and a length of up to 1400 nm. Under the influence of fluid dynamics, filoviruses are stretched as they flow, thus prolonging their circulation time and enhancing the viral transport efficiency.45 The available evidence has suggested that filiform is a more elaborate rod structure, and the retention effect of filamentous micelles in the circulation is better than that of spherical NPs.46 Inspired by filoviruses, Geng et al. evaluated the anticancer effect of the flexible material containing paclitaxel by controlling the average length of the material (Fig. 2A).47 They compared the clearance time of PEG copolymer filamentous micelles and PEGylated “stealth” vesicles at the same dose. The result showed that the circulation time of the former was up to one week, while the latter was cleared quickly in two days. Similarly, researchers have developed a uniquely shaped artificial tobacco mosaic virus (ATMV) to mimic the high aspect ratio structure advantage of TMV. ATMVs with elongated shapes are concealed in the blood until they reach the tumor site to induce tumor killing (Fig. 2B).48 Distinctly, the shape effect of the virus further enhanced the advantages of PEG as a cloaking material and achieved the synergistic impact of prolonging the blood half-life, providing a more effective passive tumor accumulation.
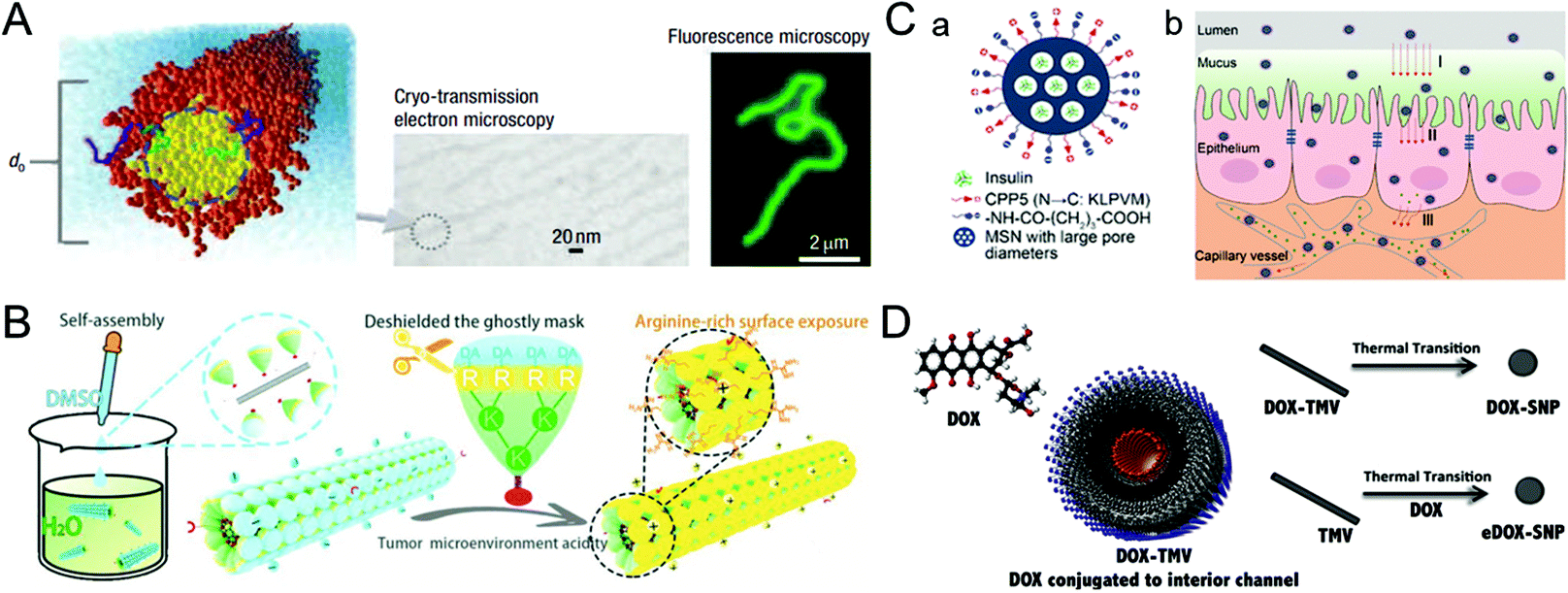 | ||
| Fig. 2 Representative diagrams of VINs for prolonging the blood circulation time. (A) Schematic illustration of filomicelles. The size and shape of the filomicelles are shown in the electron and fluorescence microscopy images. Reproduced from ref. 47 with permission from Springer Nature 2007. (B) Schematic illustration of the ATMV for tumor therapy. Reproduced from ref. 48 with permission from Wiley 2020. (C) Schematic illustration of VINs (MSN–NH2@COOH/CPP5) for improving insulin absorption. (a) Schematic diagram of the structure of VINs. (b) VINs with an electrically neutral surface can overcome the epithelial cell barrier and increase their concentration in the blood. Reproduced from ref. 52 with permission from American Chemical Society 2021. (D) Schematic diagram of 50 nm VINs coated with doxorubicin for breast cancer treatment. Reproduced from ref. 54 with permission from Elsevier 2016. | ||
In addition, adjusting the surface charge of the nanocarriers can reduce the formation of the protein corona, which is also beneficial for drug delivery.4 A study of TMV showed that virus-inspired nanosystems (VINs) were six times less capable of binding to proteins than synthetic nanosystems.49 The formation may be caused by the combination of positive and negative charges with hydrophobic and hydrophilic domains on the patching.50 Similarly, a study demonstrated that nanosystems with the amphoteric charges were found to inhibit protein crown formation more effectively than the positively, negatively, or uncharged particles.51 The reduced serum protein adsorption might result from the amphiphilic particles with less hydrophobic or electrostatic interaction domains. In another study, inspired by the process by which viruses effectively pass through epithelial tissues using electrical neutrality, VINs based on mesoporous silica nanoparticles (MSN–NH2@COOH/CPP5) were designed to improve insulin absorption by simulating the electrical neutrality of the virus surface. Owning to VINs ability to overcome the epithelial cell barrier and increase their concentration in the blood, the bioavailability of the drug is further improved (Fig. 2C).52
In terms of the size effect, generally, NPs smaller than 10 nm in diameter can be cleared rapidly by the kidneys, while NPs larger than 200 nm have the risk of activating the complement system.20 VINs with diameters in the range of 20–200 nm have been constructed for drug delivery,53 and the optimization of 50 nm (Fig. 2D) solves the problem of insufficient blood circulation time to some extent.54 In a word, nanosystems that mimic the viral membrane composition and physicochemical properties have significantly prolonged blood circulation time relative to conventional nanosystems.
3. Endothelial barrier breaking
It is well known that the low permeability of the vascular endothelium, especially the tight junctions, such as the blood–brain barrier (BBB), is another major obstacle for drug delivery.55,56 To overcome this challenge, nanocarriers need to diffuse into the dense extracellular matrix at high interstitial pressures, or cross endothelial cells using receptor-mediated endocytosis and then be excreted to the other side.6,57 In this section, taking the BBB as an example, the virus-inspired strategies to overcome the endothelial barriers are discussed.The existence of the BBB makes it harder for drugs to cure brain diseases. At present, the strategies for treating brain diseases by penetrating the BBB mainly include invasive techniques,58 receptor-mediated transport (RMT),59 and adsorption-mediated transcytosis (AMT).60 Although great efforts have been contributed for brain-targeting, there is an urgent need to develop more efficient carriers that can penetrate the BBB efficiently and safely. Inspired by the excellent ability of the rabies virus (RABV) to invade the central nervous system (CNS) by RMT,61,62 a drug carrier composed of a brain-targeting peptide called RVG29 from RABV for delivering deferoxamine was reported (Fig. 3A).63 Interestingly, the drug carriers can effectively repair the damage to dopaminergic neurons without affecting the normal function of other organs by significantly reducing oxidative stress levels of the brain. Similarly, the metal–organic framework-based nanosystems simulating the natural bullet-shaped structure and surface features of RABV were designed (Fig. 3B).64 Compared with the simple imitation of RABV individual function, they benefit from a more comprehensive RABV imitation strategy, showing an apparent enhancement of BBB penetration and better treatment of brain tumors.65 Besides, Neves et al. found that a peptide called PepH3 of the dengue virus type 2 capsid protein can be used for BBB penetration and the translocation mechanism is consistent with AMT (Fig. 3C).66 Biological distribution data in vivo showed that the radio-labeled derivative peptides exhibited high brain permeability, and PepH3 reached the equilibrium distribution concentration of BBB within 24 hours. The study suggested that PepH3 is an excellent candidate for shuttling peptides into the brain. Undoubtedly, inspired by the viral infection process, a combination of the surface receptors and physicochemical characteristics of the virus provides the main idea to break endothelial barriers for intracerebral targeted drug delivery.
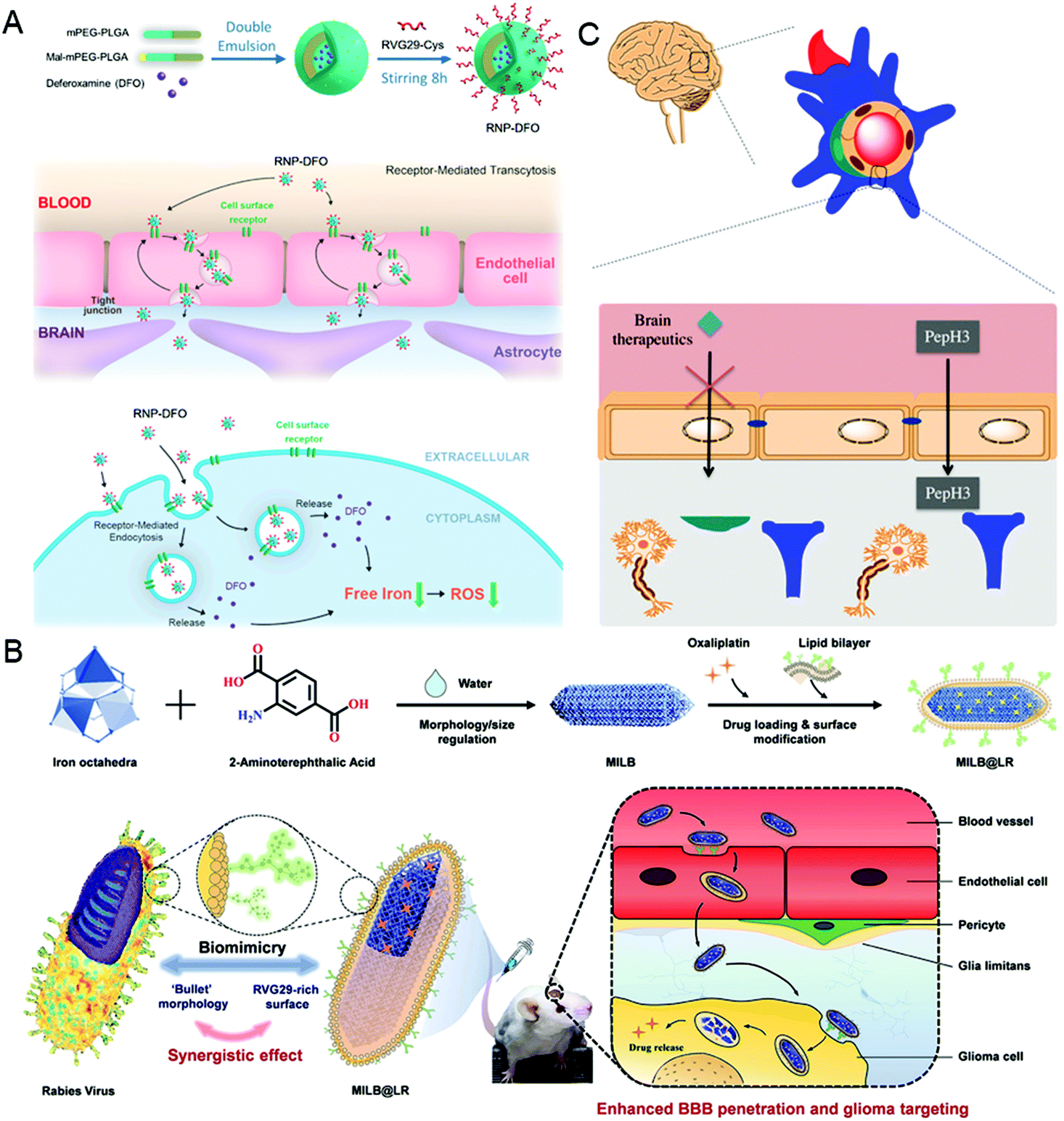 | ||
| Fig. 3 Representative diagrams of VINs for endothelial barrier breaking. (A) Schematic diagram of the RNP–DFO NPs for enhanced BBB penetration. Reproduced from ref. 63 with permission from American Chemical Society 2018. (B) Schematic diagram of MILB@LR for crossing the BBB and glioma treatment. Reproduced from ref. 64 with permission from American Chemical Society 2018. (C) Schematic diagram of PepH3 peptides translocating through the BBB. Reproduced from ref. 66 with permission from Wiley 2020. | ||
4. Enhanced cellular uptake
Viral infections mainly depend on the binding ability of viruses with the cell membranes.67 In this process, the surface morphology and the presence of polyvalent ligands of viruses are critical to them for overcoming a series of membrane barriers.8 This section focuses on the implications of enhanced cellular uptake of virus-inspired nanosystems (VINs) for improving drug delivery efficiency.4.1. Polyvalent ligands
A virus infects a cell by the recognition and attachment with the cell membrane receptor.68,69 The presence of capsid protein surface ligands is one of the main factors that determines the success of cell entry. Therefore, imitating the polyvalent ligand on the surface of the virus can greatly improve the cellular uptake efficiency of the drug carriers. It is widely studied that the HBV induces liver damage by recognizing the viral envelope protein with heparan sulfate proteoglycan on human liver cells.70 Further study showed that large proteins, especially the N-terminal myristoylated pre-S1(2–47) domain, were the key factor promoting HBV cellular targeting and entry.71 Inspired by HBV, a liposome that showed high specificity for human hepatocytes was synthesized by modifying the HBV envelope L protein on nanocarriers.72 The virus-inspired liposome modified with targeting peptides was used to deliver chemotherapeutic drugs to specifically target xenograft mice and achieve outstanding efficacy for inhibiting tumor growth. Similarly, by mimicking the envelope of the virus, a polyelectrolyte complex composed of chitosan-modified L-phe derivative copolymer and insulin fusion protein was designed.73 The structure improved the absorption and bioavailability of oral insulin through the mucous layer and intestinal epithelial penetration.In order to identify cells more accurately, Figueroa et al. constructed a nanocarrier composed of PEG polylactic acid (PEG–PLA), hydrophobic polylactic-co-glycolic acid (PLGA), and angiotensin I (Ang-I) by simulating the recognition mechanism of cells by the influenza A virus (Fig. 4A).74 The binding process is divided into two stages. In the first stage, the binding of Ang-I and angiotensin-converting enzyme (ACE) produces the secondary ligand Ang-II. In the second stage, Ang-II can specifically bind to another cell membrane receptor.7 The prepared nanocarriers cannot recognize the off-target cells that only carried the primary or secondary ligands, thus greatly improving the recognition accuracy of the target cells. After further optimization of the original VINs, the nanocarriers have a more accurate cell targeting ability and can recognize cells by three consecutive checks compared with nanocarriers lacking one or both receptors.75 However, the extracellular enzyme ligands are required in this recognition process, which has certain limitations for cells lacking the corresponding enzymes. To overcome this barrier, adenovirus (AD)-mimetic VINs were developed for enhancing the specific recognition of cells.76 In short, the Ang-II type-1 receptor (AT1r) blocker EXP3174 and the αVβ3 integrin agonist cRGDfK were attached to block-copolymers of varying lengths and then modified on the surface of NPs. When they entered the body, the VINs attached to the cell membrane through the specific binding of EXP3174 with AT1r on the cell membrane. Then the binding of cRGDfK with αVβ3 initiated the clathrin-mediated endocytosis. These studies not only provide valuable strategies for increasing targeting ability and reducing the toxicity of nanocarriers, but also put forward new insights into the development of drug delivery systems.
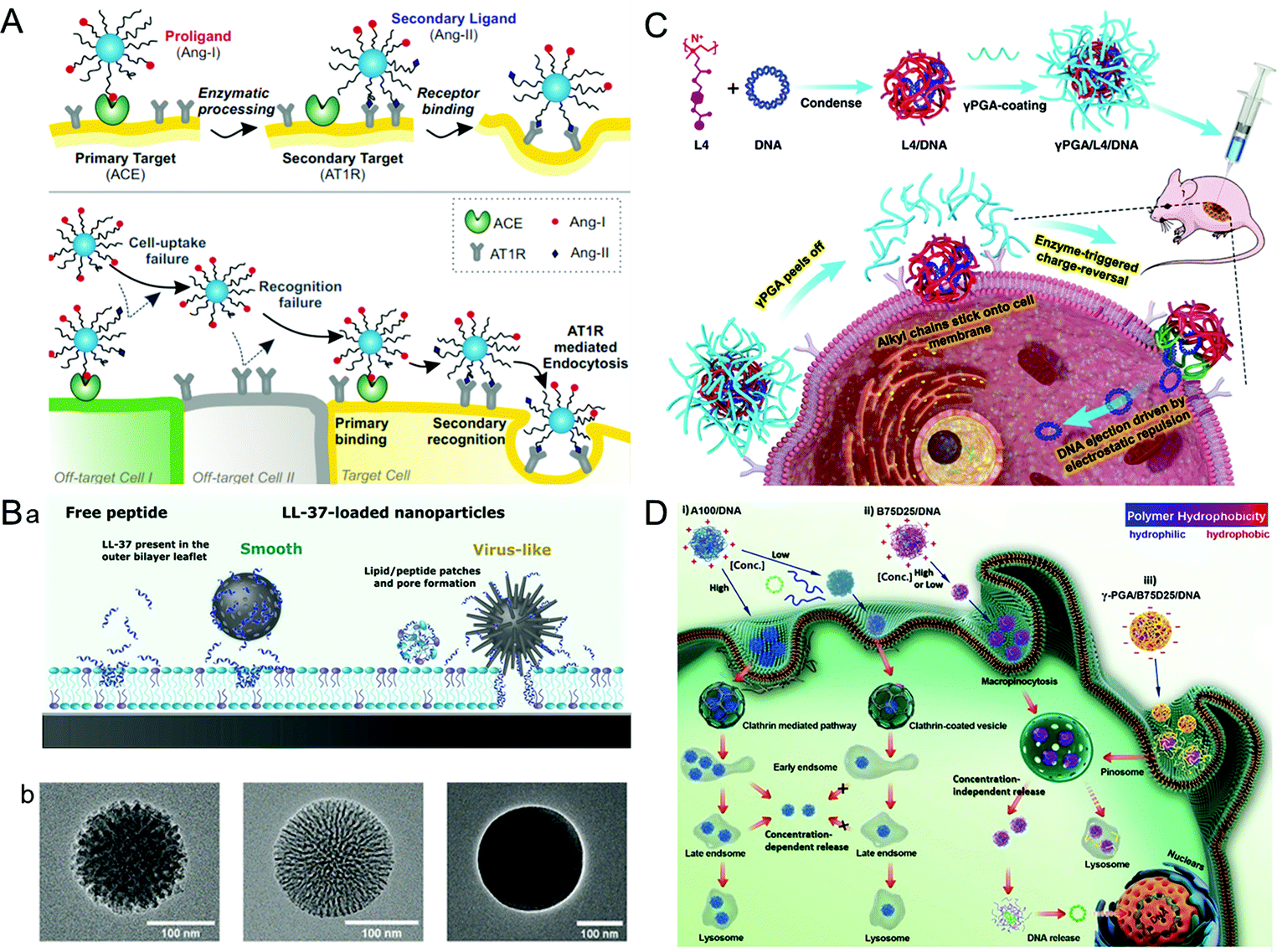 | ||
| Fig. 4 Representative diagrams of VINs for enhanced cellular uptake. (A) Schematic illustration of the concept of multivalent interactive target-cell identification by virus-mimetic NPs. Reproduced from ref. 74 with permission from PNAS 2019. (B) Schematic diagrams of the MSNs for enhancing cellular uptake. (a) Schematic illustration of the cellular uptake effects of virus-like and smooth MSNs loaded with LL-37, as well as with free LL-37. (b) TEM images of virus-like mesoporous (left), smooth mesoporous (middle), and smooth nonporous (right) NPs. Reproduced from ref. 83 with permission from American Chemical Society 2021. (C) Schematic illustration of the construction of γPGA/L4/DNA for gene transfection. Reproduced from ref. 88 with permission from Elsevier 2021. (D) Schematic illustration of gene transfection pathways of different polymers/DNA NPs. Reproduced from ref. 89 with permission from Wiley 2021. | ||
4.2. Surface morphology
The cellular uptake efficiency of drug carriers can be increased by simulating the surface morphology (e.g., rough surface and spike) of viruses, such as herpes simplex virus and HIV.77–79 For example, the modification of polyethylenimine (PEI) on the surface of the NPs can increase their surface roughness. Compared with the smooth NPs, the rough NPs exhibited better-combining ability, which greatly improved the cellular uptake efficiency.80 In another study, an organosilica nanosystem with a low Young's modulus (36 MPa) was constructed by imitating the rough surface of the virus.81 Ultrasmall SiO2 NPs were closely attached on the surface of larger mesoporous organic SiO2 NPs to explore the impact of the surface roughness and softness on cell internalization and tumor infiltration. Surprisingly, the VINs showed an 18.49-fold increase in tumor penetration compared with the hard mesoporous organosilica nanospheres. Similarly, taking advantage of viral topology and carrier-free nanosystems, a nanosystem (PEI/DNA@DNPs) was constructed by loading hydrophobic doxorubicin onto electrostatically bound PEI/DNA (PD) polymer spheres.82 These nanosystems can be used in chemotherapy and gene therapy. In addition, the spikes of the virus are also confirmed to have a significant influence on its membrane penetration. For example, a spike-like MSN with zinc ligands on its surface showed a high affinity for phosphate-rich cell membranes, and the enhanced membrane fusion was conducive to the cellular uptake of nanocarriers (Fig. 4B).83 Therefore, the surface morphology of the virus is also worthy of reference for the design of drug delivery carriers.84,854.3. Pore-mediated membrane penetration
Some viruses, such as bacteriophages, adenoviruses (AD), etc., can attach to the cell membrane through pore-mediated membrane penetration and deliver the genome directly to the host cell.86,87 Inspired by this process, a stable gene delivery system γPGA/L4/DNA was designed and constructed using cationic polymer L4, DNA and polyacrylic acid (γPGA) (Fig. 4C).88 In this complex structure, L4 with an octyl side chain can penetrate the phospholipid bilayer stably, while the strong electrostatic mutual repulsion between γPGA and DNA enables the gene to be directly delivered into the cytoplasm for transfection. The VINs avoided the interference of gene transfection due to endocytosis and prevented DNA degradation from lysosomal phagocytosis. Nevertheless, it has been found that the transfection ability of the DNA–cationic polymer complex is highly concentration-dependent. At low concentrations, most of the complex particles will spontaneously disassemble and the rest could not cause the “proton sponge effect” of endosomes, which leads to a loss of transfection ability of VINs. To overcome this problem, Zhang et al. constructed VINs with tunable hydrophobicity by systematically studying the relationship between the hydrophobicity of cationic polymers and DNA transfection capacity (Fig. 4D).89 In this study, γ-PGA was stabilized by the hydrophobic polymer through electrostatic interactions. When in contact with the cell membrane, it fell off to release DNA for dose-independent transfection. This result indicated that the VINs could infect cells independently even at low concentrations, similar to the concentration-dependent transfection of viruses.5. Endosomal escape
Enveloped viruses release their cargo by fusing with the endosomal membrane.90,91 Unlike enveloped viruses, non-enveloped viruses have two mechanisms by which they realize endosomal escape: the carpet-like mechanism resulting from membrane destruction and barrel-stave mechanism for the formation of transmembrane pores.67,92,93 In this section, virus-mimetic endosomal escape strategies for cargo release are discussed.5.1. Membrane fusion mediated endosomal escape
A number of experiments indicate that fusion proteins mediate the membrane fusion process.94 The fusion proteins undergo conformational changes to insert in the endosome with a low pH microenvironment and subsequently fuse with the exposed hydrophobic residues into the lipid bilayer, resulting in cytoplasmic transport of the genome. Inspired by the encapsulated camouflage process that most enveloped viruses utilize the phospholipids of host cells, virus-inspired nanosystems (VINs) embedded with functional polypeptides have been reported.95 In this study, the viral antigen gene was cloned into lentiviral vectors, then transfected into eukaryotic cells to express the viral antigen. Then the viral antigen was delivered to the cell surface to produce a mass of cell membrane vesicles. The results indicated that viral envelope glycoproteins could be added to the surface of liposomes to mimic the properties and conformational epitopes of natural viruses. Similarly, enlightened by the escape process of the influenza virus, a virus-like nucleic acid nanogel was constructed for glioblastoma treatment (Fig. 5A).96 In this study, the influenza virus HA2 peptide in VINs was able to fuse with the lysosomal membrane to achieve endosomal escape of VINs. In addition, the pH-sensitive fusogenic peptide can also achieve a similar result apart from endosome membrane fusion by mimicking the entire envelope camouflage strategy of the virus. For example, Chol–GALA (a cholesterylated pH-sensitive fusogenic peptide) and PEG–GALA can be used for compound modifications to enhance endosomal escape.97,98 To this end, Sasaki et al. designed a multifunctional envelope-type nanosystem Tf–MEND by encapsulating the concentrated plasmid DNA into envelope-like nanosystems containing PEG–Tf, Chol–GALA, and PEG–GALA, which can induce the interaction between liposomes and endosomes.99 In the artificial viral nanosystems, the structure of GALA changes with the decrease of the pH value of the environment, resulting in increased hydrophobicity and thus enhanced interaction with the lipid membrane. Besides, Chol–GALA and PEG–GALA can also interact with the capsule and penetrate the endosomal membrane depending on topological control, thereby synergically destabilizing the membrane structure and inducing membrane fusion.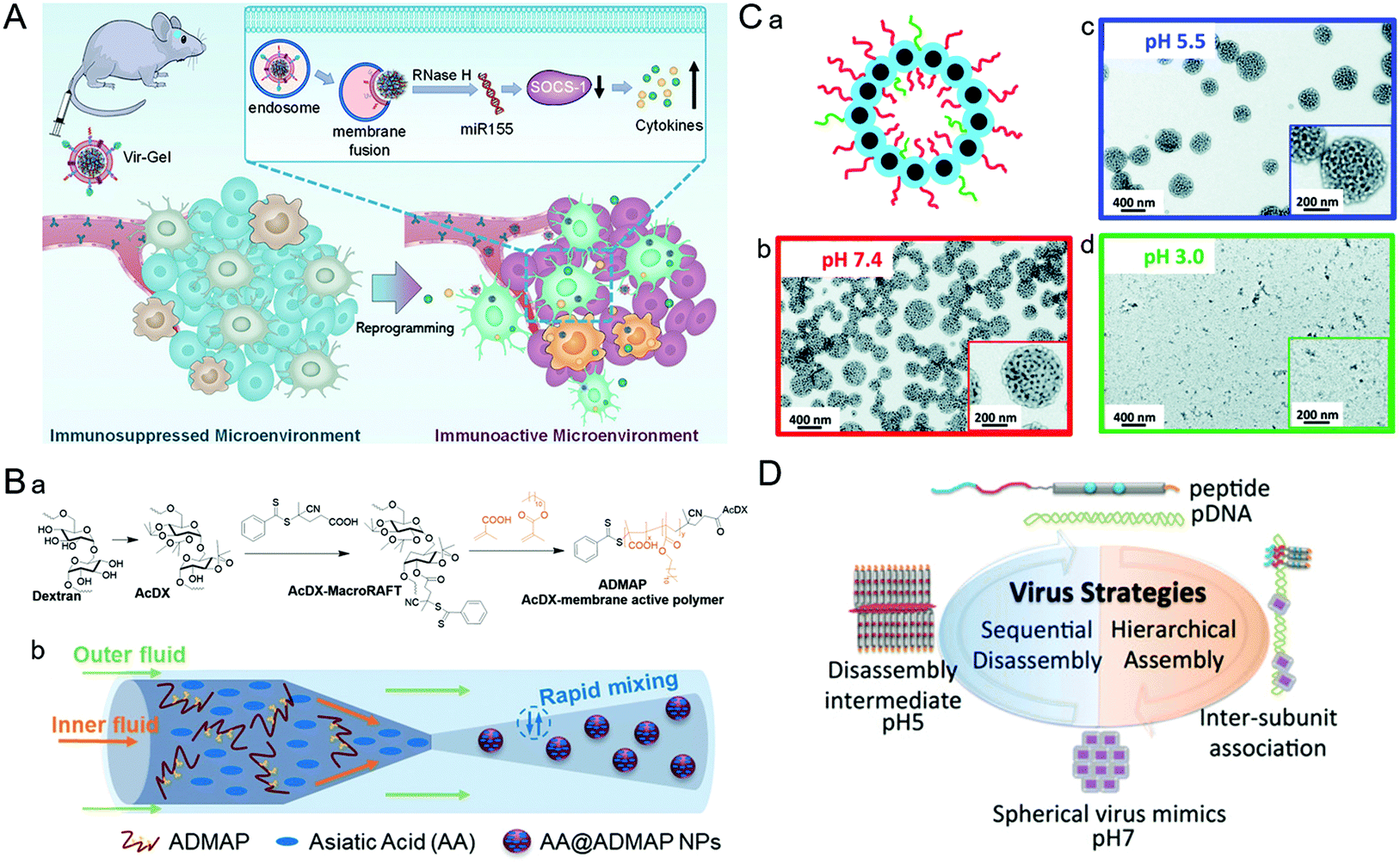 | ||
| Fig. 5 Representative diagrams of VINs for endosomal escape. (A) Schematic illustration of virus-like nanogel for glioblastoma treatment. Reproduced from ref. 96 with permission from Wiley 2021. (B) Diagrams of the synthesis route to AA@ADMAP NPs for delivering the antitumor drug. (a) The synthesis route of ADMAP for the synthesis of AA@ADMAP NPs. (b) The synthesis method of AA@ADMAP NPs. Reproduced from ref. 101 with permission from Wiley 2019. (C) Diagrams of the pH-responsive VINs for triple-negative breast cancer treatment. (a) Schematic diagram of the structure of VINs. (b–d) TEM images show the process of disassembly of VINs from pH 7.4 to 3.0. Reproduced from ref. 102 with permission from Royal Society of Chemistry 2019. (D) Schematic illustration of mimicking the sequential assembly and disassembly of a spherical virus for developing gene delivery vectors. Reproduced from ref. 110 with permission from Wiley 2020. | ||
5.2. Membrane destruction mediated endosomal escape
During the process of adenovirus (AD) escaping from the endosome, its capsid components will undergo conformational transformations with the changes of the cellular signal, causing the exposure of membrane–lytic protein VI to promote the escape from the endosome. Inspired by this, based on AD capsid pentamyl protein, a pH-sensitive bucket structure of pentameric units could be constructed artificially.100 In this study, the pentameric units disrupted the endosomal membrane by protonation and triggered its decomposition, thus facilitating the delivery of intracellular cargos. Similarly, a polymeric NP (ADMAP) mimicking the virus capsid protein was constructed for delivering antitumor drugs (Fig. 5B).101 Poly(lauryl methacrylate-co-methacrylic acid), a polymer molecule with endosomal membrane destruction function was modified on the surface of ADMAP NPs by nanoprecipitation. As pH decreases, ADMAP NPs can escape the endosome and release the drug through pH-dependent membrane destruction. In another study, inspired by the effective escape process of the dengue virus using a pH-sensitive peptide, Mable et al. synthesized dengue virus simulated NPs using biomimetic technology for triple-negative breast cancer treatment (Fig. 5C).102 The strong pH responsiveness of VINs in an acidic endosomal environment resulted in endosomal rupture and cargo release into the cytoplasm. In addition, the protonation of amino groups forced the endosome to break under high osmotic pressure, allowing the payload to escape from the endosome due to the proton sponge effect.103As an amino-rich polycation material, PEI has been known as the gold standard for proton sponge-based delivery of nucleic acids. Recently, a copolymer drug carrier using branched PEI for efficient siRNA delivery has been designed.104 In response to the acidic microenvironment in the cell, PEI cleavage resulted in endosomal membrane destruction. Meanwhile, the practical promotion of endosomal escape by disassembly of NPs was achieved. This approach provides a new potential approach to address the bottleneck of intracellular siRNA delivery. Similarly, poly(amidoamine) (PAMAM) also has a high acid-response ability to trigger endosomal escape. For example, Weiss et al. designed multifunctional MSNs modified with a PAMAM dendritic surface structure.105 Thanks to the intrinsic expansion of PAMAM during the acidification of the endosome, MSNs can eventually lead to endosomal membrane rupture and drug release through a disulfide bond mediated mechanism. These studies demonstrated that simulating the viral endosomal escape process could be helpful for boosting drug delivery.
Nevertheless, some polymers cannot destroy the endosomal membrane, and an alternative strategy is to consider adding membrane destroying polypeptides into the delivery system. For example, Cheng et al. analyzed the assembly behavior of the synthesized virus–inspired polymer (VIPER) for endosome release at pH 7.4 and pH 5.7.106 A cationic compound mellitoxin that has robust membrane-destroying ability was used in this study.107,108 The result indicated that acidic environments triggered the hydrophilic transition of poly(2-diisopropylaminoethyl methacrylate)-co-poly(pyridyl disulfide ethyl methacrylate) blocks which exposed melittin, thereby disrupting the endosomal membrane. Thus, endosomal escape in this study is facilitated by the enhancement of the polycation block and pH-sensitive block.
The interactions of protein subunits with a low aspect ratio (width over length) of spherical viruses (e.g., HIV) lead to hierarchical self-assembly of the genome. By imitating the emblematic feature, a functional spherical artificial virus was constructed for endosomal escape.109 In the presence of DNA, the interactions between synthetic polypeptides lead to the formation of the shell-like viral structure wrapping the DNA inside. The binding peptides progressively cleave and release the genome in response to pH regulation. Inspired by this, Ni et al. assembled 22 well-designed amino acid peptides into homogeneous nanodiscs (≈7 × 7 × 4 nm) by simulating the formation process of viruses (Fig. 5D).110 Briefly, in this structure, the peptide chains along the DNA backbone drive the peptides to assemble into nanodiscs. Meanwhile, hydrophobic interactions facilitate the binding between the nanodiscs, allowing DNA to be encapsulated in multilayered nanostructures. At pH 5.0, the diminishing protonation of the two histidines destroys the interaction between the nanodiscs and the peptides, thereby releasing the cargo.
6. Nuclear targeting
A practical vehicle for realizing nuclear drug delivery should be able to protect drugs from degradation by the cytoplasmic enzymes and allow specific transport through the nuclear pore complexes (NPCs).111,112 Among various nuclear delivery strategies, the so-called “Trojan horse” strategy inspired by viral infection is a representative example of efficient cytoplasmic delivery and nuclear targeting.113 This section describes several virus-inspired nuclear targeting strategies for drug delivery.After escaping from the endosome, some drugs/carriers still need to pass through the cytoplasm before reaching the nucleus.114,115 Inspired by the assembly kinetics of TMVs, baculovirus-like nanosystems with the TMV capsid protein were developed by a polypeptide-based self-assembly strategy (Fig. 6A).116 By locking the gene in artificial virus proteins, the nanosystems protected the DNA inside the carrier from being broken down by enzymes. Surprisingly, this study showed that the synergistic self-assembly process was highly similar to viruses. In another study, inspired by the stable structure of parapoxvirus, Ni et al. constructed a nanococoon system by co-assembly of 16 amino acid peptides with plasmid DNA.117 It appeared as a virus-like ellipsoid (65 × 47 nm) with repeated fringes approximately 4 nm wide. The co-assembly process used DNA as a template to combine with the peptide chain through electrostatic interaction. Meanwhile, the double-layer β-folding and its lateral association stabilize the peptide capsid and DNA in it. As a result, the nanococoon protected peptides and DNA from enzyme digestion. To achieve the goal of passing through NPCs, nuclear localization signals (NLS) are widely used for effective nuclear localization.118,119 By mimicking HIV and simian virus 40 (SV40), Kong et al. synthesized bioactive peptide-based nanocarriers pVLPs for delivering genetic cargo (Fig. 6B).120 The nanocarriers were co-assembled with DNA by electrostatic interaction between two kinds of peptides: cell-penetrating peptides with membrane penetration ability and viral peptides with nuclear targeting capability. The results showed that the NLS (PKKKRKV) of SV40 could accurately deliver the cargo to the nucleus. Similarly, the virus-mimetic light-induced nanosystems for tumor therapy were reported in another study (Fig. 6C).121 The nanocarriers were composed of PEI/DNA (PD) and a modular envelope (CCPNR). The study indicated that the PD@CCPNR system could effectively achieve nuclear targeting. Besides, a citraconic anhydride-modified nuclear signal-functionalized module further enhanced the gene delivery efficiency and improved the combined efficacy of photodynamic therapy. In short, compared with other conventional nanocarriers, the virus-inspired nanosystems (VINs) with viral polypeptides have the ability to protect the cargo from degradation and realize excellent nuclear targeting delivery. In addition, some viruses, such as AD and SV40, can recruit molecular motors near the nucleus by hijacking cell microtubule transport systems.122 Recently, inspired by this process, VINs containing modular recombinant T-Rp3 proteins have been constructed to deliver nucleic acid cargoes, including plasmid DNA and SiRNA (Fig. 6D).123 After the successful endosomal escape of VINs, T-Rp3 protein interacted with intracellular dynein and approached the NPCs.
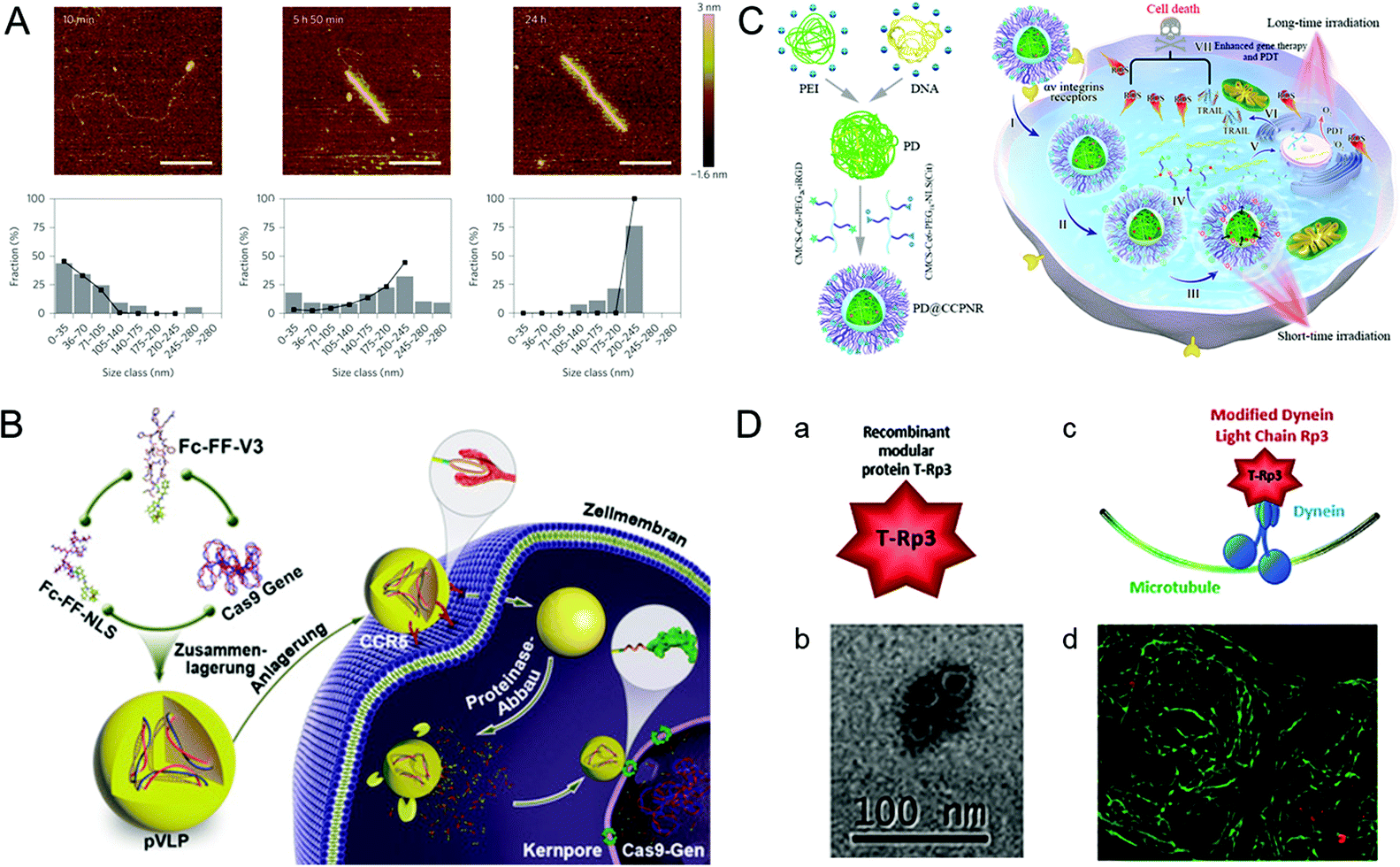 | ||
| Fig. 6 Representative diagrams of VINs for nuclear targeting. (A) Representative AFM images of VINs at different incubation times. The VINs can be used as a template for drug delivery. Reproduced from ref. 116 with permission from Springer Nature 2014. (B) Schematic illustration of pVLPs using Fc–FF–NLS to locate cell nucleus for gene transfer. Reproduced from ref. 120 with permission from Wiley 2018. (C) Schematic illustration of PD@CCPNR using PEG–NLS to locate cell nucleus for plasmid delivery. Reproduced from ref. 121 with permission from American Chemical Society 2020. (D) Schematic diagram of the VINs for nucleic acid cargo delivery. (a) and (b) The recombinant modular protein T-Rp3 and TEM image of its self-assembly. (c) and (d) The VINs (red) hijacking microtubule (green) transport system and its laser scanning confocal microscopy image. Reproduced from ref. 123 with permission from Elsevier 2018. | ||
7. Conclusions and perspective
For several years, by simulating the subtle structure of viruses, a number of nanosystems with excellent biocompatibility and high delivery efficiency have been designed and constructed. In this review, we outlined and summarized the latest progress of virus-inspired nanosystems (VINs) from the perspective of various biological barriers that need to be overcome for successful targeted drug delivery. Undoubtedly, these unique types of nanosystems provide intelligent strategies for targeted drug delivery, which broaden the application of nanotechnology for biomedicine.Although the advances in VINs have great potential in the study of targeted drug delivery, several challenges must be overcome before their further translation into clinical applications. The most crucial problem is biosafety. The prolongation of the circulation time of VINs contributes to the accumulation of carriers at the desired site, increasing the effectiveness of the drugs, but it also results in some other unsatisfactory consequences.124 On the one hand, non-specific drug distribution due to prolonged half-life will cause new toxic effects or exacerbate adverse reactions. On the other hand, the increase in the metabolism of medicines by the liver and the kidneys might be higher than the drug accumulation in the target tissues. To solve this problem, the pharmacokinetics of drug carriers in vivo should be reconsidered comprehensively. A better strategy is to increase the blood circulation of the drug while increasing its selective activation properties.125 Moreover, the immune response caused by viruses is a double-edged sword.126 The virus-induced immune response is somehow helpful for killing cancer cells, but an excessive immune response can also cause great harm to the body. Under a good balance, VINs that combine viruses with immune cells could be developed for synergistic cancer therapy.
Further improvement of the delivery efficiency of VINs could be another significant challenge. The microenvironment inside and outside the cell membrane throughout the drug delivery process poses a dilemma for carrier design. For example, in most cases, the surface charge must be shielded to prolong circulation time. For another, the disassembly activity must be restrained to stabilize the drug in the extracellular environment. To overcome this difficulty, a more comprehensive understanding of simulating the virus infection process to release the genome or drugs in a stable manner is urgently needed. For instance, one good strategy is to develop spatiotemporal controllable VINs by integrating multiple viral structures with the help of external or endogenous stimuli such as light, ultrasound, enzymes, etc.
Due to the variety of direct biological responses that underlie virus entry into cells, cargo transport, and viral DNA replication, there are particular challenges for constructing lifelike “viruses” that have universal applications. To extend the “pertinence” of viral infection to the “universality” of VIN development, testing different viral components under the basic principles is necessary. It is noteworthy that the construction of a peptide platform may allow the viroid components to display much functional diversity, extending the virus's specific infection ability. In addition, compared with the research on VINs for enhancing cellular uptake, there are few studies on nuclear targeting. In view of the importance of gene therapy, it is worth studying how to develop more nuclear targeting agents by using the nuclear targeting mechanisms of viruses.
Although VINs still face complex problems in the translation to clinical applications, we believe that, by learning from the viral infection process, utilizing the inherent merits of the viral structure, and the rational design of nanocarriers based on advances of nanotechnology, these difficulties could be overcome eventually. We predict more successful virus-inspired attempts for drug delivery shortly, which may open a bright path and expand the application of VINs in biomedical fields.
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AD | Adenovirus |
| AMT | Adsorptive-mediated transcytosis |
| Ang-I | Angiotensin I |
| AT1r | Ang-II type-1 receptor |
| ATMV | Artificial tobacco mosaic virus |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| MSN | Mesoporous silica nanoparticles |
| NLS | Nuclear localization signals |
| NPCs | Nuclear pore complexes |
| NPs | Nanoparticles |
| PEI | Polyethylenimine |
| PAMAM | Poly(amidoamine) |
| PLA | Polylactic acid |
| PLGA | Polylactic-co-glycolic acid |
| RMT | Receptor-mediated transport |
| RABV | Rabies virus |
| TMV | Tobacco mosaic virus |
| VINs | Virus-inspired nanosystems |
| VIPER | Virus-inspired polymer |
| SV40 | Simian virus 40 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (grant no. 82001959, 31630027 and 32030060), the NSFC international collaboration key project (grant no. 51861135103), and the NSFC-German Research Foundation (DFG) project (grant no. 31761133013). The authors also appreciate the support from the National Key Research & Development Program of China (grant no. 2018YFE0117800), the Fundamental Research Funds for the Central Universities (20720210102), the Key Laboratory of Biomedical Effects of Nanomaterials and Nanosafety, CAS (no. NSKF202003), the Fujian Provincial Key Laboratory of Innovative Drug Target Research (no. FJ-YW-2021KF04) and the Novel Coronavirus Prevention Emergency Scientific Research Project, Xiamen University (20720200036). S. H. is grateful for financial support from the Nanqiang Outstanding Young Talents Program from Xiamen University.References
- M. S. Draz and H. Shafiee, Theranostics, 2018, 8, 1985–2017 CrossRef CAS PubMed
.
- M. Borsa and J. M. Mazet, Nat. Rev. Immunol., 2020, 20, 592–592 CrossRef CAS PubMed
- A. Alcami and U. H. Koszinowski, Trends Microbiol., 2000, 8, 410–418 CrossRef CAS
- S. M. Figueroa, D. Fleischmann and A. Goepferich, J. Controlled Release, 2021, 329, 552–569 CrossRef PubMed
- K. Ezzat, M. Pernemalm, S. Palsson, T. C. Roberts, P. Jarver, A. Dondalska, B. Bestas, M. J. Sobkowiak, B. Levanen, M. Skold, E. A. Thompson, O. Saher, O. K. Kari, T. Lajunen, E. S. Ekstrom, C. Nilsson, Y. Ishchenko, T. Malm, M. J. A. Wood, U. F. Power, S. Masich, A. Linden, J. K. Sandberg, J. Lehtio, A. L. Spetz and S. EL Andaloussi, Nat. Commun., 2019, 10, 1–16 CrossRef CAS
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed
- D. Fleischmann, S. M. Figueroa, S. Beck, K. Abstiens, R. Witzgall, F. Schweda, P. Tauber and A. Goepferich, ACS Appl. Mater. Interfaces, 2020, 12, 34689–34702 CrossRef CAS PubMed
- D. Mudhakir and H. Harashima, AAPS J., 2009, 11, 65–77 CrossRef PubMed
- N. F. Steinmetz, EMBO Rep., 2019, 20, e48806 CrossRef
- J. Ng, S. Barral, C. D. Barrigon, G. Lignani, F. A. Erdem, R. Wallings, R. Privolizzi, G. Rossignoli, H. Alrashidi, S. Heasman, E. Meyer, A. Ngoh, S. Pope, R. Karda, D. Perocheau, J. Baruteau, N. Suff, J. A. Diaz, S. Schorge, J. Vowles, L. R. Marshall, S. A. Cowley, S. Sucic, M. Freissmuth, J. R. Counsell, R. Wade-Martins, S. J. R. Heales, A. A. Rahim, M. Bencze, S. N. Waddington and M. A. Kurian, Sci. Transl. Med., 2021, 13, eaaw1564 CrossRef CAS PubMed
- C. Yu, L. Li, P. Hu, Y. Yang, W. Wei, X. Deng, L. Wang, F. R. Tay and J. Z. Ma, Adv. Sci., 2021, 8, 2100540 CrossRef CAS PubMed
- J. T. Bulcha, Y. Wang, H. Ma, P. W. L. Tai and G. P. Gao, Signal Transduction Targeted Ther., 2021, 6, 1–24 CrossRef PubMed
- C. J. Stephens, E. J. Lauron, E. Kashentseva, Z. H. Lu, W. M. Yokoyama and D. T. Curiel, J. Controlled Release, 2019, 298, 128–141 CrossRef CAS PubMed
- J. Zabner, L. A. Couture, R. J. Gregory, S. M. Graham, A. E. Smith and M. J. Welsh, Cell, 1993, 75, 207–216 CrossRef CAS PubMed
- M. Schmid, P. Ernst, A. Honegger, M. Suomalainen, M. Zimmermann, L. Braun, S. Stauffer, C. Thom, B. Dreier, M. Eibauer, A. Kipar, V. Vogel, U. F. Greber, O. Medalia and A. Pluckthun, Nat. Commun., 2018, 9, 1–16 CrossRef PubMed
- G. Zuber, E. Dauty, M. Nothisen, P. Belguise and J. P. Behr, Adv. Drug Delivery Rev., 2001, 52, 245–253 CrossRef CAS PubMed
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed
- K. Cheng, T. Du, Y. Li, Y. Qi, H. Min, Y. Wang, Q. Zhang, C. Wang, Y. Zhou, L. Li, S. Ye, X. Zhou, S. Bi, J. Yang and L. Ren, ACS Appl. Mater. Interfaces, 2020, 12, 53682–53690 CrossRef CAS
- W. Shan, H. Zheng, G. Fu, C. Liu, Z. Li, Y. Ye, J. Zhao, D. Xu, L. Sun, X. Wang, X. L. Chen, S. Bi, L. Ren and G. Fu, Nano Lett., 2019, 19, 1719–1727 CrossRef CAS PubMed
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS
- R. H. Chen, S. S. Huang, T. T. Lin, H. S. Ma, W. J. Shan, F. Duan, J. Lv, J. D. Zhang, L. Ren and L. M. Nie, Nat. Nanotechnol., 2021, 16, 455–465 CrossRef CAS
- S. Huo, N. Gong, Y. Jiang, F. Chen, H. Guo, Y. Gan, Z. Wang, A. Herrmann and X.-J. Liang, Sci. Adv., 2019, 5, eaaw6264 CrossRef CAS PubMed
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Nat. Rev. Mater., 2021, 6, 351–370 CrossRef CAS
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021 DOI:10.1038/s41578-021-00358-0
- S. Huo, Y. Jiang, Z. Jiang, R. F. Landis, X.-J. Liang and V. M. Rotello, Nanoscale, 2018, 10, 7382–7386 RSC
- W. J. Shan, R. H. Chen, Q. Zhang, J. Zhao, B. B. Chen, X. Zhou, S. F. Ye, S. L. Bi, L. M. Nie and L. Ren, Adv. Mater., 2018, 30, 1707567 CrossRef PubMed
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed
- L. He, A. Chaudhary, X. Lin, C. Sou, T. Alkutkar, S. Kumar, T. Ngo, E. Kosviner, G. Ozorowski, R. L. Stanfield, A. B. Ward, I. A. Wilson and J. Zhu, Nat. Commun., 2021, 12, 2633 CrossRef CAS PubMed
- L. L. He, N. de Val, C. D. Morris, N. Vora, T. C. Thinnes, L. Kong, P. Azadnia, D. Sok, B. Zhou, D. R. Burton, I. A. Wilson, D. Nemazee, A. B. Ward and J. Zhu, Nat. Commun., 2016, 7, 12041 CrossRef CAS
- I. Samandoulgou, R. Hammami, R. M. Rayas, I. Fliss and J. Jean, Appl. Environ. Microbiol., 2015, 81, 7680–7686 CrossRef CAS
- I. Lostale-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef
- S. Bhaskar and S. Lim, NPG Asia Mater., 2017, 9, e371 CrossRef PubMed
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed
- R. Ni, J. L. Zhou, N. Hossain and Y. Chau, Adv. Drug Delivery Rev., 2016, 106, 3–26 CrossRef CAS PubMed
- Y. H. Chung, H. Cai and N. F. Steinmetz, Adv. Drug Delivery Rev., 2020, 156, 214–235 CrossRef CAS PubMed
- N. Ferrer-Miralles, E. Rodriguez-Carmona, J. L. Corchero, E. Garcia-Fruitos, E. Vazquez and A. Villaverde, Crit. Rev. Biotechnol., 2015, 35, 209–221 CrossRef CAS PubMed
- Y. Lu, W. Chan, B. Y. Ko, C. C. VanLang and J. R. Swartz, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12360–12365 CrossRef CAS PubMed
- M. J. Rohovie, M. Nagasawa and J. R. Swartz, Bioeng. Transl. Med., 2017, 2, 43–57 CrossRef CAS PubMed
- Z. Y. Fan, P. Zhu, Y. C. Zhu, K. Wu, C. Y. Li and H. Cheng, Curr. Opin. Biotechnol., 2020, 66, 131–139 CrossRef CAS PubMed
- B. D. Hill, A. Zak, E. Khera and F. Wen, Curr. Protein Pept. Sci., 2018, 19, 112–127 CAS
- L. E. Euliss, J. A. DuPont, S. Gratton and J. M. DeSimone, Chem. Soc. Rev., 2006, 35, 1095–1104 RSC
- S. B. Ruan, Y. Zhou, X. G. Jiang and H. L. Gao, Adv. Sci., 2021, 8, 2004025 CrossRef CAS PubMed
- J. Lin, L. Miao, G. Zhong, C.-H. Lin, R. Dargazangy and A. Alexander-Katz, Commun. Biol., 2020, 3, 205 CrossRef CAS PubMed
- A. Sen Gupta, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 255–270 Search PubMed
- K. L. Lee, S. Shukla, M. Z. Wu, N. R. Ayat, C. E. El Sanadi, A. M. Wen, J. F. Edelbrock, J. K. Pokorski, U. Commandeur, G. R. Dubyak and N. F. Steinmetz, Acta Biomater., 2015, 19, 166–179 CrossRef CAS
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed
- H. Wu, D. Zhong, Z. J. Zhang, Y. C. Li, X. Zhang, Y. K. Li, Z. Z. Zhang, X. H. Xu, J. Yang and Z. W. Gu, Adv. Mater., 2020, 32, 1904958 CrossRef CAS PubMed
- A. S. Pitek, A. M. Wen, S. Shukla and N. F. Steinmetz, Small, 2016, 12, 1758–1769 CrossRef CAS PubMed
- X. S. Liu, Y. J. Chen, H. Li, N. Huang, Q. Jin, K. F. Ren and J. Ji, ACS Nano, 2013, 7, 6244–6257 CrossRef CAS
- K. Abstiens, S. M. Figueroa, M. Gregoritza and A. M. Goepferich, Soft Matter, 2019, 15, 709–720 RSC
- Y. Zhang, M. Xiong, X. Ni, J. Wang, H. Rong, Y. Su, S. Yu, I. S. Mohammad, S. S. Y. Leung and H. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 18077–18088 CrossRef CAS
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787–796 CrossRef CAS PubMed
- M. A. Bruckman, A. E. Czapar, A. VanMeter, L. N. Randolph and N. F. Steinmetz, J. Controlled Release, 2016, 231, 103–113 CrossRef CAS PubMed
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed
- L. Tu, Z. Luo, Y.-L. Wu, S. Huo and X.-J. Liang, Cancer Biol. Med., 2021, 18, 372–387 Search PubMed
- W. A. Banks, Nat. Rev. Drug Discovery, 2016, 15, 275–292 CrossRef CAS PubMed
- C. D. Arvanitis, G. B. Ferraro and R. K. Jain, Nat. Rev. Cancer, 2020, 20, 26–41 CrossRef CAS PubMed
- G. C. Terstappen, A. H. Meyer, R. D. Bell and W. Zhang, Nat. Rev. Drug Discovery, 2021, 20, 362–383 CrossRef CAS PubMed
- M. Zheng, W. Tao, Y. Zou, O. C. Farokhzad and B. Shi, Trends Biotechnol., 2018, 36, 562–575 CrossRef CAS PubMed
- T. L. Lentz, T. G. Burrage, A. L. Smith, J. Crick and G. H. Tignor, Science, 1982, 215, 182–184 CrossRef CAS PubMed
- C. R. Fisher, D. G. Streicker and M. J. Schnell, Nat. Rev. Microbiol., 2018, 16, 241–255 CrossRef CAS PubMed
- L. H. You, J. Wang, T. Q. Liu, Y. L. Zhang, X. X. Han, T. Wang, S. S. Guo, T. Y. Dong, J. C. Xu, G. J. Anderson, Q. Liu, Y. Z. Chang, X. Lou and G. J. Nie, ACS Nano, 2018, 12, 4123–4139 CrossRef CAS PubMed
- C. Q. Qiao, R. L. Zhang, Y. D. Wang, Q. Jia, X. F. Wang, Z. Yang, T. F. Xue, R. C. Ji, X. F. Cui and Z. L. Wang, Angew. Chem. Int. Edit., 2020, 59, 16982–16988 CrossRef CAS PubMed
- M. Khongkow, T. Yata, S. Boonrungsiman, U. R. Ruktanonchai, D. Graham and K. Namdeel, Sci. Rep., 2019, 9, 8278 CrossRef PubMed
- V. Neves, F. Aires-da-Silva, M. Morais, L. Gano, E. Ribeiro, A. Pinto, S. Aguiar, D. Gaspar, C. Fernandes, J. D. G. Correia and M. A. R. B. Castanho, ACS Chem. Biol., 2017, 12, 1257–1268 CrossRef CAS PubMed
- M. Marsh and A. Helenius, Cell, 2006, 124, 729–740 CrossRef CAS PubMed
- J. Grove and M. Marsh, J. Cell Biol., 2011, 195, 1071–1082 CrossRef CAS PubMed
- C. Iadecola, J. Anrather and H. Kamel, Cell, 2020, 183, 16–27 CrossRef CAS PubMed
- C. M. Leistner, S. Gruen-Bernhard and D. Glebe, Cell. Microbiol., 2008, 10, 122–133 CAS
- H. Yan, G. C. Zhong, G. W. Xu, W. H. He, Z. Y. Jing, Z. C. Gao, Y. Huang, Y. H. Qi, B. Peng, H. M. Wang, L. R. Fu, M. Song, P. Chen, W. Q. Gao, B. J. Ren, Y. Y. Sun, T. Cai, X. F. Feng, J. H. Sui and W. H. Li, eLife, 2012, 1, e0004 CrossRef PubMed
- Q. S. Liu, M. Somiya, M. Iijima, K. Tatematsu and S. Kuroda, Biomater. Sci., 2019, 7, 322–335 RSC
- C. Liu, H. Xu, Y. Sun, X. Zhang, H. B. Cheng and S. R. Mao, J. Pharm. Sci., 2019, 108, 3408–3415 CrossRef CAS PubMed
- S. M. Figueroa, A. Veser, K. Abstiens, D. Fleischmann, S. Beck and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9831–9836 CrossRef CAS PubMed
- S. M. Figueroa, D. Fleischmann, S. Beck, P. Tauber, R. Witzgall, F. Schweda and A. Goepferich, Adv. Sci., 2020, 7, 1903204 CrossRef PubMed
- D. Fleischmann, S. Maslanka Figueroa, S. Beck, K. Abstiens, R. Witzgall, F. Schweda, P. Tauber and A. Goepferich, ACS Appl. Mater. Interfaces, 2020, 12, 34689–34702 CrossRef CAS PubMed
- G. Simmons, J. D. Reeves, A. J. Rennekamp, S. M. Amberg, A. J. Piefer and P. Bates, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4240–4245 CrossRef CAS PubMed
- K. Grunewald, P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister and A. C. Steven, Science, 2003, 302, 1396–1398 CrossRef PubMed
- P. Zhu, J. Liu, J. Bess, E. Chertova, J. D. Lifson, H. Grise, G. A. Ofek, K. A. Taylor and K. H. Roux, Nature, 2006, 441, 847–852 CrossRef CAS PubMed
- Y. T. Niu, M. H. Yu, S. B. Hartono, J. Yang, H. Y. Xu, H. W. Zhang, J. Zhang, J. Zou, A. Dexter, W. Y. Gu and C. Z. Yu, Adv. Mater., 2013, 25, 6233–6237 CrossRef CAS PubMed
- J. Tao, K. Chen, X. D. Su, L. L. Ren, J. J. Zhang, L. Bao, H. Dong, G. M. Lu, Z. G. Teng and L. H. Wang, Biomater. Sci., 2020, 8, 2227–2233 RSC
- X. T. Sun, M. H. Li, Y. Yang, H. Z. Jia and W. G. Liu, Biomater. Sci., 2018, 6, 3300–3308 RSC
- S. M. Haffner, E. Parra-Ortiz, K. L. Browning, E. Jorgensen, M. W. A. Skoda, C. Montis, X. M. Li, D. Berti, D. Y. Zhao and M. Malmsten, ACS Nano, 2021, 15, 6787–6800 CrossRef CAS PubMed
- S. Liu, J. X. Yang, H. T. Jia, H. Zhou, J. T. Chen and T. Y. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 23630–23637 CrossRef CAS PubMed
- A. Parodi, R. Molinaro, M. Sushnitha, M. Evangelopoulos, J. O. Martinez, N. Arrighetti, C. Corbo and E. Tasciotti, Biomaterials, 2017, 147, 155–168 CrossRef CAS PubMed
- F. C. Kurschus and D. E. Jenne, Immunol. Rev., 2010, 235, 159–171 CrossRef CAS PubMed
- I. J. Molineux and D. Panja, Nat. Rev. Microbiol., 2013, 11, 194–204 CrossRef CAS PubMed
- G. Wang, S. Chen, N. Qiu, B. Wu, D. Zhu, Z. Zhou, Y. Piao, J. Tang and Y. Shen, Nano Today, 2021, 39, 101215 CrossRef CAS
- Z. Zhang, N. A. Qiu, S. L. Wu, X. Liu, Z. X. Zhou, J. B. Tang, Y. P. Liu, R. H. Zhou and Y. Q. Shen, Adv. Mater., 2021, 33, 2102219 CrossRef CAS PubMed
- J. Mercer, J. E. Lee, E. O. Saphire and S. A. Freeman, Cell, 2020, 182, 786–786 CrossRef CAS PubMed
- J. M. White and G. R. Whittaker, Traffic, 2016, 17, 593–614 CrossRef CAS PubMed
- C. M. Wiethoff, H. Wodrich, L. Gerace and G. R. Nemerow, J. Virol., 2005, 79, 1992–2000 CrossRef CAS PubMed
- F. J. M. Van Kuppeveld, A. S. De Jong, W. J. G. Melchers and P. H. G. M. Willems, Trends Microbiol., 2005, 13, 41–44 CrossRef CAS PubMed
- M. Benhaim and K. K. Lee, Cell, 2018, 174, 775–777 CrossRef CAS PubMed
- P. F. Zhang, Y. X. Chen, Y. Zeng, C. G. Shen, R. Li, Z. D. Guo, S. W. Li, Q. B. Zheng, C. C. Chu, Z. T. Wang, Z. Z. Zheng, R. Tian, S. X. Ge, X. Z. Zhang, N. S. Xia, G. Liu and X. Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6129–E6138 CrossRef CAS PubMed
- X. Gao, S. Li, F. Ding, X. Liu, Y. Wu, J. Li, J. Feng, X. Zhu and C. Zhang, Adv. Mater., 2021, 33, 2006116 CrossRef CAS PubMed
- T. Kakudo, S. Chaki, S. Futaki, I. Nakase, K. Akaji, T. Kawakami, K. Maruyama, H. Kamiya and H. Harashima, Biochemistry, 2004, 43, 5618–5628 CrossRef CAS PubMed
- M. Morishita, Y. Takahashi, M. Nishikawa, R. Ariizumi and Y. Takakura, Mol. Pharmaceutics, 2017, 14, 4079–4086 CrossRef CAS PubMed
- K. Sasaki, K. Kogure, S. Chaki, Y. Nakamura, R. Moriguchi, H. Hamada, R. Danev, K. Nagayama, S. Futaki and H. Harashima, Anal. Bioanal. Chem., 2008, 391, 2717–2727 CrossRef CAS PubMed
- F. Alonso-Valenteen, S. Pacheco, D. Srinivas, A. Rentsendorj, D. Chu, J. Lubow, J. Sims, T. X. Miao, S. Mikhael, J. Y. Hwang, R. Abrol and L. K. M. Kauwe, Nucleic Acids Res., 2019, 47, 11020–11043 CrossRef CAS PubMed
- S. Wannasarit, S. Q. Wang, P. Figueiredo, C. Trujillo, F. Eburnea, L. Simon-Gracia, A. Correia, Y. P. Ding, T. Teesalu, D. F. Liu, R. Wiwattanapatapee, H. A. Santos and W. Li, Adv. Funct. Mater., 2019, 29, 1905352 CrossRef CAS
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. Ustbas Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 RSC
- C. Gallops, J. Ziebarth and Y. Wang, Polym. Ther. Delivery, 2020, 1, 1–12 Search PubMed
- L. M. P. Vermeulen, T. Brans, S. K. Samal, P. Dubruel, J. Demeester, S. C. De Smedt, K. Remaut and K. Braeckmans, ACS Nano, 2018, 12, 2332–2345 CrossRef CAS PubMed
- V. Weiss, C. Argyo, A. A. Torrano, C. Strobel, S. A. Mackowiak, A. Schmidt, S. Datz, T. Gatzenmeier, I. Hilger, C. Brauchle and T. Bein, Microporous Mesoporous Mater., 2016, 227, 242–251 CrossRef CAS
- Y. L. Cheng, R. C. Yumul and S. H. Pun, Angew. Chem., Int. Ed., 2016, 55, 12013–12017 CrossRef CAS PubMed
- Z. Tarokh, H. Naderi-Manesh and M. Nazari, Eur. J. Pharm. Sci., 2017, 99, 209–218 CrossRef CAS PubMed
- Y. Zhou, S. Q. Zhang, Z. M. Chen, Y. M. Bao, A. T. Chen, W. C. Sheu, F. Y. Liu, Z. Z. Jiang and J. B. Zhou, Adv. Sci., 2020, 7, 1901866 CrossRef CAS PubMed
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS
- R. Ni and Y. Chau, Angew. Chem. Int. Edit., 2020, 59, 3578–3584 CrossRef CAS PubMed
- M. P. Stewart, A. Sharei, X. Y. Ding, G. Sahay, R. Langer and K. F. Jensen, Nature, 2016, 538, 183–192 CrossRef CAS PubMed
- M. Nurunnabi, Z. Khatun, A. M. Badruddoza, J. R. McCarthy, Y. K. Lee and K. M. Huh, ACS Biomater. Sci. Eng., 2019, 5, 1645–1660 CrossRef CAS
- D. Bouard, N. Alazard-Dany and F. L. Cosset, Br. J. Pharmacol., 2009, 157, 153–165 CrossRef CAS PubMed
- L. Rajendran, H.-J. Knölker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29–42 CrossRef CAS PubMed
- M. G. Katz, A. S. Fargnoli, R. D. Williams and C. R. Bridges, Hum. Gene Ther., 2013, 24, 914–927 CrossRef CAS PubMed
- A. Hernandez-Garcia, D. J. Kraft, A. F. J. Janssen, P. H. H. Bomans, N. A. J. M. Sommerdijk, D. M. E. Thies-Weesie, M. E. Favretto, R. Brock, F. A. de Wolf, M. W. T. Werten, P. van der Schoot, M. C. Stuart and R. de Vries, Nat. Nanotechnol., 2014, 9, 698–702 CrossRef CAS PubMed
- R. Ni and Y. Chau, J. Am. Chem. Soc., 2014, 136, 17902–17905 CrossRef CAS PubMed
- R. Cartier and R. Reszka, Gene Ther., 2002, 9, 157–167 CrossRef CAS PubMed
- S. S. Talsma, J. E. Babensee, N. Murthy and I. R. Williams, J. Controlled Release, 2006, 112, 271–279 CrossRef CAS PubMed
- J. Kong, Y. Wang, J. Zhang, W. Qi, R. Su and Z. He, Angew. Chem., 2018, 130, 14228–14232 CrossRef
- J. Zhou, S. N. Ma, Y. X. Zhang, Y. Y. He, J. Yang, H. Zhang, K. Luo and Z. W. Gu, ACS Appl. Mater. Interfaces, 2020, 12, 22519–22533 CrossRef CAS PubMed
- I. F. de la Fuente, S. S. Sawant, M. Q. Tolentino, P. M. Corrigan and J. L. Rouge, Front. Chem., 2021, 9, 613209 CrossRef CAS PubMed
- M. T. de Pinho Favaro, U. Unzueta, M. de Cabo, A. Villaverde, N. Ferrer-Miralles and A. R. Azzoni, Eur. J. Pharm. Sci., 2018, 112, 71–78 CrossRef PubMed
- D. X. Sun, S. Zhou and W. Gao, ACS Nano, 2020, 14, 12281–12290 CrossRef CAS PubMed
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Nat. Chem., 2021, 13, 131–139 CrossRef CAS PubMed
- L. Wayteck, H. Dewitte, L. De Backer, K. Breckpot, J. Demeester, S. C. De Smedt and K. Raemdonck, Biomaterials, 2016, 77, 243–254 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |