
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium phosphate nanoparticles for potential application as enamel remineralising agent tested on hydroxyapatite discs†
Zi Hong
Mok
 a,
Petros
Mylonas
b,
Rupert
Austin
c,
Gordon
Proctor
c,
Nigel
Pitts
d and
Maya
Thanou
*e
a,
Petros
Mylonas
b,
Rupert
Austin
c,
Gordon
Proctor
c,
Nigel
Pitts
d and
Maya
Thanou
*e
aSwansea University Medical School, Swansea, UK
bSchool of Dentistry, Cardiff University, Cardiff, UK
cFaculty of Dentistry, Oral and Craniofacial Sciences, King's College London, London, UK
dReminova, Inveralmond Business Park, Auld Bond Road, Perth, UK
eSchool of Cancer and Pharmaceutical Sciences, Faculty of Life Sciences and Medicine, King's College London, London, UK. E-mail: maya.thanou@kcl.ac.uk
First published on 19th November 2021
Abstract
Calcium phosphate exhibits excellent biocompatibility, and with particle size in the nanoscale, calcium phosphate nanoparticles (CPNPs) were explored to replace the hydroxyapatite lost in the nanoporous teeth due to dental erosion. CPNPs (2% w/v) colloidally stabilised by sodium citrate were synthesised via co-precipitation. They were characterised in terms of particle size, morphology, crystallinity, Ca/P ratio and calcium ion release. To ensure uniformity of the substrate, hydroxyapatite (HA) discs were examined as an alternative substrate model to enamel. They were eroded in acetate buffer (0.5 M; pH 4.0) at various timepoints (1, 5, 10, 30 min, and 2, 4 h), and their physical differences compared to enamel were assessed in terms of surface microhardness, surface roughness and step height. The remineralisation properties of the synthesised CPNPs on eroded HA discs at different pH levels were investigated. It was established that CPNPs were heterogeneously deposited on the HA discs at pH 9.2, whereas newly precipitated minerals from CPNPs were potentially formed at pH 6.2.
1. Introduction
Dental caries is the most common disease in the world with 2.3 billion people suffering from caries of permanent teeth, and 530 million children suffering from caries of primary teeth.1 Erosive tooth wear is one of the most common dental diseases affecting both children and adults, global prevalence rates estimated between 30–50% of children and 20%–45% of adults affected.2 Dental caries develops because of prolonged plaque accumulation whilst dental erosion is caused by dietary acids or gastro-oesophageal reflux disease. Hydroxyapatite (HA) nanoparticles are useful to counter the loss of enamel nanocrystals as they have similar morphology, crystallinity, and chemical composition (Ca10(PO4)6(OH)2) to dental enamel. Pioneering work has identified the average size of hydroxyapatite crystallites on human enamel as 26 nm × 68 nm (ref. 3) or 32 nm × 37 nm,4 with pore radius of sound enamel measuring broadly between 1–30 nm.5 These hydroxyapatite crystallites are oriented in prismatic and interprismatic enamel (with smaller pores, 2–3 nm) separated by prism sheaths (with larger pores, 4–6 nm).6 Particle size and shape are known to affect the powder flowability and packing fraction, important for particle-based biomimetic remineralisation as a densely compact remineralisation layer is required. It will be useful to directly reproduce enamel prism rods with rod-shaped particles but challenging to emulate the directional ‘weave’ pattern of enamel rods. Meanwhile, spherical-shaped particles may fill the interprismatic voids, but they do not have the same specific surface area as enamel rods. Following a carious attack, HA nanoparticles, with a size of 20 nm,7 can act as a filler within nanodefects to repair small depressions on the enamel. Their small particle sizes increase the surface area for binding and stacking, then form a new biomimetic mineral coating.8 However, the random assembling, disordered orientation and weak adsorption of HA nanoparticles may not promise the full repair of enamel.7Meanwhile, amorphous calcium phosphate (ACP) is a group of prenucleation clusters in equilibrium with calcium and phosphate ions that precede HA formation. Reports have shown that these nanoclusters serve as a calcium phosphate reservoir to maintain a state of supersaturation with respect to enamel minerals, releasing ions thereby enabling ion-mediated in situ remineralisation.9 This is useful for non-cavitated lesions which are relatively intact due to the superficial remineralisation by saliva. ACP nanoparticles can bypass the superficial layer, which acts as a diffusion barrier against subsurface uptake of minerals.10 This pseudo-intact surface layer is reported to be permeable to ions too.11 ACP offers better biodegradability than HA, due to its disordered structure and higher energy state, making it more reactive for calcium and phosphate ion release on the enamel. ACP has been stabilised by casein phosphopeptides (CPP) derived from milk protein to form CPP-ACP complexes, which are reported to be 4 nm in diameter.12 ACP has also been stabilised by citrate molecules, and upon heat-induced crystallisation, they became 60–120 nm spheres or 400–1000 nm × 200–350 nm rods.13 Published studies have drawn conflicting conclusions regarding the relative efficacy of HA and ACP nanomaterials in remineralisation.14–16
Healthy, extracted human teeth have been traditionally used for dental research. They are useful for ex vivo studies, with results transferable and implementable in dental practice. However, there are a few problems encountered, such as difficulty in obtaining enough teeth for a satisfactory sample size,17 variations in tooth morphology, variable content of trace elements such as fluoride within the enamel, and presence of biofilm, organic phases, and different types of exogenous materials. In some studies, human teeth have been substituted by bovine teeth, but with conflicting conclusions.18 Though the crystallite orientation, calcium weight percentage and matrix proteins of bovine enamel resemble that of human enamel, the shear and tensile bond strength of bovine teeth, alongside the microhardness value after erosion, were found to be significantly lower.18
A simpler enamel model to represent the structure of HA could offer a testbed for repeated studies without the above-mentioned variability factors. The hydroxyapatite (HA) disc or pellet is formed by compressed HA powder, or calcium phosphate cement which sets primarily as HA. These can be used as cheap synthetic substrates to investigate demineralisation and remineralisation. They give uniform composition of calcium phosphate without the presence of organic matter, although the crystallite arrangement, thickness and calcium content are expected to be different from human and bovine enamel. Biosimilar erosion behaviour between enamel and HA discs has been established, with lower experimental variability and dissolution rate for synthetic HA than the enamel.19 The use of HA to represent a suitable biomimetic model to human teeth has also been investigated, by comparing the shear bond strength and measuring the surface roughness on HA disc by atomic force microscopy.17,20–22 More physical parameter assessments are needed for the comparison between human enamel and HA.
Novel nanomaterials in the biomineralisation and biomimicry of enamel have been studied, challenged, and scrutinised, with many variables amongst different studies. For example, a vast range of durations and frequencies of exposure to remineralisation agents has been adopted, from 3 min once a day up to 3 to 4 times a day,14,23 6 hours to represent the duration people spend sleeping,24 and even up to 7 days25 and 10 days.26 They are mainly performed under stagnant conditions, or occasionally with agitation.27,28 The remineralising agents used for remineralisation are also buffered at mainly pH 7, as the average pH of saliva is 6.7.29 A rapid rise in pH to above neutrality occurs when saliva secretion is stimulated30 or after brushing with toothpaste.31 An alkaline pH environment is usually favoured for remineralisation because it helps to precipitate ions into a calcium phosphate layer.32 On the other hand, the supersaturation of HA is limited to a pH range of 5.6 to 5.8,33 below which HA formation in saliva is unlikely, as the critical solubility pH of HA in saliva is 5.5.
In the literature, different techniques have been used to assess remineralisation. Measurement of microhardness is most often used to reflect mineral content. Qualitative visualisation has also been commonly used to observe the newly formed minerals on top of the lesion. Surface profilometry and measurement of step height have also been adopted.34 They are therefore chosen as parameters to measure the remineralisation outcome of CPNPs.
In this study, we aimed to investigate remineralisation effects on plain HA disc substrates when citrate-stabilised calcium phosphate nanoparticles are applied on their surface. The nanoparticles are characterised in terms of particle size, crystallinity, Ca/P ratio, and their ability to release calcium ions, to be potentially used as a biomimetic material. The physical differences between HA disc and enamel, including surface hardness, surface roughness and step height after controlled etching were evaluated to determine the suitability of HA disc as an alternative substrate model to human tooth. The most favourable pH for CPNPs remineralisation was also investigated in this study.
2. Experimental
2.1 Materials and reagents
Calcium chloride dihydrate (CaCl2·2H2O), trisodium orthophosphate (Na3PO4·12H2O), acetic acid (C2H4O2) and hydrochloric acid (HCl) were purchased from Fisher Scientific (Loughborough, UK). Sodium citrate dihydrate (C6H5Na3O7·2H2O) and sodium acetate trihydrate (C2H3NaO2·3H2O) were obtained from Sigma-Aldrich (St Louis, MO). Citric acid monohydrate (C6H8O7·H2O) was purchased from AnalaR (Poole, UK). Sodium hydroxide (NaOH) was obtained from Emplura (Darmstadt, Germany). Crystalline HA nanopowder (Ca5(PO4)3(OH); Ca/P = 1.67; white to faint blue powder; XRD conforms to structure; <200 nm particle size) and ACP nanopowder (Ca2P2O7·xH2O; Ca/P = 1; white powder; FTIR and XRD conforms to structure; <150 nm particle size) were also obtained from Sigma-Aldrich (St Louis, MO). HA discs (sintered, microporous, 2 mm thick and 12 mm in diameter) were ordered from Plasma Biotal (Derbyshire, UK). Silicon carbide sanding discs were purchased from Struers (VersoCit, Struers A/S; Copenhagen, Denmark).2.2 Synthesis of CPNPs
CaCl2·2H2O (350 μL; 0.21 mmol) and Na3PO4·12H2O (350 μL; 0.14 mmol) were added from automated syringe pumps to deionised water (700 μL) with a flow rate of 15 mL h−1 for 1 min 24 s. The suspension of CPNPs was stirred with a magnetic stirrer, under the speed of 2000 rpm, for a further 30 min. The apparatus set-up is illustrated in Fig. 1(A). The precipitated CPNPs in water was centrifuged by setting the relative centrifugal force to 9503g for 5 min at room temperature. The supernatant was removed, and the pellet washed by resuspending it with deionised water (1.4 mL). The process of centrifugation, supernatant removal and washing was repeated twice. Then, the final pellet was resuspended in deionised water and vortexed. To enable dispersion and control of the particle size, CPNPs (100 μL) were dispersed in sodium citrate (900 μL; 100 mM) and sonicated for 30 min at 25 °C. | ||
| Fig. 1 (A) Schematic of the equipment used during the synthesis of CPNPs. (B) Intensity size distribution of crystalline samples (CPNPs and Sigma HA) with 100 mM citrate (n = 3). (C) Cumulative calcium release as a function of time for different formulations (n = 3). (D) Particles observed on SEM (from left; ×60k): CPNPs as rods, Sigma HA and Sigma ACP as spheres. | ||
2.3 Particle size characterisation of CPNPs and other calcium phosphate NP products
The mean hydrodynamic diameter and polydispersity index of the above samples, alongside other calcium phosphate products including Sigma HA and Sigma ACP obtained from Sigma-Aldrich, were measured by the Malvern Zetasizer Nano S (Malvern, UK) at 25 °C after 24 h of synthesis. The mean zeta potential in citrate buffer (100 mM) was measured by the Malvern Zetasizer Nano ZS (Malvern, UK).A few drops of CPNPs (2% w/v) were applied on a glass slide attached to a SEM specimen stub. The sample was dried with nitrogen gas and coated with an electrically conducting layer of gold of 10 nm (Quorum Q150T Turbo-pumped Sputter Coater; East Sussex, UK). The dried particles were then viewed on the SEM (Hitachi S4000; Berkshire, UK), operated at 25 kV.
2.4 Elemental analysis of CPNPs and other calcium phosphate products
CPNPs dispersed in sodium citrate was dissolved and diluted with 5% v/v HNO3 up to 1 in 1000 dilution to give about 6 ppm calcium and 3 ppm phosphorus to meet the detection limits required for ICP-MS analysis. Calcium and phosphorus standards were used to make a calibration curve to determine the amount of calcium and phosphorus present in CPNPs. The samples were placed in the autosampler, introduced to the spray chamber by means of a peristaltic pump where they were nebulised to be analysed by the ICP-MS (PerkinElmer NexION 350D; Waltham, MA).In a separate experiment, 10 μL of CPNPs (2% w/v) in sodium citrate was mixed with gallium (10 μL; 20 mg L−1) which served as the internal standard. They were mixed well by vortexing and a 10 μL drop of the mixture was placed on an acrylic disc and dried in the 80 °C oven for 30 min. The disc was then inserted into the TXRF spectrometer (Bruker S2 PICOFOX; Karlsruhe, Germany) which had been calibrated for calcium and phosphorus analysis.
2.5 Crystallinity of CPNPs and other calcium phosphate products
The transmittance spectra of the powder sample of CPNPs were recorded, with wavenumber from 1180 cm−1 to 530 cm−1, and a total of 16 scans per sample, using the Fourier-transform infrared (FTIR) spectrometer (PerkinElmer; Waltham, MA). The transmittance spectra were converted from the absorption bands at the various wavelengths throughout the infrared region. The absorption peaks of the sample were matched to the vibrational modes of orthophosphate ion –PO43− present in HA to determine the similarities between CPNPs and HA.The Raman microscope (Renishaw inVia; Gloucestershire, UK) running in mapping scanning mode was used to scan the CPNPs using a 785 nm diode laser (100% laser power) focused using a 20/0.45 air objective, according to previously published protocols.34 The signal was acquired using a 300 lines per mm diffraction grating centred at 800 cm−1 and a charge-coupled device exposure time of 10 s. The microscope was calibrated using an internal silicon sample with a characteristic band at 520 cm−1.
The X-ray diffraction (XRD) patterns of lyophilised CPNPs (20 mg) were recorded using the X-ray diffractometer (Philips PANalytical X'Pert MPD; Eindhoven, Netherlands) with Cu Kα radiation. The sample was scanned from 10° to 70° 2θ with a 15 min counting time. The XRD spectrum of the sample was matched to that of HA to determine the overlapping peaks for similarities.
2.6 Calcium ion release study
A total of 200 mg of CPNPs was dispersed into acidified artificial saliva (10 mL) prepared as modified Tani-Zucchi's solution containing KCl (20 mM), KSCN (5.3 mM), Na2HPO4 (1.4 mM), NaHCO3 (15 mM) and lactic acid (10 mM).35 The suspension was maintained at 37 °C under shaking at 62.5 rpm. At scheduled times (30 min, 1 h, 2 h, 5 h, 17 h and 28.5 h), the supernatant (8 mL; well separated from the pellet after centrifugation at 3000 rpm for 15 min) was removed for calcium ion quantification by the ICP-OES (Thermo Scientific iCAP6300 Duo; Marietta, OH). They were mixed with yttrium (10 μL; 10 mg L−1) which served as the internal standard, and calibration curve made with calcium standards. After that, the samples were rinsed with fresh artificial saliva (8 mL to make a total of 10 mL again), and the suspensions were again shaken at 37 °C until the next timepoint.2.7 Substrate preparation
HA discs were sectioned into quarters, using the water-cooled 300 μm diamond wafering blade (Benetec XL 12205; London, UK). Enamel slabs were supplied from sectioning of the mid-buccal surfaces of previously extracted caries-free human molar teeth (REC ref 12/LO/1836), using the same blade. Each HA disc quartet and enamel slab was mounted, with surface to be ground faced downwards, in self-curing bis-acryl resin (Protemp™ 4, 3 M ESPE; Seefeld, Germany) within a custom-built silicone mould. Once cured, they were ground with successively finer silicon carbide discs of grit 500, 1200, 2000, and 4000 for 10 s, 25 s, 30 s, and 60 s respectively, using the water-cooled rotating polishing machine (Struers ApS LaboPol-30; Ballerup, Denmark) at 150 rpm and a constant pressure (10 N). For the enamel slabs, the newly ground surfaces were sonicated in deionised water for 15 min to remove the smear layer which contained organic particle debris.After the samples had been air-dried for at least 24 h at room temperature, adhesive tape was placed on the polished surface/bis-acryl resin surface such that each polished surface had an approximate 1 mm width of exposed surface, and two protected reference zones taped on either side, shown in Fig. 3(A). This allows for the comparison between eroded region and protected surface regions as the baseline.
2.8 Acetate buffer erosion to create artificial lesion
All prepared samples were assigned randomly to one of six treatment groups according to the duration of acid immersion: 1 min, 5 min, 10 min, 30 min, 2 h or 4 h; 3 HA discs and 3 enamel slabs per group. Acetate buffer (0.5 M) at pH 4 was prepared by mixing acetic acid (205 mL; 0.5 M) and sodium acetate trihydrate (45 mL; 0.5 M). Each group was immersed in the unstirred acetate buffer at room temperature and the exposure was continued up to each timepoint of the experiment without a renewal of the acetate buffer.36 All surfaces were then washed with deionised water and left to dry in air overnight.2.9 Knoop microhardness measurements
Baseline surface microhardness was measured for all polished surfaces using the Knoop microhardness tester (Struers Duramin-5; Rotherham, UK). After acid immersion and drying, PVC tapes were carefully removed, and the surfaces reassessed. The microhardness after etching was performed with five indentations that were at least 100 μm apart, selected conveniently in the eroded and protected regions, at 50 gf (equivalent to 490.3 mN) load and 10 s dwell time.2.10 Surface roughness
Profilometric measurement was conducted using a 2 μm laser spot sized red light (655 nm) from the confocal laser scanning profilometer (TaiCaan XYRIS 2000; Southampton, UK), with 0.01 μm z height resolution, vertical gauge range 0.6 mm and sensor spatial resolution (x, y) <1 μm.37 Surface roughness was measured using five representative areas, each 0.2 mm × 0.2 mm over the eroded and protected regions of the sample. Scanning occurred in a raster pattern, at a (x, y) scanning interval of 4 μm. A 25 μm Gaussian filter was applied to each scan to determine the 3D roughness (Sa) data for each sample.2.11 3D step height measurements
Two reference marks were made using an indelible pen on the bis-acryl resin of each sample, in between the PVC tapes to allow for scan localisation by the profilometer after the removal of PVC tapes. Five step height measurements were conducted, within a 3 mm × 1 mm area obtained at a (x, y) scanning interval of 10 μm, and the mean 3D step height was analysed using the MountainsMap® surface metrology software (Digital Surf; Besançon, France), according to the ISO 5436-1 standard.37Statistical analyses were conducted using OriginPro 8.5 Statistical Software (OriginPro version 8.5, OriginLab Corp.; Northampton, MA). A two-way ANOVA was conducted to examine the effect of etching time (1 min, 5 min, 10 min, 30 min, 2 h or 4 h) and substrate surfaces (protected or eroded regions of HA discs and enamel) on Knoop microhardness, surface roughness and step height. A p-value < 0.05 was considered statistically significant.
2.12 SEM imaging
A specimen from each treatment group was selected for observation under SEM at a magnification of ×15k. They were coated with an electrically conducting layer of gold and viewed on the SEM operated at 25 kV. The SEM images obtained were evaluated qualitatively to determine any visual changes between eroded and protected regions of both HA discs and enamel slabs.2.13 Exposure to CPNPs samples
Prepared and etched HA discs for 2 h were treated for 30 min with CPNPs in 100 mM citrate, at pH 6.2, 7.2, 8.2 and 9.2. The pH was adjusted by adding drops of sodium hydroxide (1 M) or hydrochloric acid (1 M) into stock sodium citrate solutions (1 M) to achieve the desired pH. The pH of the buffers created were checked and monitored with a pH meter (Fisherbrand Accumet AE150, Fisher Scientific; Leicestershire, UK).After treatment, each HA disc was placed in deionised water (10 mL) for 2 min, agitated at 62.5 rpm, to rinse off residual CPNPs. This was repeated up to 3 times. Knoop microhardness (50 gf load; 10 s dwell time) and step height were then measured after the samples had been air-dried for 24 h and the tapes removed, alongside visual inspection under the light microscope (Cole-Palmer; St Neots, UK).
Statistical analyses were conducted using the OriginPro 8.5 Statistical Software. A two-way ANOVA was conducted to examine the effect of the different substrate surfaces (baseline, etched and exposed surfaces) on Knoop microhardness and step height, assigned to the different pH environments. A p-value < 0.05 was considered statistically significant.
3. Results
3.1 Particle size and shape
Dynamic light scattering in Table 1 and Fig. 1(B) reveals the hydrodynamic diameter of CPNPs (63.9 ± 15.9 nm) was six times smaller than that of Sigma HA (393.1 ± 68.8 nm). All three CPNPs, Sigma HA and Sigma ACP were suspended in the same concentration of citrate (100 mM). Scanning electron microscope in Fig. 1(D) reveals CPNPs were rod-shaped particles, whereas Sigma HA and Sigma ACP were spherical. The zeta potential of the synthesised CPNPs (2% w/v; 1 mL) at pH 8.8 was −23.3 ± 1.3 mV.| Particles in 100 mM citrate | Mean hydrodynamic diameter, nm (n = 3) | Average polydispersity index (n = 3) |
|---|---|---|
| CPNPs | 63.9 ± 15.9 | 0.17 ± 0.01 |
| Sigma HA | 393.1 ± 68.8 | 0.24 ± 0.03 |
| Sigma ACP | Measurement was not possible due to quick sedimentation | Measurement was not possible due to quick sedimentation |
3.2 Elemental analysis of calcium phosphate nanoparticles
ICP-MS (Table 2) reveals that crystalline Sigma HA (1.59 ± 0.01) showed greater Ca/P ratios compared to amorphous Sigma ACP (0.91 ± 0.00), with that of synthesised CPNPs in between them (1.39 ± 0.00). This indicates that synthesised CPNPs was calcium deficient compared to Sigma HA. The trend was similar in TXRF. The Ca/P ratio of Sigma ACP is 1 as reported by the manufacturer,38 and this value was reflected quite closely in the results.| Calcium phosphate | Ca/P ratio | |
|---|---|---|
| ICP-MS | TXRF | |
| CPNPs | 1.39 ± 0.00 | 1.50 ± 0.16 |
| Sigma HA | 1.59 ± 0.01 | 1.77 ± 0.08 |
| Sigma ACP | 0.91 ± 0.00 | 0.84 ± 0.08 |
3.3 Crystallinity of CPNPs assessed by FTIR and Raman
The FTIR spectrum shows two main observations that distinguish crystalline HA from ACP. First is the splitting of the 570 cm−1 PO4 antisymmetric bending frequency (v4) into a well-defined doublet in crystalline HA but remains a broad singlet in ACP. In Fig. 2(A), the doublet seen in Sigma HA was also seen in CPNPs. The intensity of the 570 cm−1 peak was twice of that of the 605 cm−1 peak as it was likely that two out of the three triply degenerate states have the same value of energy upon measurement. Two further absorption bands, although weak but visible at 620 cm−1 and 870 cm−1 corresponding to apatitic hydroxyl and carbonate groups respectively, further indicate that CPNPs as carbonated HA. Meanwhile, the presence of a broad dip in Sigma HA from 820 cm−1 indicated the presence of impurities. | ||
| Fig. 2 (A) FTIR spectra of different calcium phosphate and sodium phosphate (reference) powders. (B) Raman spectra of different calcium phosphate and sodium phosphate (reference) powders. Stretching and bending of PO4 groups that contribute to the characteristic vibration bands in calcium phosphate crystals: symmetric P–O stretch/v1 stretching (ca. 960 cm−1), symmetric P–O bend/v2 bending (ca. 440 cm−1), triply degenerate antisymmetric P–O stretch/v3 stretching (ca. 1020 cm−1) and triply degenerate antisymmetric P–O bend/v4 bending (ca. 600 cm−1). | ||
Another observation that differentiates between ACP and HA samples on the FTIR spectra, that would also show on the Raman spectra, is the presence of the 960 cm−1 PO4 symmetric stretching frequency (v1) for crystalline HA, as shown in Fig. 2(B). This was apparent for the synthesised CPNPs, as for Sigma HA. Between them, it was observed that the v1 for Sigma HA on the Raman spectra had a slightly lower frequency compared to the synthesised CPNPs. Contrarily, according to Combes and Rey, the main characteristic of the ACP is the v1 PO4 band shifted to ∼950 cm−1, which is 10 cm−1 lower than that of HA.26,39 However, this could not be identified on Sigma ACP in this study, due to the band superimposition and broadening characteristic of them being amorphous. The characteristic vibrational bands of PO4 groups in HA crystals marked in the FTIR and Raman spectra are listed in Fig. 2(C).
3.4 Calcium ion release from calcium phosphate nanoparticles
An ion release study in acidified artificial saliva was adapted35 to study the ability to release Ca2+ by the different calcium phosphate agents. Fig. 1(C) shows that Sigma ACP dissolved faster, releasing more Ca2+ than Sigma HA and the synthesised CPNPs. The release of Ca2+ from Sigma ACP continued to increase after 5 h, followed only by a slight increase from the synthesised CPNPs, indicating that Ca2+ release is maintained for the latter. The Ca2+ release from Sigma HA, on the other hand, plateaued with no significant difference in the release after 5 h. The rate of release of all formulations decreased with time, as seen with the decreasing gradient of the line graph of each formulation after each timepoint.The experiment was performed with the equivalent amount of powder (200 mg), with the same relative calcium concentrations in these materials, shown in Appendix A in ESI.†
3.5 Knoop microhardness measurements of HA discs and enamel
The baseline Knoop microhardness of the polished, unetched HA discs (221.4 ± 19.4 KHN) was significantly lower than the baseline Knoop microhardness of the polished, unetched enamel slabs (348.0 ± 13.1 KHN). Taking samples used for timepoint = 1 min for example, the reference HA disc had a mean Knoop microhardness 119.9 (95% CI, 102.1 to 137.7) KHN lower than the reference enamel slab, a statistically significant difference, p < 0.0001. This showed that the HA disc, which is microporous, was not able to simulate the hardness of the enamel.Both sample types decreased in hardness with time after immersion in acetate buffer. It is also apparent that the enamel hardness decreased more rapidly than the HA disc. For example, after 5 min, the eroded HA discs only had a small decrease in mean Knoop microhardness of 26.0 (95% CI,13.2 to 38.8) KHN (p < 0.0001) from their respective references, whereas the eroded enamel slabs already had a decrease of 129.0 (95% CI, 120.9 to 137.1) KHN (p < 0.0001), as shown in Fig. 3(D).
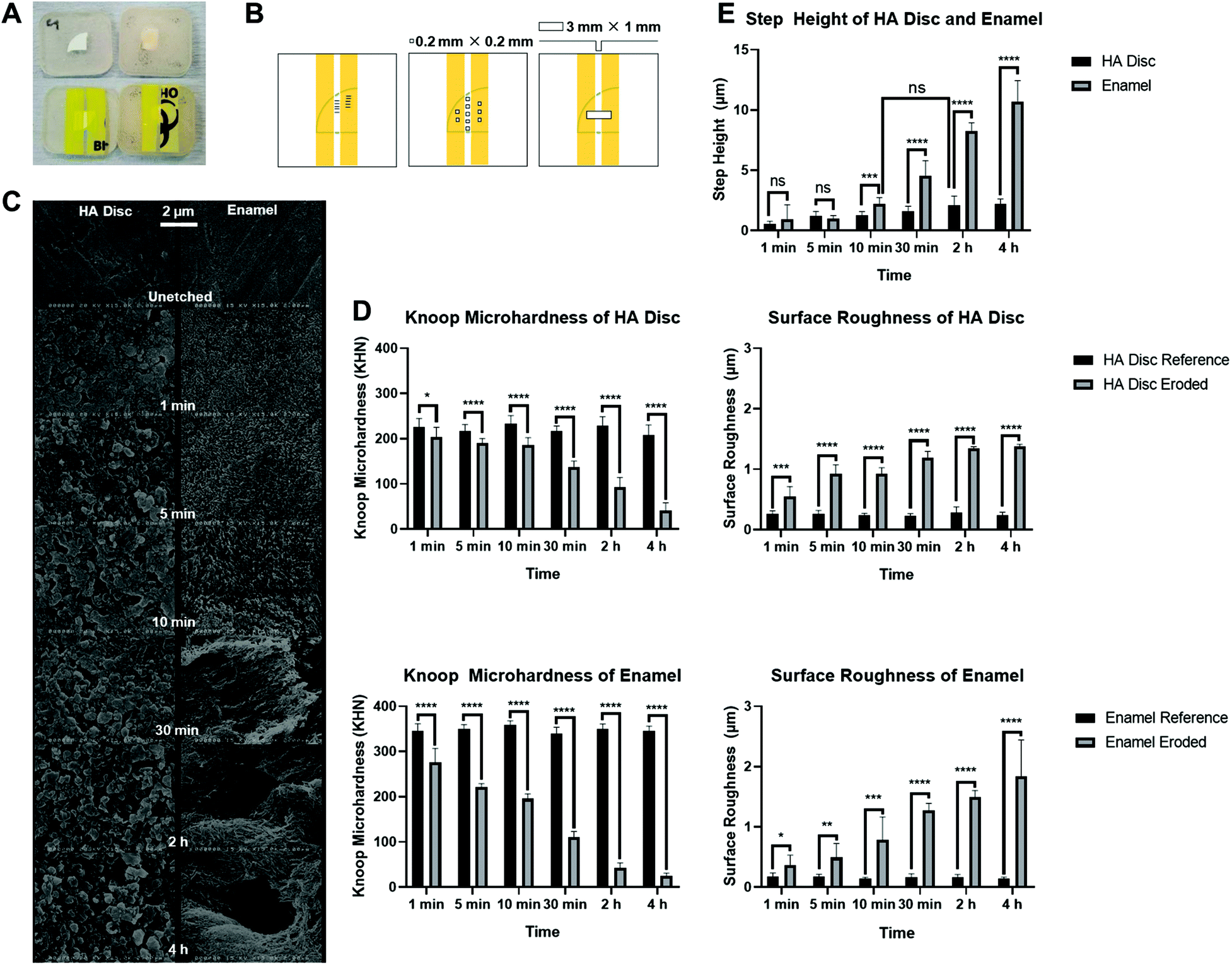 | ||
| Fig. 3 (A) Substrate preparation before etching: HA disc (top left) and enamel slab (top right) mounted in self-cured bis-acryl resin to be ground. After grinding, sides of the samples were taped, leaving about 1 mm gap that is exposed for acid etching. (B) Five Knoop microhardness indentations were measured on each etched and unetched surfaces (left), five 0.2 mm × 0.2 mm areas were measured for surface roughness on each etched and unetched surfaces (middle), five step height measurements within a 3 mm × 1 mm area were measured after etching (right). (C) SEM images of HA discs (left) and enamel slabs (right), unetched and etched at 1 min, 5 min, 10 min, 30 min, 2 h and 4 h (from top; ×15k). Both sample types looked different, and their surfaces became more damaged with longer etching time. (D) Surface Knoop microhardness for HA discs (top left) and enamel slabs (bottom left) as a function of etching time, unetched and etched at 1 min, 5 min, 10 min, 30 min, 2 h and 4 h. There were statistically significant decreases in the surface Knoop microhardness at each timepoint after etching for both HA discs and enamel slabs. Surface roughness for HA discs (top right) and enamel slabs (bottom right) as a function of etching time, unetched and etched at 1 min, 5 min, 10 min, 30 min, 2 h and 4 h. There were statistically significant increases in the surface roughness at each timepoint after etching for both HA discs and enamel slabs. (E) Step height for HA discs and enamel slabs as a function of etching time, unetched and etched at 1 min, 5 min, 10 min, 30 min, 2 h and 4 h. | ||
3.6 Surface roughness measurements of HA discs and enamel
The baseline surface roughness of the polished, unetched HA discs (0.25 ± 0.06 μm) was significantly higher than that of enamel slabs (0.16 ± 0.04 μm). Taking timepoint = 1 min for example, the reference HA disc had a mean surface roughness 0.09 (95% CI, 0.02 to 0.16) μm higher than the reference enamel slab, a statistically significant difference, p = 0.0055. The difference in baseline surface roughness showed that the grinding protocol adopted was not able to produce HA discs and enamel slabs with the same surface roughness.Both sample types increased in roughness with time after immersion in acetate buffer and had broadly similar surface roughness values with increasing erosion time. There was no significant difference in surface roughness between the sample types after erosion at timepoint 1 min (HA disc: 0.55 ± 0.17 μm and enamel: 0.37 ± 0.16 μm; p = 0.2033), 10 min (HA disc: 0.93 ± 0.10 μm and enamel: 0.79 ± 0.38 μm; p = 0.9994), 30 min (HA disc: 1.19 ± 0.10 μm and enamel: 1.27 ± 0.12 μm; p = 0.9361) and 4 h (HA disc: 1.38 ± 0.04 μm and enamel: 1.83 ± 0.61 μm; p = 0.3272), as shown in Fig. 3(D).
3.7 Etching effect on 3D step height in HA discs and enamel
In terms of step height, both HA discs and enamel slabs had significantly different erosion patterns from 10 min onwards. The enamel had a greater step height compared to HA discs, for example, an additional of 8.48 (95% CI, 6.78 to 10.19) μm after 4 h compared to the HA discs. The step height for enamel was increasing exponentially whereas there was only minimal increase for HA discs for each erosion timepoint.For HA discs, there was hardly any profile change until 2 h erosion (2.09 ± 0.78 μm), which was equivalent to 10 min erosion of enamel slabs (2.20 ± 0.51 μm), with no statistical significance between them (p = 0.6857). This indicated a similar volume of bulk loss of hydroxyapatite had occurred within the eroded regions of both substrate types at the respective timepoints. From Fig. 3(E), it is determined that at least a 2 μm step height could be useful after erosion, so that when the substrate is being remineralised, any changes in step height can become apparent.
3.8 SEM imaging
The SEM images in Fig. 3(C) allowed qualitative assessment of the eroded zone for each erosion period. Under SEM, the unetched and ground surfaces of HA disc and enamel looked similar, which were generally smooth. After etching however, the pores of HA disc were apparent, and they increased with longer etching times. On the other hand, the etched enamel revealed tightly packed and organised HA rods, which became more loosely packed with longer etching time. The prism core and the intact prism periphery of the enamel became apparent after 10 min of etching. Dissolution of HA and subsequent destruction of the prism core and periphery were also observed after 30 min of etching.3.9 Exposure of HA discs to CPNPs
A range of pH above pH 5.5, the critical pH of HA dissolution was selected to investigate the effect of pH on the remineralisation outcome. There were two pH environments that led to significant increases in Knoop microhardness after treatment. A pH of 7.2 (95.6 ± 6.4 KHN) led to a 19.6 (95% CI, 14.4 to 24.8) KHN increase from the etched surface, p < 0.0001. A pH of 9.2 (216.9 ± 132.6 KHN) led to a recovery of microhardness back to baseline, with no significance difference between them, p = 0.9918, and with a 147.1 (95% CI, 57.5 to 236.7) KHN increase from the etched surface, p = 0.0020, as shown in Fig. 4(A). | ||
| Fig. 4 (A) Knoop microhardness after treatment with CPNPs at different pH values after 2 h erosion. (B) Step height after treatment with CPNPs at different pH values after 2 h erosion. (C) Representative light microscope images for treatment with CPNPs at different pH values after 2 h erosion (left to right): pH 6.2, 7.2, 8.2 and 9.2. Growth of particles on the surface treated at pH 6.2 was observed, whereas deposition of particles on the surface treated at pH 9.2 looked relatively more homogenous. | ||
There were also two pH environments that led to significant increases in step height after treatment. A pH of 6.2 (33.7 ± 17.0 μm) led to a 36.1 (95% CI, 29.2 to 43.0) μm increase from the etched surface, p < 0.0001. A pH of 9.2 (10.0 ± 11.0 μm) led to a 12.3 (95% CI, 5.4 to 19.1) μm increase from the etched surface, p < 0.0001. The large standard deviation was due to one out of the three samples had no gain in step height and remained at −2.5 ± 0.4 μm. The increase in step height and the apparent crystal growth on the surface at pH 6.2 in Fig. 4(C) show that new minerals may have precipitated. Comparatively, the surface of pH 7.2 and 8.2 looked bare under the light microscope.
4. Discussion
4.1 Characteristics of CPNPs
The morphology of crystalline HA is affected by the pH of synthesis – rod-shaped at a pH of 11 such as for the synthesised CPNPs, spherical at a pH of 9.40 This is due to a higher concentration of OH− ions at a higher pH would adsorb on the facets of the formed crystalline nuclei, allowing crystal growth only in the facets without the OH− ions, causing particle elongation.40 Meanwhile, spherical amorphous particles (Sigma ACP) are formed by the random packing of the Posner's clusters, the prenucleation clusters in equilibrium with calcium and phosphate ions.41,42The slight discrepancy in particle size of rod-shaped particles between DLS and SEM is due to the attenuated Lorenz–Mie resonances for particles with larger aspect ratios relative to their volume equivalent spheres. For spherical particles (Sigma HA), the hydrodynamic diameter measured by the DLS is larger than the particle size measured by SEM, due to the hydration layer or solvation shell made up of water molecules and citrate ions. The agglomeration level of the particles in suspension would also increase the hydrodynamic diameter. Meanwhile, although the hydrodynamic diameter of Sigma ACP was not measurable by the DLS due to quick sedimentation, individual particles can be seen within the aggregate under SEM.
During the rearrangement of ACP clusters (Cax(PO4)y·nH2O) to form crystalline HA (Ca10(PO4)6(OH)2), additional calcium and hydroxide ions are taken up,43 alongside a local loss of water molecules filling the intercluster space.44 This changes the Ca/P ratio from ca. 1.50 for ACP to 1.67 for HA. The Ca/P ratios for crystalline HA samples were not exactly 1.67, which indicate that calcium-deficient HA are being produced in these physiological conditions, as also reported by Gómez-Morales et al.45 Meanwhile, the Ca/P ratio derived from ICP-MS was generally lower than that from TXRF, which is due to the fundamental differences in working principles between the two techniques, hence the results are not directly comparable. Apart from the Ca/P ratio, the crystallinity for synthesised CPNPs and Sigma HA is indicated by the splitting of the v4 band in the FTIR spectra into approximately the 570 cm−1 and 605 cm−1 regions (560 cm−1 and 600 cm−1 in this study), which are members of the antisymmetric bending motion.46 The crystallinity was also confirmed with the X-ray diffraction spectrum of CPNPs in Appendix B in ESI,† which shows poorly crystalline apatites.
A few factors could contribute to the differential dissolution rates, namely the crystallinity, surface area due to particle size and shape, aggregation, packing density, and homogeneity of the particles, both chemically and structurally. The Ca2+ release comparison between crystalline HA and ACP has previously been reported in a study, whereby fluoride-doped amorphous calcium phosphate releases calcium ions gradually and progressively, whereas the release from fluorapatite is negligible under the same conditions.35 Another study also showed that HA was the most stable by releasing only 3% of its calcium content in double distilled water over 21 days at 37 °C, compared to monobasic calcium phosphate, Ca(H2PO4)2 and dibasic calcium phosphate, CaHPO4.47 Low dissolution of HA as found in this study is useful for particle-mediated remineralisation. The mechanism is such that HA is able to integrate into the enamel carious lesion surface structure, similar to how HA is able to occlude dentine tubules,48 to act as a filler to repair minute pits and fissures in the enamel surface, as reported by Japanese dentifrices containing HA.49 A dimension of HA close or even smaller to that of the building blocks of enamel will be useful for such integration. 10% hydroxyapatite was shown to have a protective effect by reducing the rate of lesion progression.50 In separate experiments, 7% hydroxyapatite and a combination of hydroxyapatite (concentration unknown) and 1450 ppm fluoride were shown to increase the microhardness of demineralised enamel specimens.51,52
On the other hand, ACP is expected to be more soluble than crystalline HA and that was reflected in the results. ACP is less thermodynamically stable as it lacks crystal lattice interactions or bonds that need to be overcome for it to dissolve, leading to a higher apparent solubility compared to that of crystalline HA. For this reason, where mineral ion release is required for remineralisation, ACP is favoured due to its higher solubility and its ability to transform to HA afterwards,16 provided phase transformation can be controlled. Full-length amelogenin (rP172), leucine-rich amelogenin peptide (LRAP),53 amelogenin-inspired self-assembling peptide P11-4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 and amelogenin-derived peptide QP555 have demonstrated the ability to have high affinity towards calcium ions which acts as a nucleator facilitating de novo HA crystal formation, by stabilising amorphous calcium phosphate (ACP), regrowing enamel-like apatite crystals by directing their nucleation and growth through orientation degree.
54 and amelogenin-derived peptide QP555 have demonstrated the ability to have high affinity towards calcium ions which acts as a nucleator facilitating de novo HA crystal formation, by stabilising amorphous calcium phosphate (ACP), regrowing enamel-like apatite crystals by directing their nucleation and growth through orientation degree.
4.2 HA discs characterisations
The microhardness of the enamel complied with values found in previously reported studies.56 Supporting the work by Nozari et al., acetic acid lowered the Knoop microhardness of the enamel to 102.8 ± 37.9 KHN at 30 min timepoint.57 The acetic acid used in their study was 17 M at pH 3.1, hence the softening effect is expected to be greater compared to this study (110.1 ± 12.9 KHN at 30 min timepoint), which used a 0.5 M acetate buffer at pH 4.The damage on enamel was more apparent than that on HA discs, reflected by the lower surface hardness and greater step height difference in enamel, as firstly it was reported that dental enamel formed by biomimetic mineralisation at physiological temperature has lower crystallinity, hence more soluble than high-temperature sintered form as with the HA discs.58 Secondly, the presence of carbonate, within calcium-deficient carbonated apatite on the enamel59 causes the solubility of apatite to increase greatly with an increase in pCO2,60 making it more acid soluble than synthetic HA. Thirdly, the higher solubility was suggested to be due to surface and lattice defects and the presence of impurities on the enamel.61,62 It is also possible that the HA crystals are more densely packed in the enamel slab than in the sintered HA disc. Therefore, the effect of acetic acid will be more prominent on a dense surface due to it being more packed with apatite, as opposed to a microporous surface like HA disc, seen in the SEM results in Fig. 3(C).
The arithmetical mean height, Sa, which is the difference in height of each point compared to the arithmetical mean of the surface was used in this study to evaluate surface roughness. Unlike the previous surface microhardness test, the measured surface roughness by the profilometer was likely not to be affected by the density of HA and porosity of the structure underneath the surface. The surface roughness of enamel also continued to increase because the prism core and the intact prism periphery of the enamel became exposed after longer period of etching, as seen in the SEM results in Fig. 3(C). This is in line with the work by Zheng et al., who showed an increasingly obvious honeycomb morphology after etching, causing the surface to appear rugged with increasing erosion time.63 Therefore, it is worth noting that while HA discs provide a cheap and reproducible alternative to enamel, studies here will need to be translated to an enamel setting to confirm similar results.
4.3 Exposure of HA discs to CPNPs
A previous study found a trend for increasing rates and percentages of remineralisation by CPP-ACP as the pH decreased from 9.0 to 7.0.64 Another study found that this trend of increasing remineralisation continued to a maximum value at pH 5.5 and then decreased to a lower value at pH 4.5.65 In the present study pH 9.2 showed the greatest microhardness and increase in step height, which suggests a different mechanism of remineralisation with HA nanoparticles (particle-mediated remineralisation) compared to CPP-ACP (ion-mediated remineralisation). The remineralisation of CPP-ACP requires the breakdown of protein to release the calcium ions, whereas HA nanoparticles require the compact deposition of the crystals filling in the gaps. The latter can also dissociate into calcium and phosphate ions for HA re-precipitation.HA becomes more soluble as the pH decreases66 as expected from the effect of acid on dissociating HA and therefore more calcium ions are available for re-precipitation at a lower pH. On the other hand, HA is the least soluble at a pH of around 9;66 this should facilitate deposition of HA and dehydration without significant dissociation into ions, making a fuller and more homogenous coverage on the HA discs, as seen under the light microscope in Fig. 4(C). Future work to determine the crystallinity and stoichiometry of the new minerals needs to be performed, together with the optimal time for the minerals to reform or deposit.
5. Conclusions
Well-characterised CPNPs with a Ca/P molar ratio of 1.54 were synthesised by simple co-precipitation of calcium chloride and sodium phosphate. The formed CPNPs are crystalline but with low crystallinity, showing calcium ion release slower than that of amorphous calcium phosphate. Between ground HA disc and enamel, the changes in surface roughness after erosion were broadly similar. A 2 h etch for HA discs in acetate buffer (pH 4) produced a 2 μm step height equivalent to a 10 min etch for enamel slabs. Based on these results, within limits tested in this study, HA disc could be used to monitor changes upon remineralisation with CPNPs. Remineralisation studies have shown that a basic pH environment at pH 9.2 created a fuller and more homogenous deposition of CPNPs, whereas a slightly acidic pH environment at pH 6.2 allowed newly precipitated minerals on the surface of HA discs, thought to be formed from the dissociation of CPNPs into calcium and phosphate ions which then re-precipitated. Further studies will focus on improving the homogeneity of remineralisation by exploring salivary proteins or other physical enhancement methods to consolidate the deposition of these particles.Author contributions
Zi Hong Mok: Investigation, data curation, formal analysis, writing – original draft. Petros Mylonas: Data curation, formal analysis, writing – review & editing. Rupert Austin: Resources, software. Gordon Proctor: Supervision, visualisation, validation, writing – review & editing. Nigel Pitts: Funding acquisition, resources, supervision, visualisation, validation, writing – review & editing. Maya Thanou: Project administration, funding acquisition, resources, supervision, visualisation, validation, writing – review & editing. All involved in conceptualisation and methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by King's South East Asia Scholarship and Reminova Ltd.References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017, Lancet, 2018, 392, 1789–1858, DOI:10.1016/S0140-6736(18)32279-7.
- N. Schlueter and B. Luka, Br. Dent. J., 2018, 224, 364–370 CrossRef CAS PubMed.
- G. Daculsi and B. Kerebel, J. Ultrastruct. Res., 1978, 65, 163–172 CrossRef CAS PubMed.
- C. A. Grove, G. Judd and G. S. Ansell, J. Dent. Res., 1972, 51, 22–29 CrossRef CAS PubMed.
- G. H. Dibdin, J. Dent. Res., 1969, 48, 771–776 CrossRef CAS PubMed.
- H. Y. Gan, FB Sousa, HL Carlo, PP Maciel, MS Macena and J Han, J. Dent. Res., 2015, 94, 615–621 CrossRef CAS PubMed.
- L. Li, H. Pan, J. Tao, X. Xu, C. Mao, X. Gu and R. Tang, J. Mater. Chem., 2008, 18, 4079–4084 RSC.
- N. Roveri, E. Foresti, M. Lelli and G. Lesci, Recent Pat. Biomed. Eng., 2009, 2, 197–215 CrossRef CAS.
- E. C. Reynolds, Adv. Dent. Res., 2009, 21, 25–29 CrossRef CAS PubMed.
- L. Silverstone, Caries Res., 1977, 11(Suppl 1), 59–84 CrossRef PubMed.
- M. Hannig and C. Hannig, Nat. Nanotechnol., 2010, 5, 565–569 CrossRef CAS PubMed.
- K. J. Cross, N. L. Huq, D. P. Stanton, M. Sum and E. C. Reynolds, Biomaterials, 2004, 25, 5061–5069 CrossRef CAS PubMed.
- L. D. Esposti, S. Markovic, N. Ignjatovic, S. Panseri, M. Montesi, A. Adamiano, M. Fosca, J. v. Rau, V. Uskoković and M. Iafisco, J. Mater. Chem. B, 2021, 9, 4832–4845 RSC.
- S. Vyavhare, D. S. Sharma and V. K. Kulkarni, J. Clin. Pediatr. Dent., 2015, 39, 149–160 CrossRef CAS PubMed.
- A. Sharma, A. Rao, R. Shenoy and B. Suprabha, J. Orofacial Sci., 2017, 9, 28–33 CrossRef CAS.
- D. Skrtic, J. M. Antonucci and E. D. Eanes, Polym. Adv. Technol., 2001, 12, 369–379 CrossRef CAS.
- N. Imthiaz, G. Georgiou, D. R. Moles and S. P. Jones, Aust. Orthod. J., 2008, 24, 15–20 Search PubMed.
- G. H. Yassen, J. A. Platt and A. T. Hara, J. Oral Sci., 2011, 53, 273–282 CrossRef PubMed.
- F. C. Ronay, F. J. Wegehaupt, K. Becker, D. B. Wiedemeier, T. Attin, A. Lussi and V. Steiger-Ronay, J. Dent., 2019, 84, 89–94 CrossRef CAS PubMed.
- A. Klocke, D. Tadic, B. Kahl-Nieke and M. Epple, Dent. Mater., 2003, 19, 773–778 CrossRef CAS PubMed.
- C. Zeitz, T. Faidt, S. Grandthyll, H. Hähl, N. Thewes, C. Spengler, J. Schmauch, M. J. Deckarm, C. Gachot, H. Natter, M. Hannig, F. Müller and K. Jacobs, ACS Appl. Mater. Interfaces, 2016, 8, 25848–25855 CrossRef CAS PubMed.
- R. Shipman, B. Quelle, J. Grant and E. Lee, Conference: Experimental Biology, 2018.
- C. Poggio, M. Lombardini, P. Vigorelli and M. Ceci, Scanning, 2013, 35, 366–374 CrossRef CAS PubMed.
- M. Panich and S. Poolthong, J. Am. Dent. Assoc., 2009, 140, 455–460 CrossRef CAS PubMed.
- H. Wang, Z. Xiao, J. Yang, D. Lu, A. Kishen, Y. Li, Z. Chen, K. Que, Q. Zhang, X. Deng, X. Yang, Q. Cai, N. Chen, C. Cong, B. Guan, T. Li and X. Zhang, Sci. Rep., 2017, 7, 40701 CrossRef CAS PubMed.
- J. A. Stammeier, B. Purgstaller, D. Hippler, V. Mavromatis and M. Dietzel, MethodsX, 2018, 5, 1241–1250 CrossRef PubMed.
- A. Besinis, R. van Noort and N. Martin, Dent. Mater., 2016, 32, 385–393 CrossRef CAS PubMed.
- M. Memarpour, F. Shafiei, A. Rafiee, M. Soltani and M. H. Dashti, BMC Oral Health, 2019, 19, 92 CrossRef PubMed.
- S. Baliga, S. Muglikar and R. Kale, J. Indian Soc. Periodontol., 2013, 17, 461–465 CrossRef PubMed.
- E. A. Abou Neel, A. Aljabo, A. Strange, S. Ibrahim, M. Coathup, A. M. Young, L. Bozec and V. Mudera, Int. J. Nanomed., 2016, 11, 4743–4763 CrossRef PubMed.
- D. Mary, V. Priya and R. Gayathri, Drug Invent. Today, 2018, 10, 1731–1733 Search PubMed.
- D. Chole, Y. Jadhav, S. Kundoor, S. Bakle, A. Devagirkar and R. Deshpande, IOSR-JDMS, 2016, 15, 64–68 CrossRef.
- H. C. Margolis, Y. P. Zhang, C. Y. Lee, R. L. Kent and E. C. Moreno, J. Dent. Res., 1999, 78, 1326–1335 CrossRef CAS PubMed.
- H. Milly, F. Festy, T. F. Watson, I. Thompson and A. Banerjee, J. Dent., 2014, 42, 158–166 CrossRef CAS PubMed.
- M. Iafisco, L. Degli Esposti, G. B. Ramírez-Rodríguez, F. Carella, J. Gómez-Morales, A. C. Ionescu, E. Brambilla, A. Tampieri and J. M. Delgado-López, Sci. Rep., 2018, 8, 17016 CrossRef PubMed.
- T. Aoba, Y. Moriwaki, Y. Doi, M. Okazaki, J. Takahashi and T. Yagi, J. Oral Pathol., 1981, 10, 32–39 CrossRef CAS PubMed.
- P. Mylonas, R. S. Austin, R. Moazzez, A. Joiner and D. W. Bartlett, Dent. Mater., 2018, 34, 1391–1400 CrossRef CAS PubMed.
- Sigma-Aldrich, Calcium phosphate, amorphous nanopowder, <150 nm particle size (BET) | Sigma-Aldrich, https://www.sigmaaldrich.com/catalog/product/aldrich/693871?lang=en®ion=GB, (accessed December 8, 2020).
- C. Combes and C. Rey, Acta Biomater., 2010, 6, 3362–3378 CrossRef CAS PubMed.
- V. Rodríguez-Lugo, T. V. K. Karthik, D. Mendoza-Anaya, E. Rubio-Rosas, L. S. Villaseñor Cerón, M. I. Reyes-Valderrama and E. Salinas-Rodríguez, R. Soc. Open Sci., 2018, 5, 180962 CrossRef PubMed.
- A. S. Posner, F. Betts and N. C. Blumenthal, Prog. Cryst. Growth Charact. Mater., 1980, 3, 49–64 CrossRef CAS.
- S. Jiang, W. Jin, Y. N. Wang, H. Pan, Z. Sun and R. Tang, RSC Adv., 2017, 7, 25497–25503 RSC.
- A. Lotsari, A. K. Rajasekharan, M. Halvarsson and M. Andersson, Nat. Commun., 2018, 9, 4170 CrossRef PubMed.
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed.
- J. Gómez-Morales, M. Iafisco, J. M. Delgado-López, S. Sarda and C. Drouet, Prog. Cryst. Growth Charact., 2013, 59, 1–46 CrossRef.
- J. D. Termine and A. S. Posner, Nature, 1966, 211, 268–270 CrossRef CAS PubMed.
- S. Ali Akbari Ghavimi, B. N. Allen, J. L. Stromsdorfer, J. S. Kramer, X. Li and B. D. Ulery, Biomed. Mater., 2018, 13, 055005 CrossRef PubMed.
- B. Amaechi, S. M. Mathews, K. Ramalingam and P. K. Mensinkai, Am. J. Dent., 2015, 28, 33–39 Search PubMed.
- Sangi, Restoring mineral to the teeth, https://www.sangi-co.com/en/technology/consumer_benefits/index.html.
- A. Itthagarun, N. M. King and Y.-M. Cheung, Hong Kong Dent. J., 2010, 7, 61–66 Search PubMed.
- A. Mielczarek and J. Michalik, Am. J. Dent., 2014, 27, 287–290 Search PubMed.
- A. Ebadifar, M. Nomani and S. A. Fatemi, J. Dent. Res. Dent. Clin. Dent. Prospects, 2017, 11, 14–17 CrossRef PubMed.
- S. S. M. Dissanayake, M. Ekambaram, K. C. Li, P. W. R. Harris and M. A. Brimble, Molecules, 2020, 25 Search PubMed.
- R. Soares, I. D. N. de Ataide, M. Fernandes and R. Lambor, J. Clin. Diagn. Res., 2017, 11, ZC136–ZC141 CrossRef CAS PubMed.
- Y. Wang, D. Hu, J. Cui, Y. Zeng, X. Gan, Z. Chen, Q. Ren and L. Zhang, J. Mater. Chem. B, 2020, 8, 10373–10383 RSC.
- C. Chuenarrom, P. Benjakul and P. Daosodsai, Mat. Res., 2009, 12, 473–476 CrossRef.
- A. Nozari, A. Rahmati, Z. Shamsaei, A. P. Hashemi, M.-K. Layeghnejad and S. Zamaheni, J. Dent. Sch., Shahid Beheshti Univ. Med. Sci., 2015, 33, 66–73 Search PubMed.
- M. Iafisco, I. Foltran, S. Sabbatini, G. Tosi and N. Roveri, Bioinorg. Chem. Appl., 2012, 2012, 123953 Search PubMed.
- J. D. B. Featherstone and A. Lussi, in Dental Erosion, Karger, Basel, 2006, pp. 66–76 Search PubMed.
- H. Pan and B. W. Darvell, Cryst. Growth Des., 2010, 10, 845–850 CrossRef CAS.
- R. Chuong, J. Dent. Res., 1973, 52, 911–914 CrossRef PubMed.
- M. J. Larsen and S. J. Jensen, Arch. Oral Biol., 1989, 34, 957–961 CrossRef CAS PubMed.
- J. Zheng, F. Xiao, L. M. Qian and Z. R. Zhou, Tribol. Int., 2009, 42, 1558–1564 CrossRef CAS.
- E. C. Reynolds, J. Dent. Res., 1997, 76, 1587–1595 CrossRef CAS PubMed.
- N. Cochrane, S. Saranathan, F. Cai, K. Cross and E. Reynolds, Caries Res., 2008, 42, 88–97 CrossRef CAS PubMed.
- L. Chow, Monogr. Oral Sci., 2001, 18, 94–111 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD spectrum of CPNPs, calcium concentrations in each calcium phosphate nanomaterial, surface roughness of HA discs after remineralisation and summary of experimental steps. See DOI: 10.1039/d1nr05378g |
| This journal is © The Royal Society of Chemistry 2021 |