
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise control over supramolecular nanostructures via manipulation of H-bonding in π-amphiphiles†
Amrita
Sikder
 a,
Yujie
Xie
a,
Marjolaine
Thomas
a,
Matthew J.
Derry
b and
Rachel K.
O'Reilly
*a
a,
Yujie
Xie
a,
Marjolaine
Thomas
a,
Matthew J.
Derry
b and
Rachel K.
O'Reilly
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, Birmingham, UK. E-mail: r.oreilly@bham.ac.uk
bAston Institute of Materials Research, Aston University, Aston Triangle, Birmingham, B4 7ET, UK
First published on 30th November 2021
Abstract
Self-assembled supramolecular architectures are ubiquitous in nature. A synchronized combination of dynamic noncovalent interactions is the major driving force in forming unique structures with high-precision control over the self-assembly of supramolecular materials. Herein, we have achieved programmable nanostructures by introducing single/multiple H-bonding units in a supramolecular building block. A diverse range of nanostructures can be generated in aqueous medium by subtly tuning the structure of π-amphiphiles. 1D-cylindrical micelles, 2D-nanoribbons and hollow nanotubes are produced by systematically varying the number of H-bonding units (0–2) in structurally near identical π-amphiphiles. Spectroscopic measurements revealed the decisive role of H-bonding units for different modes of molecular packing. We have demonstrated that a competitive self-assembled state (a kinetically controlled aggregation state and a thermodynamically controlled aggregation state) can be generated by fine tuning the number of noncovalent forces present in the supramolecular building blocks. The luminescence properties of conjugated dithiomaleimide (DTM) provided insight into the relative hydrophobicity of the core in these nanostructures. In addition, fluorescence turn-off in the presence of thiophenol enabled us to probe the accessibility of the hydrophobic core in these assembled systems toward guest molecules. Therefore the DTM group provides an efficient tool to determine the relative hydrophobicity and accessibility of the core of various nanostructures which is very rarely studied in supramolecular assemblies.
Introduction
Self-assembly1–4 is one of the most fundamental characteristics of life. Nature uses self-assembly for building supramolecular materials possessing fascinating properties (self-healing, adaptive, reconfigurable and responsive) that are crucial for complex biological functions such as construction of cell membranes, protein folding and DNA double helix formation.5–7 Understanding the self-assembly processes of biological systems facilitates the fabrication of novel supramolecular materials8–13 and vice versa. Supramolecular assemblies fabricated by a hierarchical organization of the amphiphilic molecules are attracting increasing interest as platforms for building innovative materials. These materials can possess similar bioinspired dynamic properties and have been studied in the context of self-assembly and biological function with potential applications in drug delivery,14 tissue engineering,15 biosensing,16 antimicrobial activity,17 and gene delivery vehicles.18 Structurally diverse amphiphiles have been explored to find fascinating architectures.19,20 Among them, π-amphiphiles21–24 are of great interest as they can generate functional nanostructures with additional fascinating electronic and optical properties due to the presence of a π-chromophore. However, in most examples the design of amphiphiles, including π-amphiphiles, relies on inefficient and laborious trial-and-error approaches. π–π interactions25–28 are inherently weak and the overall assembly is primarily governed by the packing parameter,29,30 which depends on the ratio of hydrophobic and hydrophilic segments. This leads to immiscibility-driven uncontrolled aggregation by non-directional hydrophobic collapse which does not provide any molecular level precision. Therefore, it has been a challenging task to precisely control the size, shape and surface functional group display of a self-assembled system and the key question remains: how to control the self-assembly properties? Scientists have long envisioned that the introduction of H-bonding,31 one of the strongest noncovalent interactions, is a promising strategy to achieve a higher degree of control over supramolecular architectures due to its highly selective and directional nature.32–39In this context, Meijer and co-workers40 have reported a H-bonding mediated 1D-supramolecular assembly of amphiphilic benzene-1,3,5-tricarboxamide (BTA) derivatives where the molecules without H-bonding did not form similar morphologies, indicating the specific role of H-bonding in directing supramolecular nanostructures. In a further study41 they demonstrated the effect of the optimal position of the –COOH group in BTA amphiphiles for exhibiting polymorphism. In a different design, Ghosh and co-workers demonstrated the effect of the presence/absence and nature of the H-bonding group as well as the precise position of the H-bonding unit on the morphology and functional group display in bola-amphiphilic systems.42–44 Our group has reported a H-bonding-driven co-assembly in adenine and thymine appended amphiphilic block-copolymers and highlighted the effect of complementary multiple H-bonding in controlling the nanostructure morphology and surface chemistry.45–47
Based on these works, we aimed to further explore the effect of multiple H-bonding units on the self-assembly of π-amphiphiles. We have synthesized three amphiphiles by systematically varying the number of H-bonding units from zero up to two (Fig. 1a). Naphthalene diamide (NDI) was used as a π-chromophore as the NDI unit is synthetically highly versatile and exhibits geometrical and electronic properties.48,49 An oligo-oxy aryl group was attached to one arm of NDI to impart water solubility, whereas the other arm of NDI was attached to hydrophobic dithiomaleimide (DTM). DTMs have been reported50 as having excellent fluorescence properties along with solvatochromism. They have been successfully utilized for polymer and protein conjugation,51–53 and due to their small size and intermediate polarity, no detrimental effect on the secondary structure of the protein or polymer self-assembly was observed. It was also demonstrated that DTMs do not self-quench but enhance the emission in the aggregated state in polymer micelles.54
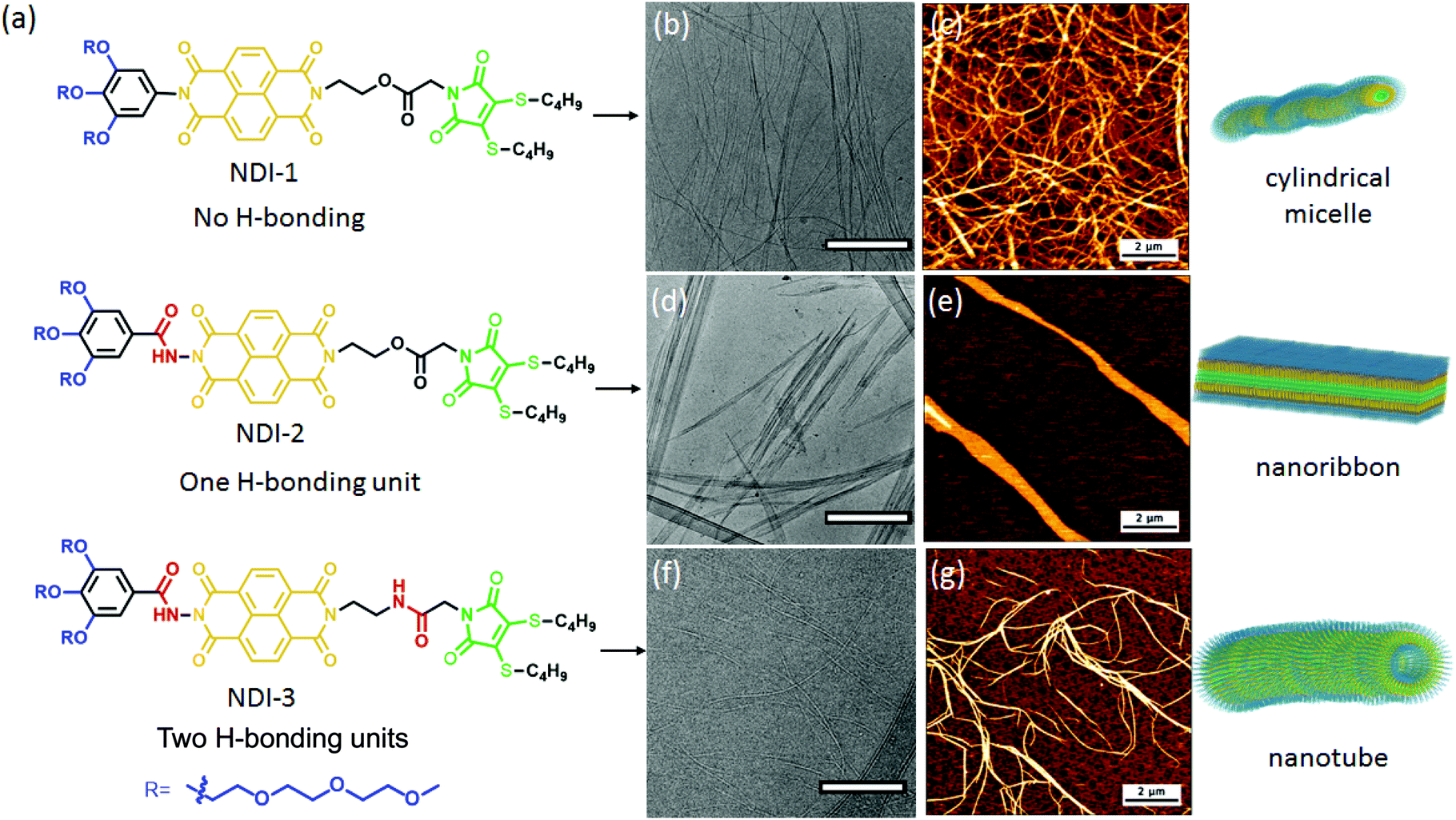 | ||
| Fig. 1 (a) Structures of NDI-amphiphiles; representative cryo-TEM (b, d, and f; scale bar-500 nm) and AFM images (c, e, and g; scale bar-2 μm) of NDI-1, NDI-2 and NDI-3, respectively. Cryo-TEM samples were prepared in water at 0.5 mM concentration and an AFM solution was prepared at 0.25 mM concentration. | ||
We envisaged that this scaffold can provide an opportunity to create versatile luminescent nanostructures by the interplay of π-stacking, H-bonding with the additional benefit of creating smart nanomaterials by virtue of solvatofluorism, and the chemico-fluorescence properties of the DTM unit. Furthermore, by taking advantage of the chemico-fluorescence behaviour of the DTM unit and monitoring the kinetics of the thiol-exchange reaction55 it should be possible to probe the accessibility of the hydrophobic core in the presence/absence of H-bonding in supramolecular assemblies.
Results and discussion
NDI-1, NDI-2, NDI-3 and water soluble maleimide DTM-1 were synthesized according to the routes shown in the ESI (Schemes S1–S4†) and characterized by NMR (Fig. S1–S8†), HRMS and CHN analysis. The self-assembly was investigated in aqueous medium and the detailed sample preparation procedure is described in the ESI.†To evaluate the self-assembly, first we investigated the morphology of the aqueous assemblies by electron microscopy. The cryogenic transmission electron microscopy (cryo-TEM) images of NDI-1 with no H-bonding (Fig. 1b and S9†) showed micrometer-long cylindrical micelles having a width of 6.5 ± 0.5 nm. The formation of one-dimensional supramolecular polymers were further confirmed by dry state TEM (Fig. S10†) and atomic force microscopy (AFM) (Fig. 1c and Fig. S11† for the height profile image). Interestingly, by introducing a single H-bonding unit (NDI-2) we could perturb the resulting nanostructure. The cryo-TEM (Fig. S12†) and dry state TEM (Fig. S13†) images of a fresh solution of NDI-2 showed spherical micelles, whereas aged solutions showed two-dimensional nanoribbons (Fig. 1d, e and S14†). Time dependent AFM images (Fig. 2 and Fig. S15†) revealed the gradual formation of nanoribbons over 12–14 days (0.5 mM, 20 °C) and once formed the nanoribbon structures were stable without any further reorganization. Morphological evolution over time was reconfirmed by a time dependent HRTEM study (Fig. S16†), whereas a time dependent DLS study showed a gradual increase in the size over time (Fig. S17†) further supporting the electron microscopy data. The time taken for complete conversion was found to be inversely proportional56 to the concentration indicating the formation of kinetically trapped spheres which subsequently transformed to energetically favourable more ordered stable nanoribbon structures.57 The width of the two-dimensional micrometer-long ribbons were estimated to be approximately 300–350 nm (exact width determination was difficult as the nanoribbons were partially folded in cryo-TEM images). The height measurement of the AFM images showed a membrane thickness of ∼ 5 nm indicating bilayer membranes (Fig. S18;† the length of fully stretched NDI-2 molecules is ≈ 3.4 nm). Intriguingly, with the introduction of the second H-bonding unit, NDI-3 formed a different morphology from the other two displaying micrometer-long rigid and hollow tubular structures as revealed by cryo-TEM (Fig. 1f and S19†), dry state TEM (Fig. S20†) and atomic force microscopy AFM images (Fig. 1g and Fig. S21† for the height profile image). The width of the nanotubes was measured to be 15 ± 1 nm with a wall thickness of 5.3 ± 0.5 nm from cryo-TEM. Fluorescent supramolecular polymers were visualized by confocal microscopy showing micrometer-long green nanostructures (Fig. S22†) further supporting the morphology obtained by electron microscopy. Although NDI-2 was transformed from initial spheres to thermodynamically stable nanoribbons, no such morphological evolution (monitored for 90 days) was observed for NDI-1 and NDI-3 as evidenced by time dependent AFM images (Fig. S23 and S24†). The sizes of the nanostructures obtained by different measurements are summarized in Table S1 in the ESI.†
 | ||
| Fig. 2 Time dependent AFM images obtained from an aqueous solution of NDI-2. (0.5 mM solution was aged at 20 °C and diluted twice before drop-casting on mica plates). | ||
Small-angle X-ray scattering (SAXS) patterns were also recorded (Fig. 3a) to analyse the self-assembly behaviour of these NDI-amphiphiles in aqueous solution (NDI-2 aged solution). In support of TEM and AFM observations, initial observations indicated that the SAXS patterns recorded for NDI-1 and NDI-3 were similar, and each was significantly different from that of NDI-2. Indeed, NDI-1 and NDI-3 were successfully fitted to a well-established worm-like micelle model58,59 (see overlaid data on the experimental SAXS data in Fig. 3a). It is important to note that the SAXS analysis performed was not sensitive to the hollow nature of the NDI-3 nanotubes due to the very small lumen (estimated by TEM analysis to be <4 nm). From these SAXS data fits, the mean width and mean length of the elongated NDI-1 and NDI-3 nanoparticles were determined, as well as the volume occupied by the hydrophilic (Vhydrophilic) and hydrophobic (Vhydrophobic) segments of the assembled amphiphiles was determined. The mean widths of both NDI-1 and NDI-3 were similar (8.5 nm and 8.8 nm, respectively), but the apparent mean length of these nano-objects appeared to be significantly different (292 nm and >1500 nm, respectively). However, the accessible q-range did not allow for accurate determination of this mean length, which may account for the discrepancy between SAXS and TEM analyses. The hydrophilic/hydrophobic balance was also similar in each case, with SAXS fitting indicating 85 vol% and 83 vol% hydrophobic character in the NDI-1 and NDI-3 nano-assemblies, respectively. Interestingly, an additional interaction peak at q ∼ 0.076 Å−1 was visible in SAXS data recorded for NDI-1 cylindrical micelles, which corresponds to a length scale of ∼8.2 nm. This may represent interactions between neighbouring nanoparticles with a mean cylinder–cylinder centre-to-centre distance of ∼16.4 nm,60 but this could not be confirmed by modelling due to the complexities of interactions between non-spherical nano-objects which require analysis at a wide range of concentrations (NDI-1 can only be homogeneously dispersed up to 3 mM concentration). Instead, this peak was accounted for in the model by adding a Gaussian peak (see the ESI† for details). In the case of NDI-3, nanoparticle aggregation was evident as indicated by increased scattering intensities at low q values, which was accounted for by introducing a power law (see the ESI† for details). Attempts to model SAXS data obtained for the more complex nanoribbon morphology adopted by NDI-2 amphiphiles were unsuccessful.
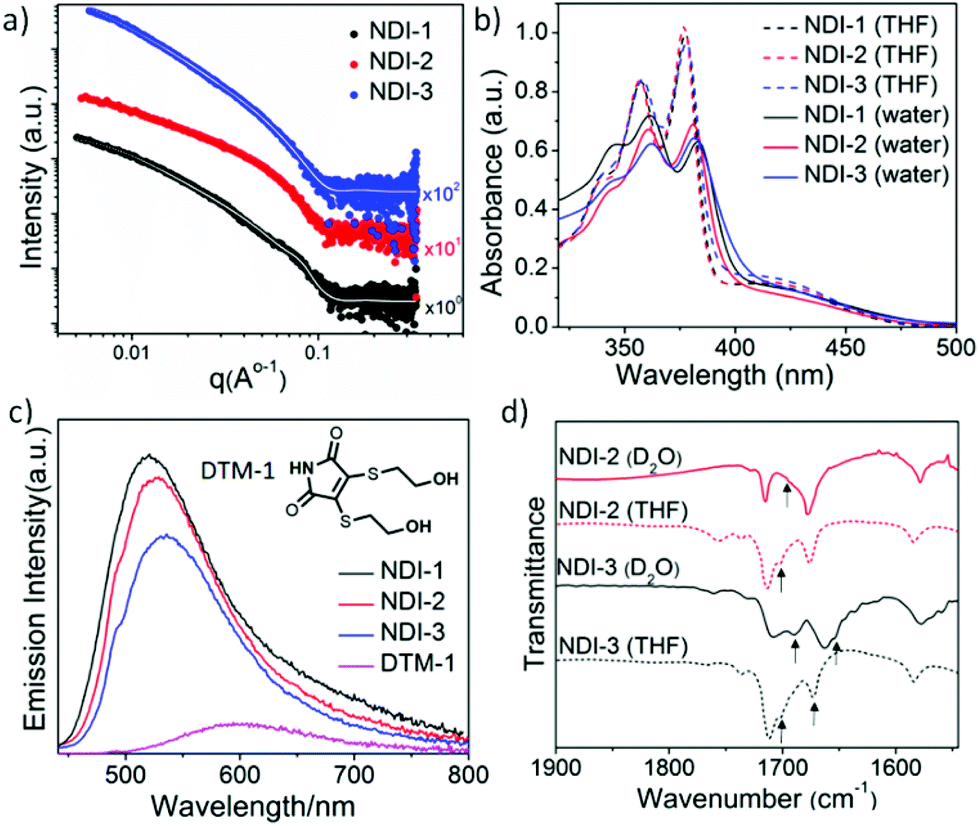 | ||
| Fig. 3 (a) Small-angle X-ray scattering patterns and the corresponding data fits (white lines) for a 3 mM aqueous solution; (b) solvent dependent UV/Vis spectra (l = −0.1 cm); (c) comparison of fluorescence spectra (λex = −420 nm, slit-2); and (d) stack plot of solvent dependent IR spectra; UV/Vis, fluorescence and IR spectra were taken in 0.5 mM solution (aged for 3 weeks at 20 °C prior to taking spectra). | ||
Distinctly different morphologies for NDI-1, NDI-2 and NDI-3, having a comparable hydrophobic–hydrophilic ratio, indicated the strong influence of H-bonding on the self-assembly of these π-amphiphiles. Furthermore, it was observed that the stability of the nanostructure can be altered by tuning the number of noncovalent forces involved in the self-assembly process. While morphological transformation was observed for NDI-2, NDI-3, having two H-bonding units, and NDI-1, lacking any H-bonding unit, formed thermodynamically stable nanostructures which were unaltered over time. A possible explanation could be that NDI-1, with no intermolecular H-bonding, self-assembles by relatively weaker π–π stacking, hence it has inadequate force for achieving efficient kinetic trapping. On the other hand, NDI-3, by virtue of having two H-bonding units, exerts strong noncovalent forces that lead to static, stable assemblies.61,62 In addition, the increased solubility of NDI-3 in aqueous medium due to the presence of two additional amide bonds provides adequate time to adopt preferential packing, leading to a thermodynamically stable state without kinetic trapping. But, for NDI-2, the presence of one H-bonding unit enables an intermediate supramolecular interaction (an initial hydrophobic effect leading to spherical micelles but over time the hydrogen bonding drives the reorganisation into ribbons); thus the kinetic control becomes an important factor in regulating the assembly.63,64
To elucidate the molecular packing in different nanostructures we probed the nature of the intermolecular interaction by UV/Vis spectra using aged solutions having a stable morphology (Fig. 3b). In a good solvent THF, sharp absorption bands (λmax = 378, 358 and 340 nm corresponding to the π–π* transition) were noted indicating monomeric NDI,65 whereas in H2O, a red-shifted absorption band, together with a significant hypochromic shift, was observed, indicating π–π stacking in water for all the amphiphiles. Comparison of the fluorescence data (Fig. 3c) of NDI-amphiphiles with water-soluble DTM-1 provided more insight into molecular packing in the assembled state. The emission of DTM-1 in water was mostly quenched with a 95 nm red shift compared to the emission of DTM-1 in THF (Fig. S25†).50 Quenching of emission is attributed to H-bonding induced proton transfer between the protic solvent water and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of maleimide.66 In contrast, the aqueous solution of NDI-1/2/3 showed intense emission compared to the aqueous solution of DTM-1 and the emission maxima were rather blue-shifted to 520 nm, 525 nm and 534 nm relative to the emission spectra in THF for NDI-1, NDI-2 and NDI-3, respectively (Fig. 3c, for the emission spectra in THF see ESI Fig. S26†). Significant emission in water confirms that NDI-amphiphiles in all the nanostructures’ (cylindrical micelles/nanoribbons/nanotubes) maleimide moiety remain in a hydrophobic microenvironment and are shielded from water. It is known that the emission wavelength and the intensity of the DTM moiety are closely related to the microenvironment67,68 and the different values of blue-shifted spectra for the NDI 1/2/3 assembly indicate the degree of hydrophobicity of the maleimide microenvironment; this varies (NDI-1 having the most hydrophobic core while NDI-3 has the least hydrophobic core) as a consequence of H-bonding. Intermolecular H-bonding in aqueous medium was confirmed by a solvent dependent FT-IR study (Fig. 3d). Peaks at 1701 cm−1 and 1674 cm−1 (CO stretching of imide carbonyl also appears at the same position) in the FTIR spectra of the THF solution of NDI-3 are assigned to the nonbonded CO stretching of hydrazide and amide, respectively. The FT-IR spectrum of NDI-1 (Fig. S27†) devoid of the H-bonding moiety allowed the peak assignment. The amide peak shifted to 1655 cm−1 and the hydrazide peak shifted to 1689 cm−1 in D2O confirming the intermolecular H-bonding between the amides and hydrazides, respectively, in aqueous solution. Similarly, for NDI-2 the sharp band at 1701 cm−1 shifted to 1687 cm−1 in D2O confirming the intermolecular H-bonding between the hydrazides. H-bonding was further supported by monitoring the N–H stretching band (Fig. S28†), which showed a significant shift (from 3500 cm−1 to 3410 cm−1 for NDI-3 and 3502 cm−1 to 3375 cm−1 for NDI-2) going from THF to D2O. A relatively broad peak was observed for NDI-3 as both N–H from the amide and hydrazide groups merged into a single broad peak. Time dependent UV/Vis spectroscopy was performed in order to have more insight into molecular packing during the morphological transformation of NDI-2. The absorption band at 425 nm (Fig. S29†) was found to decrease with time and finally saturate after 13 days. This can be attributed to the rearrangement of the NDI-chromophore from the initially formed slipped stacked (J-aggregate) into a more ordered cofacial stack (H-aggregate) leading to a nanoribbon structure.69–71 The time dependent fluorescence spectra (Fig. S30†) showed a decrease in the emission intensity of maleimide emission with a concomitant blue shift (6 nm) reconfirming chromophoric rearrangement during the morphological transformation.
O group of maleimide.66 In contrast, the aqueous solution of NDI-1/2/3 showed intense emission compared to the aqueous solution of DTM-1 and the emission maxima were rather blue-shifted to 520 nm, 525 nm and 534 nm relative to the emission spectra in THF for NDI-1, NDI-2 and NDI-3, respectively (Fig. 3c, for the emission spectra in THF see ESI Fig. S26†). Significant emission in water confirms that NDI-amphiphiles in all the nanostructures’ (cylindrical micelles/nanoribbons/nanotubes) maleimide moiety remain in a hydrophobic microenvironment and are shielded from water. It is known that the emission wavelength and the intensity of the DTM moiety are closely related to the microenvironment67,68 and the different values of blue-shifted spectra for the NDI 1/2/3 assembly indicate the degree of hydrophobicity of the maleimide microenvironment; this varies (NDI-1 having the most hydrophobic core while NDI-3 has the least hydrophobic core) as a consequence of H-bonding. Intermolecular H-bonding in aqueous medium was confirmed by a solvent dependent FT-IR study (Fig. 3d). Peaks at 1701 cm−1 and 1674 cm−1 (CO stretching of imide carbonyl also appears at the same position) in the FTIR spectra of the THF solution of NDI-3 are assigned to the nonbonded CO stretching of hydrazide and amide, respectively. The FT-IR spectrum of NDI-1 (Fig. S27†) devoid of the H-bonding moiety allowed the peak assignment. The amide peak shifted to 1655 cm−1 and the hydrazide peak shifted to 1689 cm−1 in D2O confirming the intermolecular H-bonding between the amides and hydrazides, respectively, in aqueous solution. Similarly, for NDI-2 the sharp band at 1701 cm−1 shifted to 1687 cm−1 in D2O confirming the intermolecular H-bonding between the hydrazides. H-bonding was further supported by monitoring the N–H stretching band (Fig. S28†), which showed a significant shift (from 3500 cm−1 to 3410 cm−1 for NDI-3 and 3502 cm−1 to 3375 cm−1 for NDI-2) going from THF to D2O. A relatively broad peak was observed for NDI-3 as both N–H from the amide and hydrazide groups merged into a single broad peak. Time dependent UV/Vis spectroscopy was performed in order to have more insight into molecular packing during the morphological transformation of NDI-2. The absorption band at 425 nm (Fig. S29†) was found to decrease with time and finally saturate after 13 days. This can be attributed to the rearrangement of the NDI-chromophore from the initially formed slipped stacked (J-aggregate) into a more ordered cofacial stack (H-aggregate) leading to a nanoribbon structure.69–71 The time dependent fluorescence spectra (Fig. S30†) showed a decrease in the emission intensity of maleimide emission with a concomitant blue shift (6 nm) reconfirming chromophoric rearrangement during the morphological transformation.
A proposed model of molecular packing to rationalize the observed microscopic image and spectroscopic results is shown in Scheme 1. NDI-1 molecules exert a head-to-head orientation ensuring π–π stacking, finally leading to flexible cylindrical micelles where the core consists of a hydrophobic butane chain substituted DTM moiety and the hydrophilic oligo-oxy chains remain in the shell (correlating closely with the observed thickness of the cylinders obtained by TEM), whereas NDI-2 forms a bilayer structure by utilizing intermolecular H-bonding between the hydrazides, along with π–π interaction among the NDI-chromophores. On the other hand, NDI-3, having an additional amide bond along with the hydrazide bond, ensures intermolecular H-bonding among the hydrazide groups and the amide groups together with π–π interactions in a bilayer arrangement similar to NDI-2, but the additional amide bonding present in NDI-3 introduces a secondary directional supramolecular interaction in the self assembly that might have resulted in folding of the bilayer membrane into nanotubes.
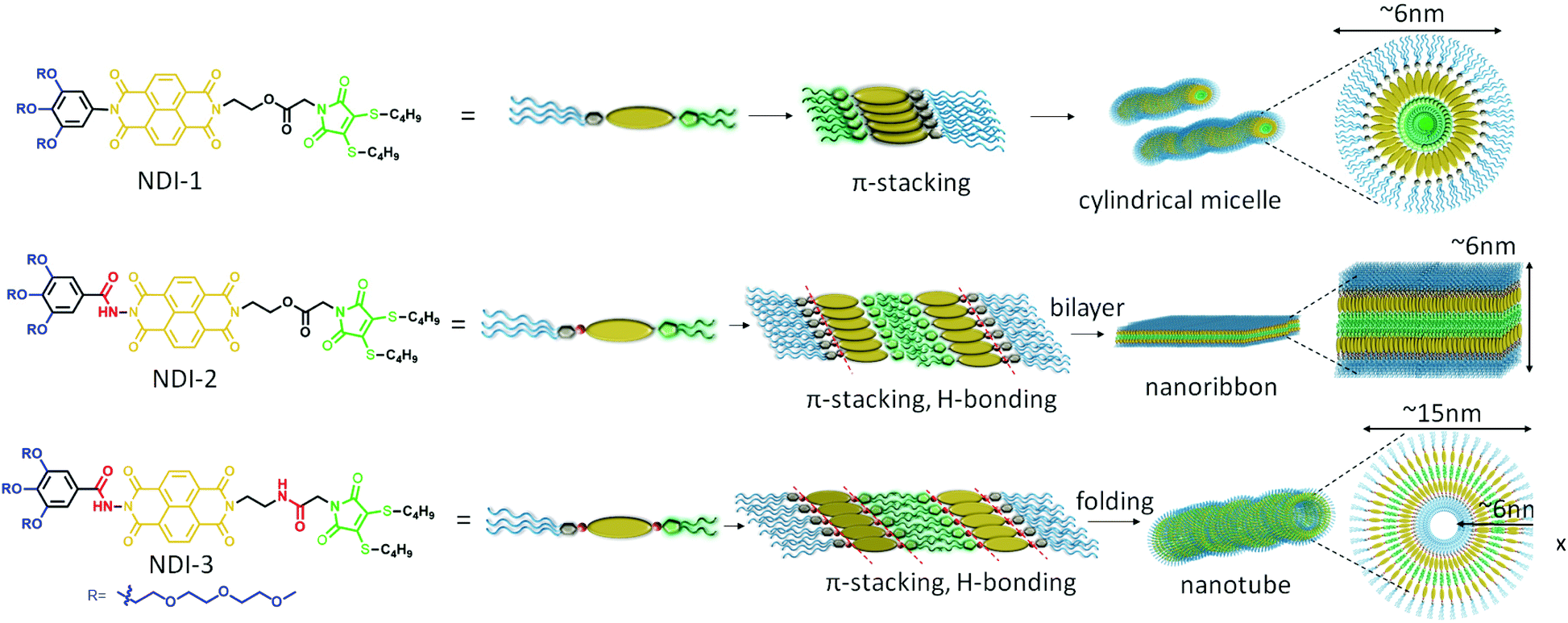 | ||
| Scheme 1 Schematic representation of proposed molecular packing for NDI-1, NDI-2 and NDI-3. | ||
In order to evaluate the effect of H-bonding and morphological variation, different self-assembly properties were studied. Concentration dependent UV/Vis studies (Fig. S31†) estimated the critical aggregation constant (CAC) to be lower than 5 × 10−6 M for all the assemblies, making them a potential material for biological study.14,18 Unfortunately, the exact CAC could not be determined as below 5 × 10−6 M, and the absorbance was unable to be detected by a UV/Vis spectrometer. The thermal stability was evaluated by a temperature dependent UV/Vis study (Fig. S32†), which revealed lower critical solution temperatures (LCSTs) between 73 and 76 °C for the assemblies, attributed to the dehydration of oligo-oxy ethylene chains.72 The quantum yields (Φf) of the NDI-amphiphiles and free dye DTM-1 in THF were calculated to be between 13 and 15% (Fig. S33†), whereas in water the quantum yields decreased (Φf = ∼ 7%) for all the NDI-amphiphiles. On the other hand, the quantum yield of DTM-1 in water was found to be 0.1% and this result corroborates well with our previous finding of fluorescence quenching in polar protic solvents.42 In NDI-assemblies, the DTM moiety prefers to remain inside the hydrophobic microenvironment and therefore should produce higher quantum yields compared to the free maleimide dye in water. However, the Φf values were lower than in THF indicating that the hydrophobic pocket is not completely insulated from aqueous environments, which is the case for macromolecular self-assemblies.51 The possibility of aggregation-induced quenching was eliminated by performing a set of control experiments with NDI-1. NDI-1 was found to form reverse micelles (Fig. S34†) in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 cyclohexane/dichloromethane. Intermolecular π–π stacking was confirmed by UV/Vis spectroscopy (Fig. S35†) and the Φf value was found to be ∼ 13%. Similar Φf values were obtained in organic solvents for both free and aggregated dye molecules confirming that the quenching observed in aqueous media is likely due to the ingress of water molecules.
:5 cyclohexane/dichloromethane. Intermolecular π–π stacking was confirmed by UV/Vis spectroscopy (Fig. S35†) and the Φf value was found to be ∼ 13%. Similar Φf values were obtained in organic solvents for both free and aggregated dye molecules confirming that the quenching observed in aqueous media is likely due to the ingress of water molecules.
DTM can undergo thiol-exchange reactions with externally added thiols, resulting in the elimination of the original thiol ligands and the addition of two new thiol ligands, and with the addition of aromatic thiols it is possible to turn off the fluorescence.55 Indeed, thiophenol-containing DTM molecules are known to exert drastically reduced emission and by performing a thiol-exchange reaction on an emissive DTM-functional moiety present in a supramolecular polymer with thiophenol, it should be possible to achieve an ON-to-OFF switching of fluorescence (Fig. 4a). The rate of reaction can provide additional insight into the accessibility of the hydrophobic core in these nanostructures. To demonstrate this, an excess amount of thiophenol was added to an aqueous solution of NDI-1/2/3 and reduction in the emission was monitored over time. The reaction kinetics (Fig. 4b) were found to be rather slow in the assembled state (t1/2 = 120, 45 and 32 min for NDI-1/2/3, respectively) in comparison to the monomeric state in THF (t1/2 = 5.5 min), suggesting a different extent of penetration of the guest molecule for different nanostructures. Furthermore, an increase in the reaction rate in the presence of H-bonding suggested that along with increasing the overall polarity of the molecular system, the intermolecular H-bonding in aqueous medium loosens the π-stacking,42 making access easier through the rigid π-wall to the hydrophobic core for an external ligand. This corroborates well with the trends observed from fluorescence spectroscopy.
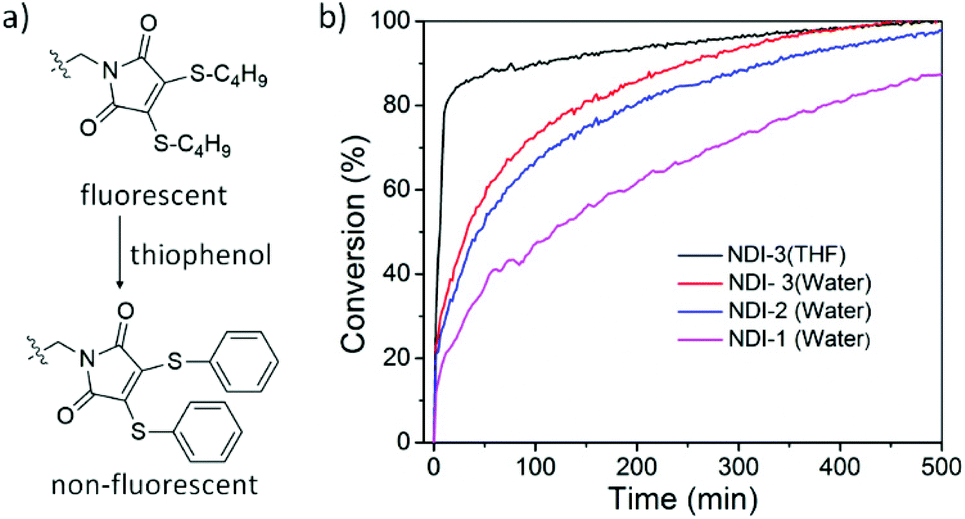 | ||
| Fig. 4 (a) Representative thiol exchange reaction with thiophenol; (b) conversion vs. time as measured by (concentration 0.5 mM; aged for three weeks at 20 °C). The emission of the solution was monitored during the reaction performed at 20 °C by fluorescence spectroscopy; 10 μL of 1 M thiophenol dissolved in DMF was added to 2 mL of NDI-maleimide aqueous solution. | ||
To characterize the particle morphology after the thiol exchange reaction, AFM images were taken with the thiophenol-incubated final solutions. 2D-nanosheets with different aspect ratios (Fig. S36†) were observed for all the three thiophenol modified amphiphiles confirming the bilayer structure. We propose that the formation of 2D sheets is likely as a consequence of increased π–π interaction due to the presence of two additional benzene rings. However, as a relatively larger area of the nanosheets generated by thiophenol modified NDI-2 and NDI-3 compared to NDI-1 further highlights the role of H-bonding in self-assembly.
Conclusions
In summary, we have demonstrated the impact of H-bonding on regulating the nanostructure of supramolecular polymers in aqueous medium and its effect on the accessibility of the hydrophobic core in the resultant nanostructures. We have prepared three new near identical π-amphiphiles with a varying number of H-bonding units. Distinctly different supramolecular assemblies for each revealed the decisive role of H-bonding units along with π–π interaction in determining the final nanostructure. NDI-1 without any H-bonding unit formed cylindrical micelles, whereas NDI-2 having a single hydrazide bonding generated 2D nanoribbon structures. The addition of another H-bonding unit led to folding of the nanoribbons to turn into nanotubes. Intriguingly, NDI-1 and NDI-3 having zero and two H-bonding units, respectively, formed stable nanostructures, whereas NDI-2 – having a single H-bond – initially formed kinetically trapped spherical micelles which further undergo a morphological transformation to flat nanoribbons. This observation demonstrated the possibility of forming kinetically trapped assemblies by fine tuning the number of noncovalent forces present in the supramolecular building blocks. Spectroscopy studies provided detailed insights into molecular packing. Promising features of these supramolecular assemblies such as very low CAC, thermal stability, polarity sensitive fluorescence and chemico-fluorescence responsive properties by a thiol-exchange reaction have been illustrated. Furthermore, the kinetics of a thiol-exchange reaction provided insight into the accessibility of the hydrophobic core in different nanostructures, which is rarely studied in a supramolecular assembly. We believe that these findings will open new directions/avenues to explore the self-assembled functional aqueous materials of π-amphiphiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS and ROR acknowledge funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant (agreement no 897666) and the University of Birmingham.References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular biology of the cell, Garland Science, New York, 4th edn, 2002 Search PubMed.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- H. Watson, Essays Biochem., 2015, 59, 43–70 CrossRef PubMed.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- C. C. Piras, P. Slavik and D. K. Smith, Angew. Chem., Int. Ed., 2020, 59, 853–859 CrossRef CAS PubMed.
- Supramolecular chemistry: From molecules to nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012 Search PubMed.
- A. Sikder, S. Chakraborty, P. Rajdev, P. Dey and S. Ghosh, Acc. Chem. Res., 2021, 54, 2670–2682 CrossRef CAS PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- X. Yan, Y. R. Chen, Y. F. Song, J. Ye, M. Yang, B. B. Xu, J. Y. Zhang, X. Wang and J. K. Yu, Front. Bioeng. Biotechnol., 2020, 8, 847 CrossRef PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- X. Li, H. Bai, Y. Yang, J. Yoon, S. Wang and X. Zhang, Adv. Mater., 2019, 31, 1805092 Search PubMed.
- K. Miyata, N. Nishiyamaa and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed.
- R. Matmour, I. De Cat, S. J. George, W. Adriaens, P. Leclère, P. H. H. Bomans, N. A. J. M. Sommerdijk, J. C. Gielen, P. C. M. Christianen, J. T. Heldens, J. C. M. van Hest, D. W. P. M. Löwik, S. De Feyter, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2008, 130, 14576–14583 CrossRef CAS PubMed.
- S. K. Albert, M. Golla, N. Krishnan, D. Perumal and R. Varghese, Acc. Chem. Res., 2020, 53, 2668–2679 CrossRef CAS PubMed.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- M. R. Molla and S. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 26672–26683 RSC.
- X. Feng, W. Pisula, L. Zhi, M. Takase and K. Müllen, Angew. Chem., 2008, 120, 1727–1730 CrossRef.
- J. P. Hill, W. Jin, A. Kosaka, T. I. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- L. Chen, X. Dou, W. Pisula, X. Yang, D. Wu, G. Floudas, X. Feng and K. Müllen, Chem. Commun., 2012, 48, 702–704 RSC.
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS PubMed.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 151683 Search PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- A. Sikder and S. Ghosh, Mater. Chem. Front., 2019, 3, 2602–2616 RSC.
- Hydrogen bonded supramolecular structures, ed. Z.-T. Li and L.-Z. Wu, Springer, 2015 Search PubMed.
- A. T. ten Cate, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 3801–3808 CrossRef CAS PubMed.
- J.-F. Xu, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Org. Lett., 2014, 16, 4016–4019 CrossRef CAS PubMed.
- J. Liu, M. J. G. Schotman, M. M. R. M. Hendrix, X. Lou, P. P. M. S. Román, I. K. Voets and R. P. Sijbesma, J. Polym. Sci., 2021, 59, 1162–1170 CrossRef CAS.
- T. T. T. Myllymäki, Nonappa, H. Yang, V. Liljeström, M. A. Kostiainen, J.-M. Malho, X. X. Zhub and O. Ikkala, Soft Matter, 2016, 12, 7159–7165 RSC.
- K. Bertula, Nonappa, T. T. T. Myllymäki, H. Yang, X. X. Zhu and O. Ikkala, Polymer, 2017, 126, 177–187 CrossRef CAS.
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- N. M. Matsumoto, R. M. Lafleur, X. Lou, K.-C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. Van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed.
- P. Rajdev, M. R. Molla and S. Ghosh, Langmuir, 2014, 30(8), 1969–1976 CrossRef CAS PubMed.
- A. Sikder, A. Das and S. Ghosh, Angew. Chem., 2015, 127, 6859–6864 CrossRef.
- A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1606–1611 CrossRef CAS PubMed.
- Z. Hua, A. Pitto-Barry, Y. Kang, N. Kirby, T. R. Wilks and R. K. O'Reilly, Polym. Chem., 2016, 7, 4254–4262 RSC.
- Z. Hua, R. Keogh, Z. Li, T. R. Wilks, G. Chen and R. K. O'Reilly, Macromolecules, 2017, 50, 3662–3670 CrossRef CAS PubMed.
- Z. Hua, J. R. Jones, M. Thomas, M. C. Arno, A. Souslov, T. R. Wilks and R. K. O'Reilly, Nat. Commun., 2019, 10, 5406–5413 CrossRef PubMed.
- S. V. Bhosale, C. H. Janiab and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- M. A. Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef CAS PubMed.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O'Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
- M. P. Robin, J. E. Raymond and R. K. O'Reilly, Mater. Horiz., 2015, 2, 54–59 RSC.
- K. Renault, J. W. Fredy, P.-Y. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS PubMed.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- M. P. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518–9524 CrossRef CAS PubMed.
- M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC.
- Time taken for complete morphological transformation as observed by AFM: ∼14 days for 0.5 mM, ∼10 days for 0.25 mM solution and ∼5 days for 0.1 mM solution.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- Neutrons, X-rays and light: scattering methods applied to soft condensed matter, ed. T. Zemb and P. Lindner, Elsevier, 2002, 391–420 Search PubMed.
- L. J. Prins, E. E. Neuteboom, V. Paraschiv, M. Crego-Calama, P. Timmerman and D. N. Reinhoudt, J. Org. Chem., 2002, 67, 4808–4820 CrossRef CAS PubMed.
- T. F. A. de Greef, G. B. W. L. Ligthart, M. Lutz, A. L. Spek, E. W. Meijer and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 5479–5486 CrossRef CAS PubMed.
- Y. Tidhar, H. Weissman, S. G. Wolf, A. Gulino and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 6068–6075 CrossRef CAS PubMed.
- E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 8819–8821 CrossRef CAS PubMed.
- H. Shao, T. Nguyen, N. C. Romano, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2009, 131, 16374–16376 CrossRef CAS PubMed.
- M. Staniforth, W. D. Quan, T. N. V. Karsili, L. A. Baker, R. K. O'Reilly and V. G. Stavros, J. Phys. Chem. A, 2017, 121, 6357–6365 CrossRef CAS PubMed.
- A. B. Mabire, Q. Brouard, A. Pitto-Barry, R. J. Williams, H. Willcock, N. Kirby, E. Chapman and R. K. O'Reilly, Polym. Chem., 2016, 7, 5943–5948 RSC.
- S. Jimaja, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2021, 12, 105–112 RSC.
- M. Kumar and S. J. George, Chem. – Eur. J., 2011, 17, 11102–11106 CrossRef CAS PubMed.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS PubMed.
- E. C. C. Goh and H. Stoever, Macromolecules, 2002, 35, 9983–9989 CrossRef CAS.
- V. Grande, B. Soberats, S. Herbst, V. Stepanenkoa and F. Wurthner, Chem. Sci., 2018, 9, 6904–6911 RSC.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, additional experimental details and spectroscopy and microscopy data. See DOI: 10.1039/d1nr04882a |
| This journal is © The Royal Society of Chemistry 2021 |