
Observation of quantum-confined exciton states in monolayer WS2 quantum dots by ultrafast spectroscopy†
Shu-Wen
Zheng
a,
Lei
Wang
 *a,
Hai-Yu
Wang
*a,
Chen-Yu
Xu
a,
Yang
Luo
b and
Hong-Bo
Sun
c
*a,
Hai-Yu
Wang
*a,
Chen-Yu
Xu
a,
Yang
Luo
b and
Hong-Bo
Sun
c
aState Key Laboratory of Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: haiyu_wang@jlu.edu.cn
bChangchun Institute of Optics, Fine Mechanics and Physics, Chinese Academy of Sciences, Changchun 130033, China
cState Key Laboratory of Precision Measurement Technology and Instruments, Department of Precision Instrument, Tsinghua University, Haidian, Beijing 100084, China
First published on 17th September 2021
Abstract
Monolayer transition metal dichalcogenide quantum dots (TMDC QDs) could exhibit unique photophysical properties, because of both lateral quantum confinement effect and edge effect. However, there is little fundamental study on the quantum-confined exciton dynamics in monolayer TMDC QDs, to date. Here, by selective excitations of monolayer WS2 QDs in broadband transient absorption (TA) spectroscopy experiments, the excitation-wavelength-dependent ground state bleaching signals corresponding to the quantum-confined exciton states are directly observed. Compared to the time-resolved photophysical properties of WS2 nanosheets, the selected monolayer WS2 QDs only show one ground state bleaching peak with larger initial values for the linear polarization anisotropy of band-edge excitons, probably due to the expired spin–orbit coupling. This suggests a complete change of the band structure for monolayer WS2 QDs. In the femtosecond time-resolved circular polarization anisotropy experiments, a valley depolarization time of ∼100 fs is observed for WS2 nanosheets at room temperature, which is not observed for monolayer WS2 QDs. Our findings suggest a strong state-mixing of band-edge valley excitons responsible for the large linear polarization in monolayer WS2 QDs, which could be helpful for understanding the exciton relaxation mechanisms in colloidal monolayer TMDC QDs.
Introduction
Two-dimensional (2D) transition metal dichalcogenides (TMDCs) (such as MoS2, WS2, WSe2, etc.) are promising building blocks for fabricating next-generation electronic, optoelectronic and valleytronic devices due to their extraordinary properties.1–3 First of all, 2D TMDCs have optical band gaps in the visible to near infrared band, which are suitable for photodetectors and photocatalysis.4 Secondly, unlike their indirect-band gap bulk counterparts, monolayer TMDCs further exhibit direct band gaps and optical transparency and are promising candidates in thin film transistors and solar cells.5,6 Last but not least, strong spin orbit coupling and lack of inversion symmetry in 2D TMDCs result in valley-selective optical properties, providing a new degree of freedom to control the materials properties.7–9 Therefore, 2D TMDCs also possess huge potential for valleytronics.The lateral dimension of the 2D TMDCs can be further reduced into nanosheets or quantum dots (QDs), which provides a new method to adjust the electronic band structures of 2D TMDCs.10,11 2D TMDC nanosheets and QDs are expected to not only have the intrinsic properties of the TMDC materials, but also may exhibit different photophysical properties of their film materials, because of the additional quantum confinement effect in lateral dimension and the edge effect, which could extend their applications in catalysis and biological aspects.12 For example, the good dispersion and chemical stability of 2D TMDC QDs are suitable for biological imaging and cancer physiotherapy.13,14 Besides, a large number of edge active sites of TMDC QDs are applicable to the field of catalysis.15,16 Wu's group has successfully synthesized MoS2 and WS2 QDs through the solvothermal treatment, and characterize their steady-state absorption and photoluminescence properties, where the strong fluorescence of TMDC QDs under UV light irradiations has been reported.10 Son's group and Cheon's group have developed a new method to produce highly uniform monolayer WS2 QDs, showing well-defined photoluminescence spectra without ensemble heterogeneity, and observed that solvents have a strong influence on the optical properties of TMDC QDs.11,17 However, there is little fundamental study on the quantum-confined exciton dynamics in monolayer TMDC QDs to date, which would hinder the further development of the optoelectronic applications based on 2D TMDC QDs.
In this paper, we use broadband femtosecond transient absorption (TA) spectroscopy to selectively excite monolayer WS2 QDs in the visible region. The excitation-wavelength-dependent ground state bleaching signals corresponding to the quantum-confined exciton states are directly observed in monolayer WS2 QDs. Compared to multiple-exciton-state WS2 nanosheets, only one ground state bleaching peak with large initial values for the linear polarization anisotropy for band-edge excitons of selected monolayer WS2 QDs implies a complete change of the band structure. We further find that the valley depolarization time of WS2 nanosheets is ∼100 fs at room temperature, and the valley polarization for monolayer WS2 QDs is not observed. Our findings could be helpful for understanding the exciton relaxation mechanisms in colloidal monolayer TMDC QDs.
Experimental
Sample preparation
The WS2 nanosheets and monolayer WS2 QDs were synthesized by a modified solvothermal method.10 Typically, 0.1 g of WS2 powder (2 μm, 99% Sigma-Aldrich) was added into 10 ml N-methylpyrrolidone (NMP) first, and it was sonicated using a cell disruptor for 3 hours with an output power of 300 W. In order to avoid high temperature during the ultrasonic treatments, ice bath conditions were required. When the sonication procedure was completed, the supernatant of WS2 dispersion was centrifuged at 1500 rpm for 45 minutes, and the top 3/4 of the dispersion was collected. Then, it was further centrifuged at 200 rpm for 45 minutes, and the top 1/2 of this dispersion was labeled as WS2 nanosheets. The dispersion of WS2 nanosheets was transferred into a flask and vigorously stirred for 6 hours at 140 °C. Finally, the resulting supernatant was centrifuged for 5 minutes at 3500 rpm to obtain monolayer WS2 QDs.Results and discussion
Fig. 1a shows the transmission electron microscopy (TEM) images of WS2 QDs, whose average size is 2.8 ± 0.9 nm, and the d-spacing is 2.7 Å corresponding to the (101) face of WS2. The thickness of WS2 QDs is ∼1 nm, according to the height profile shown in AFM images (Fig. S1a†). This suggests that most of the WS2 QDs are monolayers10 and their lateral dimension is comparable with the exciton Bohr radius of TMDC monolayers.18 For WS2 nanosheets, the thickness is ∼3 nm and the lateral size is ∼200 nm, according to the scanning electron microscopy (SEM) image (Fig. S2†), which is consistent with the AFM image of WS2 nanosheets (Fig. S1b†). Besides, we characterize the X-ray powder diffraction (XRD) spectra of WS2 nanosheets and QDs (Fig. S3†). WS2 nanosheets have a characteristic XRD peak at 2θ = 14.4° for the (002) reflection, while WS2 QDs exhibit no apparent XRD peak. The disappearance of the (002) reflection in the XRD patterns of TMDCs could occur when the layer number reduces from multilayer to monolayer.19 This further indicates that the synthesized WS2 nanosheets are multilayer, and WS2 QDs are mainly monolayer. The steady-state absorption spectrum of WS2 nanosheets shows four absorption peaks, which locates at 634 nm, 527 nm, 457 nm and 416 nm, respectively (Fig. 1b). The absorption peaks at 634 nm and 527 nm for WS2 nanosheets are attributed to the direct transitions at the K point, while the other absorption peaks at 457 nm and 416 nm are assigned to the indirect transitions at band-nesting areas between Γ and Q valleys in band structures.20 But all the typical exciton absorption peaks for WS2 nanosheets also disappear in monolayer WS2 QDs, which only show a smooth and structureless absorption curve in the UV-vis-near infrared spectral range. This could be attributed to the size-distribution effect for monolayer WS2 QDs, and the change of band structure induced by quantum confinement.21Fig. 1c shows the PL spectra of monolayer WS2 QDs under different excitation wavelengths (there is no PL signals for WS2 nanosheets). As the excitation wavelength increases from 350 nm to 380 nm, the PL intensity of monolayer WS2 QDs increases and its maximum value locates at 445 nm. When the excitation wavelength is further increased from 380 nm to 470 nm, the PL peaks of monolayer WS2 QDs gradually shift from 450 nm to 530 nm, exhibiting an excitation wavelength-dependent behavior.10 The multiple-peak PL of monolayer WS2 QDs could be related to the vibronic coupling states that might come from edge bonds.11 The PL excitation (PLE) spectra of monolayer WS2 QDs (Fig. 1d) also could reflect the exciton fine structures of corresponding PL peaks. The two PLE peaks at 320 nm and 360 nm in the UV range are responsible for the relatively stronger blue emissions (440–480 nm) of monolayer WS2 QDs. For the relatively weaker green and red emissions (500–620 nm) of monolayer WS2 QDs, the PLE spectra are broad and the contributions of the PLE peak at 320 nm and 360 nm become smaller and smaller, as the detection wavelengths gradually red-shift. This implies the size-distribution effect for monolayer WS2 QDs. | ||
| Fig. 1 (a) High resolution transmission electron microscopy (HRTEM) image of monolayer WS2 QDs. The inset of panel (a) is the size distribution of monolayer WS2 QDs. (b) Steady-state absorption spectra of the WS2 nanosheet and monolayer WS2 QDs in NMP. (c) Steady-state PL spectra of monolayer WS2 QDs in NMP under different excitation wavelengths. The vertical dashed line indicates the PL peak at 445 nm. (d) Corresponding PLE spectra for monolayer WS2 QDs in NMP probed at different emission wavelengths. The vertical dashed lines indicate the PLE peaks at 320 nm and 360 nm, respectively. | ||
In order to reveal the underlying quantum-confined effects in monolayer WS2 QDs, selective-excitation TA experiments are performed. Fig. 2 shows the TA spectra of monolayer WS2 QDs under different excitation wavelengths. We use 470 nm, 570 nm, and 610 nm pump lights to selectively excite the relatively large WS2 QDs (considering our sample has a size distribution of 2.8 ± 0.9 nm). The relatively small WS2 QDs may have the main absorption peaks in the spectral range of <400 nm, which are beyond our probe window. The state filling caused by the transition of ground electrons to the excited states will lead to the generation of ground state bleaching (GSB) signals, which are the negative signals in our TA spectra. The positive signals are typically assigned to the excited-state absorption (ESA) signals.22,23 All the observed GSB peaks for monolayer WS2 QDs are located around the chosen excitation wavelengths, demonstrating that there are quantum-confined states.24 Since the pump fluences for those TA experiments are under control (all pump fluences are ∼25 μJ cm−2), the decreased intensity of the GSB signals observed in Fig. 2 with increasing excitation wavelength indicates a smaller number of nanoparticles for larger WS2 QDs. Instead, the TA spectra of WS2 nanosheets show the narrower GSB signals for the band-edge A-exciton (632 nm) and B-exciton (525 nm) under the same excitation conditions pumped by 610 nm pulses (Fig. S4†). The difference between TA spectra of monolayer WS2 QDs and WS2 nanosheets indicates a dramatic change of the band structure at the K point for WS2 QDs.
 | ||
| Fig. 2 TA spectra of monolayer WS2 QDs under (a) 470 nm, (b) 570 nm, and (c) 610 nm excitation, respectively, probed at different delay times. The values of ΔOD are equal to the changes of absorption, when it is excited with and without pump light. OD, optical density. (d) Normalized dynamics of band-edge excitons probed at 460 nm, 550 nm and 583 nm for monolayer WS2 QDs at 470 nm, 570 nm, and 610 nm excitation wavelengths, respectively, in comparison with that probed at 630 nm for WS2 nanosheets under 610 nm excitation. The shadow areas in panel (a–c) are from the scattering of excitation lights. | ||
Fig. 2d compares the band-edge exciton dynamics between WS2 nanosheets and monolayer WS2 QDs. For WS2 nanosheets, the A-exciton resonant excitation at 610 nm results in the photogeneration of exciton distribution close to the K point in an indirect band gap structure, in which those excitons have to experience fast non-radiative recombination processes. For the band-edge exciton dynamics of selected monolayer WS2 QDs, although those quantum-confined exciton states are obtained from the size-selective excitations, their decay rates are almost the same, and remarkably slower than that of WS2 nanosheets. This indicates that those relatively large WS2 QDs with a direct band gap structure could undergo the same exciton relaxation pathway and exhibit the resulting radiative recombination behaviors. The dynamics of exciton decay of monolayer WS2 QDs and WS2 nanosheets can be well fitted by a multi-exponential decay function: I(t) = Ai![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τi). For WS2 nanosheets, there are three lifetimes, 0.57 ± 0.01 ps (46%), 4.9 ± 0.1 ps (41%) and 703 ± 30 ps (13%). For monolayer WS2 QDs, for example those under 470 nm excitation, the fitted lifetimes are 1.4 ± 0.1 ps (37%), 12 ± 1 ps (50%) and 184 ± 14 ps (13%). All the best-fitted parameters shown in Fig. 2d are presented in Table S1.† The first two lifetime components are usually attributed to the trapping or scattering processes of TMDCs.25 The slowest lifetime component in WS2 nanosheets could be assigned to the indirect recombination of electrons and holes. For monolayer WS2 QDs, the slowest lifetime component could be related to the radiative recombination processes,26 which is consistent with the multi-exponential fitting results of time-resolved PL dynamics of monolayer WS2 QDs (Fig. S5†), where the fastest PL decay component is ∼0.16 ns.
exp(−t/τi). For WS2 nanosheets, there are three lifetimes, 0.57 ± 0.01 ps (46%), 4.9 ± 0.1 ps (41%) and 703 ± 30 ps (13%). For monolayer WS2 QDs, for example those under 470 nm excitation, the fitted lifetimes are 1.4 ± 0.1 ps (37%), 12 ± 1 ps (50%) and 184 ± 14 ps (13%). All the best-fitted parameters shown in Fig. 2d are presented in Table S1.† The first two lifetime components are usually attributed to the trapping or scattering processes of TMDCs.25 The slowest lifetime component in WS2 nanosheets could be assigned to the indirect recombination of electrons and holes. For monolayer WS2 QDs, the slowest lifetime component could be related to the radiative recombination processes,26 which is consistent with the multi-exponential fitting results of time-resolved PL dynamics of monolayer WS2 QDs (Fig. S5†), where the fastest PL decay component is ∼0.16 ns.
We further investigate the linearly and circularly polarized exciton dynamics for monolayer WS2 QDs and WS2 nanosheets. Fig. 3a and c present the TA spectra of monolayer WS2 QDs and WS2 nanosheets, respectively, under the conditions that the polarization direction of probe lights is parallel (I∥) or perpendicular (I⊥) to that of excitation light. The linear polarization anisotropy is defined as r = (I∥ − I⊥)/(I∥ + 2I⊥).27Fig. 3b and Fig. S6† show the initial anisotropy spectra of WS2 nanosheets and monolayer WS2 QDs, respectively. When the overlapping areas between the negative GSB and positive ESA signals in the TA spectra appear around the crossing points of ΔOD = 0, it will lead to infinite anisotropy values in the anisotropy spectra, which should be ignored. This indicates a strong linear polarization for the band-edge exciton state of selected monolayer WS2 QDs, which are much larger than that of WS2 nanosheets. Since the maximum theoretical anisotropy is 0.4 for a system with random distribution of linear polarization orientation,8 which has been verified here by the linearly polarized anisotropy TA experiments for the aqueous solution of rhodamine B under 520 nm excitation too (it is r ≈ 0.4 probed at 0.2 ps, and r = 0.34 probed at 1 ps; the grey dashed line in Fig. 3d shows its anisotropic dynamics, which demonstrates that there is no artifactual anisotropy component in our measurement system), it is quite interesting that there is a remarkably high initial anisotropy value of 0.59 at ∼1 ps (probed at 510 nm; the black line in Fig. 3d and it might be larger if probed at 0.2 ps) for monolayer WS2 QDs, for example, under 520 nm excitation. Such large initial linear polarization anisotropy for band-edge excitons of selected monolayer WS2 QDs could be a result of band structure change, like expired spin–orbit coupling, since the lateral quantum confinement effect in monolayer WS2 QDs would enforce a strong state-mixing on the original ‘A-exciton’ and ‘B-exciton’ states observed in WS2 nanosheets or monolayer WS2 films. This would lead to the ‘degenerate states’ at the band edge of monolayer WS2 QDs, which corresponds to the observed broad GSB peak and exhibits large initial values for the linear polarization anisotropy as a coherent superposition of valley excitons.28–32 This might be further affected by the signal superposition of monolayer WS2 QDs with similar sizes, or the other factors like the so-called solvent-QD interactions.16 The time-resolved PL anisotropy dynamics of monolayer WS2 QDs is measured, which presents a single exponential decay of 1.3 ns, corresponding to a rotational diameter of ∼1.4 nm (see details in Fig. S7†).33 It is worth noting that the initial PL anisotropy value for monolayer WS2 QDs is ∼0.16 probed at 0.4 ns, in agreement with the anisotropic value of ∼0.19 for the band-edge exciton state obtained from the TA experiment recorded at 400 ps. This suggests that there is actually a minor effect of signal superposition for monolayer WS2 QDs with different sizes on the linear polarization anisotropy values. In addition, the linear polarization dynamics for the ESA part (probed at 630 nm; the red line in Fig. 3d) of monolayer WS2 QDs shows a switching process from the positive value of 0.27 at ∼1 ps to a negative value of −0.45 after 100 ps. Its decay trend is also different from that observed at 510 nm (Fig. S8†). Together with the calculated rotational diameter, which is slightly larger than the physical thickness of monolayer WS2 QDs, both the facts mentioned above imply the influence of the solvent on rotational diffusion behavior and linear polarization of monolayer WS2 QDs. Furthermore, according to our observations, there could be two electronic states in monolayer WS2 QDs. One is responsible for the size-dependent PL of monolayer WS2 QDs, which is the intrinsic state corresponding to the GSB peak; and the other could be affected by the solvent environment, which is assigned to the edge state, corresponding to the ESA part in the red-side of the GSB signal. On the other hand, the initial linear polarization anisotropy for the band-edge A-exciton of WS2 nanosheets under 610 nm excitation is only ∼0.1, maintained for ∼10 ps. Then, it gradually increases to 0.33 at ∼150 ps. Finally, it begins to decay very slowly (probed at 630 nm; the blue line in Fig. 3d). The complex linear anisotropic dynamics indicates the heterogeneity of WS2 nanosheets, which may contain small monolayers attached to larger and thicker multilayer nanosheets.
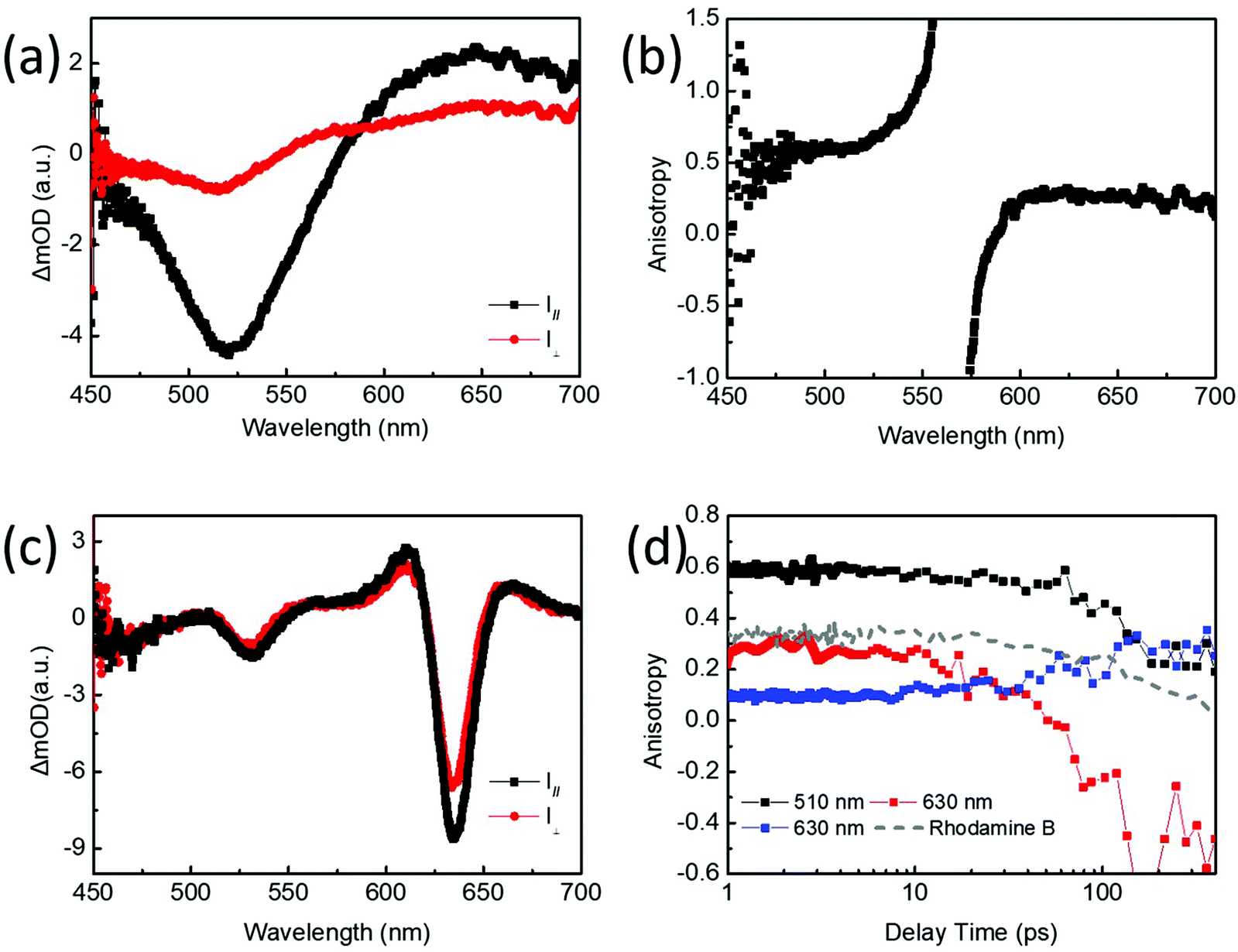 | ||
| Fig. 3 (a) Parallel and perpendicular TA spectra of monolayer WS2 QDs under 520 nm excitation probed at 1 ps. (b) Anisotropic spectrum of monolayer WS2 QDs probed at 1 ps. (c) Parallel and perpendicular TA spectra of WS2 nanosheets under 610 nm excitation probed at 0.28 ps. (d) Anisotropic dynamics of monolayer WS2 QDs under 520 nm excitation probed at 510 nm (black line) and 630 nm (red line); anisotropic dynamics of WS2 nanosheets under 610 nm excitation probed at 630 nm (blue line). The grey dashed line represents the anisotropy dynamics of rhodamine B under 520 nm excitation probed at 560 nm. | ||
TA spectra of monolayer WS2 QDs and WS2 nanosheets under the same circularly polarized (SCP) and opposite circularly polarized (OCP) conditions for the pump and probe lights are presented in Fig. 4. The degree of valley polarization is defined as 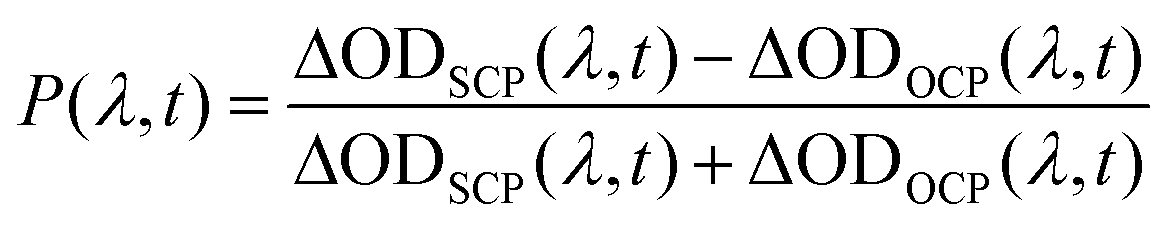 . There is no distinction between the SCP and OCP signals for monolayer WS2 QDs in the circularly polarized TA spectra and corresponding dynamics (Fig. 4a and b). WS2 nanosheets show larger SCP signals than the OCP signals at the band-edge A-exciton state (Fig. 4c). These SCP and OCP dynamics of A-exciton for WS2 nanosheets are presented in Fig. S9.†Fig. 4d displays the valley polarization dynamics of WS2 nanosheets and monolayer WS2 QDs. For WS2 nanosheets, the valley depolarization lifetime is 108 ± 6 fs, which could be mainly attributed to the electron–hole exchange interaction and phonon-assisted intervalley scattering.34 For monolayer WS2 QDs, the circular polarization response is not observed here too, which could be mainly due to the change of the band structure, like the expired spin–orbit coupling. Besides, the lateral quantum confinement for WS2 QDs enhances the solvent-QD interactions, which might further shorten the possible valley depolarization time, and make it beyond our detection limit of ∼35 fs. Under our current experimental conditions, only a small part of the monolayer WS2 QDs corresponding to the pump pulses are selectively excited, which absorbs parts of pump lights, and the remaining excitation pulses will strongly interact with the solvents, generating undesired nonlinear signals within the initial probe times (the shadow area in Fig. 4b; this disturbance is much smaller in WS2 nanosheets, since they exhibit larger absorption under their A-exciton resonance excitation than that of monolayer WS2 QDs). This hinders the direct observation of the initially linear and circular depolarization processes of monolayer WS2 QDs. Higher quality of monolayer WS2 QDs with better lateral size distributions would be helpful for this investigation in the future.
. There is no distinction between the SCP and OCP signals for monolayer WS2 QDs in the circularly polarized TA spectra and corresponding dynamics (Fig. 4a and b). WS2 nanosheets show larger SCP signals than the OCP signals at the band-edge A-exciton state (Fig. 4c). These SCP and OCP dynamics of A-exciton for WS2 nanosheets are presented in Fig. S9.†Fig. 4d displays the valley polarization dynamics of WS2 nanosheets and monolayer WS2 QDs. For WS2 nanosheets, the valley depolarization lifetime is 108 ± 6 fs, which could be mainly attributed to the electron–hole exchange interaction and phonon-assisted intervalley scattering.34 For monolayer WS2 QDs, the circular polarization response is not observed here too, which could be mainly due to the change of the band structure, like the expired spin–orbit coupling. Besides, the lateral quantum confinement for WS2 QDs enhances the solvent-QD interactions, which might further shorten the possible valley depolarization time, and make it beyond our detection limit of ∼35 fs. Under our current experimental conditions, only a small part of the monolayer WS2 QDs corresponding to the pump pulses are selectively excited, which absorbs parts of pump lights, and the remaining excitation pulses will strongly interact with the solvents, generating undesired nonlinear signals within the initial probe times (the shadow area in Fig. 4b; this disturbance is much smaller in WS2 nanosheets, since they exhibit larger absorption under their A-exciton resonance excitation than that of monolayer WS2 QDs). This hinders the direct observation of the initially linear and circular depolarization processes of monolayer WS2 QDs. Higher quality of monolayer WS2 QDs with better lateral size distributions would be helpful for this investigation in the future.
 | ||
| Fig. 4 (a) TA spectra of monolayer WS2 QDs probed at 1 ps delay times with SCP and OCP configurations under 520 nm excitation. (b) The SCP and OCP dynamics of A exciton probed at 520 nm of monolayer WS2 QDs. The shadow area shows the disturbance from the scattering of excitation lights. (c) TA spectra of WS2 nanosheets probed at 0.25 ps in SCP and OCP configurations under 610 nm excitation. (d) Valley polarization dynamics of monolayer WS2 QDs under 520 nm excitation and WS2 nanosheets under 610 nm excitation. | ||
Conclusions
In conclusion, we have directly observed the quantum-confined exciton states in monolayer WS2 QDs. Compared to WS2 nanosheets, the selected monolayer WS2 QDs only show one GSB peak for band-edge excitons and exhibit larger initial values for linear polarization anisotropy, all indicating a transition of the band structure for monolayer WS2 QDs due to expired spin orbit coupling. The lost spin–orbit coupling in monolayer WS2 QDs may be relevant to the changes in atomic arrangements (like curving in solution) or symmetry (like the broken C3-rotation symmetry), under strong quantum confinement in the lateral dimension. We also suggest that there could be a strong state-mixing of band-edge valley excitons in monolayer WS2 QDs, which is responsible for the resulting large linear polarization. Instead, in femtosecond time-resolved circular polarization anisotropy experiments, no obvious valley polarization is observed for monolayer WS2 QDs, and an ultrafast valley depolarization time of ∼100 fs for WS2 nanosheets is recorded at room temperature. It needs more theoretical and time-resolved experimental investigation in the future for the electronic structure of monolayer WS2 QDs, which could be affected by the coherent state-mixing due to the quantum confinement effects and the additional solvent-QD interactions.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China and the National Natural Science Foundation of China (NSFC) under Grants #2017YFB1104300, #21773087, #21603083, #61927814, #21903035 and #22073037.References
- X. Zong, H. Hu, G. Ouyang, J. Wang, R. Shi, L. Zhang, Q. Zeng, C. Zhu, S. Chen, C. Cheng, B. Wang, H. Zhang, Z. Liu, W. Huang, T. Wang, L. Wang and X. Chen, Light: Sci. Appl., 2020, 9, 114 CrossRef CAS PubMed.
- P. Zhang, C. Bian, J. Ye, N. Cheng, X. Wang, H. Jiang, Y. Wei, Y. Zhang, Y. Du, L. Bao, W. Hu and Y. Gong, Sci. China Mater., 2020, 63, 1548–1559 CrossRef CAS.
- Y. Liu, Y. Gao, S. Zhang, J. He, J. Yu and Z. Liu, Nano Res., 2019, 12, 2695–2711 CrossRef CAS.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- D. Wickramaratne, F. Zahid and R. K. Lake, J. Chem. Phys., 2014, 140, 124710 CrossRef PubMed.
- C. Hu, X. Wang and B. Song, Light: Sci. Appl., 2020, 9, 88 CrossRef CAS PubMed.
- M. M. Glazov, E. L. Ivchenko, G. Wang, T. Amand, X. Marie, B. Urbaszek and B. L. Liu, Phys. Status Solidi B, 2015, 252, 2349–2362 CrossRef CAS.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- D. Xiao, G. B. Liu, W. Feng, X. Xu and W. Yao, Phys. Rev. Lett., 2012, 108, 196802 CrossRef PubMed.
- S. Xu, D. Li and P. Wu, Adv. Funct. Mater., 2015, 25, 1127–1136 CrossRef CAS.
- H. Jin, M. Ahn, S. Jeong, J. H. Han, D. Yoo, D. H. Son and J. Cheon, J. Am. Chem. Soc., 2016, 138, 13253–13259 CrossRef CAS PubMed.
- L. Wang, Q. Li, H.-Y. Wang, J.-C. Huang, R. Zhang, Q.-D. Chen, H.-L. Xu, W. Han, Z.-Z. Shao and H.-B. Sun, Light: Sci. Appl., 2015, 4, e245–e245 CrossRef.
- L. Yuwen, J. Zhou, Y. Zhang, Q. Zhang, J. Shan, Z. Luo, L. Weng, Z. Teng and L. Wang, Nanoscale, 2016, 8, 2720–2726 RSC.
- Y. Yong, T. Bao, M. Zu, L. Yan, W. Yin, C. Ge, D. Wang, Z. Gu, Y. Zhao and X. Cheng, ACS Nano, 2013, 9, 12451–12463 CrossRef PubMed.
- X. Zhao, X. Ma, J. Sun, D. Li and X. Yang, ACS Nano, 2016, 10, 2159–2166 CrossRef CAS PubMed.
- X. Ren, L. Pang, Y. Zhang, X. Ren, H. Fan and S. Liu, J. Mater. Chem. A, 2015, 3, 10693–10697 RSC.
- H. Jin, B. Baek, D. Kim, F. Wu, J. D. Batteas, J. Cheon and D. H. Son, Nano Lett., 2017, 17, 7471–7477 CrossRef CAS PubMed.
- W. X. Yin, X. Bai, P. Chen, X. Y. Zhang, L. Su, C. Y. Ji, H. M. Gao, H. W. Song and W. W. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 43824–43830 CrossRef CAS PubMed.
- H. S. Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. Rao, Angew. Chem., Int. Ed. Engl., 2010, 49, 4059–4062 CrossRef CAS PubMed.
- V. Štengl, J. Tolasz and D. Popelková, RSC Adv., 2015, 5, 89612–89620 RSC.
- C. Han, Y. Zhang, P. Gao, S. Chen, X. Liu, Y. Mi, J. Zhang, Y. Ma, W. Jiang and J. Chang, Nano Lett., 2017, 17, 7767–7772 CrossRef CAS PubMed.
- L. Wang, H.-Y. Wang, H.-T. Wei, H. Zhang, Q.-D. Chen, H.-L. Xu, W. Han, B. Yang and H.-B. Sun, Adv. Energy Mater., 2014, 4, 1301882 CrossRef.
- H. Wang, H.-Y. Wang, Q.-D. Chen, H.-L. Xu, H.-B. Sun, F. Huang, W. Raja, A. Toma and R. Zaccaria, Laser Photonics Rev., 2018, 12, 1700176 CrossRef.
- L. Wang, H. Y. Wang, Y. Wang, S. J. Zhu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, W. Han, H. L. Xu, B. Yang and H. B. Sun, Adv. Mater., 2013, 25, 6539–6545 CrossRef CAS PubMed.
- H. Y. Shi, R. S. Yan, S. Bertolazzi, J. Brivio, B. Gao, A. Kis, D. Jena, H. G. Xing and L. B. Huang, ACS Nano, 2013, 7, 1072–1080 CrossRef CAS PubMed.
- A. Granados Del Aguila, S. Liu, T. T. H. Do, Z. Lai, T. H. Tran, S. R. Krupp, Z. R. Gong, H. Zhang, W. Yao and Q. H. Xiong, ACS Nano, 2019, 13, 13006–13014 CrossRef CAS PubMed.
- Y. Wang, L. Wang, H. Y. Wang, B. R. Gao and H. B. Sun, J. Phys. Chem. Lett., 2020, 11, 1483–1489 CrossRef CAS PubMed.
- K. F. Mak, D. Xiao and J. Shan, Nat. Photonics, 2018, 12, 451–460 CrossRef CAS.
- Z. Ye, D. Sun and T. F. Heinz, Nat. Phys., 2016, 13, 26–29 Search PubMed.
- B. Zhu, H. Zeng, J. Dai, Z. Gong and X. Cui, Anomalously robust valley polarization and valley coherence in bilayer WS2, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11606 CrossRef CAS PubMed.
- G. Wang, X. Marie, B. L. Liu, T. Amand, C. Robert, F. Cadiz, P. Renucci and B. Urbaszek, Phys. Rev. Lett., 2016, 117, 187401 CrossRef CAS PubMed.
- A. M. Jones, H. Yu, N. J. Ghimire, S. Wu, G. Aivazian, J. S. Ross, B. Zhao, J. Yan, D. G. Mandrus, D. Xiao, W. Yao and X. Xu, Nat. Nanotechnol., 2013, 8, 634–638 CrossRef CAS PubMed.
- L. Wang, S. J. Zhu, H. Y. Wang, Y. F. Wang, Y. W. Hao, J. H. Zhang, Q. D. Chen, Y. L. Zhang, W. Han, B. Yang and H. B. Sun, Adv. Opt. Mater., 2013, 1, 264–271 CrossRef.
- R. Schmidt, G. Berghauser, R. Schneider, M. Selig, P. Tonndorf, E. Malic, A. Knorr, S. Michaelis de Vasconcellos and R. Bratschitsch, Nano Lett., 2016, 16, 2945–2950 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04868f |
| This journal is © The Royal Society of Chemistry 2021 |