
Well-defined Co9S8 cages enable the separation of photoexcited charges to promote visible-light CO2 reduction†
Xiahui
Lin
,
Zidong
Xie
,
Bo
Su
,
Mei
Zheng
,
Wenxin
Dai
 ,
Yidong
Hou
,
Zhengxin
Ding
*,
Wei
Lin
*,
Yuanxing
Fang
* and
Sibo
Wang
*
,
Yidong
Hou
,
Zhengxin
Ding
*,
Wei
Lin
*,
Yuanxing
Fang
* and
Sibo
Wang
*
State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fujian, Fuzhou, 350002, China. E-mail: zxding@fzu.edu.cn; wlin@fzu.edu.cn; yxfang@fzu.edu.cn; sibowang@fzu.edu.cn
First published on 30th September 2021
Abstract
Exploring affordable cocatalysts with high performance for boosting charge separation and CO2 activation is an effective strategy to reinforce CO2 photoreduction efficiency. Herein, well-defined Co9S8 cages are exploited as a nonprecious promoter for visible-light CO2 reduction. The Co9S8 cages are prepared via a multistep strategy with ZIF-67 particles as the precursor and fully characterized by physicochemical techniques. The hollow Co9S8 cocatalyst with a high surface area and profuse catalytically active centers is discovered to accelerate separation and transfer of light-induced charges, and strengthen concentration and activation of CO2 molecules. In a hybrid photosensitized system, these Co9S8 cages efficiently promote the deoxygenative reduction of CO2 to generate CO, with a high yield rate of 35 μmol h−1 (i.e., 35 mmol h−1 g−1). Besides, this cocatalyst is also of high stability for the CO2 photoreduction reaction. Density functional theory (DFT) calculations reveal that the Ru(bpy)32+ photosensitizer is strongly absorbed on the Co9S8 (311) surface through forming four Co–C bonds, which can serve as the “bridges” to ensure quick electron transfer from the excited photosensitiser to the active Co9S8 cocatalyst, thus promoting the separation of photoexcited charges for ehannced CO2 reduction performance.
 Yuanxing Fang | Yuanxing Fang went to the University of Sussex, United Kingdom, to study chemistry, and obtained his MChem degree in 2012 and PhD degree in 2017. He went to the State Key Laboratory of Photocatalysis on Energy and Environment at Fuzhou University, P. R. China, as a Postdoctoral fellow in 2017. In 2019, he was appointed as Associated Professor in the College of Chemistry, Fuzhou University, and promoted to Special Professor in 2021. His longstanding interests are the developments of photocatalytic and photoelectrochemical devices for energy and environmental applications, such as water splitting, CO2 reduction, organosynthesis and others. He has published more than 45 peer-reviewed publications, which have garnered more than 2000 citations and an H-index of 21. |
During the past few decades, a great deal of strategies have been proposed to improve CO2 photoreduction efficiency with inspiring progress realized.11–16 Therein, cocatalyst engineering is verified to be a preferred approach. This is because cocatalysts can enable capture and activation of CO2 molecules, accelerate separation and transfer kinetics of charge carriers, and provide catalytically active sites to selectively operate the redox reactions.17,18 The noble metal nanoparticles are the classic CO2 reduction cocatalysts with high catalytic performance,19–22 and alternatively, the transition metal ions/complexes of cobalt and nickel can serve as efficient promoters for CO2 photoreduction in cooperation with specific light-sensitizers.23–30 Comparatively, considering the requirements of large-scale execution (i.e., low price, high abundance, easy separation/retrieval), exploring heterogeneous cocatalysts composed of cost-affordable elements for CO2 photoreduction is highly desirable.
Besides the chemical composition, designing catalytic materials with favored architectures is also of vital importance for photocatalytic CO2 reduction.31 As a group of multifunctional catalysts for energy storage and conversion,32,33 hollow structures are actively employed for CO2 photoreduction,34–40 owing to their inherent advantages, such as a shortened perpendicular path for charge transfer to prevent their recombination, plentiful surface-active sites to boost CO2 capture and redox catalysis, and strengthened interior reflection/scattering to enhance incident light utilization.41–43
With all the aforementioned concerns in mind, in this work, the well-defined Co9S8 cages are applied as a cocatalyst coupled with Ru(bpy)32+ (abbreviated as Ru, bpy = 2′2-bipyridine) as a photosensitizer for CO2 reduction under visible light. The dodecahedral Co9S8 cage is created via a multi-step method as depicted schematically in Fig. 1. Starting with Co2+ and 2-methylimidazole as the raw materials, the zeolitic imidazolate framework (ZIF)-67 dodecahedron is first prepared through a reported precipitation method with some adjustments (step I).44,45 Next, the ZIF-67 particle is converted to a CoSx cage by a hydrothermal sulfidation reaction (step II). Finally, the Co9S8 cage is harvested after annealing the CoSx intermediate under a N2 atmosphere at 550 °C (step III).
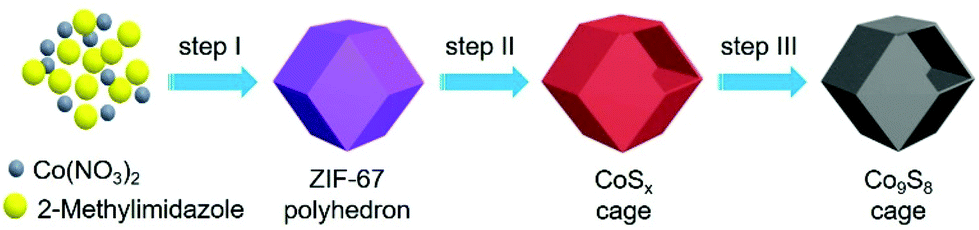 | ||
| Fig. 1 Schematic illustration of the preparation processes of dodecahedral Co9S8 cages: (I) self-assembly, (II) sulfidation, and (III) annealing in N2. | ||
The Co9S8 cocatalyst is revealed by diverse photo-/electro-chemical tests to augment CO2 activation and promote separation of light-excited charges. In a tandem photochemical system, the Co9S8 cocatalyst enables the deoxygenative reduction of CO2 by visible light, with a CO formation rate of 35 μmol h−1, under the cooperation of Ru(bpy)32+ as a photosensitizer. Indeed, the reduction ability of the Co9S8 catalyst in photocatalysis has been demonstrated by hydrogen evolution reactions;46–50 however, its talent for CO2 photoreduction has been seldom exploited so far.
Production of the ZIF-67 precursor was confirmed by powder X-ray diffraction (XRD) and energy-dispersive X-ray (EDX) characterization (Fig. S1, ESI†). The field emission scanning electron microscopy (FESEM) images show that the as-synthesized ZIF-67 particles present a morphology of nearly monodisperse dodecahedra with high uniformity (Fig. 2a and b).
 | ||
| Fig. 2 (a and b) FESEM images of ZIF-67 dodecahedra. (c–e) FESEM images and (f) TEM image of CoSx cages. | ||
After the sulfidation treatment, the ZIF-67 precursor was entirely transformed into an amorphous cobalt sulfide (CoSx) product with a Co/S ratio of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.42 (Fig. S2, ESI†). The FESEM images indicate that the CoSx intermediate can inherit the dodecahedral shape of the parental ZIF-67 particles perfectly (Fig. 2c). From the magnified FESEM images (Fig. 2d and e), it is found that the CoSx particles have a relatively rougher surface than the ZIF-67 precursor, and the observed clear cavity of the broken CoSx dodecahedra implies their hollow structure. The empty interior of the well-defined CoSx cages is then confirmed visually by the TEM image (Fig. 2f). The possible formation process of CoSx cages is clarified as follows. During the hydrothermal sulfidation reaction, the thioacetamide is decomposed to release the sulfide ions to react with the cobalt ions on the ZIF-67 particle surface, yielding an outermost layer of CoSx. Further formation of the CoSx shell continues via the reactions between inward diffusing sulfide ions and outward diffusing metal ions, finally producing the polyhedral CoSx cages.46,51
:3.42 (Fig. S2, ESI†). The FESEM images indicate that the CoSx intermediate can inherit the dodecahedral shape of the parental ZIF-67 particles perfectly (Fig. 2c). From the magnified FESEM images (Fig. 2d and e), it is found that the CoSx particles have a relatively rougher surface than the ZIF-67 precursor, and the observed clear cavity of the broken CoSx dodecahedra implies their hollow structure. The empty interior of the well-defined CoSx cages is then confirmed visually by the TEM image (Fig. 2f). The possible formation process of CoSx cages is clarified as follows. During the hydrothermal sulfidation reaction, the thioacetamide is decomposed to release the sulfide ions to react with the cobalt ions on the ZIF-67 particle surface, yielding an outermost layer of CoSx. Further formation of the CoSx shell continues via the reactions between inward diffusing sulfide ions and outward diffusing metal ions, finally producing the polyhedral CoSx cages.46,51
The CoSx material was treated by annealing in N2 to attain the Co9S8 product. To monitor the conversion procedure, thermogravimetry (TG) analysis was conducted. As shown in Fig. 3a, the result of TG indicates that the CoSx intermediate undergoes gradual weight loss during the thermal treatment due to sulphur volatilization, and finally forms a stable product at 550 °C, which is confirmed to be the Co9S8 phase (JCPDS card no.: 19-0364) by XRD analysis (Fig. 3b). No impurity XRD peaks are discerned, signifying the high phase purity of the Co9S8 material. The Raman spectrum of the Co9S8 sample presents a distinct vibrational peak at around 640 cm−1 together with a set of bands below 750 cm−1 (Fig. 3c), indexing to the characteristic Raman peaks of the Co9S8 phase.52,53 The EDX spectrum shows that the Co9S8 product is only composed of Co and S elements with a molar ratio of about 9:7.63 (Fig. 3d), which is close to the theoretical value.
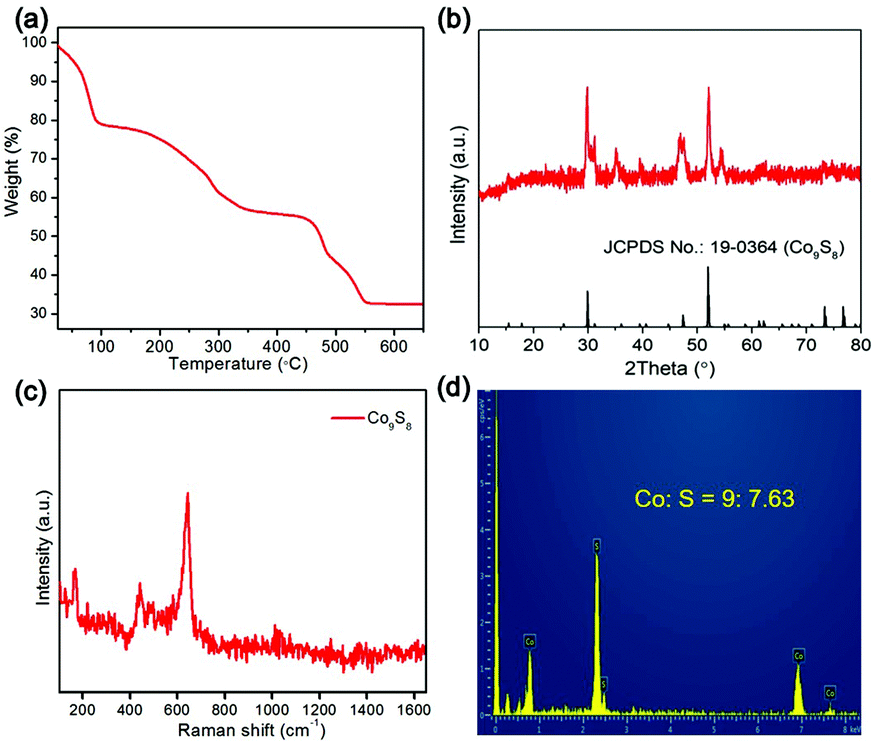 | ||
| Fig. 3 (a) TG curve of CoSx cages in in N2. (b) XRD pattern, (c) Raman spectrum and (d) EDX spectrum of Co9S8 cages. | ||
The morphology and microstructures of the Co9S8 product were researched by FESEM and TEM. As can be seen from the FESEM images (Fig. 4a and b), the Co9S8 particles well preserve the pristine polyhedral construction of the CoSx intermediate without perceptible agglomeration or fractures. The careful FESEM analysis discloses that the surface of Co9S8 particles experiences slight shrinkage after the thermal treatment (Fig. 4c). The structural features of Co9S8 cages revealed by TEM are consistent with the results of FESEM (Fig. 4d and e). The crystal features of Co9S8 cages were then checked by high-resolution TEM (HRTEM) analysis. As shown in Fig. 4f, the HRTEM image displays strong lattice fringes with an interlayer d-spacing of 0.28 nm determined by the corresponding inverse fast Fourier transformation (IFFT) image and line scans (Fig. 4g and h), which is assigned to the (222) crystal plane of the cubic Co9S8 phase. The Co9S8 product is verified to be a polycrystalline material, as revealed by the selected area electron diffraction (SAED) pattern (Fig. 4i), in which the clear diffraction fringes are attributed to the interplanar spacings of the cubic Co9S8 phase.
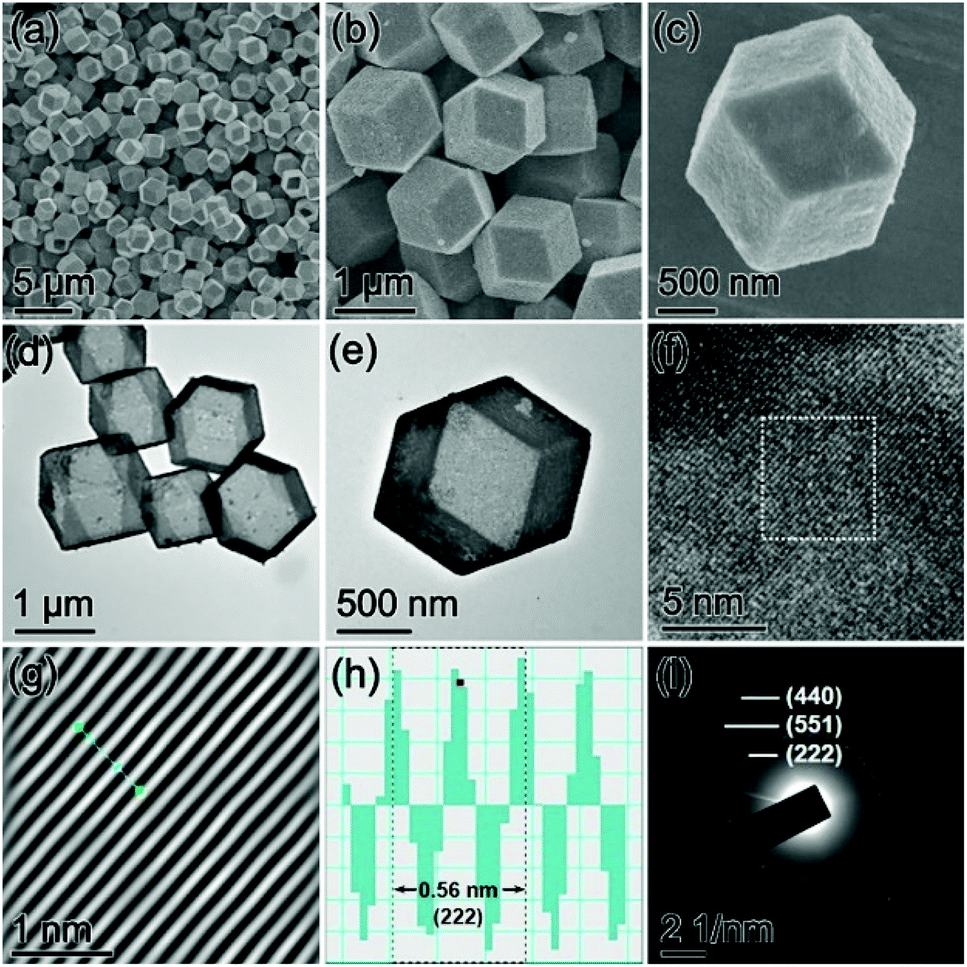 | ||
| Fig. 4 (a–c) FESEM images, (d and e) TEM images, (f) HRTEM image, (g and h) the corresponding inverse fast Fourier transformation (IFFT) image and line scans of the dotted square region in (f), and (i) SAED pattern of Co9S8 cages. | ||
The surface chemical states of elements of the Co9S8 material were studied by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 5a, the high-resolution Co 2p spectrum is fitted into two sets of spin–orbit doublets and satellite (Sat.) peaks. The first doublet located at 778.3 and 779.4 eV and the second doublet with binding energies of 793.4 and 795.5 eV are attributed to Co 2p3/2 and Co 2p1/2, respectively. The presence of these doublets indicates the coexistence of Co3+ and Co2+, which is in agreement with the results of previous work.54–56 In the XPS spectrum of S 2p (Fig. 5b), the fitted peaks with binding energies of about 162.1 and 163.1 eV are assigned to the Co–S species, while the other two peaks centered at 163.7 and 164.9 eV are attributable to the S–O bonds,46 implying the surface oxidation of the Co9S8 sample that is generally detected during XPS measurements.
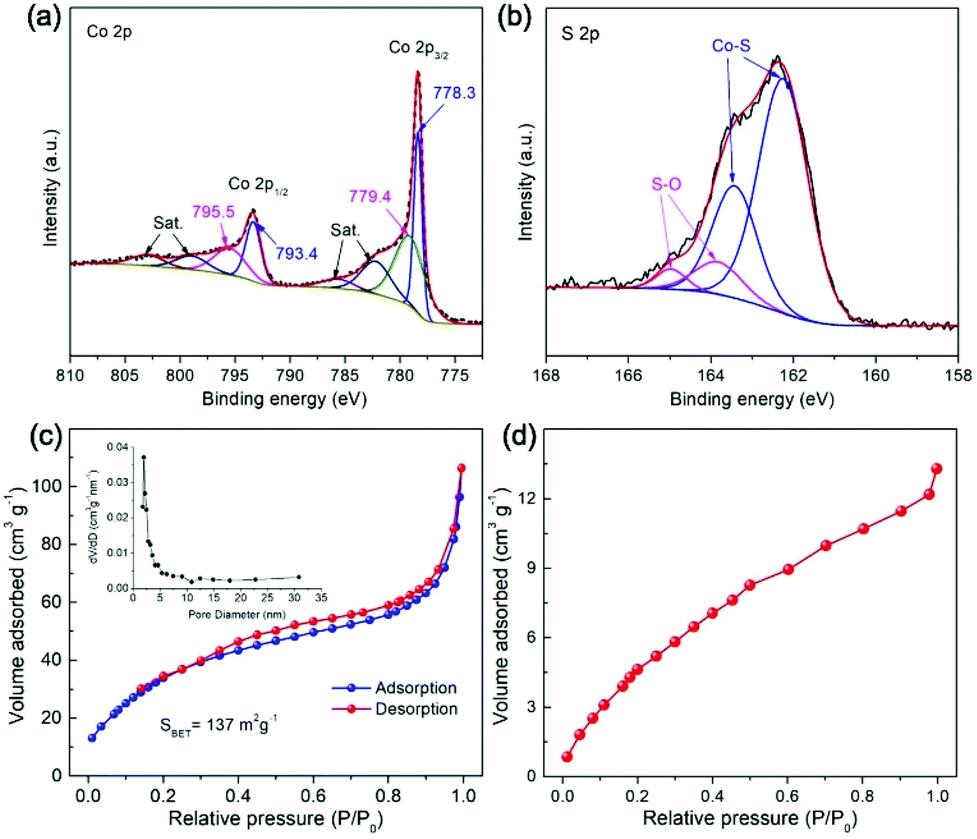 | ||
| Fig. 5 (a) Co 2p and (b) S 2p XPS spectrum, (c) N2 adsorption–desorption isotherms, and (d) CO2 adsorption isotherm of Co9S8 cages. | ||
The textural properties of Co9S8 cages were examined by N2 sorption measurements. As displayed in Fig. 5c, classified type-II N2 adsorption–desorption isotherms together with a type H3 hysteresis loop are observed, suggesting the formation of mesopores in the material, which can be further confirmed by the corresponding pore size distribution plot (inset, Fig. 5c). Generation of the porous structure of Co9S8 cages is positive to facilitate mass transportation for heterogeneous photocatalytic applications. The Co9S8 material is measured to have a high specific Brunauer–Emmett–Teller (BET) surface area of about 137 m2 g−1, which should be mainly contributed by the well-defined hollow configuration. Such a high surface area will enable the capture and concentration of CO2 molecules, as reflected by the CO2 adsorption isotherm, which gives a maximum CO2 uptake of about 13.5 cm3 g−1 at 0 °C and 1 atm (Fig. 5d). These textural features of Co9S8 cages are beneficial to afford rich exposed catalytically active sites and boost mass transport for heterogeneous CO2 fixation reactions.
We then evaluated the performance of Co9S8 cages by the visible-light CO2 reduction reaction conducted in a classic hybrid system,2,25,54,57–61 engaging Ru(bpy)32+ as the light-harvester and H2O/acetonitrile/TEOA mixture as the reaction medium. Fig. 6a presents the CO2 reduction activity of the Co9S8 cocatalyst as a function of reaction time. As can be seen, the system manifests a high photocatalytic performance in the initial first 1 h of the reaction, showing a CO2-to-CO conversion rate of 35 μmol h−1, coupled with a H2-releasing rate of 12 μmol h−1. In addition to CO and H2, no detectable formation of hydrocarbon products is observed in the liquid phase. On further continuing the reaction, generation of the products increases progressively, but with a decreased rate, which should result from degradation of the ruthenium photosensitizer.57,62,63 The total yield of CO reaches 65 μmol after photoreaction for 7 h, corresponding to a catalytic turnover number (TON) of about 54 relative to the cocatalyst.
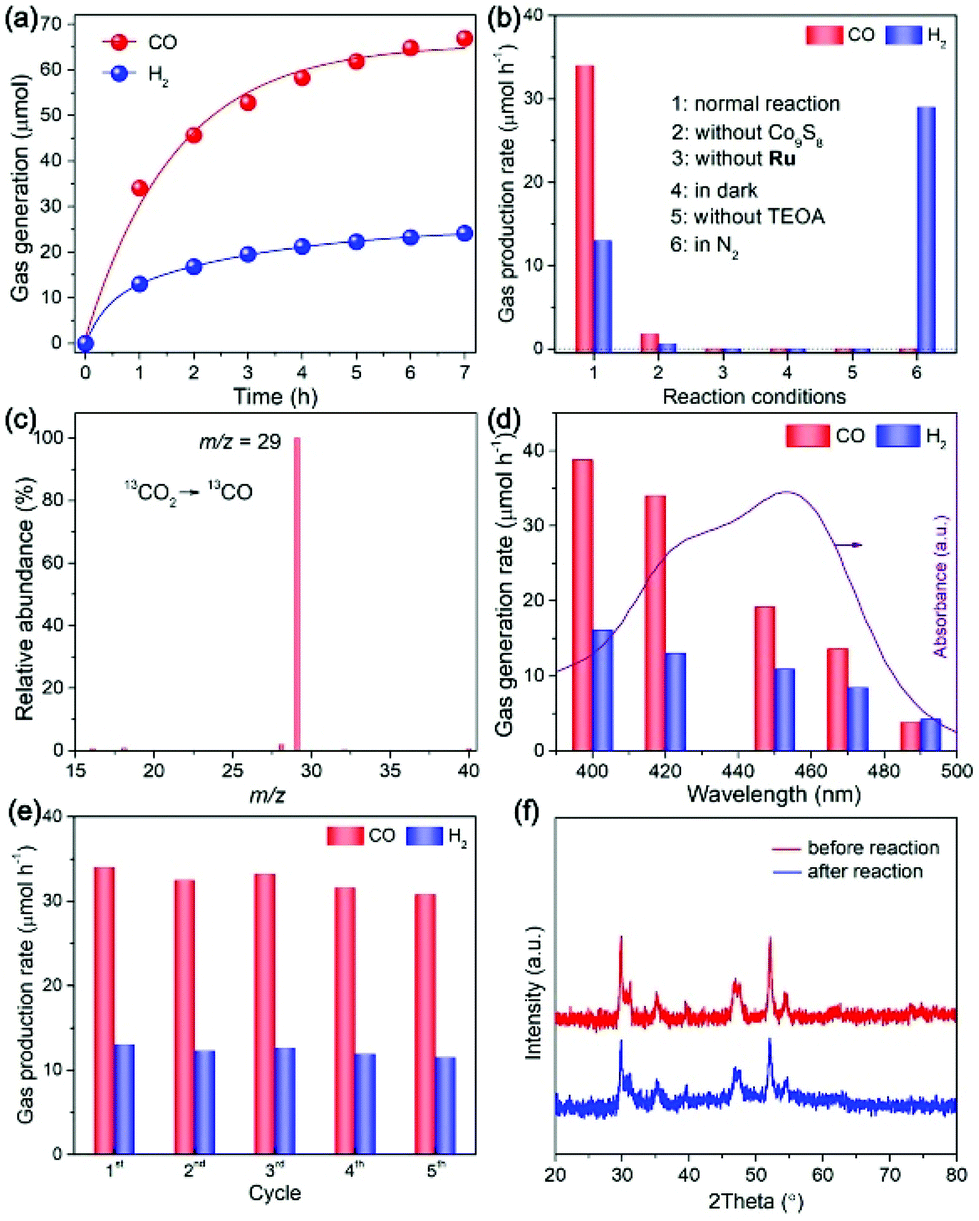 | ||
| Fig. 6 (a) CO2 photoreduction activity of the Co9S8 cocatalyst as a function of reaction time. (b) Performance of the CO2 reduction system under various conditions. (c) Mass spectrum of CO yielded from the 13CO2 isotope reaction. (d) CO and H2 generation by visible light with different wavelengths. (e) Stability tests of the Co9S8 cocatalyst. (f) XRD patterns of the Co9S8 cocatalyst before and after CO2 reduction reactions. | ||
The working mechanism of the CO2 reduction system was inspected by controlling the reaction conditions. As shown in Fig. 6b, compared to that of the normal reaction (column 1), the evolution of CO/H2 reduces significantly once the Co9S8 cocatalyst is omitted from the system, highlighting its critical role in promoting the CO2 reduction reaction. No product is detected without the introduction of a photosensitizer (column 3) or visible light irradiation (column 4), which indicates that the CO2 reduction reaction is a visible-light-sensitized process. It is revealed that TEOA is essential to achieve the CO2 reduction photocatalysis, as the reaction would be completely terminated without its participation (column 5), which matches well with the results of reported studies in analogous photochemical systems.2,25,57,62,64 When employing N2 as the gas feedstock to substitute CO2 for running the reaction (column 6), the only product generated is H2, suggesting that the CO product should be derived from the CO2 gas.
To directly track the origin of CO, we performed a 13C-labelled reaction with 13CO2 as the reactant and analyzed the produced CO by mass spectroscopy (MS). As indicated in Fig. 6c, the MS spectrum manifests a predominant peak with a m/z value of 29, which is attributed distinctly to 13CO. This observation firmly validates the source of CO generation, that is, the CO2 feedstock.
The CO2 photoreduction reactions were also initiated with light irradiation of different wavelengths through applying specific long-pass cut-off filters. As revealed in Fig. 6d, the formation of CO/H2 diminishes gradually under light irradiation of longer wavelengths, because of the decline in the incident photons when extending the wavelength of the light filter. These findings indicate that the CO2 reduction is motivated by light excitation of the photosensitizer.
To examine the stability of the Co9S8 cocatalyst, after CO2 photoreduction reactions, it was separated, washed, and re-added into fresh reaction mixtures for repeated operations. No apparent decrease in CO/H2 production is detected during the stability tests (Fig. 6e), pointing to its high activity stability. The results of XRD, FTIR, Raman, and XPS tests for the Co9S8 cocatalyst before and after photoreaction expose its high stabilities in the crystal, chemical and surface structures in the photocatalytic CO2 reduction system (Fig. 6f, and Fig. S3, ESI†).
In order to gain an understanding of the high performance of the CO2 photoreduction system mediated by Co9S8 cages, we carried out photo-/electro-chemical measurements. To demonstrate the function of Co9S8 in the CO2 reduction reaction, linear sweep voltammetry (LSV) was conducted. As shown in Fig. 7a, the Co9S8 catalyst delivers a more positive onset potential and a higher cathodic current in CO2-saturated solution than those under N2-saturated conditions. These outcomes verify that the Co9S8 cages can activate CO2 molecules and drive their reduction reaction.63,65
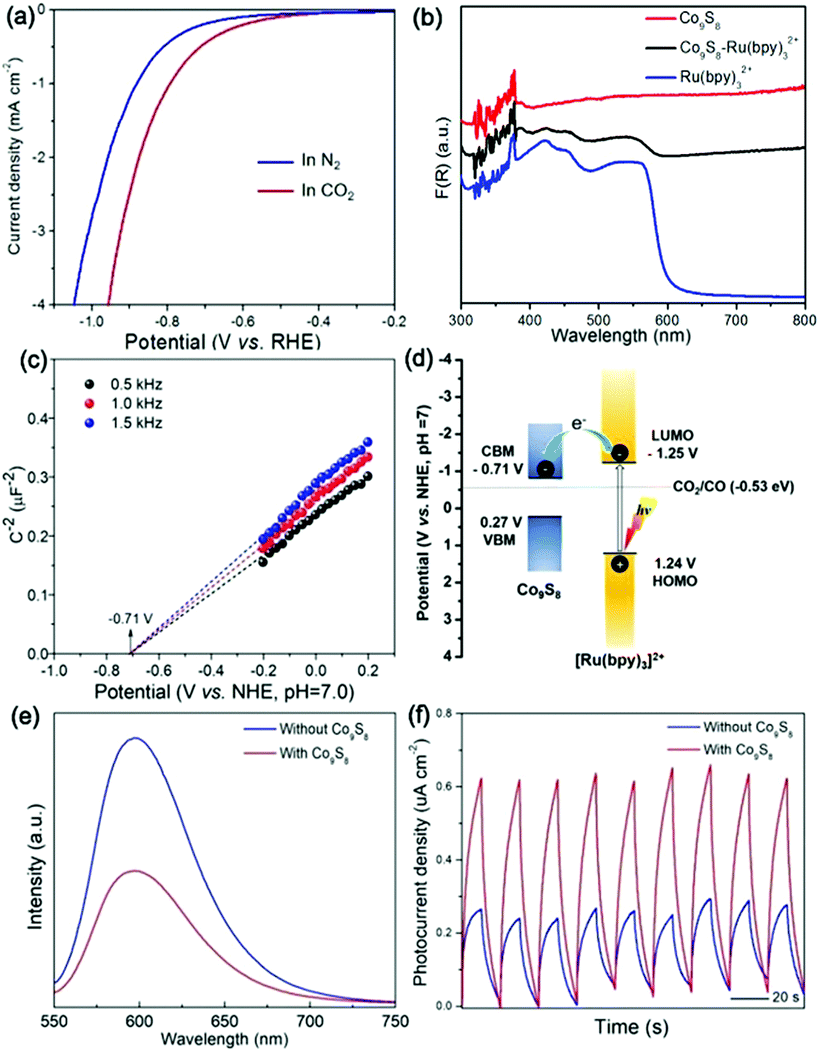 | ||
| Fig. 7 (a) LSV curve of the Co9S8 cocatalyst under N2- and CO2-saturated solutions. (b) DRS spectra of Co9S8, Ru(bpy)32+ and Ru(bpy)32+/Co9S8 hybrid. (c) Mott–Schottky plots. (d) Schematic illustration of energy levels and electron transfer from the ruthenium photosensitizer to the Co9S8 cocatalyst. (e) PL spectra and (f) transient photocurrent generation of reaction systems with and without the Co9S8 cocatalyst. | ||
UV–vis diffuse reflectance spectra (DRS) indicate that the Co9S8 cocatalyst and Ru(bpy)32+ photosensitizer show strong visible light absorption (Fig. 7b), while their hybrid possesses an optical harvesting ability. By using UV-vis DRS and the Tauc curve of Co9S8 (Fig. S4, ESI†), its bandgap energy is measured to be 0.98 eV.37,47 To define the conduction band bottom (CBM) and valence band maximum (VBM) positions, the flat band potential of Co9S8 was estimated by electrochemical Mott–Schottky plots. As shown in Fig. 7c, the derived CBM potential of the Co9S8 cocatalyst is about −0.71 V (vs. NHE, pH = 7.0), which integrated with the bandgap energy determines its VBM position at about 0.27 V (vs. NHE, pH = 7.0). Regarding the band structure of Co9S8, the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) levels of Ru(bpy)32+, and the redox potential for reducing CO2 to CO (Fig. 7d),26,62,66 the excited electrons of the photosensitizer are encouraged thermo-dynamically to travel to the cocatalyst for driving the CO2 reduction reaction.
To monitor the separation and transport of light-excited charges in the photochemical system, room temperature photoluminescence (PL) was implemented. As revealed in Fig. 7e, the system involving Co9S8 reveals a quenched PL emission compared to the system free of the cocatalyst, suggesting the prohibited recombination of light-stimulated charges of the former.31,40 Simultaneously, the time-resolved PL (TRPL) spectra (Fig. S5, ESI†) reveal that the average emission lifetime of the reaction mixture with the cocatalyst (399 ns) is much shorter than that of the mixture without its presence (450 ns), indicating that quick electron transfer exists between the photosensitizer and the cocatalyst. Consistently, the photocurrent generation of the Co9S8-mediated system is higher than that of the system without its existence (Fig. 7f), which mirrors the boosted transport of charge carriers induced by the metal sulfide cocatalyst.26,47,67 All the above results show that the Co9S8 cocatalyst can expedite separation and transfer of light-excited charges and promote activation of CO2 molecules, thus reinforcing the CO2 photoreduction efficiency.
Density functional theory (DFT) calculations were carried out to gain some insights into the interaction between the Ru(bpy)32+ photosensitizer and Co9S8 cocatalyst. The results prove that the Ru(bpy)32+ complex can be strongly adsorbed on the Co9S8 (311) surface, in which the carbon atoms of Ru(bpy)32+ and the surface Co atoms of Co9S8 form four Co–C bonds with a length of about 2.1 Å (Fig. 8). The chemical bonds between the photosensitizer and the cocatalyst may serve as the “bridges” to guarantee quick electron migration from the excited photosensitiser to the active Co9S8 cocatalyst to run the CO2 reduction reaction.
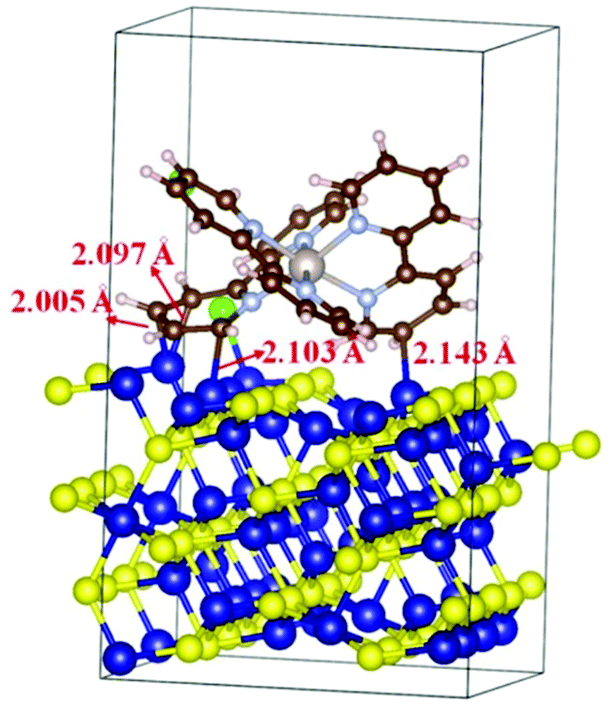 | ||
| Fig. 8 The structure of Ru(bpy)3Cl2 adsorbed on the Co9S8 (311) surface with an adsorption energy of −4.5 eV. The Co, S, Ru, N, C, H, and Cl atoms are denoted by blue, yellow, pink, gray, brown, white, and green, respectively. | ||
Finally, we have proposed a probable mechanism of the CO2 photoreduction reaction catalyzed by the caged Co9S8 cocatalyst. Being stimulated by visible light illumination, the ground state photosensitizer Ru(bpy)32+ moves to the excited state of Ru(bpy)32+*. Such an excited state will react with the electron donor TEOA through a reductive quenching process, leading to the formation of the photosensitizer in a reduced state of Ru(bpy)3+.26,64 The excited electrons of the Ru(bpy)3+ species will delocalize and move to the porous Co9S8 cocatalyst to run the CO2-to-CO conversion reaction,60,62,65 during which the Co9S8 cages not only strengthen the adsorption and activation of CO2 molecules but also push the separation and migration of light-generated charges to support the reaction.
In summary, well-defined dodecahedral Co9S8 cages have been fabricated and applied as a new cocatalyst for CO2 photoreduction with visible light. Owing to the high surface area and plentiful catalytically reactive locations, the hollow C9S8 cocatalyst effectively boosts CO2 activation and prevents charge recombination. In a classic light-sensitized system, such a non-noble-metal cocatalyst can powerfully promote the conversion of CO2 to CO with high efficiency and good stability. Inspired by this contribution, future studies about the sulfide semiconductors with narrow bandgaps and the hybrids of Co9S8/semiconductor for photocatalytic CO2 reduction may be anticipated.
Author contributions
Xiahui Lin: conceptualization, methodology, feasibility analysis, validation, writing-original draft, and writing-review and editing. Zidong Xie: methodology, feasibility analysis, and validation. Bo Su: methodology, validation, and writing-review and editing. Mei Zheng: theoretical calculation. Wenxin Dai: feasibility analysis. Yidong Hou: writing-review and editing. Zhengxin Ding: writing-review and editing. Wei Lin: theoretical calculation and supervision. Yuanxing Fang: writing-review and editing and supervision. Sibo Wang: validation, writing-review and editing, and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (U1805255, 22075047, 21802030, 21872033, and 21973014) and the Natural Science Foundation of Fujian Province of China (2020J01446).Notes and references
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- K. Niu, Y. Xu, H. Wang, R. Ye, H. L. Xin, F. Lin, C. Tian, Y. Lum, K. C. Bustillo, M. M. Doeff, M. T. M. Koper, J. Ager, R. Xu and H. Zheng, Sci. Adv., 2017, 3, e1700921 CrossRef PubMed.
- S. C. Shit, I. Shown, R. Paul, K.-H. Chen, J. Mondal and L.-C. Chen, Nanoscale, 2020, 12, 23301–23332 RSC.
- L. Huang, B. Li, B. Su, Z. Xiong, C. Zhang, Y. Hou, Z. Ding and S. Wang, J. Mater. Chem. A, 2020, 8, 7177–7183 RSC.
- Y. Fang, Y. Zheng, T. Fang, Y. Chen, Y. Zhu, Q. Liang, H. Sheng, Z. Li, C. Chen and X. Wang, Sci. China: Chem., 2020, 63, 149–181 CrossRef CAS.
- W. Zhang, A. R. Mohamed and W.-J. Ong, Angew. Chem., 2020, 59, 22894–22915 CrossRef CAS PubMed.
- W. Zhang, R. Huang, L. Song and X. Shi, Nanoscale, 2021, 13, 9075–9090 RSC.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- G. Lin, L. Sun, G. Huang, Q. Chen, S. Fang, J. Bi and L. Wu, Sustainable Energy Fuels, 2021, 5, 732–739 RSC.
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed.
- G. Zhang, Z. Wang and J. Wu, Nanoscale, 2021, 13, 4359–4389 RSC.
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- W.-J. Ong, L. K. Putri and A. R. Mohamed, Chem. – Eur. J., 2020, 26, 9710–9748 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- Y. Ma, Y. Fang, X. Fu and X. Wang, Sustainable Energy Fuels, 2020, 4, 5812–5817 RSC.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- S. Wang, J. Lin and X. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14656–14660 RSC.
- L. Wei, J. Lin, S. Xie, W. Ma, Q. Zhang, Z. Shen and Y. Wang, Nanoscale, 2019, 11, 12530–12536 RSC.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS PubMed.
- C. Tsounis, R. Kuriki, K. Shibata, J. J. M. Vequizo, D. Lu, A. Yamakata, O. Ishitani, R. Amal and K. Maeda, ACS Sustainable Chem. Eng., 2018, 6, 15333–15340 CrossRef CAS.
- Y.-X. Chen, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, Sustainable Energy Fuels, 2020, 4, 2249–2255 RSC.
- K. Zhao, S. Zhao, C. Gao, J. Qi, H. Yin, D. Wei, M. F. Mideksa, X. Wang, Y. Gao, Z. Tang and R. Yu, Small, 2018, 14, 1800762 CrossRef PubMed.
- Q. Li, F. Lin, F. Liu and X. Wang, Chem. Commun., 2019, 55, 3903–3906 RSC.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y.-J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- X. Lin, S. Wang, W. Tu, H. Wang, Y. Hou, W. Dai and R. Xu, ACS Appl. Energy Mater., 2019, 2, 7670–7678 CrossRef CAS.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- J. Di, C. Chen, C. Zhu, P. Song, M. Duan, J. Xiong, R. Long, M. Xu, L. Kang, S. Guo, S. Chen, H. Chen, Z. Chi, Y.-X. Weng, H. Li, L. Song, M. Wu, Q. Yan, S. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CrossRef CAS.
- R. Xu, H. Xu, S. Ning, Q. Zhang, Z. Yang and J. Ye, Trans. Tianjin Univ., 2020, 26, 470–478 CrossRef CAS.
- C. Qiu, S. Bai, W. Cao, L. Tan, J. Liu, Y. Zhao and Y.-F. Song, Trans. Tianjin Univ., 2020, 26, 352–361 CrossRef CAS.
- B. Su, L. Huang, Z. Xiong, Y. Yang, Y. Hou, Z. Ding and S. Wang, J. Mater. Chem. A, 2019, 7, 26877–26883 RSC.
- S. Wang, Y. Wang, S. L. Zhang, S. Q. Zang and X. W. Lou, Adv. Mater., 2019, 31, 1903404 CrossRef CAS PubMed.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H.-M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef.
- Y. Wang, S. Wang, S. L. Zhang and X. W. Lou, Angew. Chem., 2020, 59, 11918–11922 CrossRef CAS PubMed.
- Y. Wang, S. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef CAS PubMed.
- S. Wang, B. Y. Guan, Y. Lu and X. W. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS PubMed.
- S. Wang, Y. Wang, S.-Q. Zang and X. W. Lou, Small Methods, 2020, 4, 1900586 CrossRef CAS.
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1902868 CrossRef CAS PubMed.
- L. Wang, J. Wan, Y. Zhao, N. Yang and D. Wang, J. Am. Chem. Soc., 2019, 141, 2238–2241 CrossRef CAS PubMed.
- S. Wang, B. Y. Guan and X. W. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- P. Zhang, S. Wang, B. Y. Guan and X. W. Lou, Energy Environ. Sci., 2019, 12, 164–168 RSC.
- Z. Wang, S. A. Monny and L. Wang, ChemNanoMat, 2020, 6, 881–888 CrossRef CAS.
- X. Liu, M. Ye, S. Zhang, G. Huang, C. Li, J. Yu, P. K. Wong and S. Liu, J. Mater. Chem. A, 2018, 6, 24245–24255 RSC.
- H. Hu, L. Han, M. Yu, Z. Wang and X. W. Lou, Energy Environ. Sci., 2016, 9, 107–111 RSC.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- S. Wang, B. Y. Guan, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2018, 140, 15145–15148 CrossRef CAS PubMed.
- B. Qiu, Q. Zhu, M. Du, L. Fan, M. Xing and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2684–2688 CrossRef CAS PubMed.
- G. Zhang, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Angew. Chem., 2020, 59, 8255–8261 CrossRef CAS PubMed.
- T. P. Yendrapati, J. Soumya, S. Bojja and U. Pal, J. Phys. Chem. C, 2021, 125, 5099–5109 CrossRef CAS.
- P. Tan, Y. Liu, A. Zhu, W. Zeng, H. Cui and J. Pan, ACS Sustainable Chem. Eng., 2018, 6, 10385–10394 CrossRef CAS.
- L. Shen, L. Yu, H. B. Wu, X.-Y. Yu, X. Zhang and X. W. Lou, Nat. Commun., 2015, 6, 6694 CrossRef CAS PubMed.
- L.-L. Feng, G.-D. Li, Y. Liu, Y. Wu, H. Chen, Y. Wang, Y.-C. Zou, D. Wang and X. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 980–988 CrossRef CAS PubMed.
- S. Zhang, D. Zhai, T. Sun, A. Han, Y. Zhai, W.-C. Cheong, Y. Liu, C. Su, D. Wang and Y. Li, Appl. Catal., B, 2019, 254, 186–193 CrossRef CAS.
- S. Wang, B. Y. Guan and X. W. Lou, Energy Environ. Sci., 2018, 11, 306–310 RSC.
- S. Wang, Y. Hou and X. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4327–4335 CrossRef CAS PubMed.
- W. Wei, W. Chen and D. G. Ivey, Chem. Mater., 2008, 20, 1941–1947 CrossRef CAS.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- W. Yang, H.-J. Wang, R.-R. Liu, J.-W. Wang, C. Zhang, C. Li, D.-C. Zhong and T.-B. Lu, Angew. Chem., 2021, 60, 409–414 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- J. Nai, S. Wang and X. W. Lou, Sci. Adv., 2019, 5, eaax5095 CrossRef CAS PubMed.
- Q. Mu, W. Zhu, G. Yan, Y. Lian, Y. Yao, Q. Li, Y. Tian, P. Zhang, Z. Deng and Y. Peng, J. Mater. Chem. A, 2018, 6, 21110–21119 RSC.
- P. Niu, Z. Pan, S. Wang and X. Wang, ChemSusChem, 2021, 14, 1302–1307 CrossRef CAS PubMed.
- S. Wang, Z. Ding and X. Wang, Chem. Commun., 2015, 51, 1517–1519 RSC.
- Y. Su, Z. Song, W. Zhu, Q. Mu, X. Yuan, Y. Lian, H. Cheng, Z. Deng, M. Chen, W. Yin and Y. Peng, ACS Catal., 2021, 11, 345–354 CrossRef CAS.
- P. Niu, Z. Pan, S. Wang and X. Wang, ChemCatChem, 2021, 13, 3581–3587 CrossRef CAS.
- X. Lin, Y. Gao, M. Jiang, Y. Zhang, Y. Hou, W. Dai, S. Wang and Z. Ding, Appl. Catal., B, 2018, 224, 1009–1016 CrossRef CAS.
- B. Li, W. Wang, J. Zhao, Z. Wang, B. Su, Y. Hou, Z. Ding, W.-J. Ong and S. Wang, J. Mater. Chem. A, 2021, 9, 10270–10276 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data. See DOI: 10.1039/d1nr04812k |
| This journal is © The Royal Society of Chemistry 2021 |