
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSources of variability in nanoparticle uptake by cells†
Christoffer
Åberg
 *,
Valeria
Piattelli
,
Daphne
Montizaan
and
Anna
Salvati
*
*,
Valeria
Piattelli
,
Daphne
Montizaan
and
Anna
Salvati
*
Groningen Research Institute of Pharmacy, University of Groningen, A. Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: christoffer.aberg@rug.nl; a.salvati@rug.nl
First published on 5th October 2021
Abstract
Understanding how nano-sized objects are taken up by cells is important for applications within medicine (nanomedicine), as well as to avoid unforeseen hazard due to nanotechnology (nanosafety). Even within the same cell population, one typically observes a large cell-to-cell variability in nanoparticle uptake, raising the question of the underlying cause(s). Here we investigate cell-to-cell variability in polystyrene nanoparticle uptake by HeLa cells, with generalisations of the results to silica nanoparticles and liposomes, as well as to A549 and primary human umbilical vein endothelial cells. We show that uptake of nanoparticles is correlated with cell size within a cell population, thereby reproducing and generalising previous reports highlighting the role of cell size in nanoparticle uptake. By repeatedly isolating (using fluorescence-activated cell sorting) the cells that take up the most and least nanoparticles, respectively, and performing RNA sequencing on these cells separately, we examine the underlying gene expression that contributes to high and low polystyrene nanoparticle accumulation in HeLa cells. We can thereby show that cell size is not the sole driver of cell-to-cell variability, but that other cellular characteristics also play a role. In contrast to cell size, these characteristics are more specific to the object (nanoparticle or protein) being taken up, but are nevertheless highly heterogeneous, complicating their detailed identification. Overall, our results highlight the complexity underlying the cellular features that determine nanoparticle uptake propensity.
Introduction
How nanomaterials interface with biological systems is a very active field, impinging on applications such as nanomedicine1–3 and on nanosafety.4–7 One of the key facets for such applications is the understanding of how nanomaterials interact with (human) cells. While nanomaterials have been shown to cause effects on cells stemming from their interactions at cell membrane level,8–10 likely the majority of their potential effects requires that they are internalised by cells. A range of in vitro models, including three-dimensional and co-culture models11–14 as well as organ-on-a-chip devices,15,16 of varying degrees of faithfulness to the physiological situation, have been advanced to study nanoparticle–cell interactions. In comparison, adherent mono cell cultures are a much simpler model, which do not represent the full complexity and heterogeneity of tissue. Nevertheless, even when using such a simple cell system, it is a common observation that when it comes to nanoparticle accumulation, the variability between cells is surprisingly large.17–19 Understanding the variability within these simpler systems is therefore crucial towards understanding the heterogeneity in actual tissue.The potential sources of this variability are manifold and the situation is further complicated by the fact that likely there are several mechanisms at play and their various contributions will depend upon the detailed experimental circumstances. For example, we have previously studied in detail, both experimentally18 and theoretically,20,21 the variability induced (in proliferating cell systems) by the cell-division cycle. The cell-division cycle causes a correlation between the time since last division and nanoparticle load, such that recently divided cells have fewer nanoparticles than cells that divided earlier. This effect is, however, most readily apparent at time-scales comparable to the cell population doubling time and largely irrelevant for shorter exposure times (unless the divided cells are explicitly identified and studied). Furthermore, it does not explain the full variability between cells.
Another source of variability could stem from the transport of nanoparticles in the extracellular medium as they are carried around by hydrodynamic flows (at least just after the nanoparticle-containing medium is added) and diffusion before meeting a cell and, potentially, being taken up. These processes depend not only on the exposure conditions, but also on nanoparticle properties (including size, shape and agglomeration state).22–27 Together with the inherent thermodynamic fluctuations in concentration, transport in the extracellular medium will lead to some degree of variability in the local concentration each cell is exposed to. Additionally, the (presumed) stochastic nature of the uptake process itself, whether it stems from a particle finding a receptor or adsorbing to a location where an uptake event takes place, will lead to variability. That is, if there is a certain probability that a cell takes up a nanoparticle, then some cells will take up fewer nanoparticles than other cells purely by chance. Under the assumption that uptake events are independent, one can explicitly write down the variability (Poisson statistics) expected due to this mechanism17,19 which again explains part, but not all, of the variability between cells.17
Another driver behind the variability could be cell size, which has long been posited,18 and later shown in progressively more detail,19,28–30 to play a role in nanoparticle uptake, such that larger cells take up more nanoparticles and smaller cells fewer. Naturally, cell “size” is not very specific and could potentially encompass a range of related but distinct concepts, including cell volume,29 outer cell surface area and even the cross-sectional area one can observe in microscopy.19 For nanoparticle uptake, the most reasonable would appear to be outer cell surface area, and more specifically the outer cell surface area that is accessible to nanoparticles, though we note the difficulty in defining the cell surface area nanoparticles engage with.21 For adherent cell cultures, the exposed cell surface will, furthermore, depend on details of the cell culture, such as cell density (with more sparse cells exposing a larger area) and how well cells spread on the substrate. In the following “cell size” should be interpreted in a loose sense and to encompass these complications.
Aside from cellular characteristics, nanoparticle properties (amongst others size,31,32 shape,32,33 surface charge34 and biomolecular corona composition35,36) also affect overall uptake rate. To what extent nanoparticle properties affect cell-to-cell variability is, however, not known. The various mechanisms driving cell-to-cell variability will also act in concert. For example, the variability induced by the cell-division cycle will couple to the variability due to cell size (since cells about to divide are larger than recently divided cells) and/or the variability due to differing numbers of cell membrane receptors (since cells express different receptors depending upon their position along the cell-division cycle37). Current evidence suggests only minor effects on these particular couplings,18,21 but there nevertheless may be situations where they are important.
Recently it has been suggested that cell size combined with stochastic uptake not only plays a role in, but is sufficient to describe the cell-to-cell variability in nanoparticle uptake between cells (for short exposure times where the cell-division cycle is irrelevant).19 In this context, we here use flow cytometry to study the variability in polystyrene nanoparticle uptake by HeLa cells, with generalisations of the results to silica nanoparticles and liposomes, as well as to A549 and primary human umbilical vein endothelial cells. We reproduce the large variability previously observed and also generalise the observation that nanoparticle uptake is correlated with cell size. Beyond confirming cell size as one of the factors giving rise to cell-to-cell variability, we then seek to identify the underlying cause(s). To this end, we employ an approach that, complementary to previous studies that focus on the cell population as a whole, is focussed more on the outliers, the idea being that their properties may more clearly shine light on the underlying cause of cell-to-cell variability. Specifically, we use fluorescence-activated cell sorting to isolate the HeLa cells that take up the most or least number of polystyrene nanoparticles, respectively, reculturing the cells and repeating the isolation procedure several times. We thereby aim at acquiring cell samples which exhibit heritable traits associated with, respectively, low and high nanoparticle uptake. We then study uptake, both of nanoparticles and a protein, as well as RNA expression in these subpopulations. Based on these results, we can show that cell size is not the only cell characteristic that drives cell-to-cell variability, but that other characteristics also play a role.
Experimental
Materials
Carboxylated polystyrene nanoparticles of 40 and 100 nm diameter were purchased from ThermoFisher (Life Technologies, Eugene, Oregon, USA) with the following fluorescent labels: yellow/green (ex 505 nm, em 515 nm), orange (ex 540 nm, em 560 nm) and far-red (ex 660 nm, em 680 nm). 50 nm silica plain nanoparticles were purchased from Kisker Biotech fluorescently labelled in red (ex 569 nm, em 585 nm). For the experiment shown in ESI Fig. S4 and S10,† green fluorescently labelled (ex 485 nm, em 510 nm) sicastar-greenF 50 and 100 nm silica plain nanoparticles purchased from Micromod were used. Liposomes were made using 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG) and cholesterol (Avanti Polar Lipids) and were labelled with sulforhodamine B from ThermoFisher. The nanoparticles were stored at 4 °C and were used without further modification or purification.Transferrin from human serum conjugated to Alexa Fluor 546 (ex 546 nm, em 573 nm) and Alexa Fluor 647 (ex 650 nm, em 665 nm) were purchased from Molecular Probes (Life Technologies, Eugene, Oregon, USA). The dry protein was resuspended in water to a concentration of 5 mg ml−1 and stored at 4 °C.
CellTrace far red cell proliferation kit (ex 630 nm, em 661 nm), CellMask deep red (ex 649 nm, em 666 nm) and CellMask orange (ex 554 nm, em 567 nm) were purchased from Molecular Probes (Life Technologies, Eugene, Oregon, USA). For CellTrace, the main stock was stored at −20 °C. Before use, a frozen vial was defrosted and diluted with 20 μl DMSO to make a 1 mM stock solution. The stock solution was aliquoted and stored at −20 °C.
Liposome preparation
Liposomes were prepared by freeze and thaw and extrusion as previously described.38 Briefly, DOPG and cholesterol were mixed in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in chloroform, after which a dried film was obtained by evaporation of the solvent with a nitrogen stream and incubation under vacuum overnight. The dried lipid film was hydrated with a 25 mM sulforhodamine B solution in Phosphate Buffered Saline (PBS) to a final lipid concentration of 10 mg ml−1. Then, the suspension was subjected to 8 freeze and thaw cycles with liquid nitrogen and a water bath at 37 °C. Unilamellar liposomes were obtained by extrusion 21 times through a 100 nm pore polycarbonate membrane using an Avanti mini extruder. Free sulforhodamine B was removed using a Zeba Spin Desalting Column with a 7 K MWCO (ThermoFisher). Dynamic light scattering results has confirmed that this method allows obtaining liposomes with a hydrodynamic diameter of around 130 nm and with a very low polydispersity.38
:1 molar ratio in chloroform, after which a dried film was obtained by evaporation of the solvent with a nitrogen stream and incubation under vacuum overnight. The dried lipid film was hydrated with a 25 mM sulforhodamine B solution in Phosphate Buffered Saline (PBS) to a final lipid concentration of 10 mg ml−1. Then, the suspension was subjected to 8 freeze and thaw cycles with liquid nitrogen and a water bath at 37 °C. Unilamellar liposomes were obtained by extrusion 21 times through a 100 nm pore polycarbonate membrane using an Avanti mini extruder. Free sulforhodamine B was removed using a Zeba Spin Desalting Column with a 7 K MWCO (ThermoFisher). Dynamic light scattering results has confirmed that this method allows obtaining liposomes with a hydrodynamic diameter of around 130 nm and with a very low polydispersity.38
Cell culture
HeLa cells (adherent culture of human cervical adenocarcinoma) were purchased from American Type Culture Collection (ATTC; Manassas, Virginia, USA) at passage “unknown + 5” (CCL-2TM, lot no. 61647128). A549 cells (adherent culture of human lung epithelial cancer) were also purchased from ATTC, at passage 82 (CCL-185, lot no. 58016241). Primary human umbilical vein endothelial cells (HUVECs) from pooled donors were purchased from Lonza (Allendale, NJ, USA) at passage 1 (lot no. 394986). The HeLa cells, A549 cells and HUVECs were banked locally after 5, 13 and 2 passages, respectively. HeLa and A549 cells were cultured under standard conditions (37 °C, 5% CO2) in complete medium (cMEM), composed of Minimal Essential Medium containing Earle's salt and L-glutamine (MEM, Gibco, Life technologies, Paisley, UK) supplemented with 10% v/v Fetal Bovine Serum (FBS, Gibco, Life Technologies, Brazil, EU-approved). HUVECs were cultured under standard conditions (37 °C, 5% CO2) in Endothelial Cell Growth Medium 2 Ready-to-use (ECGM-2, from PromoCell, Germany; note that this contains 2% FBS). For the sorting experiments with HeLa cells and for the subsequent experiments on the sorted cells (see below), the cMEM included 1% penicillin (10000 units per ml) and 1% streptomycin (10000 μg ml−1, from Gibco, Life Technologies); the other experiments were performed without antibiotics. Dulbecco's Phosphate Buffered Saline (PBS; Gibco, Life Technologies, Paisley, UK) was used to wash the cells and a solution of 0.05% trypsin–EDTA (Gibco, Life Technologies) to detach them. Mycoplasma tests were performed once a month and never showed a positive result. Generally, experiments on HeLa and A549 cells were performed using cells subcultured between 10 and 25 times (counted from bringing cells into culture from cryopreservation). However, for the sorting experiments, the cells by necessity had to be subcultured more times (between 49 and 56). For the primary HUVECs, cells were used at passage 3 to a maximum of 7.
Nanoparticle characterisation
Dispersions of the 40 nm carboxylated polystyrene nanoparticles at a final concentration of 50 μg ml−1 in water, PBS and cMEM were characterized in terms of size and ζ potential using a Malvern ZetaSizer Nano ZS (Malvern Instruments). Three measurements were recorded for each sample.Flow cytometry
When estimating the fraction of fluorescence due to nanoparticles adhered to the outside of the cells, cells were pre-incubated at 4 °C for 30 min to block active processes, exposed to the nanoparticles for 2 h at 4 °C and then washed and harvested for flow cytometry as described below.
000 cells per well in 24-well plates (Greiner Bio-One) (50000 cells per well for the experiments of ESI Fig. S10 and S11†). For HUVECs, wells were precoated by incubation with a cold solution of 100 μg ml−1 rat-tail collagen type-I (Corning, NY, USA) for 1 h at room temperature, washed three times with PBS and air-dried for 20 min before adding the cell suspension. One day after seeding, the cells were labelled with CellTrace as described above or not (depending upon experiment), after which the cells were exposed to nanoparticles or transferrin for different periods of time. After exposure, the medium with nanoparticles or transferrin was removed and the cells were washed once with cMEM and twice with PBS. Cells were detached using a 0.05% trypsin–EDTA solution for 5 min at 37 °C. Detached cells were spun down at 300 g for 5 min, the supernatant was removed and the cell pellet was resuspended in 100 μl of PBS.
During the measurement, the tubes were stored at room temperature in darkness and analysed one by one at the flow cytometer. The same experimental procedure was used for each experiment and typically 15000–20000 single cells were recorded for each sample. Single cells were differentiated from debris and multiple cells by identifying the main population in side scattering vs. forward scattering and side scattering area vs. side scattering width (BD FACSArray) or forward scattering area vs. forward scattering height (CytoFLEX S) plots.
Fluorescence imaging
HeLa cells were seeded on a glass coverslip at a density of 50000 cells per well in a 24 well plate and used one day after seeding. To measure nanoparticle uptake by microscopy, cells were exposed to 50 μg ml−1 40 nm carboxylated polystyrene nanoparticles in cMEM for 5 h, after which the nanoparticle dispersion was discarded and the cells were labelled with a 1:1000 dilution of CellMask far red in cMEM for 15 min at 37 °C and with 5% CO2. To measure the size of CellTrace-labelled cells, cells were first exposed to a 2 μM solution of CellTrace deep red in serum free MEM for 20 min at 37 °C and with 5% CO2. Then cells were washed 2 times with cMEM and kept for 15 min at 37 °C and with 5% CO2. Finally, cells were exposed to a 1:1000 dilution of CellMask orange in cMEM for 10 min at 37 °C and with 5% CO2. To measure the size of the cells after sorting (see below), the high uptakers, low uptakers and unsorted cells subcultured a comparable number of times were seeded as described above. One day after seeding, cells were labelled with a 1:1000 dilution of CellMask far red in cMEM for 10 min at 37 °C and with 5% CO2.
In all cases, after staining, cells were washed once with cMEM and once with PBS, and then fixed by incubation for 15–20 min in 4% formaldehyde at room temperature or on a plate heater at 37 °C in darkness. Nuclei were stained by incubation for 5–10 min with a 0.2 μg ml−1 solution of DAPI in water. Finally coverslips were mounted on a glass slide using Mowiol 4–88 (Calbiochem). The cells were observed using a Leica AF600 epifluorescence microscope (excitation/emission 340–380/450–490 nm for DAPI, 460–500/512–542 nm for nanoparticles, 541–551/565–605 nm for CellMask orange and 590–650/662–738 nm for CellMask far red and CellTrace deep red). The images were analysed using ImageJ39,40 by outlining a cell in the CellMask channel and subsequently quantifying the total fluorescence intensity within the outline in the nanoparticle or CellTrace channel.
Fluorescence activated sorting
All media used during the sorting process contained 1% penicillin (10000 units per ml) and 1% streptomycin (10000 μg ml−1, from Gibco, Life Technologies), in order to reduce the risk of contamination during the lengthy sorting process. Two sorting experiments were performed, one with a 5% threshold and one with a 1%.
000 cells per well of a 6-well plate; Greiner Bio-One), and in some cases also in flasks.
For each sorting session, a dispersion of yellow/green nanoparticles in cMEM (100 μg ml−1 for the experiment with 5% threshold and 50 μg ml−1 for the 1%) with 1% penicillin/streptomycin was freshly prepared and the sorted cells and the unsorted control sample were exposed to it for 2 h as described above. In order to avoid aging of the samples during the lengthy sorting procedure, the first half of the samples to be sorted were first exposed and later the second half (with a fresh dispersion).
For the sorting procedure, the samples were harvested in the same way as for flow cytometry analysis (above) but after being spun down, the cells were instead re-suspended in a solution of MEM including 2% FBS, 5 mM EDTA (Gibco, Life Technologies) and 1% penicillin/streptomycin. The EDTA was included to reduce cell agglomeration in the cell suspension that had to be kept at fairly high concentration to achieve an efficient sort. (EDTA was introduced after we noticed some clogging of the sorter with the first half of samples for the first sorting session with the 5% threshold.) For the same reason, the dispersions were filtered by using test tubes with cell strainer caps (Falcon, Corning; mesh size 35 μm) in order to reduce the presence of cell clumps that could interfere with the sorting.
For each subsequent sorting session after the first, the unsorted cells were always used as a reference to define the cells with the lowest/highest fluorescence. Thus, the fluorescence distribution of the unsorted cells exposed to nanoparticles was first assessed and the fluorescence value that delimited the 1% or 5% of cells with the lowest/highest nanoparticle fluorescence was recorded. Then the fluorescence distribution of the sorted cell subpopulation was assessed and the proportion of cells below/above the fluorescence threshold quantified (for later use). Next, the sorting procedure started proper. Thus, the sorted cell subpopulation was sorted (again), initially using the fluorescence threshold determined from the unsorted cells to define the 1% or 5% of cells with lowest/highest nanoparticle fluorescence. As each (sub)sample was sorted, the fluorescence distribution corresponding to the cells currently being sorted was quantified continuously. We observed a decrease in the overall fluorescence signal throughout the sorting procedure, and therefore shifted the fluorescence threshold continuously. We did so by ensuring that the fluorescence threshold always contained the same proportion of cells as initially quantified (using the threshold defined by the unsorted cells at the start of the sorting procedure). We applied this step starting on the 4th sorting session for the sorting experiment with the 5% threshold, while it was done throughout the whole sorting experiment with the 1% threshold. We used unsorted cells seeded in cell culture flasks (area 175 cm2) as a reference during the first few sorting sessions (during the first two sessions for both sorted cell subpopulations and the third session for the subpopulation exhibiting low fluorescence for the experiment with the 5% threshold); for all other sorting sessions, we used unsorted cells seeded in plates at equivalent densities (to save on material). In these cases, sorted cells were additionally seeded in plates in order to define the proportion of cells below/above the fluorescence thresholds under conditions matched as closely as possible and this proportion was subsequently used throughout the rest of the sorting procedure as already described.
For the sorting experiment with the 1% threshold, the purity of the sorted sample was checked at the end of each sorting procedure, by assessing the fluorescence of a smaller number of cells from the sorted subpopulation (with the same instrument used to sort the cells).
For the sorting experiment with the 5% threshold, the cells exhibiting the lowest nanoparticle fluorescence were sorted 5 times, while the cells exhibiting the highest were sorted 6 times; for the sorting experiment with the 1% threshold, both cell subpopulations were sorted 5 times.
There are several technical and interpretational issues with these experiments, which dictated our detailed choices and interpretation of the results. These are detailed in the ESI† to keep the main text brief.
Statistical analysis
To quantify the degree of correlation between two variables, we used Spearman's correlation coefficient,41 except when we were interested in a linear correlation, in which case we used Pearson's correlation coefficient.Statistical differences between groups were assessed using the non-parametric rank-based Mann–Whitney U test for two groups and the Kruskal–Wallis H test for more than two groups; statistical differences where other factors (time or concentration) could play a role were assessed using an extension of the non-parametric rank-based Friedman test to multiple observations per cell,41 using time or concentration as a blocking factor and replicate samples as multiple observations. A significance level of 5% was used. The tests used are specified in the relevant figure captions.
Transcriptomics
These differentially expressed genes were analysed per sample with STRING including all interactions of medium confidence or higher (version 11.0, https://string-db.org/).44
Results and discussion
To investigate the variability in nanoparticle uptake we used 40 nm carboxylated polystyrene nanoparticles (of various fluorescent labels) as a model system, because they are fairly monodisperse18,45–49 and also disperse well in cell media18,45 (ESI Table S1 and ESI Fig. S1†). This is important to minimise the characteristics of the particles as a source of heterogeneity in nanoparticle uptake as much as possible. Furthermore, a large amount of previous data on how these particles interact with cells aid interpretation.18,45,50–52 In particular, previous reports (including from us) confirm the uptake of polystyrene nanoparticles into cells by a combination of flow cytometry,18,45,52–54 fluorescence (confocal) microscopy,18,31,45,52,53,55 single-particle tracking56–58 and electron microscopy.59 Additionally, carboxylated polystyrene nanoparticles appear to be benign to cells even after 72 h-long exposure times, given a lack of effect on cell proliferation,18,50 cellular ATP levels,18 cell membrane permeability,49,60 cell viability46,49,61 and cell cycle phase distributions.18,50 As our main cell model, we chose (certified) HeLa (adenocarcinomic human cervical epithelial) cells, which remain a widely used cell model of reference in cell biology in general,62–64 as well as for nanoparticle uptake in particular.35,55,65–68 Observations were generalised in A549 (carcinomic human alveolar basal epithelial) cells and primary human umbilical vein endothelial cells (HUVECs), as well as using a panel of nanoparticles of different materials and size, consisting of 100 nm carboxylated polystyrene nanoparticles, 50 nm silica nanoparticles, 100 nm silica nanoparticles and 100 nm liposomes as a model for common nanomedicines. We also used transferrin as a model for protein uptake.We used flow cytometry69,70 to measure nanoparticle uptake,54 because it allows both measuring individual cells, a necessity in order to investigate cell variability, and because it is high-throughput, a necessity for good statistics. In principle, flow cytometry reports on the overall fluorescence due to the nanoparticles associated with cells, that is, both the nanoparticles internalised by cells as well as those adsorbed to the outer cell membrane. However, we have previously utilised a methodology that allowed us to measure the number of nanoparticles adsorbed to the outside of the cell separately, and thereby demonstrated that after some 2–3 h, the majority of the fluorescence stems from the nanoparticles within the cells;52 similar results were found here in a more limited assessment (ESI Fig. S2†). It should also be noted that while the data is quantitative, it is (at least in practice) in arbitrary units. Furthermore, the fluorescence measurements show, in absolute terms, inter-experimental variability and hence we show representative results rather than averaging over repeated experiments.
We started by confirming previous observations of a large variability in nanoparticle uptake.17,19,28,45,71,72 HeLa cells were exposed to 40 nm polystyrene nanoparticles and we assessed the fluorescence intensity (due to the nanoparticles) of some 16000 HeLa cells using flow cytometry. We observe a well-defined distribution with the majority of cells exhibiting a certain fluorescence and some spread around it (Fig. 1a). However, it must be stressed that Fig. 1a is presented in logarithmic scale, so what appears to be a somewhat narrow distribution is actually very wide in linear scale, with the most extreme cells exhibiting an almost 100-fold difference in fluorescence. A similar wide variability is also observed when A549 cells and HUVECs were exposed to 40 nm polystyrene nanoparticles (ESI Fig. S3†) and also for HeLa cells exposed to 100 nm polystyrene nanoparticles, 50 nm silica nanoparticles, 100 nm silica nanoparticles and 100 nm liposomes (ESI Fig. S4†).
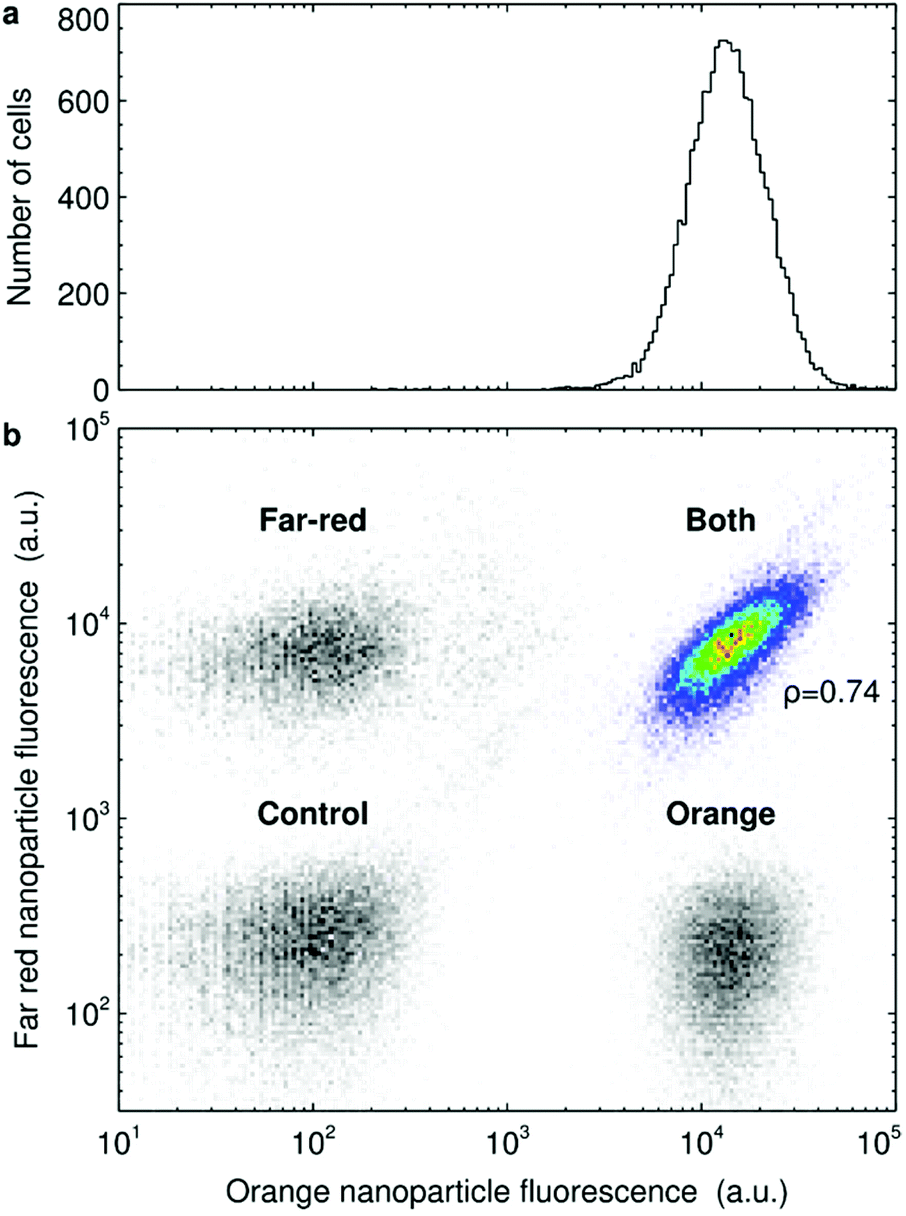 | ||
| Fig. 1 Cell-to-cell variability in 40 nm polystyrene nanoparticle uptake by HeLa cells. (a) Cells were exposed to 5 μg ml−1 of 40 nm orange polystyrene nanoparticles for 5 h and the fluorescence of the cells was assessed by flow cytometry. The panel shows the number of cells which exhibit a given nanoparticle fluorescence. (b) Cells were exposed to either 40 nm far-red nanoparticles, 40 nm orange nanoparticles or to both nanoparticle colours at the same time. The nanoparticle concentration was 5 μg ml−1 for each colour (so the cells exposed to both colours were exposed to 10 μg ml−1 nanoparticles in total) and the exposure lasted for 5 h. Afterwards the fluorescence of the cells was assessed by flow cytometry. The panel shows all the different samples (including also control cells) as heat maps. The correlation between the two variables was quantified in terms of Spearman's correlation coefficient, ρ, as indicated in the figure. ESI Fig. S6† shows similar results for other exposure times. N.B. nanoparticle fluorescence is presented in log scale. | ||
Part of the variability could stem from the technique itself (e.g., due to cells being differently oriented as they pass the excitation beam and the resulting fluorescence is detected). However, it is clear that the large variability exemplified by Fig. 1a is not mainly due to the technique, since markers with known low cell-to-cell variability show a narrow distribution (e.g., cellular DNA staining with dyes such as propidium iodide are usefully reported in linear scale70). Furthermore, when cells exposed to larger (micron-sized) particles are analysed using flow cytometry, then one can explicitly count the number of particles per cell and in this way confirm a large cell-to-cell variability in uptake.71 Similarly, using a different technique, ion beam analysis, it has been shown that cells exhibit a large variability in gold nanoparticle uptake.72 Fluorescence imaging studies also confirm a large variability, both for the same 40 nm polystyrene nanoparticles used here,29 as well as other nanoparticles,19 and we reproduce this observation in ESI Fig. S5.†
Next we wished to exclude the variability due to fluctuations in the local concentration each cell is exposed to. We note that cells were exposed to the nanoparticle dispersion diluted and “pre-mixed” after removal of the original medium (rather than a concentrated nanoparticle dispersion being added to the original medium which was then mixed) in order to promote a uniform exposure.54 Nevertheless, some degree of statistical fluctuations in concentration at a microscopic scale are unavoidable. To test the extent of the variability due to local concentration fluctuations, we reasoned that co-exposure to nanoparticles of two different colours, but otherwise nominally the same, would be a useful test. Statistical fluctuations on the extracellular side will then be different for the two nanoparticles, whereas the cells and uptake process(es) are kept the same (part, but not all, of the variability due to the technique will also be different). We thus exposed HeLa cells to two differently coloured 40 nm polystyrene nanoparticles (Fig. 1b; heat map in colour). It is clear that the cellular fluorescence of the two colours is positively correlated (“the cloud is diagonal”), though there is also some variability super-imposed upon this trend (it is a “cloud”, not a line). This means that cells that, say, take up more nanoparticles of one colour typically also take up more nanoparticles of the other colour, with some variation. The correlation is established already after 1 h and remains over longer exposure times (ESI Fig. S6†). To assert that the results are not due to cross-talk between the two colours (that is, fluorescence from one of the nanoparticles being visible also when assessing the other) we also included three controls (grey scale heat maps): non-exposed cells and cells exposed to only one of the colours. The fact that the fluorescence distributions of cells exposed only to one colour overlaps with non-exposed cells confirms the lack of cross-talk.
We observe similar results for HeLa cells co-exposed to 40 nm polystyrene nanoparticles and fluorescently labelled transferrin protein (ESI Fig. S7†). That is, cells that, say, take up more nanoparticles typically also take up larger amounts of transferrin. The results thus show that there is a part of the variability in nanoparticle uptake that is shared with transferrin accumulation, suggesting that part of the variability stems from the cells themselves and the uptake process.
We next tested the cell variability induced by cell size.18,19,28–30 To quantify cell size using flow cytometry, we labelled the cells with the CellTrace dye, which binds to free amines inside and on the surface of cells. Using fluorescence microscopy we can measure cell size directly and by doing so on CellTrace-labelled cells independently assess the relationship between CellTrace fluorescence and cell size. Such measurements confirm a strong linear correlation between cell size and CellTrace fluorescence (ESI Fig. S8†), justifying our usage of CellTrace as a proxy for cell size.
When subsequently assessing the relationship between 40 nm polystyrene nanoparticle uptake and CellTrace fluorescence in HeLa cells, we observe a clear positive correlation (Fig. 2a). More direct measurements of cell size using fluorescence microscopy gives the same outcome (ESI Fig. S5†). In line with previous reports in literature,19,28–30 these results confirm that cell size does play a role in the variability in nanoparticle uptake. We corroborate this correlation also for A549 cells and primary HUVECs exposed to 40 nm polystyrene nanoparticles (ESI Fig. S9†), as well as for HeLa cells exposed to a series of different nanoparticles, including 100 nm polystyrene nanoparticles, 50 nm and 100 nm silica nanoparticles and 100 nm liposomes (ESI Fig. S10†). However, in all cases there is still a large degree of variability not explained by cell size alone (correlation coefficients often below 0.5). To illustrate this further, we selected a fairly restricted number of cells of a “small”, “medium” and “large” size based on their CellTrace fluorescence (Fig. 2a; vertical lines) and studied the nanoparticle fluorescence distribution of these cells separately (Fig. 2b). These distributions are shifted with respect to each other, but they nevertheless overlap to a significant degree, even with these fairly stringent selection criteria (N.B. that we explicitly defined these subpopulations such that their sizes do not overlap). We corroborated this finding also in the other cells (ESI Fig. S9†) and for all the nanoparticles tested (ESI Fig. S10†). Fluorescence microscopy measurements also show a significant variability beyond the correlation between nanoparticle uptake and cell size (ESI Fig. S5†). For transferrin (ESI Fig. S11†), we observed somewhat of a positive correlation between its accumulation and cell size, but this correlation was weak.
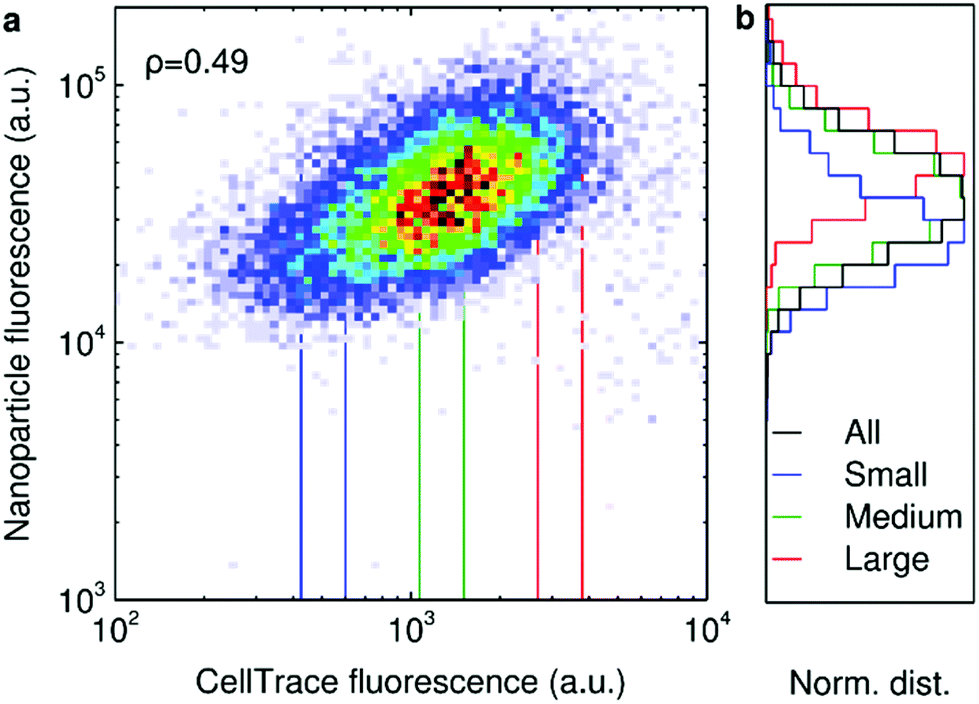 | ||
| Fig. 2 40 nm polystyrene nanoparticle uptake as a function of HeLa cell size. Cells labelled with CellTrace were exposed to 40 nm orange polystyrene nanoparticles (10 μg ml−1 for 5 h) and the fluorescence of the cells assessed by flow cytometry. (a) Cellular nanoparticle fluorescence vs. cell size measured in terms of CellTrace fluorescence. The correlation between the two variables was quantified in terms of Spearman's correlation coefficient, ρ, as indicated in the figure. (b) The distribution of cellular nanoparticle fluorescence for the total cell population as well as three subpopulations corresponding to small, medium and large cells. The distributions have been normalised such that the peak corresponds to 1 in order to present all results on the same scale. The subpopulations were defined in terms of the CellTrace fluorescence as indicated in panel a (vertical lines). | ||
Previous literature has suggested that cell size combined with stochastic uptake is sufficient to describe the cell-to-cell variability in nanoparticle uptake between cells.19 Our results are certainly consistent with cell size being important, though whether it is sufficient to describe the cell-to-cell variability is less clear. Regardless, the underlying cause(s) remain unidentified. Perhaps the most immediate possibility is the number of membrane receptors, which most certainly vary between cells and which may be highly correlated with cell size, and hence could be the underlying origin of the cell size dependence. However, one could also imagine other possibilities, including the expression of various proteins involved in endocytic mechanisms (e.g., clathrin) and, indeed, even cell surface area itself.
To explore such questions, we reasoned that an identification of the heritable cell traits that contribute to cell-to-cell variability in nanoparticle uptake would be useful. While not all cell characteristics that could potentially play a role in nanoparticle uptake are heritable, we expect that many of them are at least partly heritable. A prime example would be the number of cell membrane receptors, which likely is heritable, while also exhibiting some degree of phenotypic variability. In order to investigate if there are such heritable traits that contribute to the variability in nanoparticle uptake, we attempted to separate two HeLa subpopulations, those cells that take up very few 40 nm polystyrene nanoparticles and those that take up many, from the full cell population. One should keep in mind that selecting the absolute lowest or highest uptakers may not be representative of the cell population at large; e.g., the low uptakers could be particularly unhealthy cells, while the high uptakers could be otherwise anomalous. Nevertheless, we considered this was a complementary approach to previous studies. We performed the isolation using fluorescence-activated cell sorting, whereby the fluorescence of each cell of the population is measured and the cells are collected in different tubes post-measurement depending upon the (nanoparticle) fluorescence they exhibit. In order to further enrich the two cell subpopulations (and in contrast to a previous study73), we repeated this sorting procedure 5 times, on each occasion keeping only the low or high uptakers. The repeated exposure may be an issue for nanoparticles that cause adverse effects on the cells, but we deem that to be less of an issue for the polystyrene nanoparticles used here, for which no major effects have been reported.18,46,49,50,60,61 Using fairly stringent criteria to define the “low” and “high” uptakers implies that we collected rather few cells and thus it took some time (around 10–20 days) for the cell (sub)populations to recover and grow. Overall, enriching the two subpopulations 5 times thus took us around 2 months. We did so twice, once defining the subpopulations as the 5% of cells that took up the least or most nanoparticles, and the other as the 1%. There are several technicalities with these sets of experiments which dictated the detailed choices we made and which we feel are important to note. However, to keep the main text reasonably brief, we include the majority of these in the ESI† and discuss only the main ones here.
We thus exposed HeLa cells to 40 nm polystyrene nanoparticles for 2 h and quantified the distribution of nanoparticle fluorescence (Fig. 3a). We could thereby define the nanoparticle fluorescence thresholds that correspond to the 1% of cells that exhibit the lowest nanoparticle fluorescence (blue) and the 1% of cells that exhibit the highest (red). These fluorescence thresholds (dotted lines) were subsequently used to sort the cells into the two subpopulations. In order to examine the accuracy of the sorting procedure, the fluorescence of cells from the two sorted subpopulations was assessed post-sorting. For the cells sorted for low uptake (Fig. 3b), we found that the majority of cells had clearly been correctly collected (left peak). However, there is also a small number of cells (right peak) that have been incorrectly identified. The results were the same for the high uptakers (Fig. 3c), with the majority of cells (corresponding to the right peak) correctly identified, together with a minority (left peak) incorrectly included. In both cases, the fluorescence thresholds used to sort the cells (dotted lines) do not exactly correspond to the separation of the two peaks, which may be due to a number of effects (see the ESI† for a discussion). With this in mind, we estimate the purity of the subpopulations as being the proportion of cells in the main peaks, resulting in around 94% and 79% for the low and high uptakers, respectively (Fig. 3b and c).
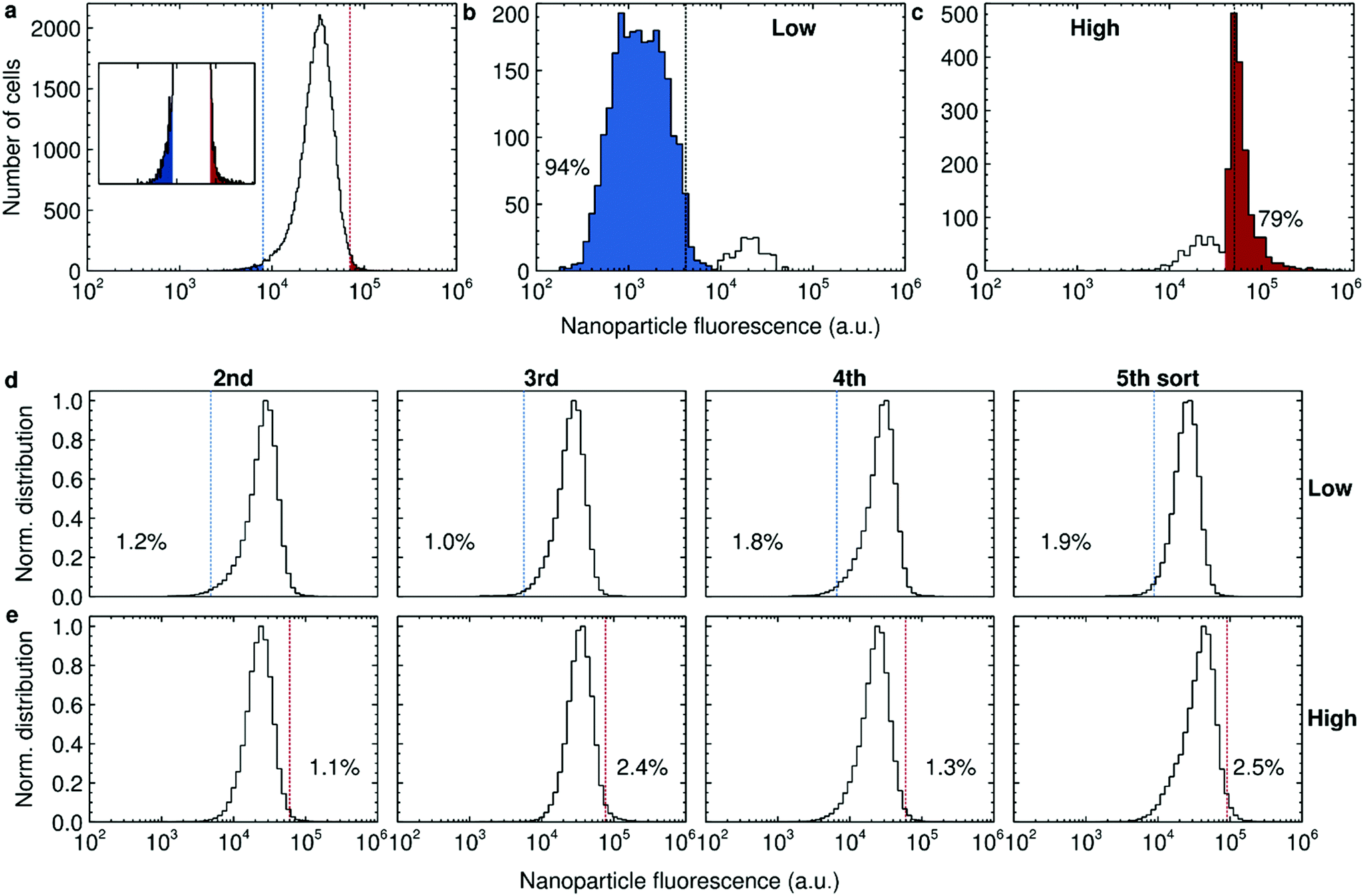 | ||
| Fig. 3 Separation of the 1% of HeLa cells exhibiting the lowest and highest uptake of 40 nm polystyrene nanoparticles. (a) Beginning of the sorting experiment. Nanoparticle fluorescence distribution of cells exposed to 50 μg ml−1 of the nanoparticles for 2 h. Dotted lines show the fluorescence thresholds that correspond to the 1% of cells taking up the least and most nanoparticles, respectively. Inset shows an enlarged view (with the same x scale). (b and c) Accuracy of the sorting procedure for low (panel b) and high (panel c) uptakers, respectively. After the first sorting session, the fluorescence of the sorted cells was assessed and compared to the thresholds used to sort the cells (dotted lines). The proportion of cells within the main peaks are also reported. The fluorescence thresholds in panel a and b–c (dotted lines) do not correspond, because they were continuously adjusted to accommodate for a decrease in fluorescence throughout the sorting procedure. See the Experimental section for further discussion of this point. (d and e) Enrichment upon each subsequent sorting session for low (panel d) and high (panel e) uptakers, respectively. At the beginning of each sorting session, the fluorescence of unsorted cells was assessed and used to set an “absolute” fluorescence reference (dotted lines). Subsequently, the fluorescence of the sorted cells was measured (distribution) and the proportion of cells within the limits quantified (percentages). The distributions have been normalised such that the peak corresponds to 1 in order to represent all results on the same scale. | ||
Since the purity was reasonably high, we continued amplifying the cells until there were enough cells to perform a second sorting procedure. For enriching the subpopulations, we can imagine two alternative approaches: (i) take (for example) the 1% lowest uptakers each time, regardless of their actual absolute uptake rates; or (ii) take (for example) all cells exhibiting an absolute uptake rate lower than a certain rate, which remains fixed throughout the experiment. We opted for the second approach (but, as will transpire, the first approach would effectively have been equivalent) and hence we needed an external reference to define the fluorescence threshold that corresponded to our original definition of low and high uptakers. Since extended subculturing could potentially lead to phenotypic changes, we decided to use unsorted cells subcultured for the same length of time (and roughly the same number of times) as a reference, rather than “fresh” cells.
We performed the sorting 4 more times using the 1% threshold (Fig. 3d and e). Interestingly, even though the accuracy of the sorting procedure is quite high (Fig. 3b and c) we did not observe any clear enrichment (percentages in Fig. 3d and e). We also could not observe any major difference in overall nanoparticle uptake during the sorting procedure (ESI Fig. S12†). Furthermore, we did not observe any obvious phenotypic differences by microscopy during day-to-day handling of the sorted and unsorted cells.
We concluded the sorting procedure after 5 sorts. Fluorescence microscopy quantification of cell size of the resulting cell populations did not show any substantial differences (Fig. 4a) and we also could not observe any obvious phenotypic differences (Fig. 4b). When we used the 5% threshold (ESI Table S2†) we found a larger enrichment, in particular for the high uptakers, but it nevertheless never went above 20%. The most immediate interpretation of these results would be that the characteristics of the HeLa cells that are responsible for the variability in 40 nm polystyrene nanoparticle uptake within the cell population are not heritable and, specifically, that cell size is not one of these characteristics.
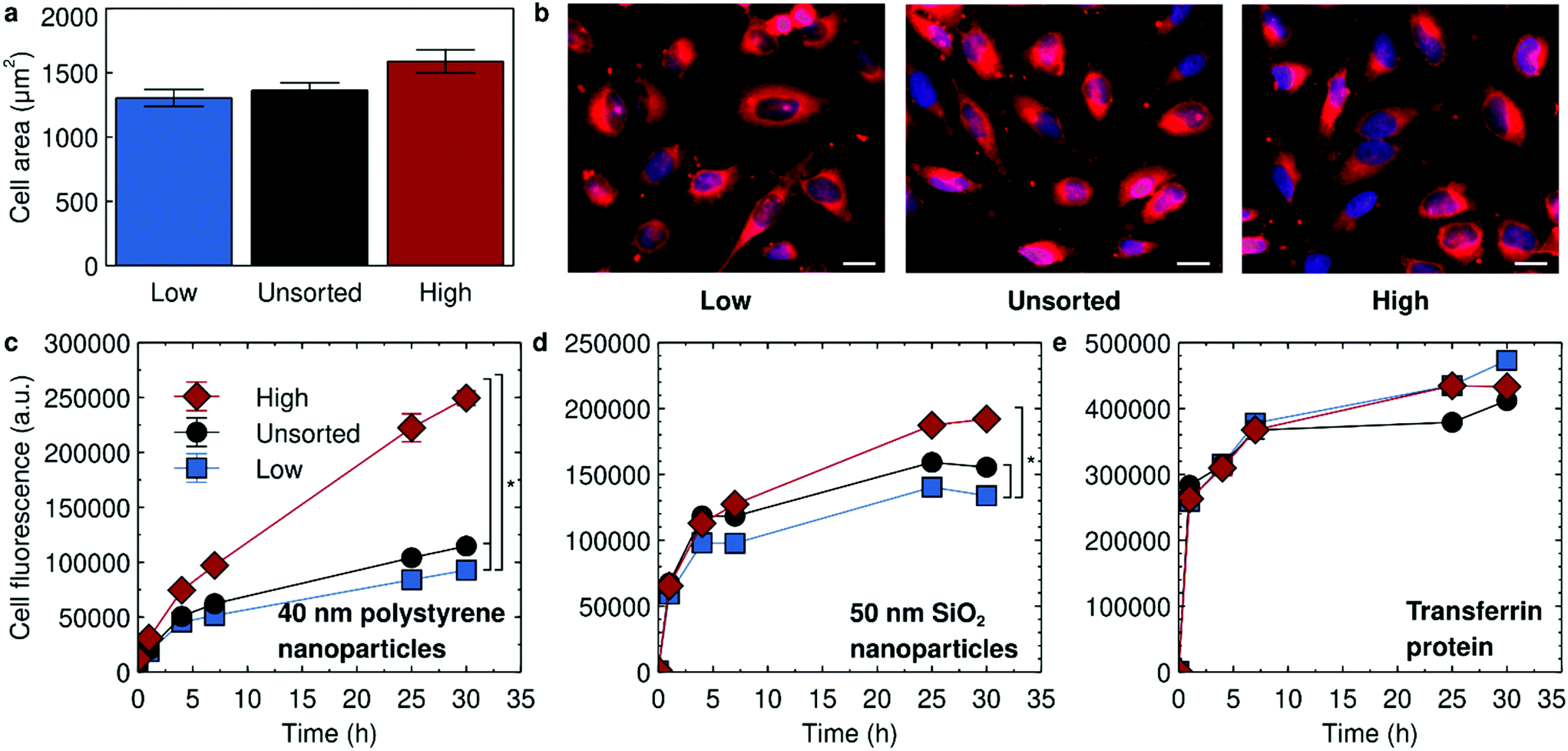 | ||
| Fig. 4 Characterisation of the sorted HeLa cells. Cells were sorted 5 times with the threshold set to separate the 1% of cells exhibiting the lowest and highest nanoparticle uptake, respectively. Unsorted cells subcultured an equivalent number of times were used as controls. (a and b) Fluorescence microscopy of the low and high uptakers, together with unsorted cells cultured in an equivalent fashion, after conclusion of the sorting (followed by cryopreservation and subsequent reculturing). Cells were labelled with a cell membrane stain (CellMask far red) and then imaged using fluorescence microscopy. (a) Quantification of cell area from cell outlines. The bars show the mean area with the error bar representing standard error of the mean (over 106, 102 and 101 cells, respectively). A Kruskal–Wallis test does not show a statistically significant difference between the three different cell populations. (b) Representative images. Scale bar corresponds to 20 μm. (c–e) Nanoparticle and protein uptake in sorted HeLa cells. The experiment was performed on all cell cultures simultaneously, which implies that it had passed 9 and 7 days after the last sorting session for the low and high uptakers, respectively (the two cultures ended up being sorted on different days). See the main text and Experimental section for details. The cells were exposed to (c) 10 μg ml−1 of 40 nm yellow/green polystyrene nanoparticles (d) 100 μg ml−1 of 50 nm SiO2 nanoparticles and (e) 15 μg ml−1 transferrin for the indicated time periods and the fluorescence of the cells assessed by flow cytometry. Datapoints and error bars represent the mean and standard deviation over two replicate samples. A Friedman test with time as blocking factor shows a statistically significant difference between the three different types of cells for both the 40 nm polystyrene and 50 nm SiO2 nanoparticles. Comparison between individual curves (taking into account multiple comparisons) gives the statistically significant differences indicated by asterisks in the figure. See also ESI Fig. S14† for a repeat experiment performed a week later. | ||
Based on these results, we next wanted to confirm that, indeed, the sorted HeLa cells did not exhibit any difference in nanoparticle uptake. We consequently exposed the sorted cells, as well as unsorted cells cultured an equivalent amount of time as control, to the same 40 nm polystyrene nanoparticles. Note that the sorted cells essentially do not exhibit any residual nanoparticle fluorescence from the sorting procedure (ESI Fig. S13†) before the exposure. To our surprise, we observed a clear difference in uptake behaviour between the different cells sorted with the 1% threshold, with the high uptakers indeed taking up significantly more 40 nm polystyrene nanoparticles, followed by the unsorted cells and finally the low uptakers (Fig. 4c). The same ranking remained for at least 2 weeks after the sorting was concluded (ESI Fig. S14;† the experiment shown in Fig. 4c was instead performed around one week after the sorting). Thus, it appears that there are indeed cellular traits that determine nanoparticle uptake.
When we investigated uptake of another nanoparticle (50 nm silica), different from the one used to define the sorted subpopulations (40 nm polystyrene), we initially observed the same ranking (Fig. 4d). This suggests that some parts of the endocytic machinery may be common to uptake of the two different nanoparticles (possibly because of their nano-size). However, interestingly, the effect seemed weaker than for the nanoparticle used to define the high and low uptakers (cf.Fig. 4c and d). Furthermore, the ranking disappeared a week later (ESI Fig. S14†). Thus, other aspects of the uptake must be specific to the nanoparticle used for sorting the cells (polystyrene) because otherwise we would expect an equally large and consistent effect of the sorting on the other nanoparticle (silica).
The ranking observed for 40 nm polystyrene nanoparticles was not observed when exposing the sorted HeLa cells to transferrin (Fig. 4e, ESI Fig. S14†). Though transferrin is recycled and its intracellular level thus reflect both its uptake and export, the lack of an effect is consistent with transferrin and 40 nm polystyrene nanoparticles not sharing the same internalisation mechanism in HeLa cells, at least not to a significant extent.
When using the 5% threshold, the cells were cryopreserved after the sorting procedure and then brought back into culture to study their uptake behaviour. We found similar results for these cells (ESI Fig. S15†) compared to the cells sorted with a 1% threshold (Fig. 4c–e), for the 40 nm polystyrene nanoparticles used to define the cell subpopulations, for 50 nm silica nanoparticles and for transferrin.
In order to investigate the source of the differences at a biomolecular level, we next performed RNA sequencing of all three HeLa cell samples: high and low uptakers acquired using the 1% threshold and unsorted cells. Overall, the majority of genes found expressed was present in all three samples, though some genes were only found expressed in two or even one sample (Fig. 5a). For the genes that were found in all three samples, we calculated the change in the high and low uptakers, respectively, compared to the unsorted cells (Fig. 5b).
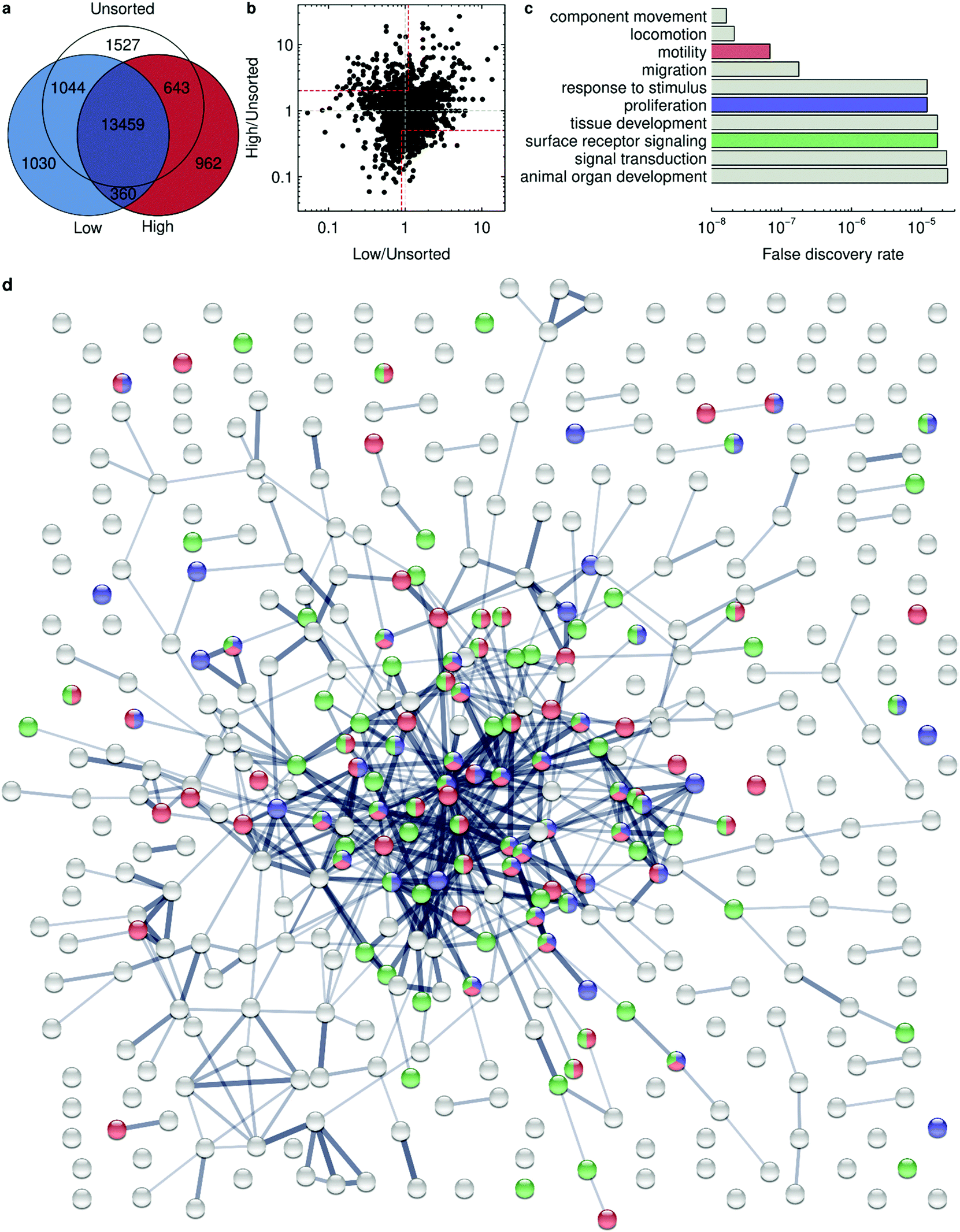 | ||
| Fig. 5 RNA sequencing analysis of HeLa cells sorted for high and low 40 nm polystyrene nanoparticle uptake. Cells were sorted 5 times with the threshold set to separate the 1% of cells exhibiting the lowest and highest nanoparticle uptake, respectively. Unsorted cells subcultured an equivalent number of times were used as control. RNA sequencing was performed on the three samples and the expressed genes identified. (a) Venn diagram showing the expressed genes that are common between the different samples. N.B. The diagram is not to scale (because the large overlap would make it hard to read). (b) Expression levels of genes in high and low uptakers, respectively, compared to unsorted cells (of all genes found expressed in all three samples). Each dot represents one gene. (Grey dashed lines) Same expression as in unsorted cells. (Areas limited by red dashed lines.) Genes of particular interest for high uptake. Note the log-scale. (c) False discovery rate of the top-10 pathways from Gene Ontology biological processes for high uptake. The descriptions have been shortened to fit in the figure and read unabridged: regulation of cellular component movement; regulation of locomotion; regulation of cell motility; regulation of cell migration; regulation of response to stimulus; regulation of cell population proliferation; tissue development; cell surface receptor signaling pathway; regulation of signal transduction; animal organ development. (d) Clustering analysis for high uptake showing protein interactions using STRING. Colors are consistent with panel c. (Red) Genes involved in regulation of cell motility. (Blue) Genes involved in regulation of cell population proliferation. (Green) Genes involved in cell surface receptor signaling pathway. Line thickness represents the confidence of the interactions with a minimum of medium confidence (0.400). For reasons of legibility and to more clearly show the cluster, we have omitted the names of the genes; the full picture is available as ESI.† | ||
We next attempted to identify the genes responsible for high and low 40 nm polystyrene nanoparticle uptake separately. Thus, for high uptake we selected genes that were upregulated in the high uptakers and either downregulated or unchanged in the low uptakers (upper-left area indicated in dotted red in Fig. 5b), together with the genes that were downregulated in the high uptakers and either upregulated or unchanged in the low uptakers (lower-right). For low uptake we made the opposite selection. In both cases, we set thresholds to define “unchanged” with some margin to allow for experimental error (dotted red vertical lines in Fig. 5b). Where possible we also included genes that were missing in one or two of the samples (which cannot be represented in Fig. 5b since either or both changes are not well-defined), as described in detail in the Experimental section. The top-20 expressed genes from these two lists are shown in Table 1 (low) and Table 2 (high); the full lists are available as ESI.† We observe that both for low and high uptake, there is a clear difference in gene expression levels compared to control, and the differentially expressed genes are also different for low and high uptake.
| Name | Description | Unsorted (rpma) | Low (rpma) | High (rpma) | Low fold changeb | High fold changeb |
|---|---|---|---|---|---|---|
| a Reads per million. b With respect to unsorted cells. c Gene was not found expressed in unsorted cells, so fold change is undefined. | ||||||
| LRRIQ3 | Leucine rich repeats and IQ motif containing 3 | 0 | 2.172 | 0 | Infc | — |
| NEK11 | NIMA related kinase 11 | 0 | 2.172 | 0 | Infc | — |
| RBBP4P2 | RB binding protein 4 pseudogene 2 | 0.255 | 2.172 | 0 | 8.508 | 0 |
| RGS11 | Regulator of G protein signaling 11 | 0.255 | 2.172 | 0 | 8.508 | 0 |
| LRP2BP | LRP2 binding protein | 0.255 | 2.172 | 0 | 8.508 | 0 |
| AP003469.4 | Novel transcript | 0.255 | 2.172 | 0 | 8.508 | 0 |
| UNC5D | Unc-5 netrin receptor D | 3.063 | 18.677 | 3.023 | 6.097 | 0.987 |
| RPS4XP13 | Ribosomal protein S4X pseudogene 13 | 0.511 | 3.04 | 0 | 5.956 | 0 |
| HSP90B2P | Heat shock protein 90 beta family member 2, pseudogene | 1.787 | 10.424 | 1.512 | 5.834 | 0.846 |
| PRSS36 | Serine protease 36 | 0.766 | 3.909 | 0.756 | 5.105 | 0.987 |
| ADAP2 | ArfGAP with dual PH domains 2 | 2.553 | 12.162 | 0.756 | 4.765 | 0.296 |
| RNF125 | Ring finger protein 125 | 0.766 | 3.475 | 0 | 4.538 | 0 |
| STK32A | Serine/threonine kinase 32A | 0.766 | 3.475 | 0.756 | 4.538 | 0.987 |
| RN7SL689P | RNA, 7SL, cytoplasmic 689, pseudogene | 0.766 | 3.475 | 0 | 4.538 | 0 |
| INHBE | Inhibin subunit beta E | 0.766 | 3.475 | 0.756 | 4.538 | 0.987 |
| PRPH | Peripherin | 0.511 | 2.172 | 0 | 4.254 | 0 |
| C3orf33 | Chromosome 3 open reading frame 33 | 1.021 | 4.343 | 0.756 | 4.254 | 0.74 |
| HYKK | Hydroxylysine kinase | 1.021 | 4.343 | 0.756 | 4.254 | 0.74 |
| GLI1 | GLI family zinc finger 1 | 1.021 | 4.343 | 0.756 | 4.254 | 0.74 |
| KHSRPP1 | KH-type splicing regulatory protein pseudogene 1 | 0.511 | 2.172 | 0 | 4.254 | 0 |
| Name | Description | Unsorted (rpma) | Low (rpma) | High (rpma) | Low fold changeb | High fold changeb |
|---|---|---|---|---|---|---|
| a Reads per million. b With respect to unsorted cells. c Gene was not found expressed in unsorted cells, so fold change is undefined. | ||||||
| SERTAD4 | SERTA domain containing 4 | 0 | 0 | 3.779 | — | Infc |
| FLI1 | Fli-1 proto-oncogene, ETS transcription factor | 0 | 0 | 4.535 | — | Infc |
| CD33 | CD33 molecule | 0 | 0 | 3.779 | — | Infc |
| LINC01234 | Long intergenic non-protein coding RNA 1234 | 0 | 0 | 3.779 | — | Infc |
| POSTN | Periostin | 0 | 0 | 7.558 | — | Infc |
| CPEB1 | Cytoplasmic polyadenylation element binding protein 1 | 0 | 0 | 3.779 | — | Infc |
| RPL15P18 | Ribosomal protein L15 pseudogene 18 | 0 | 0 | 3.779 | — | Infc |
| PGK1P2 | Phosphoglycerate kinase 1, pseudogene 2 | 0 | 0 | 3.779 | — | Infc |
| AC125238.2 | Zinc finger protein 207 (ZNF207) pseudogene | 0 | 0 | 3.779 | — | Infc |
| KCNMA1 | Potassium calcium-activated channel subfamily M alpha 1 | 0 | 0 | 4.535 | — | Infc |
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase | 0 | 0 | 4.535 | — | Infc |
| MAGED1 | MAGE family member D1 | 0 | 0 | 3.779 | — | Infc |
| AC006262.2 | Novel transcript | 0.255 | 0 | 7.558 | 0 | 29.611 |
| HHAT | Hedgehog acyltransferase | 0.255 | 0 | 6.803 | 0 | 26.65 |
| CACNA1G | Calcium voltage-gated channel subunit alpha1 G | 0.766 | 0.434 | 14.361 | 0.567 | 18.754 |
| CARD11 | Caspase recruitment domain family member 11 | 0.255 | 0 | 4.535 | 0 | 17.767 |
| SH3RF3 | SH3 domain containing ring finger 3 | 0.255 | 0 | 4.535 | 0 | 17.767 |
| LRRC15 | Leucine rich repeat containing 15 | 0.766 | 0.434 | 12.093 | 0.567 | 15.793 |
| POPDC3 | Popeye domain containing 3 | 0.511 | 0 | 7.558 | 0 | 14.806 |
| SEMA5A | Semaphorin 5A | 0.255 | 0 | 3.779 | 0 | 14.806 |
From the two lists of genes, we attempted to identify differences in expressed pathways by performing a gene ontology analysis (see Experimental section for details). This analysis does not take into account whether the genes are up- or downregulated but can nevertheless give an idea of the relevant pathways. For low uptake, we do not observe any major clusters of genes (see ESI†). However, for high uptake, we find a strong clustering of genes related to cell motility (strongly overlapping with genes classified as regulating cellular component movement, locomotion and cell migration), regulation of cell population proliferation, and cell surface receptor signaling pathways (Fig. 5d, depicted in red, blue and green respectively; see also ESI† for a complete picture).
We may speculate that the differential expression of genes related to cell motility (and also cell migration and locomotion) is because the genes involved with this process include elements of the actin machinery and other parts of the cytoskeleton. Indeed, it is known that actin and microtubules play a role in cell internalisation pathways in general,74–76 and in nanoparticle uptake specifically.77 For instance, blocking actin and microtubule polymerization with, respectively, cytochalasin D and nocodazole, strongly reduces the uptake of negatively charged liposomes, but not zwitterionic ones.78 Similarly, blocking actin with cytochalasin D reduces uptake under the same conditions considered here (40 nm carboxylated polystyrene nanoparticles in HeLa cells) as well as in several other cell types,79 confirming a role of actin in the uptake of these nanoparticles. This result is also consistent with some effect of the sorting on both the polystyrene and silica nanoparticles, as a perturbation to the endocytic machinery is expected to affect both particles.
Another speculative idea is that the presence of genes involved with cell proliferation is due to the cell division cycle playing a role even in the short exposure scenario (2 h) we used to define these cells. Also, it is perhaps not surprising to find a differential expression of cell surface receptor signalling pathways, as different receptors are presumably involved in nanoparticle uptake. This may also explain (at least in part) the smaller effect of the sorting on the silica nanoparticles (Fig. 4d and ESI Fig. S14 and S15†). Indeed, different surface receptors are likely to be involved in the uptake of the different particles because of their different size, material, surface characteristics and consequently also different corona. Similarly, the absence of clathrin-mediated endocytosis in the top-10 expressed pathways is consistent with the observation that there is no effect on the sorting on transferrin accumulation levels (Fig. 4e and ESI Fig. S14 and S15†).
Further studies would be required to confirm these speculative interpretations. Overall, however, it seems clear that there is a differential expression of genes for the low and high uptakers. Furthermore, it is clear that several different processes are involved, rather than a well-defined subset.
Conclusions
Multiple mechanisms contribute to cell-to-cell variability in nanoparticle uptake, several of which were investigated here. We observe a correlation between cell size and polystyrene nanoparticle, silica nanoparticle and liposome accumulation in HeLa cells, A549 cells and HUVECs, consistent with previous reports on the same 40 nm polystyrene nanoparticles used here29 and quantum dots.19 These results highlight cell size (independent of its exact definition) as a central cell property that correlates with nanoparticle uptake. Nevertheless, we show that size alone cannot explain the variability observed and that other factors contribute to it.After sorting HeLa cells based on high and low 40 nm polystyrene nanoparticle uptake, we observe that the high and low uptakers do, indeed, take up more and fewer nanoparticles, respectively. Still, the effect is far smaller than the selection limits we applied, suggesting that the cell features that determine nanoparticle uptake are only partly heritable. Furthermore, the fact that cells sorted for high and low uptake take up more and fewer nanoparticles, respectively, would be consistent with the selection acting on cell size (or the selection acting on some other property that correlates with cell size). However, we could not detect any major difference in cell size between the high and low uptakers. Furthermore, the effect that the high uptakers take up more particles than the low uptakers appears to be the strongest for the polystyrene nanoparticles that were used to define the low and high uptakers. Thus, beyond cell size, other cell characteristics also appear to matter and they appear to be specific to the object being taken up. Potential characteristics include various receptors and other molecules involved in cellular internalisation mechanisms with which the nanoparticle engages. In the case of nanoparticles, naturally these features should also be related to the biomolecular corona surrounding the nanoparticles.80–84 The presence of a corona is known to affect overall uptake,52,85–88 but in the present context the more important question is which individual biomolecules are present in the corona and able to engage with the cells. Indeed, since corona composition is fairly specific to a given particle (while also depending on the biological fluid and its concentration89) it could be part of the reason for the somewhat specific response to a given nanoparticle. Furthermore, we here used fairly monodisperse nanoparticles so while the heterogeneity among the particles could be a source of variability, we expect it to be small and certainly smaller than the wide distribution we observe over cells. However, there is an additional effect of variability due to the corona, which is known to not be identical for each nanoparticle even within the same nanoparticle sample.80,90,91 The corona could therefore also be a source of the observed variability in cell uptake.
Subsequent RNA sequencing of the sorted cells shows genetic (or at least phenotypic) differences between high and low uptakers, as well as with unsorted cells. However, we were not able to identify one or a few pathways that were clearly up- or downregulated and could be connected to nanoparticle uptake. Thus, these results suggest that the cell characteristics contributing to a high or low uptake are multiple and heterogeneous.
From a methodological point of view, the repeated sorting is complementary to other more direct approaches. In future, it would be interesting to explore variations of the procedure (e.g., different thresholds for the sorting procedure) or different systems (e.g., different nanoparticles and/or different biomolecular coronae80), but the length of the experiments make them less amenable for routine experimentation. A variation of this procedure for more advanced in vitro systems,11–16 could also be interesting to capture the underlying heterogeneity in a more realistic scenario. For example, we have recently shown that in liver slices, nanoparticles preferentially accumulate in macrophages and uptake remains heterogeneous, even between macrophages.47 The sources of variability between the macrophages (and other cell types), however, remains to be investigated. Single-cell RNA sequencing, as applied previously,73 could also be an interesting extension to better identify the highly heterogeneous pathways.
In summary, cell size is clearly correlated with the large cell-to-cell variability in nanoparticle uptake. Our results for 40 nm polystyrene nanoparticle uptake by HeLa cells, however, suggest that other cell characteristics also play a role. Furthermore, these characteristics are heterogeneous, but nevertheless specific to a given particle, and only partly heritable. From a broader perspective, cell-to-cell variability in vivo will lead to a wide range of intracellular doses. This variability may need to be addressed, whether to achieve a uniform therapeutic outcome in all relevant cells (in nanomedicine) or to avoid overloading particular high-accumulating cells (in nanosafety).
Author contributions
C. Å. and A. S. designed research; V. P. performed all experiments except as otherwise noted; D. M. assisted in experimental design of the cell sorting experiments and performed the transcriptomic work; A. S. performed the experiments generalising the observations to other cell types and nanoparticles, as well as the fluorescence imaging; all authors analysed and interpreted data; C. Å. wrote the paper with comments and input from all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme under grant agreement no. 637614 (NanoPaths) to A. S. The cell sorting experiments were performed at the Central Flow Cytometry Unit of the University Medical Center Groningen, while the RNA sequencing was performed by the group of P. Lansdorp at the European Research Institute of the Biology of Ageing, University Medical Center Groningen. The authors would like to thank G. Mesander for help with the cell sorting experiments; C. Reker-Smit for sample preparation and particle characterisation and S. de Weerd for liposome preparation; D. Spierings and K. Hoekstra-Wakker from the group of P. Lansdorp for suggestions and technical help with sample preparation for RNA sequencing, respectively; and V. Guryev (European Research Institute of the Biology of Ageing, University Medical Center Groningen) for technical help and suggestions on the analysis of the RNA sequencing.Notes and references
- J. Wolfram and M. Ferrari, Nano Today, 2019, 25, 85–98 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- P. Couvreur, Adv. Drug Delivery Rev., 2013, 65, 21–23 CrossRef CAS PubMed.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823–839 CrossRef PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- P. Rivera Gil, G. Oberdörster, A. Elder, V. Puntes and W. J. Parak, ACS Nano, 2010, 4, 5527–5531 CrossRef CAS PubMed.
- E. Valsami-Jones and I. Lynch, Science, 2015, 350, 388–389 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- G. Bhabra, A. Sood, B. Fisher, L. Cartwright, M. Saunders, W. H. Evans, A. Surprenant, G. Lopez-Castejon, S. Mann, S. A. Davis, L. A. Hails, E. Ingham, P. Verkade, J. Lane, K. Heesom, R. Newson and C. P. Case, Nat. Nanotechnol., 2009, 4, 876–883 CrossRef CAS PubMed.
- A. Sood, S. Salih, D. Roh, L. Lacharme-Lora, M. Parry, B. Hardiman, R. Keehan, R. Grummer, E. Winterhager, P. J. Gokhale, P. W. Andrews, C. Abbott, K. Forbes, M. Westwood, J. D. Aplin, E. Ingham, I. Papageorgiou, M. Berry, J. Liu, A. D. Dick, R. J. Garland, N. Williams, R. Singh, A. K. Simon, M. Lewis, J. Ham, L. Roger, D. M. Baird, L. A. Crompton, M. A. Caldwell, H. Swalwell, M. Birch-Machin, G. Lopez-Castejon, A. Randall, H. Lin, M.-S. Suleiman, W. H. Evans, R. Newson and C. P. Case, Nat. Nanotechnol., 2011, 6, 824–833 CrossRef CAS PubMed.
- S. G. Klein, T. Serchi, L. Hoffmann, B. Blömeke and A. C. Gutleb, Part. Fibre Toxicol., 2013, 10, 31 CrossRef CAS PubMed.
- P. E. C. Leite, M. R. Pereira, G. Harris, D. Pamies, L. M. G. dos Santos, J. M. Granjeiro, H. T. Hogberg, T. Hartung and L. Smirnova, Part. Fibre Toxicol., 2019, 16, 1–20 CrossRef CAS PubMed.
- A. I. Astashkina, C. F. Jones, G. Thiagarajan, K. Kurtzeborn, H. Ghandehari, B. D. Brooks and D. W. Grainger, Biomaterials, 2014, 35, 6323–6331 CrossRef CAS PubMed.
- J. Lee, G. D. Lilly, R. C. Doty, P. Podsiadlo and N. A. Kotov, Small, 2009, 5, 1213–1221 CAS.
- S. K. Mahto, V. Charwat, P. Ertl, B. Rothen-Rutishauser, S. W. Rhee and J. Sznitman, Nanotoxicology, 2015, 9, 381–395 CrossRef CAS PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- H. D. Summers, P. Rees, M. D. Holton, M. R. Brown, S. C. Chappell, P. J. Smith and R. J. Errington, Nat. Nanotechnol., 2011, 6, 170–174 CrossRef CAS PubMed.
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62–68 CrossRef CAS PubMed.
- P. Rees, J. W. Wills, M. R. Brown, C. M. Barnes and H. D. Summers, Nat. Commun., 2019, 10, 2341 CrossRef PubMed.
- C. Åberg, J. A. Kim, A. Salvati and K. A. Dawson, EPL, 2013, 101, 38007 CrossRef.
- C. Åberg, J. A. Kim, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2017, 12, 600–603 CrossRef PubMed.
- L. K. Limbach, P. Wick, P. Manser, R. N. Grass, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2007, 41, 4158–4163 CrossRef CAS PubMed.
- J. G. Teeguarden, P. M. Hinderliter, G. Orr, B. D. Thrall and J. G. Pounds, Toxicol. Sci., 2007, 95, 300–312 CrossRef CAS PubMed.
- J. M. Cohen, J. G. Teeguarden and P. Demokritou, Part. Fibre Toxicol., 2014, 11, 20 CrossRef PubMed.
- G. M. DeLoid, J. M. Cohen, G. Pyrgiotakis and P. Demokritou, Nat. Protoc., 2017, 12, 355–371 CrossRef CAS PubMed.
- V. Sharma, K. Park and M. Srinivasarao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4981–4985 CrossRef CAS PubMed.
- N. Feliu, X. Sun, R. A. Alvarez Puebla and W. J. Parak, Langmuir, 2017, 33, 6639–6646 CrossRef CAS PubMed.
- M. J. Ware, B. Godin, N. Singh, R. Majithia, S. Shamsudeen, R. E. Serda, K. E. Meissner, P. Rees and H. D. Summers, ACS Nano, 2014, 8, 6693–6700 CrossRef CAS PubMed.
- E. Panet, T. Mashriki, R. Lahmi, A. Jacob, E. Ozer, M. Vecsler, I. Lazar and A. Tzur, Nat. Nanotechnol., 2017, 12, 598–600 CrossRef CAS PubMed.
- J. Khetan, M. Shahinuzzaman, S. Barua and D. Barua, Biophys. J., 2019, 116, 347–359 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- L. Boselli, H. Lopez, W. Zhang, Q. Cai, V. A. Giannone, J. Li, A. Moura, J. M. de Araujo, J. Cookman, V. Castagnola, Y. Yan and K. A. Dawson, Commun. Mater., 2020, 1, 1–12 CrossRef.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailänder, Macromol. Biosci., 2008, 8, 1135–1143 CrossRef CAS PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- V. Francia, K. Yang, S. Deville, C. Reker-Smit, I. Nelissen and A. Salvati, ACS Nano, 2019, 13, 11107–11121 CrossRef CAS PubMed.
- E. Boucrot and T. Kirchhausen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7939–7944 CrossRef CAS PubMed.
- K. Yang, B. Mesquita, P. Horvatovich and A. Salvati, Acta Biomater., 2020, 106, 314–327 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- W. J. Conover, in Practical nonparametric statistics, John Wiley & Sons, Inc., New York, 3rd edn, 1999, ch. 5, pp. 269–427 Search PubMed.
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson and T. R. Gingeras, Bioinformatics, 2013, 29, 15–21 CrossRef CAS PubMed.
- S. Anders, P. T. Pyl and W. Huber, Bioinformatics, 2015, 31, 166–169 CrossRef CAS PubMed.
- D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N. T. Doncheva, J. H. Morris, P. Bork, L. J. Jensen and C. von Mering, Nucleic Acids Res., 2019, 47, D607–D613 CrossRef CAS PubMed.
- A. Salvati, C. Åberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine: Nanotechnology, Biology and Medicine, 2011, 7, 818–826 CrossRef CAS PubMed.
- F. Bertoli, D. Garry, M. P. Monopoli, A. Salvati and K. A. Dawson, ACS Nano, 2016, 10, 10471–10479 CrossRef CAS PubMed.
- R. Bartucci, C. Åberg, B. N. Melgert, Y. L. Boersma, P. Olinga and A. Salvati, Small, 2020, 16, 1906523 CrossRef CAS PubMed.
- J. A. Varela, M. Bexiga, C. Åberg, J. C. Simpson and K. A. Dawson, J. Nanobiotechnol., 2012, 10, 39 CrossRef CAS PubMed.
- M. G. Bexiga, J. A. Varela, F. Wang, F. Fenaroli, A. Salvati, I. Lynch, J. C. Simpson and K. A. Dawson, Nanotoxicology, 2011, 5, 557–567 CrossRef CAS PubMed.
- J. A. Kim, C. Åberg, G. de Cárcer, M. Malumbres, A. Salvati and K. A. Dawson, ACS Nano, 2013, 7, 7483–7494 CrossRef CAS PubMed.
- J. A. Kim, A. Salvati, C. Åberg and K. A. Dawson, Nanoscale, 2014, 6, 14180–14184 RSC.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- A. Salvati, I. Nelissen, A. Haase, C. Åberg, S. Moya, A. Jacobs, F. Alnasser, T. Bewersdorff, S. Deville, A. Luch and K. A. Dawson, NanoImpact, 2018, 9, 42–50 CrossRef.
- P. Sandin, L. W. Fitzpatrick, J. C. Simpson and K. A. Dawson, ACS Nano, 2012, 6, 1513–1521 CrossRef CAS PubMed.
- J. A. Varela, C. Åberg, J. C. Simpson and K. A. Dawson, Small, 2015, 11, 2026–2031 CrossRef CAS PubMed.
- S. Deville, R. Penjweini, N. Smisdom, K. Notelaers, I. Nelissen, J. Hooyberghs and M. Ameloot, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 2411–2419 CrossRef CAS PubMed.
- C. Åberg, J. A. Varela, L. W. Fitzpatrick and K. A. Dawson, Sci. Rep., 2016, 6, 34457 CrossRef PubMed.
- M. Bramini, D. Ye, A. Hallerbach, M. Nic Raghnaill, A. Salvati, C. Åberg and K. A. Dawson, ACS Nano, 2014, 8, 4304–4312 CrossRef CAS PubMed.
- F. Wang, L. Yu, M. P. Monopoli, P. Sandin, E. Mahon, A. Salvati and K. A. Dawson, Nanomedicine: Nanotechnology, Biology and Medicine, 2013, 9, 1159–1168 CrossRef CAS PubMed.
- M. Nic Raghnaill, M. Bramini, D. Ye, P.-O. Couraud, I. A. Romero, B. Weksler, C. Åberg, A. Salvati, I. Lynch and K. A. Dawson, Analyst, 2014, 139, 923–930 RSC.
- C. Collinet, M. Stoter, C. R. Bradshaw, N. Samusik, J. C. Rink, D. Kenski, B. Habermann, F. Buchholz, R. Henschel, M. S. Mueller, W. E. Nagel, E. Fava, Y. Kalaidzidis and M. Zerial, Nature, 2010, 464, 243–249 CrossRef CAS PubMed.
- J. C. Simpson, B. Joggerst, V. Laketa, F. Verissimo, C. Cetin, H. Erfle, M. G. Bexiga, V. R. Singan, J.-K. Hériché, B. Neumann, A. Mateos, J. Blake, S. Bechtel, V. Benes, S. Wiemann, J. Ellenberg and R. Pepperkok, Nat. Cell Biol., 2012, 14, 764–774 CrossRef CAS PubMed.
- B. Snijder, R. Sacher, P. Ramo, E.-M. Damm, P. Liberali and L. Pelkmans, Nature, 2009, 461, 520–523 CrossRef CAS PubMed.
- X. Jiang, C. Röcker, M. Hafner, S. Brandholt, R. M. Dörlich and G. U. Nienhaus, ACS Nano, 2010, 4, 6787–6797 CrossRef CAS PubMed.
- S. Schöttler, K. Klein, K. Landfester and V. Mailänder, Nanoscale, 2016, 8, 5526–5536 RSC.
- A. Panarella, M. G. Bexiga, G. Galea, E. D. O’ Neill, A. Salvati, K. A. Dawson and J. C. Simpson, Sci. Rep., 2016, 6, 28865 CrossRef CAS PubMed.
- C. Carrillo-Carrion, A. I. Bocanegra, B. Arnaiz, N. Feliu, D. Zhu and W. J. Parak, ACS Nano, 2019, 13, 4631–4639 CrossRef CAS PubMed.
- H. M. Shapiro, Practical Flow Cytometry, John Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2003 Search PubMed.
- A. Longobardi Givan, Flow Cytometry: First Principles, John Wiley & Sons, Ltd, 2nd edn, 2001 Search PubMed.
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, Small, 2011, 7, 3341–3349 CrossRef CAS PubMed.
- J. C. G. Jeynes, C. Jeynes, M. J. Merchant and K. J. Kirkby, Analyst, 2013, 138, 7070–7074 RSC.
- H. D. Mitchell, L. M. Markillie, W. B. Chrisler, M. J. Gaffrey, D. Hu, C. J. Szymanski, Y. Xie, E. S. Melby, A. Dohnalkova, R. C. Taylor, E. K. Grate, S. K. Cooley, J. E. McDermott, A. Heredia-Langner and G. Orr, ACS Nano, 2016, 10, 10173–10185 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed.
- O. L. Mooren, B. J. Galletta and J. A. Cooper, Annu. Rev. Biochem., 2012, 81, 661–686 CrossRef CAS PubMed.
- L. Johannes, R. G. Parton, P. Bassereau and S. Mayor, Nat. Rev. Mol. Cell Biol., 2015, 16, 311–321 CrossRef CAS PubMed.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- D. Montizaan, K. Yang, C. Reker-Smit and A. Salvati, Nanomedicine: Nanotechnology, Biology and Medicine, 2020, 102300 CrossRef CAS PubMed.
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, PLoS One, 2011, 6, e24438 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- V. Forest, M. Cottier and J. Pourchez, Nano Today, 2015, 10, 677–680 CrossRef CAS.
- L. Treuel and G. Nienhaus, Biophys. Rev., 2012, 4, 137–147 CrossRef CAS PubMed.
- P. del Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
- M. S. Ehrenberg, A. E. Friedman, J. N. Finkelstein, G. Oberdörster and J. L. McGrath, Biomaterials, 2009, 30, 603–610 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- X. Cheng, X. Tian, A. Wu, J. Li, J. Tian, Y. Chong, Z. Chai, Y. Zhao, C. Chen and C. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 20568–20575 CrossRef CAS PubMed.
- P. Chandran, J. E. Riviere and N. A. Monteiro-Riviere, Nanotoxicology, 2017, 11, 507–519 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- V. Forest and J. Pourchez, Nano Today, 2016, 11, 700–770 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary tables and figures; differentially expressed genes in HeLa cells that take up low numbers of 40 nm polystyrene nanoparticles; differentially expressed genes in HeLa cells that take up high numbers of 40 nm polystyrene nanoparticles; clustering analysis for low 40 nm polystyrene nanoparticle uptake in HeLa cells, showing protein interactions using STRING; clustering analysis for high 40 nm polystyrene nanoparticle uptake in HeLa cells showing protein interactions using STRING; further discussion of the sorting experiments. See DOI: 10.1039/d1nr04690j |
| This journal is © The Royal Society of Chemistry 2021 |