
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiparametric nanoparticle-induced toxicity readouts with single cell resolution in HepG2 multicellular tumour spheroids†
Suainibhe
Kelly
a,
Maria H.
Byrne
b,
Susan J.
Quinn
 b and
Jeremy C.
Simpson
*a
b and
Jeremy C.
Simpson
*a
aCell Screening Laboratory, UCD School of Biology & Environmental Science, University College Dublin, Dublin 4, Ireland. E-mail: jeremy.simpson@ucd.ie
bUCD School of Chemistry, University College Dublin, Dublin 4, Ireland
First published on 18th October 2021
Abstract
The use of nanomaterials as therapeutic delivery vehicles requires their careful pre-clinical evaluation. Of particular importance in this regard is measurement of cellular toxicity, ideally assessing multiple parameters in parallel from various relevant subcellular organelles. In recent years it has become evident that in vitro monolayer-grown cells do not always accurately predict any toxicity response seen in vivo, and so there is a need for more sophisticated in vitro cell models, employing a greater depth of characterisation. In this work we present an automated high-content screening microscopy approach for quantifying nanoparticle-induced toxicity in a three-dimensional multicellular tumour spheroid (MCTS) cell model. As a proof-of-principle, we perform a comparative toxicity profile study of carboxylate- versus amine-modified polystyrene nanoparticles in HepG2 spheroids. Following treatment with these nanoparticle types, we demonstrate that several hundred spheroids, of various sizes, can be morphologically profiled in a single well using automated high-content image analysis. This provides a first level of information about spheroid health in response to nanoparticle treatment. Using a range of fluorescent reporters assessing membrane permeability, lysosome function and mitochondrial activity, we also show that nanoparticle-induced toxicity information can be obtained from individual cells with subcellular resolution. Strikingly, our work demonstrates that individual cells do not all behave in a consistent manner within a spheroid structure after exposure to nanoparticles. This highlights the need for toxicity studies to not only assess an appropriate number of spheroids, but also the importance of extracting information at the subcellular level.
Introduction
Nanomedicine describes the use of nano-sized materials for applications including diagnostics, therapeutics and the use of nanoparticles (NPs) as drug delivery systems.1 The use of NPs in this context is an attractive solution to counter several key limitations in the development of therapeutics, such as off-target toxicity, low tissue and cell-type specificity and limited efficacy.2 The wide range of applications associated with the emerging use of NPs in the therapeutic context is largely due to their versatility. They are available in a range of sizes, shapes and materials, and they can be conjugated to therapeutics, as well as modified to bind to specific targets. All of these features make them highly therapeutically attractive, particularly in targeting cancer.3The pre-clinical development of therapeutics largely hinges on the use of in vitro toxicity assays, traditionally conducted in cells grown using two-dimensional (2D) culture methods. Although 2D cell culture models have been widely used to investigate cellular toxicity, in recent years the limitations of these methods have been under scrutiny. To overcome limitations associated with 2D cell culture models, more complex models are needed, as they have the potential to both better predict toxicity and reduce the use of animals in testing. Indeed, a number of studies have recently emerged that report differences in cellular toxicity profiles for the same cell types depending on whether they were grown as monolayers or as three-dimensional assemblies. In some instances, these differences seem to be linked to differential NP uptake rates, in turn manifesting in altered IC50 values, for example in the case of NPs delivering the anti-cancer therapeutic paclitaxel to breast cancer cells.4 Interestingly, in response to the addition of zinc oxide NPs, the levels of reactive oxygen species produced by colorectal cancer Caco-2 cells was also found to be significantly different between the two cell growth conditions.5 Three-dimensional (3D) cell culture methods have been under development for several years,6 and now many types of 3D cell culture models exist, including multicellular tumour spheroids (MCTS)7 and organoids.8 Several methods have now been optimised for the generation of complex 3D cell culture models.6,9 Notable examples include patient-derived tumour cells grown in ultra-low attachment plates,10 primary cell lines to grow organoids and co-culture models,11 microfluidic devices and the hanging drop method for growth of MCTS.7,12 These models have been shown to more accurately represent the complex in vivo environment compared to traditional 2D culture methods.13 While the use of monolayer cells for studying NP-induced toxicity remains useful, such systems do not recapitulate certain important features that are found in vivo, most notably complex cell–cell interactions and intercellular communication mechanisms. These features are particularly important in the context of nanomedicine, in which it is essential that we better understand how the therapeutic to be delivered is transferred across multiple cellular layers, and the effects caused during this process. Therefore, such models represent an attractive model to investigate toxicity effects in vitro, prior to moving to in vivo systems.
In recent years there has been a move towards the increased use of 3D cell culture models for the testing of efficacy, penetration and toxicity of a wide range of NPs. As mentioned above, this work has started to reveal that cells grown in 3D, compared to when grown as 2D monolayers, can exhibit different responses on exposure to NPs.4,5,13–15 It is therefore important that we understand the mechanisms of toxicity induced by NPs, not only for nanomedicine development, but also in the wider context of nano-pollution.16 Overcoming hepatotoxicity is a constant hurdle in the development of therapeutics.17 For this reason, MCTS derived from liver cells are popular for investigating toxicity in vitro, with cell lines such as HepG212 and HepaRG18 commonly used in the development of 3D cell culture models and consequently used in the investigation of both drugs and nanomedicines. The cytotoxicity of several NP types has been investigated in 3D in vitro models, using end-point assays such as the MTT assay19,20 and the Alamar Blue assay,15,21 which have been used to detect changes in cell viability.
Although end-point toxicity assays remain commonly used, those that are based on colourimetric or luminescent readouts suffer from a number of key limitations. For example, there is increasing evidence that the unique properties of NPs increase the likelihood that they will interfere with analytical readout, namely optical interference in the assay.22–25 Furthermore, such plate reader-based assays do not provide any information with respect to morphological changes to the 3D cell assembly. In the case of MCTS, NP- or nanomedicine-induced cell death has been seen to cause changes in spheroid shape, or spheroid shrinkage.14,26–28 This additional information at the spheroid-level can be insightful, but becomes even more powerful when combined with fluorescence. In recent years, there has been a move in this direction, as this allows both morphological and functional information relating to toxicity to be assessed. For example, Oliveira and colleagues analysed the penetration of solid lipid NPs into an MCF7 spheroid model, and used fluorescent doxorubicin presence at individual confocal slices to report on delivery and cytotoxicity.29 Confocal fluorescence imaging has also been used to provide a functional readout of cytotoxicity in mesothelioma MSTO cell spheroids, by measurement of cleaved caspase-3 activity in response to paclitaxel-loaded NPs.30 However, it should be noted that individual confocal slices were selected for this analysis.
Fluorescence microscopy is at its most powerful when precise and systematic quantification of the images is made. When automated, this approach is generally termed high-content screening (HCS) microscopy. HCS is an attractive approach to employ for the study of nanoparticle–cell interactions as it brings consistency of sample preparation, imaging, and quantification to the assays employed,31 potentially enabling the rapid and parallel evaluation of multiple NP types or formulations. However, the deployment of HCS (and subsequent high-content analysis) of 3D cell assemblies remains challenging with respect to unified sample preparation and processing, imaging the full depth of the structure, as well as problems associated with analysis of vast data sets.32 As such, image analysis solutions used to date either rely on information from a small number of selected confocal slices or maximum projections of several slices. Critically, both of these analysis approaches limit the amount of information that can be deduced from 3D structures, resulting in a lack of morphological information such as volume, shape and surface area. In addition, they cannot capture complete information from the individual cells that constitute a spheroid.
In this work we present a method for the optimised production and processing of HepG2 spheroids on scale compatible with HCS microscopy. We use these spheroids in a multiparametric in vitro toxicity assay, using modified polystyrene NPs as a model to demonstrate our system. Importantly, our approach not only facilitates detailed quantitative readout from several thousand spheroids in parallel, but using a volumetric-based image analysis approach it provides information about individual cells within each spheroid. By applying quantitative profiling of cell nuclei, lysosomes and mitochondria, we propose that our system provides a new paradigm for assessment of NP-induced toxicity in 3D cell models.
Experimental
Cell culture
The human hepatocellular carcinoma HepG2 cell line (ATCC HB-8065) was maintained in culture in Minimum Essential Media (MEM) (Thermo Fisher, Ireland) with 10% Foetal Bovine Serum (FBS) (Thermo Fisher, Ireland) and 2% L-glutamine (Thermo Fisher, Ireland), together termed complete MEM medium (cMEM), at 37 °C with 5% CO2.Spheroid characterisation
Reduced growth factor Matrigel (MTG) basement membrane (Corning, New York, USA) was allowed to defrost on ice overnight. A solution with a concentration of 4 mg mL−1 MTG was prepared in cold phenol red-free, serum-free MEM using pre-chilled tips while keeping all solutions on ice. 15 μL of the 4 mg mL−1 MTG/MEM solution was used to coat the wells of CellCarrier Ultra 96-well microplates (PerkinElmer, Massachusetts, USA). Plates were centrifuged at 4 °C, 900 rpm for 20 minutes then incubated for 30 minutes at 37 °C. HepG2 cells were subcultured and resuspended in the required amount of phenol red-free cMEM to produce a solution of 1 × 104 cells per μL. HepG2 cells were seeded into the 96-well plates at a density of 3 × 104 cells per well and incubated at 37 °C for 1 hour. A 2% solution of MTG and phenol red-free, serum-free MEM was prepared and added to the 96-well plate. The final volume in each well was 60 μL. The next day the medium was replaced with fresh phenol red-free cMEM.NP characterisation
Spectroscopic measurements were recorded at room temperature in serum-free medium. UV-visible absorption spectra were recorded using a Cary 50 scanning spectrometer and emission spectra were recorded using a Varian Cary Eclipse Fluorescence Spectrophotometer working in fluorescence mode. Emission spectra were recorded in a 3 cm quartz cell and were optically dilute at the excitation wavelength. Samples for transmission electron microscopy (TEM) analysis were prepared by dropping a small volume (7 μL) of (0.025 mg mL−1) NP suspension on a formvar/carbon coated copper TEM grid (Ted Pella, Inc., California, USA) and allowing the sample to evaporate at room temperature in air. TEM images were recorded using a Philips Tecnai F20 electron microscope that was operated at an accelerating voltage of 200 kV. Dynamic light scattering (DLS) and zeta potential measurements were carried out on a Malvern Zetasizer Nano-ZS, equipped with a 4 mW He–Ne laser operating at 632.8 nm; measurements were taken at 173°. Data were plotted using Origin Pro Software.NP toxicity assays in spheroids
Spheroids were prepared as described above with the modification that cells were seeded at 2.5 × 104 cells per well. On the third day of growth samples were prepared for NP treatment. Spheroids were washed in serum-free, phenol red-free MEM twice and incubated for 15 minutes at 37 °C. Amine-modified 100 nm fluorescently-labelled NPs (NH2-NPs) (L9904, Sigma-Aldrich, New Jersey, USA) and carboxylated 100 nm fluorescently-labelled NPs (COOH-NPs) (F8803, Thermo Fisher, Ireland) were vortexed for 3 minutes and sonicated for 2 minutes. A 200 μg mL−1 NP solution was prepared in phenol red-free, serum-free MEM and vortexed for 2 minutes. The MEM was removed from the spheroids and 60 μL of the 200 μg mL−1 NP solution was added. The spheroids were incubated at 37 °C for 1 hour. Following this, 60 μL phenol red-free 20% FBS MEM was added to the spheroids, bringing the final concentration of NPs on the spheroids to 100 μg mL−1. Spheroids were incubated for 24, 48 and 72 hours. Spheroids were treated with 20 μM staurosporine (STS) (S6942, Sigma-Aldrich, New Jersey, USA) made up in phenol red-free cMEM for 24 hours.Spheroid processing, fixing and staining
After 5 days of growth the spheroids were processed as previously described,33 with certain modifications. Spheroids were washed in PBS twice and fixed for 1 hour in 3% paraformaldehyde (PFA). Following fixation, the samples were washed twice in PBS and quenched for 30 minutes in 0.5 M glycine at 37 °C with gentle agitation. Spheroids were permeabilised in a solution containing 0.2% Triton X-100, 0.3 M glycine and 20% DMSO in PBS for 30 minutes 37 °C. Spheroids were washed twice in PBS for 5 minutes each. Blocking buffer was prepared containing 0.2% Triton X-100, 1% bovine serum albumin (BSA) (Sigma-Aldrich, New Jersey, USA), and 10% DMSO in PBS. Spheroids were incubated in the blocking buffer for 1 hour and 30 minutes at 37 °C with gentle agitation. Antibody buffer containing 0.3% Tween 20, 1% BSA and 5% DMSO in PBS was prepared. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300 dilution of the primary antibody (mouse anti-LAMP1, H4A3, DSHB, Iowa, USA; or mouse anti-β-catenin, 610153, BD Transduction Laboratories, New Jersey, USA) in the antibody buffer was prepared, and the spheroids were incubated in the primary antibody solution overnight at 37 °C. Spheroids were washed in a washing buffer containing 0.2% Tween 20 and 1% BSA in PBS five times for 5 minutes. Spheroids were incubated overnight in the antibody buffer containing a 1:300 dilution of the secondary antibody (A-11031, goat anti-mouse Alexa Fluor 568, Thermo Fisher, Ireland) and 1:5000 Hoechst 33342 (ImmunoChemistry Technologies, Bloomington, USA). Spheroids were washed five times in the washing buffer for 5 minutes. Spheroids were kept in PBS for image acquisition.
:300 dilution of the primary antibody (mouse anti-LAMP1, H4A3, DSHB, Iowa, USA; or mouse anti-β-catenin, 610153, BD Transduction Laboratories, New Jersey, USA) in the antibody buffer was prepared, and the spheroids were incubated in the primary antibody solution overnight at 37 °C. Spheroids were washed in a washing buffer containing 0.2% Tween 20 and 1% BSA in PBS five times for 5 minutes. Spheroids were incubated overnight in the antibody buffer containing a 1:300 dilution of the secondary antibody (A-11031, goat anti-mouse Alexa Fluor 568, Thermo Fisher, Ireland) and 1:5000 Hoechst 33342 (ImmunoChemistry Technologies, Bloomington, USA). Spheroids were washed five times in the washing buffer for 5 minutes. Spheroids were kept in PBS for image acquisition.
Spheroids treated with NPs and STS were processed, fixed and permeabilised as described above. NP- and STS-treated spheroids were incubated in 100 μg mL−1 DNase-free RNase (A3832, VWR International, Pennsylvania, USA) made up in 2× SSC buffer for 20 minutes at 37 °C. Following incubation spheroids were incubated in the antibody buffer containing 1:300 anti-LAMP1 primary antibody overnight. Spheroids were washed as above. Spheroids were incubated overnight in the antibody buffer containing 1:300 secondary antibody (A21235, goat anti-mouse Alexa Fluor 647, Thermo Fisher, Ireland) and 1:5000 Hoechst 33342 at 37 °C. Spheroids were washed in the washing buffer and stained with propidium iodide (PI) (P1470, Sigma-Aldrich, New Jersey, USA) or TOTO-3 (T3604, Thermo Fisher, Ireland) for 30 minutes and 15 minutes, respectively at 37 °C. Following incubation spheroids were washed several times with PBS and stored in PBS for image acquisition.
Live cell staining
Spheroids were treated with NPs and STS as described previously. Spheroids were stained with Hoechst 33342 (1:1000), LysoTracker Deep Red (1:20000) (L12492, Thermo Fisher, Ireland) and Tetramethylrhodamine methyl (TMRM) ester perchlorate (1:10000) (T668, Thermo Fisher, Ireland) made up in phenol red-free cMEM and incubated for 1 hour prior to image acquisition.
Image acquisition and analysis
All imaging was carried out on an Opera Phenix High Content Screening System (PerkinElmer, Massachusetts, USA) using either a 20×/1.0 NA or 63×/1.15 NA water immersion objective. Sequential acquisition between channels was used in all imaging to eliminate crosstalk. A 405 nm laser line was used to excite Hoechst 33342, a 488 nm laser line was used to excite the NPs, a 561 nm laser line was used to excite the PI, TMRM or Alexa Fluor 568, and a 640 nm laser line was used to excite the LysoTracker Deep Red or Alexa Fluor 647. Images were acquired on one of four parallel sCMOS cameras (16 bit, 4.4 megapixels, 2100 × 2100 resolution, 6.5 μm pixel size). Typically, 30–40 slices were taken at an interval of 1.5 μm. Complete image stacks were used in all analyses. Image analysis was performed using Harmony Image Analysis software v4.8 (PerkinElmer, Massachusetts, USA). Two different types of image analysis were used, maximum projection image analysis (Fig. 1 only) and volumetric image analysis (all other figures) (see ESI† for details of image analysis pipelines). Briefly, each spheroid was identified as a distinct ‘object’ from the confocal stack using the 405 nm/Hoechst 33342 channel, which also facilitated identification and segmentation of each cell nucleus. This channel was selected for identification of the spheroids as the Hoechst 33342 dye was used in all experiments. In addition, Hoechst 33342 is well-known to be present in residual amounts in the cytoplasm of cells, and therefore can be used differentially to identify not only the cell nuclei but also the boundary of each cell. This channel was used to calculate spheroid morphological measurements including volume, sphericity and cross-sectional area (volumetric analysis), area, roundness, length and width (maximum projection analysis). Spheroids were categorised into size classes from small to extra-large, the nuclei were identified in each spheroid. PI intensity was quantified using the 561 nm channel, this was used to identify nuclei showing toxicity. These nuclei were counted, and morphological measurements were made. The NPs were identified and segmented using the 488 nm channel. The lysosomes were identified and segmented using the 647 nm channel, the number of lysosomes was calculated, as well as their volume. TMRM was used to stain the mitochondria and evaluate mitochondrial membrane potential in response to NP toxicity. The 568 nm channel was used to measure TMRM intensity in spheroids as an indication of mitochondrial function. All data are shown as mean and standard deviation from at least 6 replicate wells, unless otherwise indicated. All statistical analysis was made using a one-way ANOVA and post-hoc Tukey test.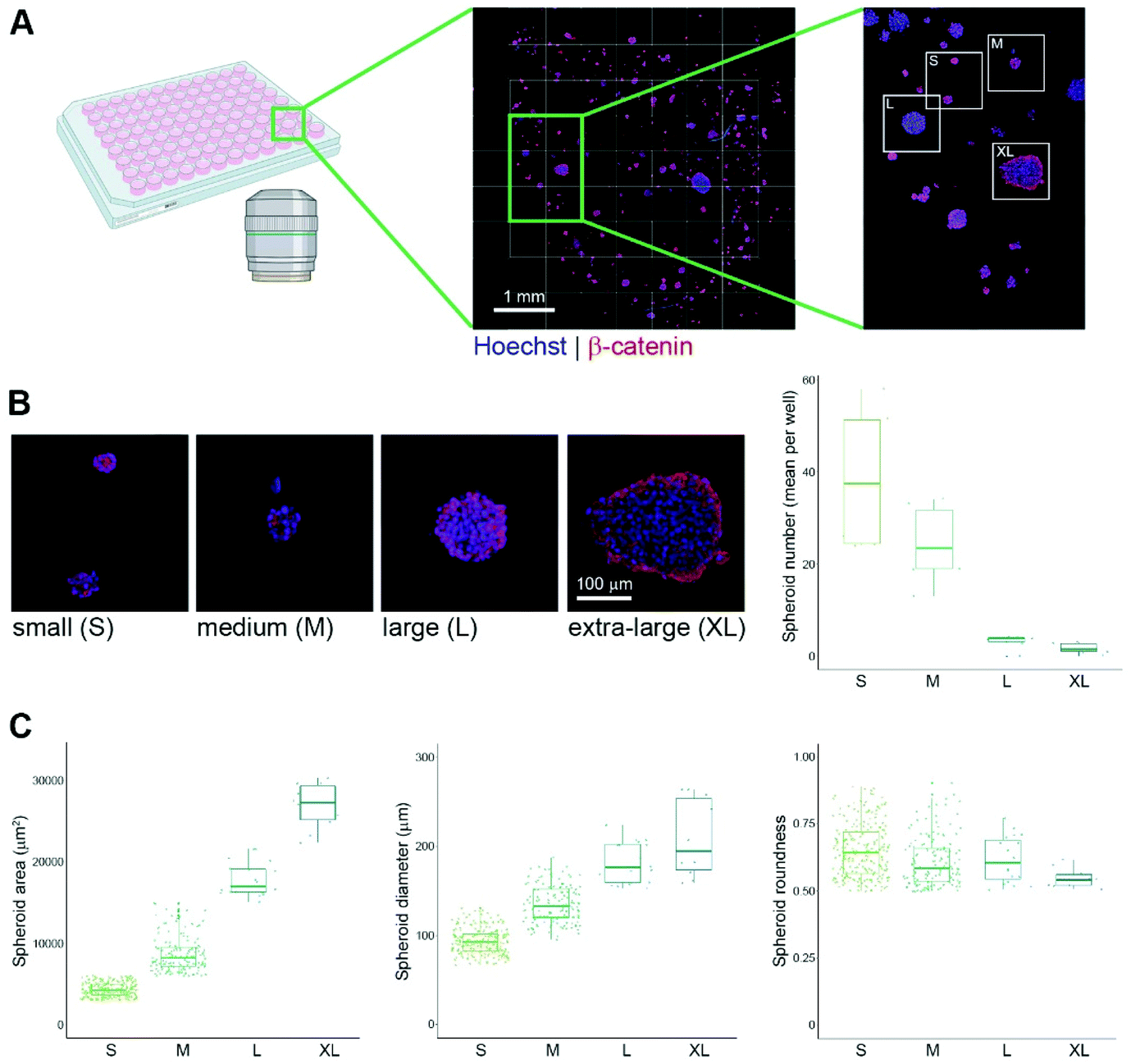 | ||
| Fig. 1 Generation and automated imaging of differently-sized HepG2 spheroids. (A) HepG2 cells were seeded in Matrigel in 96-well plates and grown for 5 days to facilitate spheroid formation. Following immunostaining for β-catenin (red), and nuclear staining with Hoechst 33342 (blue) spheroids were imaged on a fully automated confocal microscope. (B) Example images showing each spheroid size class and corresponding boxplots showing the number of each size class in a typical experiment. (C) Analysis of the spheroid population in a typical experiment based on maximum intensity projections of the confocal stack, showing spheroid maximum cross-sectional area, diameter and roundness. Data in (B and C) are from 6 replicate wells; total numbers of spheroids analysed were 229 (small), 142 (medium), 18 (large) and 9 (extra-large). Boxplots show median value and quartiles for each spheroid class. Artwork in panel A was created in BioRender. | ||
Results
In vitro toxicity studies are usually performed in a cell line of liver origin,17,34 as this organ is primarily the site of accumulation of toxins, and has highly adapted response mechanisms. In addition, it is becoming increasingly clear that NPs used in a therapeutic context also accumulate in this organ.35 In order to create a suitable in vitro model for the study of NP-induced toxicity, which would facilitate the generation of spheroid-level, single cell and subcellular resolution information from a 3D cell assembly, we adapted a method utilised previously in our laboratory.36 This method uses Matrigel as a support in which to grow MCTS structures, and has the advantage that potentially several hundred MCTS can be grown in each well of a 96-well plate, in a format compatible for high-resolution automated microscopy. We selected HepG2 liver cells for our model, and plated these cells with Matrigel into optical quality 96-well plates. The cells were grown for up to 5 days and then immunostained for β-catenin, a constituent of cell–cell adherens junctions, as well as being counter-stained with Hoechst 33342 to label the cell nuclei. Sample wells were imaged on a fully automated confocal high-content screening microscope, revealing a range of MCTS sizes in each well (Fig. 1A). This variety of spheroid sizes in each well was advantageous, as it allowed us to directly compare spheroids of different properties, but all in the same well. Using automated image analysis software, we initially categorised the MCTS into four size classes based on their area determined from maximum projection images of the entire confocal stack. These sizes were denoted as small (3000–6000 μm2), medium (>6000–15000 μm2), large (>15000–220000 μm2) and extra-large (>220000–350000 μm2). This allowed us to assess the frequency of each size class in any well of interest. As expected, the small class of MCTS was the most prevalent, and the extra-large class was the least prevalent (Fig. 1B). We then expanded our image analysis approach to measure a number of spheroid-level properties for the population, including their cross-sectional area, maximum diameter and roundness (in the xy planes) (Fig. 1C). As expected, this analysis revealed a strong correlation between spheroid area and diameter, underlining the accuracy of the automated measurement protocols. Importantly, this initial automated analysis allowed us to remove spheroids from analysis that did not meet certain criteria, for example those that were deemed too small to be considered as assembled spheroids, and those that displayed a low roundness (less than 0.5). This helped ensure that we were analysing a similar population within each size class.
One significant weakness of most toxicity studies performed to date is that they only consider information from a single optical plane. Our approach purposefully wanted to extract the maximum information from the cells in the spheroid models as we reasoned that toxicity effects at the single cell level were unlikely to be similar across all the cells that constitute a spheroid. We extended our initial approach to image the entire volume of the spheroids in the wells, typically collecting up to 40 confocal slices at a height separation of 1.5 μm (Fig. 2A). We then refined our image analysis strategy to use volumetric tools to identify the MCTS in each well. Spheroids were identified based on their individual cell nuclei, and this information was used to calculate the total volume of each spheroid, which formed the basis of their size classification. These sizes were small (50000–110000 μm3), medium (>110000–200000 μm3), large (>200000–800000 μm3) and extra-large (>800000–1800000 μm3) (Fig. 2B and C). Identification (segmentation) of each nucleus allowed us to count the number of cells in each spheroid (Fig. 2D), and this correlated well with measurement of spheroid volume and surface area for each size class (Fig. 2D). To provide additional consistency of spheroid measurement, we excluded all spheroids that displayed a sphericity value of <0.5 from subsequent analysis.
 | ||
| Fig. 2 Volumetric analysis of HepG2 spheroids. (A) Example stack of confocal images acquired for each field of view. (B) Example showing volumetric-based detection of each spheroid. The Hoechst 33342 channel is used to identify each spheroid, a volumetric measurement is made and this then forms the basis of spheroid size classification. Subsequently each cell nucleus is segmented as an individual object. (C) Boxplot showing the distribution of spheroids to each size class based on their volume. S, small; M, medium; L, large; XL, extra-large. (D) Analysis of the spheroid population in a typical experiment based on volumetric measurements from the confocal stack, showing number of nuclei per spheroid, surface area, maximum cross-sectional area and sphericity. Data in (C and D) are from 6 replicate wells; total numbers of spheroids analysed were 363 (small), 158 (medium), 113 (large) and 8 (extra-large). Boxplots show median value and quartiles for each spheroid class. | ||
Given the wide range of nanomaterials currently under development for therapeutic use, our experimental approach was focused on developing a robust and automated high-throughput system for assessing nanoparticle-induced toxicity in our HepG2 spheroids. As model NPs, we selected commercially available fluorescently-labelled polystyrene spheres, with a nominal size of 100 nm. This size is both compatible with cellular uptake mechanisms, and appropriate for encapsulation of any potential therapeutic. Furthermore, we tested two variants of this particle type, one with a carboxylate (COOH) modification and the other amine-modified (NH2). Amine-modified NPs carry a net positive charge, and although a matter of debate, there is evidence to suggest that such NPs can induce cellular damage and toxicity after internalisation, most likely through the proton sponge effect.37 Prior to their use, we characterised the physico-chemical properties of these NP types, initially by checking their fluorescence absorption and emission characteristics. Analysis of these spectra revealed that both NP types carried a fluorophore that is excited by blue light, with maximum excitation values at 505 nm and 481 nm, for the COOH and NH2-modified NPs, respectively. Their corresponding emission peaks were 515 nm and 535 nm, respectively (Fig. 3A). These two NP types would be compatible with the 488 nm laser excitation line present on our automated confocal screening microscope.
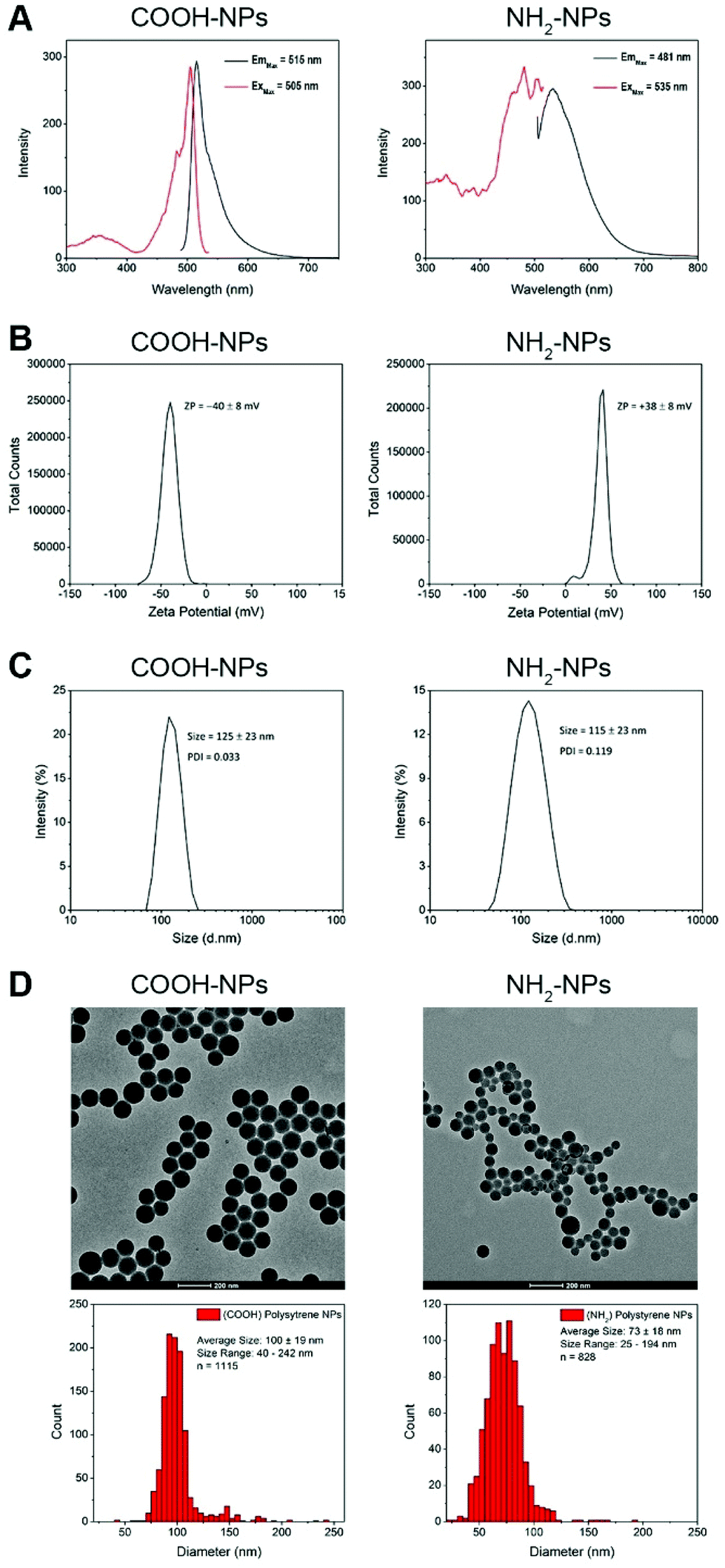 | ||
| Fig. 3 Characterisation of COOH-NPs and NH2-NPs. (A) Excitation (red) and emission (black) spectra recorded in serum-free medium. (B) Zeta potential and (C) dynamic light scattering measurements recorded in deionised water at 25 °C. (D) TEM images and corresponding size distribution of the NPs. | ||
We next wanted to measure the charge on the two NP types. Measurement of the zeta-potential of the NPs revealed that the COOH-NPs carried a charge of −40 ± 8 mV, whereas the NH2-NPs had a charge of +38 ± 8 mV (Fig. 3B). These values were in line with our expectations and importantly confirmed that the NH2-NPs were likely to induce the proton sponge effect once internalised into cells. Finally, we measured the size of the two NP types. Dynamic light scattering (DLS) experiments revealed that the COOH-NPs had a mean diameter of 125 ± 23 nm, whereas the NH2-NPs measured 115 ± 23 nm (Fig. 3C). While DLS provides an indicative hydrodynamic size of the NPs, it does not reveal the full range of NP sizes present in the population. To examine this, we subjected the NPs to examination by transmission electron microscopy (TEM) (Fig. 3D). The mean size for the COOH-NPs was measured at 100 ± 19 nm, which was consistent with that recorded by DLS, but it also revealed that this NP type was highly homogeneous. By contrast, the NH2-NPs displayed a broader size range across their population, with a mean size of 73 ± 18 nm, somewhat smaller than that recorded by DLS. This slightly higher heterogeneity in the population was evident in the TEM images, as well as being reflected in the higher polydispersity index calculated during the DLS (Fig. 3C). Nevertheless, both NP types fulfilled our criteria in terms of fluorescence properties, charge and being of a biologically-relevant size.
We prepared HepG2 spheroids as described above, and exposed them to 100 μg mL−1 NP solutions of either the COOH-NPs or the NH2-NPs for up to 72 h. As a positive control for our assay, we incubated the spheroids for 24 h with staurosporine, a protein kinase C inhibitor that induces apoptosis. Following fixation, Hoechst 33342 was added to stain all cell nuclei. Automated confocal imaging of several wells containing the spheroids was carried out, and the spheroids were subjected to volumetric image analysis, allocating them to the four size classes, as defined previously. Under control conditions, the most well-represented size class was ‘large’, containing 41% of the spheroids. Treatment with staurosporine resulted in a small decrease in the prevalence of large and extra-large spheroids, presumably due to the activity of this metabolite inhibiting cell growth. Treatment with the NH2-NPs however, had a profound effect on the size distribution of the spheroid population, resulting in only 19% and 4% of the spheroids being classified as ‘large’ and ‘extra-large’, respectively, after 72 h of incubation (Fig. 4). This suggested that the amine-modified NPs were influencing spheroid growth, and presumably causing toxicity, which then inhibited their further growth. By contrast, similar incubations with the COOH-NPs had no effect on the size profile of the spheroids compared to untreated control spheroids, which while not formally excluding any toxicity from this NP type, did suggest that their effects on cells were different from those seen with the NH2-NPs (Fig. 4). Our experimental approach utilising high-resolution confocal microscopy was designed in such a way to be able to measure nanoparticle-induced toxicity at the level of individual cells. To this end, we incubated the spheroids with propidium iodide (PI), a widely-used fluorescent stain that is impermeant to intact cells, but which is able to enter the nucleus of dead cells and bind to the DNA. Visualisation of the spheroids using either a volumetric or 3-plane view confirmed that the spheroids were well formed, and that NPs could be seen both surrounding the spheroids and in a number of the cells (Fig. 5A). What was also noticeable, particularly in spheroids that had been exposed to the NH2-NPs for 72 h, was that these spheroids seemed less compact, and small groups of cells close to the main body of the spheroid were often visible (Fig. 5A, arrows). We interpreted this as a toxicity effect on these spheroids, resulting in the assemblies becoming less stable, resulting in cells being lost from the main spheroid body. When individual confocal planes were examined (Fig. 5A), it was striking that dead cells (identified by the presence of strong PI staining) were not evenly distributed through each spheroid, emphasising the importance of employing a volumetric approach to study toxicity, rather than just considering gross measurements of the spheroid. Our volumetric-based image analysis pipeline allowed us to detect and segment every cell nucleus in each spheroid (Fig. 2B), and in turn measure the intensity of PI in each nucleus. We defined a cut-off intensity value, allowing us to score each nucleus as either PI-negative or PI-positive, and thereby calculate the number of PI-positive nuclei in each spheroid size class. This analysis revealed that in small spheroids, treated with staurosporine, ca. 60% of the nuclei were classified as dead/dying. In the wells where the spheroids had been incubated with NH2-NPs, we observed that ca. 45% of the nuclei were also PI-positive after 48 h and 72 h. However, the spheroids incubated with COOH-NPs contained few cell nuclei (ca. 10%) that were PI-positive (Fig. 5B). Visualisation of the same experiment using boxplot graphs to depict the median and quartile values for every spheroid analysed showed similar overall results, but importantly revealed heterogeneity of the toxicity response across the population, underlining the importance of analysing a large number of spheroids (Fig. 5C). Examination of the data from the population of medium spheroids revealed a very similar trend with more than 40% of the nuclei in these spheroids being observed as PI-positive (Fig. 5D and E). When we analysed the large spheroid population, we also observed a time-dependent increase in the percentage of PI-positive nuclei (Fig. S1A and B†), however this was not the case for the spheroids designated as extra-large. In this case only a small increase in PI-positive nuclei was seen in the presence of the NH2-NPs, compared to control cells (Fig. S1C and D†). We interpret this as reduced penetration ability of the NPs into these large cellular structures.
 | ||
| Fig. 4 Effect on NP incubation on spheroid size classes. HepG2 spheroids were incubated with either NH2- or COOH-NPs for increasing lengths of time. Graph shows the proportion of spheroids in each size class for each condition. Data are from 3 replicate wells per treatment; total numbers of spheroids analysed were 563 (control, C), 653 (staurosporine (STS)-treated), 586 (NH2-NPs/24 h), 507 (NH2-NPs/48 h), 338 (NH2-NPs/72 h), 680 (COOH-NPs/24 h), 677 (COOH-NPs/48 h) and 655 (COOH-NPs/72 h). | ||
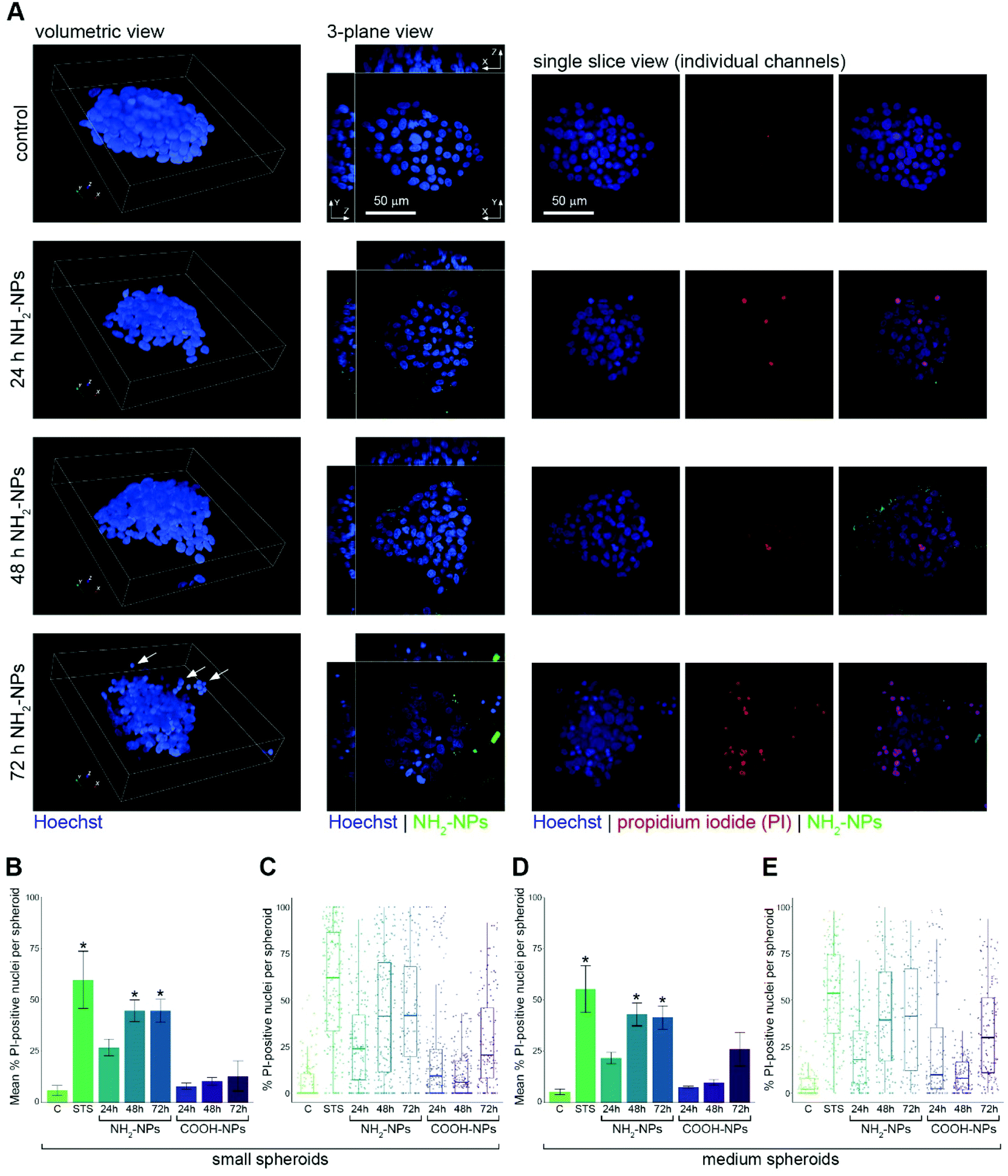 | ||
| Fig. 5 Single cell analysis of spheroid nuclei following treatment with NPs. (A) Representative images of HepG2 spheroids, either untreated (control), or incubated with NH2-NPs for increasing lengths of time as indicated. Hoechst-stained nuclei are shown in blue, NH2-NPs in green, and propidium iodide (PI) in red. Arrows point to cells that seem to have detached from the main spheroid assembly. (B) Graph showing mean% of PI-positive nuclei per small spheroid after the various treatments as indicated. (C) Boxplot showing median% of PI-positive nuclei value and quartiles in small spheroids. Data in (B and C) are from 3 replicate wells per treatment; total number of spheroids analysed was 1515. (D) Graph showing mean% of PI-positive nuclei per medium spheroid after the various treatments as indicated. (E) Boxplot showing median% of PI-positive nuclei value and quartiles in medium spheroids. Data in (D and E) are from 3 replicate wells per treatment; total number of spheroids analysed was 1049. C, control; STS, staurosporine. Asterisks denote p < 0.01 compared to control samples. | ||
To gain a deeper understanding of the toxicity effects on the spheroids, we wanted to further exploit the single cell resolution imaging data obtained. Imaging a large number of confocal planes allowed us to identify spheroids of different sizes sitting at different heights in the Matrigel (Fig. 6A). Our volumetric image analysis routines were used to detect all nuclei in each spheroid, and using the information from the PI colour channel we were able to classify each nucleus as either PI-positive or PI-negative. For each of these classifications we measured the volume of each nucleus. The PI-negative nuclei in the small spheroids showed a high degree of volume consistency, irrespective of whether their associated spheroids had been incubated with amine- or carboxylate-modified NPs (Fig. 6B). By contrast, the PI-positive nuclei in the same spheroids showed a marked reduction in volume, following incubation with the NH2-NPs (Fig. 6C). Analysis of the medium spheroids revealed a very similar pattern to that seen in the small spheroids, with a more than 50% reduction in nucleus volume in the cells exposed to NH2-NPs (Fig. 6D and E). To further validate these observations, we measured the surface area of both the PI-negative and PI-positive nuclei, as the algorithm used to make these measurements works independently of that measuring volume. The surface area measurements obtained followed a very similar pattern to those of volume, showing a significant reduction in nucleus size in the presence of NH2-NPs (Fig. S2†). Visual examination of our image data suggested that the PI-positive nuclei were often seen in the cells at the periphery of the spheroids. We wondered therefore whether this observation might be linked to the inability of the NPs to penetrate into the more central cells in the spheroid. We randomly selected 20 medium spheroids, 10 of which had been incubated with NH2-NPs and 10 with COOH-NPs, for 72 h in both cases. Cells in these spheroids were classified as either ‘outer’ or ‘inner’, depending on whether they were in contact with the medium or only other cells (Fig. S3A and B†). Quantification of the NPs associated with these two classes of cells revealed that for the amine-modified NPs, on average 73% were found in outer cells, and 27% in inner cells. The carboxylate-modified NPs showed a slightly greater ability to penetrate into inner cells, with values of 58% in outer cells and 42% in inner cells (Fig. S3C†). This suggests that these NP types are able to access cells that are located centrally within the spheroids. Interestingly however, preliminary analysis did not find a direct correlation between PI intensity and NP fluorescence intensity at the level of individual cells (not shown). This suggests that the amount of NP loading in each cell per se, is not necessarily the only determinant of whether cell death pathways will be initiated. Further work will be needed to understand this observation.
 | ||
| Fig. 6 Single cell analysis of spheroid nuclei volume following treatment with NPs. (A) Representative images of HepG2 spheroids incubated with NH2-NPs for 72 h. Three example slices from the confocal stack are shown. Hoechst-stained nuclei are shown in blue, NH2-NPs in green, and propidium iodide (PI) in red. Central panels show the cell nuclei detected from the slice view, and the right-hand panels show the classification of the nuclei as either PI-negative (green) or PI-positive (red). (B) Graph showing volumes of PI-negative nuclei in small spheroids after the various treatments as indicated. (C) Graph showing volumes of PI-positive nuclei in small spheroids after the various treatments as indicated. (D) Graph showing volumes of PI-negative nuclei in medium spheroids after the various treatments as indicated. (E) Graph showing volumes of PI-positive nuclei in medium spheroids after the various treatments as indicated. Data are from 3 replicate wells per treatment; total number of small spheroids analysed was 1515; total number of medium spheroids analysed was 1049. C, control; STS, staurosporine. Asterisks denote p < 0.01 compared to control samples. | ||
The effects that we observed on the cell nuclei represent late stages in cell death, and so we were keen to explore whether our approach could also be applied to study other nanoparticle-induced toxicity phenotypes on additional organelles. Uptake studies on polystyrene NPs in monolayer-grown cells, have revealed that they utilise the endogenous trafficking machinery to track through the endosomal system to lysosomes.38,39 We prepared HepG2 spheroids and exposed them to COOH- or NH2-NPs for 72 h. After this time, we assessed cell death by propidium iodide treatment, and also immunostained for the lysosomal associated membrane protein-1 (LAMP1) (Fig. 7A). Confocal stacks of small and medium spheroids were acquired and volumetric analysis was carried out. We first counted the mean number of LAMP1 (lysosome) structures per spheroid, normalised to the number of cells in each spheroid. This revealed a small, although statistically non-significant decrease in lysosome number in spheroids treated with the amine-modified NPs (Fig. 7B). However, when we measured the volume of individual lysosomes, we noted a significant increase in this parameter in spheroids exposed to NH2-NPs, compared to control spheroids (Fig. 7C). This increase in lysosome volume was clearly visible in the associated images, and the effect correlated well with those cells that displayed a strong PI intensity (Fig. 7A). Finally, we wanted to test our automated high-content imaging approach in the context of measuring functional changes to the cells in response to the presence of NPs. TMRM is a membrane-permeable dye that can be used to assess mitochondrial membrane potential. Specifically, healthy mitochondria become rapidly labelled with this reporter, whereas cells that are metabolically compromised display comparatively low TMRM intensity. We incubated our spheroids with either COOH- or NH2-NPs for 72 h, and then added TMRM. Following confocal imaging, untreated spheroids displayed a strong mitochondrial pattern, with elongated and interconnected mitochondria present throughout the cytoplasm of cells. By contrast, in the spheroids incubated with the NH2-NPs, the TMRM intensity was weak, more diffuse in nature, and the few mitochondria that could be seen, appeared more vesiculated (Fig. 7D). Quantitative analysis of the spheroids revealed no significant change in mitochondrial number (Fig. 7E), however when their intensity was measured, there was a dramatic decrease in TMRM intensity in spheroids treated with NH2-NPs (Fig. 7F). Taken together, this suggests that in this spheroid model, amine-modified NPs elicit a toxic response to cells via functional changes to both lysosomes and mitochondria.
 | ||
| Fig. 7 NP-induced effects on lysosomes and mitochondria in HepG2 spheroids. (A) Representative images of HepG2 spheroids, either untreated (control), or incubated with COOH-NPs or NH2-NPs for 72 h. One example slice from the confocal stack is shown for each treatment. Hoechst-stained nuclei are shown in blue, lysosomes (LAMP1) in green, and propidium iodide (PI) in red. Arrow points to cells that seem to have detached from the main spheroid assembly. (B) Graph showing mean number of distinct lysosomes detected per spheroid (normalised to cell number) after the various treatments as indicated. (C) Graph showing mean individual lysosome volume detected per cell and per spheroid after the various treatments as indicated. (D) Representative images of HepG2 spheroids, either untreated (control), or incubated with NH2-NPs for 72 h. One example slice from the confocal stack is shown for each treatment. Hoechst-stained nuclei are shown in blue and TMRM-labelled mitochondria in red. (E) Graph showing mean number of distinct mitochondria detected per spheroid (normalised to cell number) after the various treatments as indicated. (F) Graph showing mean mitochondria (TMRM) intensity per spheroid after the various treatments as indicated. Total number of spheroids analysed for the LAMP1 experiments (B and C) was 62 (94897 lysosomes); total number of spheroids analysed for the TMRM experiments (E and F) was 148 (151692 mitochondria). C, control; STS, staurosporine. Asterisks denote p < 0.01 compared to control samples. | ||
Discussion
In this work we detail a robust strategy for automated high-throughput profiling of NP-induced toxicity in a liver cell spheroid model. Unlike other studies reported, our approach not only provides information at the spheroid level, but extracts information from every cell within the spheroid. Importantly, our work also highlights that individual cells within a 3D assembly show a differential response to nanoparticle exposure, using amine-modified NPs as a model system to induce cytotoxicity. This information is therefore of value in the design of nanomedicines targeting solid tumours, increasing our understanding of how the penetration of such therapeutics correlates with cytotoxicity.Fluorescence microscopy is arguably the most powerful method available to us to extract multi-parametric functional information from individual cells. The greatest challenge, however, is capturing the required level of detail from cells growing as spheroids. The approach that we have taken is to employ a series of well-established fluorescent reporters, which together can provide a more holistic view of cytotoxicity, both in terms of the range of organelles assessed, as well as at a higher level of resolution to that seen in previous studies. Indeed, similar reagents to the ones that we selected were successfully used in a model of miniature Hep3B spheroids to assess toxicity in response to a panel of chemical compounds; although in this case measurements were not made from single cells. Using Hoechst 33342, TMRM and calcein AM, Joshi and colleagues quantitatively evaluated mitochondrial membrane potential, oxidative stress and cell membrane integrity at the spheroid level.40 We selected propidium iodide (PI) to measure late stages of cell death, a dye that is comparatively well-established in flow cytometry cytotoxicity studies and is considered highly specific.41 PI was also used recently to detect toxicity in HepG2 spheroids, specifically incubated with silver oxide and zinc oxide NPs.15 As expected, we saw a time-dependent increase in the number of PI-positive nuclei in our spheroids, peaking at ca. 50% after 72 h of incubation with amine-modified NPs, the model used in our study. Furthermore, by analysing each individual cell nucleus, not only were we able to measure PI intensity, but also quantify changes to nuclear morphology, including parameters such as volume and surface area.
One particularly striking feature of our experiments was the heterogeneity observed, both across the spheroid population, and within each individual spheroid. Indeed, this ability to quantitatively measure toxicity from several hundred spheroids in parallel, is a unique feature of the high-content imaging approach that we adopted. Other microscopy studies that have used PI to measure NP-induced toxicity in spheroids have typically prepared a very small number of spheroids, and often used only a single confocal slice for analysis.42 Given that MCTS form by a self-assembly process it is unsurprising that they show such heterogeneity across their population, and we would argue that it is essential to sample a large number of spheroids in order to make any valid statement about toxicity. Another advantage of our approach was that we quantified the number of PI-positive nuclei in each spheroid using volumetric analysis, thereby ensuring that our quantification was not biased due to the selection of any one confocal plane. At the level of an individual spheroid, we observed that while cells at the edge of the structure were in general more likely to display cytotoxic effects, presumably because they were exposed to higher levels of the amine-modified NPs, dying cells further inside the spheroid were often found in various locations. This presumably relates to NP access deeper inside the assembly, and we suggest that our analysis approach aligns well with other studies wishing to accurately quantify NP penetration into solid tumours.43,44
While PI intensity represents one means of assessing cytotoxicity, NPs and the therapeutics they carry, are known to elicit wider effects on cells. Indeed, we and others have previously shown in monolayer cells that amine-modified NPs cause mitochondrial fragmentation and swelling.45 By employing imaging of TMRM we have also been able to demonstrate that a similar effect occurs in HepG2 cells growing as spheroids, providing an additional independent measure of toxicity. TMRM provides the specific advantage of not only allowing us to identify the mitochondria for volumetric analysis, but also provides a readout of mitochondrial membrane potential. Therefore, from a single colour channel, we have been able to quantify at the subcellular level, both changes to mitochondrial morphology and to their function.
NPs enter cells by a range of endocytic mechanisms, and intracellular trafficking pathways ultimately result in most NP types accumulating in lysosomes.38,39 Although the mechanisms that mediate transfer of NPs through the intracellular environment were not the focus of this study, we were curious to see whether we could also use our approach to detect changes in the endomembrane system of cells exposed to NPs. To this end, we also visualised LAMP1, a well-established marker of acidic late endosomes and lysosomes. Our HCS approach revealed that amine-modified NPs result in a decrease in detected lysosomal number per cell, and that these lysosomes become significantly swollen, by more than two-fold. By contrast, the presence of equivalent-sized carboxylated NPs had no such effect. To our knowledge this is the first observation of this phenomenon in a MCTS model, although it has been reported previously in monolayer cells. The molecular basis for NP-induced swelling has not been clearly elucidated, but has been attributed to the proton sponge effect.37 A recent study utilising cationic gold nanorods in monolayer H1975 cancer cells suggests that this phenomenon is caused by a combination of calcium influx and release of chloride ions from lysosomes in the presence of a strong cationic charge.46 Interestingly we also observed a strong correlation, in individual cells, between swollen lysosomes and a strong PI signal in the nucleus. Future studies could utilise HCS of spheroids to combine analysis of lysosome swelling with NP delivery to this compartment,36 allowing better links between intracellular trafficking and the functional consequences to the cell to be established.
We believe that our approach provides an unprecedented level of subcellular detail with respect to studying and understanding NP-induced toxicity, however it has the additional advantage that large-scale spheroid-level data are also gathered. Spheroid size and morphology are features that have been used by many others as a readout of NP toxicity.14,26–28 While these previous studies have been well-conducted, they suffer from the common problem that they consider a relatively low number of spheroids in their analysis. For example, spheroid production using the hanging-drop method, or in U-bottomed ultra-low attachment plates, results in the formation of only a single spheroid per well. By contrast, the approach that we describe here facilitates the production of several hundred spheroids in a single well. This allows for monitoring of phenotypes from spheroids of different sizes, but all treated in the same well and in the same manner, providing perspective across a large population, rather than from a small number of individually selected spheroids. An additional innovation of our work is the use of volumetric analysis. We believe that this strategy is important, as not only does it minimise the inherent bias caused by analysis of individually selected confocal planes, but it ensures that data are gathered and used from the entire 3D cellular assembly. Given that MCTS can adopt a variety of sizes and morphologies during their growth, we suggest that volumetric analysis approaches should become the standard for toxicity studies performed in spheroids. However, one major limitation is that such an imaging approach requires access to expensive automated imaging equipment, and the resulting image datasets that are generated can be of the order of more than 1 Terabyte in size for a single 96-well plate. In turn, such datasets require powerful computers and image processing software to perform the analysis.
Overall, in addition to our detailed readouts gathered from individual cells, our spheroid size-based population analysis over time strongly suggested that the presence of amine-modified NPs results in a reduction of spheroid size, whereas carboxylated NPs did not cause this effect. Further evidence for this came from visual analysis of the spheroids, where it was frequently possible to observe small groups of cells detaching from the main spheroid assembly. Together, we believe that this work proposes and demonstrates a new paradigm for the assessment of NP-induced toxicity in MCTS, reconciling information from three scales; namely a population of spheroids, individual cells in a spheroid, and also subcellular information from selected markers.
Conclusions
In summary, we present a high-throughput approach employing high-content screening microscopy and advanced automated image analysis to measure NP-induced cytotoxicity at multiple scales from hundreds of spheroids in parallel. We propose that confocal HCS microscopy is a powerful approach that should be routinely employed in NP toxicity studies. It aligns well with other optical techniques, such as light sheet microscopy47 and photothermal microscopy,43 but crucially can provide quantitative data on a massive scale. HCS and associated image analysis offers the possibility of gaining a deeper insight into the mechanisms that drive cytotoxicity, providing multiple layers of information only restricted by the choice of fluorophores available.Author contributions
The study was conceived and supervised by JCS. The cell-based experiments were carried out by SK and the characterisation of the nanoparticles by MHB. The work was supervised by JCS and SJQ. The original draft was prepared by JCS and SK. All authors contributed to the final review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to all members of the lab for helpful advice, in particular Margaritha Mysior, Alannah Chalkley and Elena Garcia for advice on the image analysis pipelines. The Simpson lab acknowledges the support of an infrastructure research grant from Science Foundation Ireland (SFI) (16/RI/3745), the support through the SFI Centre for Research in Medical Devices (CURAM), co-funded by the European Regional Development Fund (13/RC/2073), and support from the UCD College of Science. Maria H. Byrne acknowledges funding from the SFI Synthesis and Solid State Pharmaceutical Centre (SSPC), co-funded under the European Regional Development Fund (12/RC/2275_P2).References
- F. Farjadian, A. Ghasemi, O. Gohari, A. Roointan, M. Karimi and M. R. Hamblin, Nanomedicine, 2019, 14, 93–126 CrossRef CAS PubMed.
- J. P. Martins, J. das Neves, M. de la Fuente, C. Celia, H. Florindo, N. Günday-Türeli, A. Popat, J. L. Santos, F. Sousa, R. Schmid, J. Wolfram, B. Sarmento and H. A. Santos, Drug Delivery Transl. Res., 2020, 10, 726–729 CrossRef PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- J. Zhao, H. Lu, S. Wong, M. Lu, P. Xiao and M. H. Stenzel, Polym. Chem., 2017, 8, 3317–3326 RSC.
- Z. Wu, R. Guan, M. Tao, F. Lyu, G. Cao, M. Liu and J. Gao, RSC Adv., 2017, 7, 12437–12445 RSC.
- D. Liu, S. Chen and M. Win Naing, Biotechnol. Bioeng., 2021, 118, 542–554 CrossRef CAS PubMed.
- S. W. Lee, S. Hong, B. Jung, S. Y. Jeong, J. H. Byeon, G. S. Jeong, J. Choi and C. Hwang, Biotechnol. Bioeng., 2019, 116, 3041–3052 CrossRef CAS PubMed.
- S. A. Sloan, J. Andersen, A. M. Paşca, F. Birey and S. P. Paşca, Nat. Protoc., 2018, 13, 2062–2085 CrossRef CAS PubMed.
- D. Lv, Z. Hu, L. Lu, H. Lu and X. Xu, Oncol. Lett., 2017, 14, 6999–7010 Search PubMed.
- Z. Zhang, H. Wang, Q. Ding, Y. Xing, Z. Xu, C. Lu, D. Luo, L. Xu, W. Xia, C. Zhou and M. Shi, PLoS One, 2018, 13, e0194016 CrossRef PubMed.
- S. Tsai, L. McOlash, K. Palen, B. Johnson, C. Duris, Q. Yang, M. B. Dwinell, B. Hunt, D. B. Evans, J. Gershan and M. A. James, BMC Cancer, 2018, 18, 335 CrossRef PubMed.
- T. Hurrell, A. A. Ellero, Z. F. Masso and A. D. Cromarty, Toxicol. In Vitro, 2018, 50, 86–94 CrossRef CAS PubMed.
- J. Lee, G. D. Lilly, R. C. Doty, P. Podsiadlo and N. A. Kotov, Small, 2009, 5, 1213–1221 CAS.
- H. Ma, Q. Jiang, S. Han, Y. Wu, J. Cui Tomshine, D. Wang, Y. Gan, G. Zou and X.-J. Liang, Mol. Imaging, 2012, 11, 487–498 CrossRef CAS PubMed.
- E. Elje, E. Mariussen, O. H. Moriones, N. G. Bastús, V. Puntes, Y. Kohl, M. Dusinska and E. Rundén-Pran, Nanomaterials, 2020, 10, E545 CrossRef PubMed.
- K. Kik, B. Bukowska and P. Sicińska, Environ. Pollut., 2020, 262, 114297 CrossRef CAS PubMed.
- J. A. Kyffin, P. Sharma, J. Leedale, H. E. Colley, C. Murdoch, P. Mistry and S. D. Webb, Toxicol. In Vitro, 2018, 48, 262–275 CrossRef CAS PubMed.
- S. C. Ramaiahgari, S. Waidyanatha, D. Dixon, M. J. DeVito, R. S. Paules and S. S. Ferguson, Toxicol. Sci., 2017, 160, 189–190 CrossRef CAS PubMed.
- H. Katifelis, A. Lyberopoulou, I. Mukha, N. Vityuk, G. Grodzyuk, G. E. Theodoropoulos, E. P. Efstathopoulos and M. Gazouli, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S389–S398 CrossRef CAS PubMed.
- P. Pellen-Mussi, S. Tricot-Doleux, C. Neaime, N. Nerambourg, F. Cabello-Hurtado, S. Cordier, F. Grasset and S. Jeanne, J. Nanosci. Nanotechnol., 2018, 18, 3148–3157 CrossRef CAS PubMed.
- M. Dubiak-Szepietowska, A. Karczmarczyk, T. Winckler and K.-H. Feller, Toxicology, 2016, 370, 60–69 CrossRef CAS PubMed.
- A. Kroll, M. H. Pillukat, D. Hahn and J. Schnekenburger, Arch. Toxicol., 2012, 86, 1123–1136 CrossRef CAS PubMed.
- K. J. Ong, T. J. MacCormack, R. J. Clark, J. D. Ede, V. A. Ortega, L. C. Felix, M. K. M. Dang, G. Ma, H. Fenniri, J. G. C. Veinot and G. G. Goss, PLoS One, 2014, 9, e90650 CrossRef PubMed.
- R. Guadagnini, B. H. Kenzaoui, L. Walker, G. Pojana, Z. Magdolenova, D. Bilanicova, M. Saunders, L. Juillerat-Jeanneret, A. Marcomini, A. Huk, M. Dusinska, L. M. Fjellsbø, F. Marano and S. Boland, Nanotoxicology, 2015, 9, 13–24 CrossRef CAS PubMed.
- C. Andraos, I. J. Yu and M. Gulumian, Int. J. Toxicol., 2020, 39, 397–421 CrossRef CAS PubMed.
- A. S. Mikhail, S. Eetezadi and C. Allen, PLoS One, 2013, 8, e62630 CrossRef CAS PubMed.
- X. Wang, X. Zhen, J. Wang, J. Zhang, W. Wu and X. Jiang, Biomaterials, 2013, 34, 4667–4679 CrossRef CAS PubMed.
- A. Hornung, M. Poettler, R. P. Friedrich, B. Weigel, S. Duerr, J. Zaloga, I. Cicha, C. Alexiou and C. Janko, Anticancer Res., 2016, 36, 3093–3101 CAS.
- M. S. Oliveira, B. Aryasomayajula, B. Pattni, S. V. Mussi, L. A. M. Ferreira and V. P. Torchilin, Int. J. Pharm., 2016, 512, 292–300 CrossRef CAS PubMed.
- H. Lei, S. C. Hofferberth, R. Liu, A. Colby, K. M. Tevis, P. Catalano, M. W. Grinstaff and Y. L. Colson, J. Thorac. Cardiovasc. Surg., 2015, 149, 1417–1424 CrossRef CAS PubMed ; discussion 1424-1425.e1.
- D. J. Brayden, S.-A. Cryan, K. A. Dawson, P. J. O'Brien and J. C. Simpson, Drug Discovery Today, 2015, 20, 942–957 CrossRef CAS PubMed.
- T. H. Booij, L. S. Price and E. H. J. Danen, SLAS Discovery, 2019, 24, 615–627 CAS.
- E. Nürnberg, M. Vitacolonna, J. Klicks, E. von Molitor, T. Cesetti, F. Keller, R. Bruch, T. Ertongur-Fauth, K. Riedel, P. Scholz, T. Lau, R. Schneider, J. Meier, M. Hafner and R. Rudolf, Front. Mol. Biosci., 2020, 7, 20 CrossRef PubMed.
- S. D. Collins, G. Yuen, T. Tu, M. A. Budzinska, K. Spring, K. Bryant and N. A. Shackel, in Hepatocellular Carcinoma, ed. J. E. E. Tirnitz-Parker, Codon Publications, Brisbane (AU), 2019 Search PubMed.
- P. Bevilacqua, S. Nuzzo, E. Torino, G. Condorelli, M. Salvatore and A. M. Grimaldi, Nanomaterials, 2021, 11, 780 CrossRef CAS PubMed.
- M. B. Cutrona and J. C. Simpson, Small, 2019, 15, e1902033 CrossRef PubMed.
- L. M. P. Vermeulen, S. C. De Smedt, K. Remaut and K. Braeckmans, Eur. J. Pharm. Biopharm., 2018, 129, 184–190 CrossRef CAS PubMed.
- A. Panarella, M. G. Bexiga, G. Galea, E. D. O’ Neill, A. Salvati, K. A. Dawson and J. C. Simpson, Sci. Rep., 2016, 6, 28865 CrossRef CAS PubMed.
- V. Francia, D. Montizaan and A. Salvati, Beilstein J. Nanotechnol., 2020, 11, 338–353 CrossRef CAS PubMed.
- P. Joshi, S.-Y. Kang, A. Datar and M.-Y. Lee, Curr. Protoc. Toxicol., 2019, 79, e66 CrossRef PubMed.
- N. Atale, S. Gupta, U. C. S. Yadav and V. Rani, J. Microsc., 2014, 255, 7–19 CrossRef CAS PubMed.
- I. García, M. Henriksen-Lacey, J. Calvo, D. J. de Aberasturi, M. M. Paz and L. M. Liz-Marzán, Bioconjugate Chem., 2019, 30, 242–252 CrossRef PubMed.
- E. Darrigues, Z. A. Nima, D. A. Nedosekin, F. Watanabe, K. M. Alghazali, V. P. Zharov and A. S. Biris, Sci. Rep., 2020, 10, 3362 CrossRef CAS PubMed.
- K. Bromma, A. Alhussan, M. M. Perez, P. Howard, W. Beckham and D. B. Chithrani, Cancers, 2021, 13, 1465 CrossRef CAS PubMed.
- M. G. Bexiga, C. Kelly, K. A. Dawson and J. C. Simpson, Nanomedicine, 2014, 9, 1651–1664 CrossRef CAS PubMed.
- D. U. Lee, J.-Y. Park, S. Kwon, J. Y. Park, Y. H. Kim, D. Khang and J. H. Hong, Nanoscale, 2019, 11, 19980–19993 RSC.
- G. Lazzari, D. Vinciguerra, A. Balasso, V. Nicolas, N. Goudin, M. Garfa-Traore, A. Fehér, A. Dinnyés, J. Nicolas, P. Couvreur and S. Mura, Eur. J. Pharm. Biopharm., 2019, 142, 195–203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04460e |
| This journal is © The Royal Society of Chemistry 2021 |