
Photoenhanced cytosolic protein delivery based on a photocleavable group-modified dendrimer†
Yafei
Li‡
,
Yang
Zhou‡
,
Tianyi
Wang
,
Kaiqi
Long
 ,
Yaming
Zhang
and
Weiping
Wang
*
,
Yaming
Zhang
and
Weiping
Wang
*
State Key Laboratory of Pharmaceutical Biotechnology & Department of Pharmacology and Pharmacy, Li Ka Shing Faculty of Medicine & Dr. Li Dak-Sum Research Centre, The University of Hong Kong, Hong Kong SAR, China. E-mail: wangwp@hku.hk
First published on 17th September 2021
Abstract
Numerous recently developed therapies have highlighted the advantages of using proteins as therapeutics. However, in many protein delivery systems, the complicated carrier designs, low loading content, and off-targeting effects have limited their clinical applications. Here we report a photoresponsive protein-binding moiety and use it to prepare a simple nanoscale protein delivery system with high delivery efficiency and photoenhanced cellular uptake of proteins. The carrier was prepared by modifying a photocleavable molecule, DEACM, onto the surface of a cationic dendrimer, poly(amidoamine). DEACM simultaneously contributed to protein binding, self-assembly, and photocontrollability of the system. The multi-functional DEACM enabled the simplicity of the protein delivery system, which does not require complex organic synthesis or protein modification. The high delivery efficiency, high serum tolerance, and photoenhanced cellular uptake have been proved with functional proteins, presenting the potential for delivering protein therapeutics.
Introduction
Proteins, an important class of biomacromolecules, perform complex and indispensable functions for organism growth and maintenance.1 Over the past decades, there has been emerging interest in using active proteins for various biological applications, such as cancer therapy,2 gene therapy,3 and vaccination.4 Extracellular delivery of protein therapeutics, such as insulin and nimotuzumab, has made great progress in the clinic.5 However, development on cytosolic protein delivery focusing on intracellular targets is still challenging, mainly due to their large molecular weights, low cellular affinity, and poor endosomal escape ability.6 Moreover, the potential risks of off-targeting delivery still exist in many current protein delivery systems.7 Therefore, developing strategies for precise and efficient intracellular protein delivery is highly desired in the pharmaceutical industry.8Nanoscale delivery systems are a commonly used platform for protein delivery. They could load proteins through either chemical conjugation or physical encapsulation.9 Chemical conjugation, such as covalent conjugation with polyethylene glycol10 and polyethyleneimine (PEI),11 could achieve tight protein binding with carrying materials, but it might affect protein functionalities and increase production costs.12 The encapsulation systems, such as polymersomes,13,14 lipid nanoparticles,15,16 and silica nanoparticles,17,18 can carry the entrapped protein to the designated sites. However, the low encapsulation efficiency, especially for proteins with large molecular weights, has limited their overall performance.19 To address these issues, co-assembled protein delivery systems through physical adsorption are developed.20,21 In these systems, polymers or ultra-small nanoparticles modified with protein binding moieties could co-assemble with proteins into nanoparticles with a high loading content. The commonly used binding moieties include positively charged groups,22,23 boronic acid derivates,24–26 guanidine derivates,27,28 and fluoroalkanes.29 These moieties could provide tight protein binding ability, whereas they lack stimuli responsiveness to actively control protein release. For an efficient protein delivery system, the interactions between carriers and proteins should be tight and controllable.
To achieve controlled protein delivery, stimuli-responsive moieties are usually incorporated into the carriers. Smart materials responsive to internal or external triggers such as pH,15 ultrasound,30 and light irradiation,31 could provide precise control for targeted protein delivery and release. For example, Hentzen et al.32 reported a dendritic polymer composed of photocleavable nitrobenzyl-guanidine conjugates. Guanidine contributed for protein binding and the nitrobenzyl moiety enabled photo-disassociation of this conjugate. Through this design, spatiotemporally controlled protein release and endosomal escape had been achieved.
To simplify the complexity of stimuli-responsive protein delivery systems, exploration of protein binding molecules with stimuli responsiveness provides one solution. Here, for the first time, we report the protein binding ability of a photoresponsive group, 7-diethylamino-4-hydroxymethylcoumarin (DEACM). It enabled the simple design of a photoresponsive protein delivery system without complicated synthesis and protein modification. The protein binding and photo-triggered dissociation of this system were proved with functional proteins, indicating its applicability for protein-based therapies.
DEACM is a photocleavable coumarin derivative, widely applied in controlled drug release and targeted drug delivery.33–35 Its simple synthesis and high biosafety have been evaluated in many drug delivery systems.36 In this protein delivery system, besides the photoresponsiveness, DEACM also contributed to the hydrophobicity for self-assembly and the protein binding ability of the system, resulting in high protein loading and photoenhanced cellular uptake. Poly(amidoamine) dendrimers (PAMAM), a tree-like polymer with multiple amino groups, supported the easy modification of DEACM on the surface. The PAMAM–DEACM conjugates were able to form nanocomplexes with proteins. Through the physical adsorption of hyaluronic acid and albumin, the protein nanocomplexes achieved high stability in physiological solutions and maintained a high delivery efficiency in serum-containing medium. Upon visible light irradiation, the protein nanocomplexes were demonstrated to undergo fast disassociation and enhanced cellular uptake, providing a simple yet efficient strategy for controlled protein delivery.
Results
DEACM contributed to protein binding and nanocomplex formation
The PAMAM–DEACM conjugate was prepared by conjugating generation-five PAMAM (128 amino groups on the surface) with DEACM through a two-step synthesis (Fig. S1A†). According to the feeding molar ratios of DEACM to amino groups on PAMAM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 2:10, 3:10, 4:10, and 6:10), the synthesized PAMAM–DEACM were denoted as PD0.1, PD0.2, PD0.3, PD0.4 and PD0.6, respectively. The successful conjugation was confirmed by 1H NMR spectra (Fig. S1B†) and the grafting ratios were calculated with UV-Vis absorption spectra (Fig. S2†).
:10, 2:10, 3:10, 4:10, and 6:10), the synthesized PAMAM–DEACM were denoted as PD0.1, PD0.2, PD0.3, PD0.4 and PD0.6, respectively. The successful conjugation was confirmed by 1H NMR spectra (Fig. S1B†) and the grafting ratios were calculated with UV-Vis absorption spectra (Fig. S2†).
PAMAM–DEACM could form nanocomplexes in water with a model protein, bovine serum albumin (BSA), simply through the flash nanoprecipitation method (Fig. 1A).23 The size distribution of free BSA, mixture of PAMAM and BSA (PAMAM/BSA), and nanocomplexes were measured by dynamic light scattering (DLS, Fig. 1B and Table S1†). It should be noticed that pure PAMAM could not form nanocomplexes with BSA. However, after conjugating with DEACM even though with a very low feeding ratio (PD0.1), the conjugates and BSA could form stable nanocomplexes of around 140 nm in water, exhibiting the excellent protein binding ability of the DEACM group.
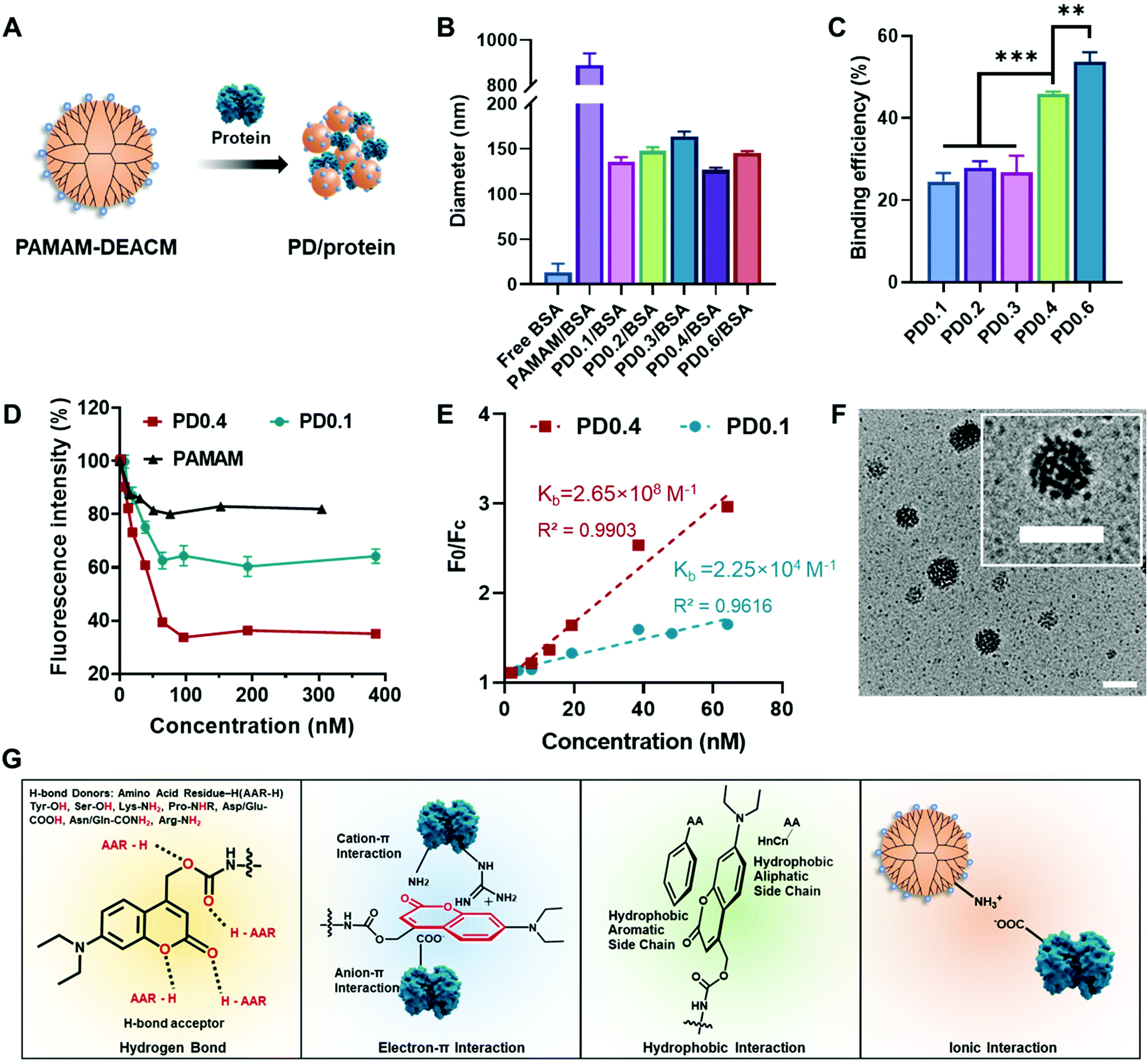 | ||
| Fig. 1 DEACM-modified dendrimer binds to protein. (A) Schematic illustration of the co-assembled PD/protein nanocomplex. (B) DLS results of free BSA, PAMAM/BSA mixture and PD/BSA nanocomplexes. (C) Protein binding efficiency of a series of PAMAM–DEACM. (D) Fluorescence change of rBSA (100 nM) with different concentrations of PAMAM–DEACM or PAMAM. (E) The Stern–Volmer curve for the interaction of PAMAM–DEACM with rBSA (100 nM). F0 is the fluorescence intensity of rBSA, Fc is the fluorescence intensity of PD/rBSA. λex = 548 nm, λem = 586 nm. (F) TEM images of PD0.4/BSA. Scale bar: 200 nm. (G) Possible interactions between PAMAM–DEACM and proteins: hydrogen bond, electron–π interaction, hydrophobic interaction, and ionic interaction. Data analysis: n = 3, ***p < 0.001, **p < 0.01. | ||
Rhodamine-labelled BSA (rBSA) was synthesized to evaluate the interaction between PAMAM–DEACM conjugates and proteins. The protein binding efficiency of the conjugates was calculated with the absorbance at 560 nm (Fig. 1C). With more DEACM groups per PAMAM, the conjugates exhibited higher protein binding efficiency, which also indicated the role of DEACM in protein loading. Furthermore, we conducted the titration experiment, as the fluorescence of rBSA would be quenched due to the attachment of the conjugates (Fig. 1D). The quench phenomenon of PD0.4 was much more significant than PD0.1 and pure PAMAM, indicating that the nanocomplexes formed by PD0.4 were more compacted in water than PD0.1 and pure PAMAM.37 According to an established method,38,39 we fitted the fluorescence intensity changes (Fig. 1E) and obtained the binding constant Kb of PD0.4 as 2.65 × 108 M−1, which was much larger than the Kb of PD0.1 (2.25 × 104 M−1).
In another experiment, the fluorescence of DEACM was measured and analyzed. Rhodamine chromophore worked as the fluorescence acceptor and DEACM worked as the donor. The fluorescence resonance energy transfer (FRET) phenomenon between PD0.4 and rBSA also demonstrated the short distance between each other and the formation of condensed nanocomplexes (PD/rBSA) in water (Fig. S3A†). Considering the balance between protein binding and photoresponsiveness, PD0.4, but not PD0.6, was used for further studies. The nanocomplexes formed by PD0.4 and BSA were denoted as PD/BSA, which showed a spherical and raspberry-like morphology under transmission electron microscopy (TEM, Fig. 1F).
Based on the above results and literature studies, the possible interactions between DEACM and proteins were proposed and are shown in Fig. 1G. The oxygen atoms from DEACM may contribute the binding force via hydrogen bonds.38,40 The uneven electronic cloud distribution of the coumarin ring made it possible to bind with the guanidinium groups, ammonium groups, or carboxyl groups on proteins.41 The hydrophobic structure of DEACM resulted in an organized π–π stacking or hydrophobicity-guided self-assembling behavior of the protein complexes in aqueous solutions.42,43 Also, the remaining amino groups on PAMAM could provide additional ionic interactions with proteins.26
Surface coating stabilized the protein nanocomplexes under physiological conditions
Protein nanocomplexes for biomedical applications would face a complicated microenvironment in vivo. Therefore, preparing a stable protein delivery system capable of tolerating physiological conditions is highly demanded. Although PD0.4 could form stable protein complexes with BSA in water, PD/rBSA showed a dramatic fluorescence increment once added into solutions containing 5% (w/v) BSA (Fig. S3B†). The increment is because the competition of free BSA with rBSA would lower the FRET effect. PD/rBSA also showed fast fluorescence decrement in phosphate-buffered saline (PBS) because of an aggregation behavior of the nanocomplexes in buffers with a high ionic concentration.44To increase the stability of nanomedicines, surface coating is generally needed.45 In this study, biodegradable hyaluronic acid (HA) and BSA protecting layers were subsequently coated on the surface to stabilize the protein nanocomplexes (BH-PD/protein, Fig. 2A). The negatively charged HA was spontaneously adsorbed onto the positively charged nanocomplexes during preparation. Meanwhile, HA endowed the nanocomplexes with a targeting ability for CD44 receptors, which are usually overexpressed on cancer cell surfaces, such as A549 and HeLa cells.46 The BSA coating provided an extra steric protection for stabilization in physiological solutions.47 The low cost, ready availability, and high biocompatibility of the coating method make it widely applicable for surface decorations.48
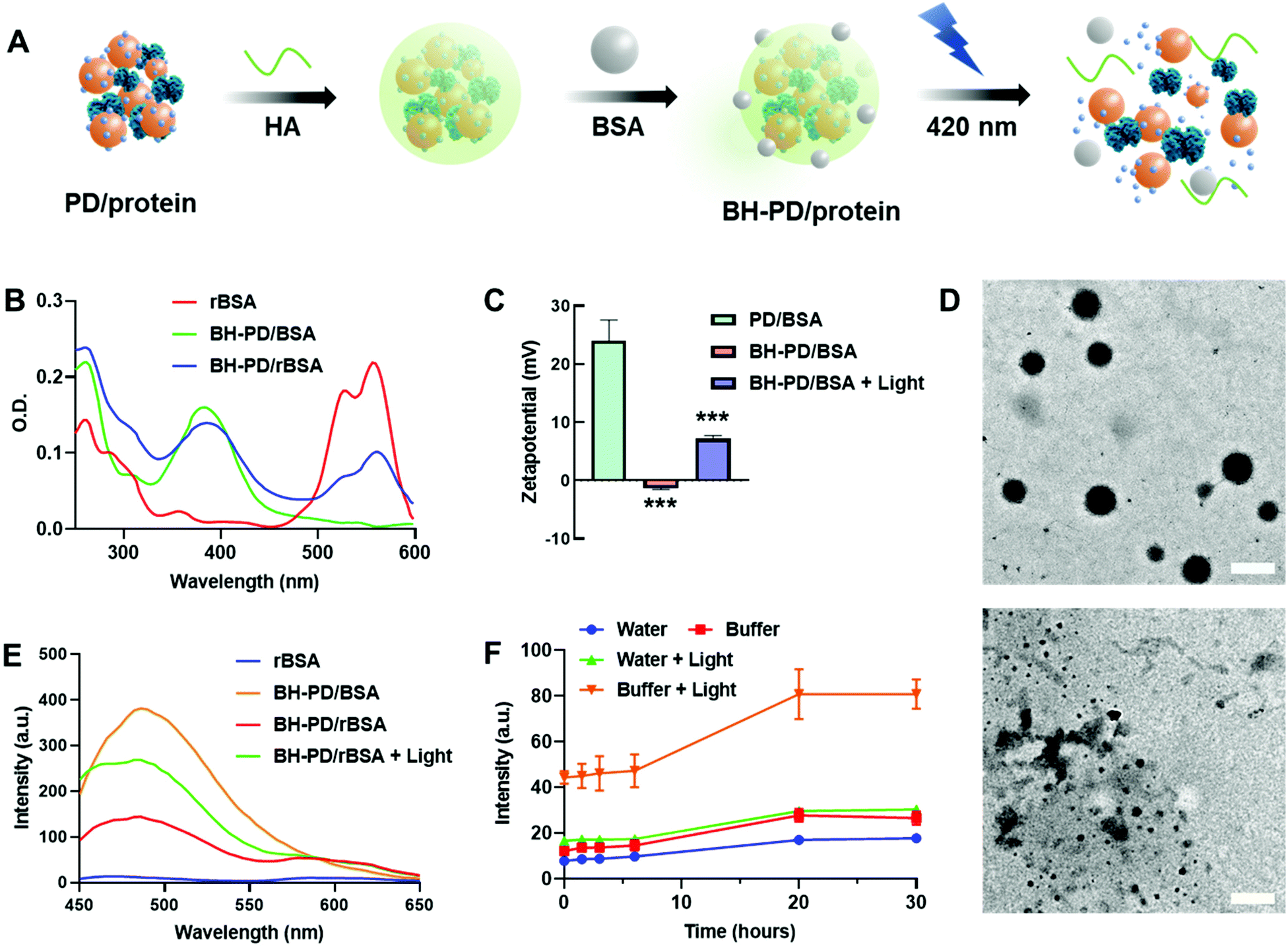 | ||
| Fig. 2 Optimization and characterization of protein nanocomplexes. (A) Schematic illustration of HA and BSA coating and the photoinduced disassociation of the protein nanocomplex. (B) Absorption spectra of rBSA, BH-PD/BSA and BH-PD/rBSA nanocomplexes in water. (C) Zeta potential and (D) TEM images of BH-PD/BSA nanocomplexes before and after light irradiation. Scale bar: 200 nm. (E) Fluorescence spectra of rBSA, BH-PD/BSA, BH-PD/rBSA, and BH-PD/rBSA after light irradiation in water. (F) Fluorescence intensity at 474 nm of BH-PD/rBSA in water and in PBS containing 5% (w/v) BSA before and after light irradiation. λex = 405 nm, λem = 474 nm. Irradiation condition: 420 nm, 50 mW cm−2, 2 min. Data analysis: n = 3, ***p < 0.001. | ||
After the coating and purification of BH-PD/rBSA nanocomplexes through centrifugation, the absorption spectrum was recorded (Fig. 2B), which indicated the successful self-assembly of PD0.4 with rBSA. After coating, the zeta potential dropped from +24.0 mV to −1.32 mV. Meanwhile, the diameter of the coated nanocomplexes increased from 126.5 nm to 139.2 nm. In darkness, BH-PD/rBSA nanocomplexes showed good tolerance to 5% (w/v) BSA solution or PBS buffer (Fig. S4A†). When incubated in complete medium containing 10% (v/v) fetal bovine serum (FBS) at 37 °C, the size kept almost the same for 48 h (Fig. S4B†), representing the good stability of the nanocomplexes.
Photocleavage of DEACM triggered protein release
The photoresponsiveness of the system was investigated by analyzing the photocleavage rate of the PD0.4 conjugate with HPLC (Fig. S5†). It showed over 75% cleavage within 2 min under 420 nm light irradiation at 50 mW cm−2, which released ∼37% DEACM with the generation of some by-products.49 The sensitive photoresponsiveness provided the foundation for the photo-controlled protein release of the nanocomplexes. After light irradiation, the size of BH-PD/BSA increased to over 800 nm, and the zeta potential increased to +7.13 mV (Fig. 2C), indicating the photo-induced disruption of the nanostructure. Under TEM, the spherical morphology of BH-PD/BSA nanocomplexes could be observed, while disordered polymer and protein aggregates were noticed after light irradiation (Fig. 2D).The emission spectra of BH-PD/rBSA were measured to monitor the photo-induced protein release. After light irradiation, due to the cleavage of DEACM and the reduced FRET effect, the fluorescence intensity of DEACM increased immediately (Fig. 2E). In PBS buffer containing 5% (w/v) BSA, after photo-induced structure disruption, the nanocomplexes showed greater fluorescence increment than the groups in water over time (Fig. 2F). It might be because that the competition of free BSA with rBSA over the binding with DEACM accelerated the release of rBSA. This result exhibited the photo-triggered protein release from the nanocomplexes in physiological solutions. Also, in darkness over 24 h, BH-PD/rBSA only showed a slight fluorescence increment, indicating the stability of the nanocomplexes under physiological conditions.
Light irradiation enhanced the cellular uptake of the encapsulated protein
We then evaluated the intracellular protein delivery performance of the nanocomplexes using rBSA as the model protein. The red fluorescence of rhodamine was monitored to assess the protein uptake. The A549 cancer cell line with overexpressed CD44 receptors was chosen in this study. As shown in Fig. 3A, the PAMAM/rBSA mixture showed a slight rBSA-uptake increment compared to free rBSA, while BH-PD0.4/rBSA exhibited the highest rBSA delivery efficiency among all groups in darkness. With light irradiation, the BH-PD0.4/rBSA group showed further improved cellular uptake. The increased uptake is presumably because after the dissociation of the protein nanocomplexes, the photocleaved conjugates might still bind with proteins temporarily, which had an increased surface charge that facilitated cellular uptake. To confirm the hypothesis, the cellular uptake of rBSA mixed with free DEACM and PAMAM was investigated (Fig. S6†). The mixture of rBSA with DEACM or PAMAM could not sufficiently improve the protein delivery efficiency compared to BH-PD/rBSA nanocomplexes with or without light irradiation, demonstrating the unique intracellular protein delivery property of the protein nanocomplexes. Another BH-PD/rBSA nanocomplex uptake analysis was conducted using different light irradiation strategies (Fig. S7†). After incubation with BH-PD/rBSA nanocomplexes for 2 h and washing out the non-internationalized nanocomplexes, light irradiation did not increase the fluorescence signal, which rules out the interference of the rBSA release-induced fluorescence increment on cellular uptake evaluation. Moreover, the two groups both showed lower fluorescence than the group where cells were incubated with light-activated nanocomplexes for 2 h. This result further confirms that light irradiation enhanced the cellular uptake of rBSA.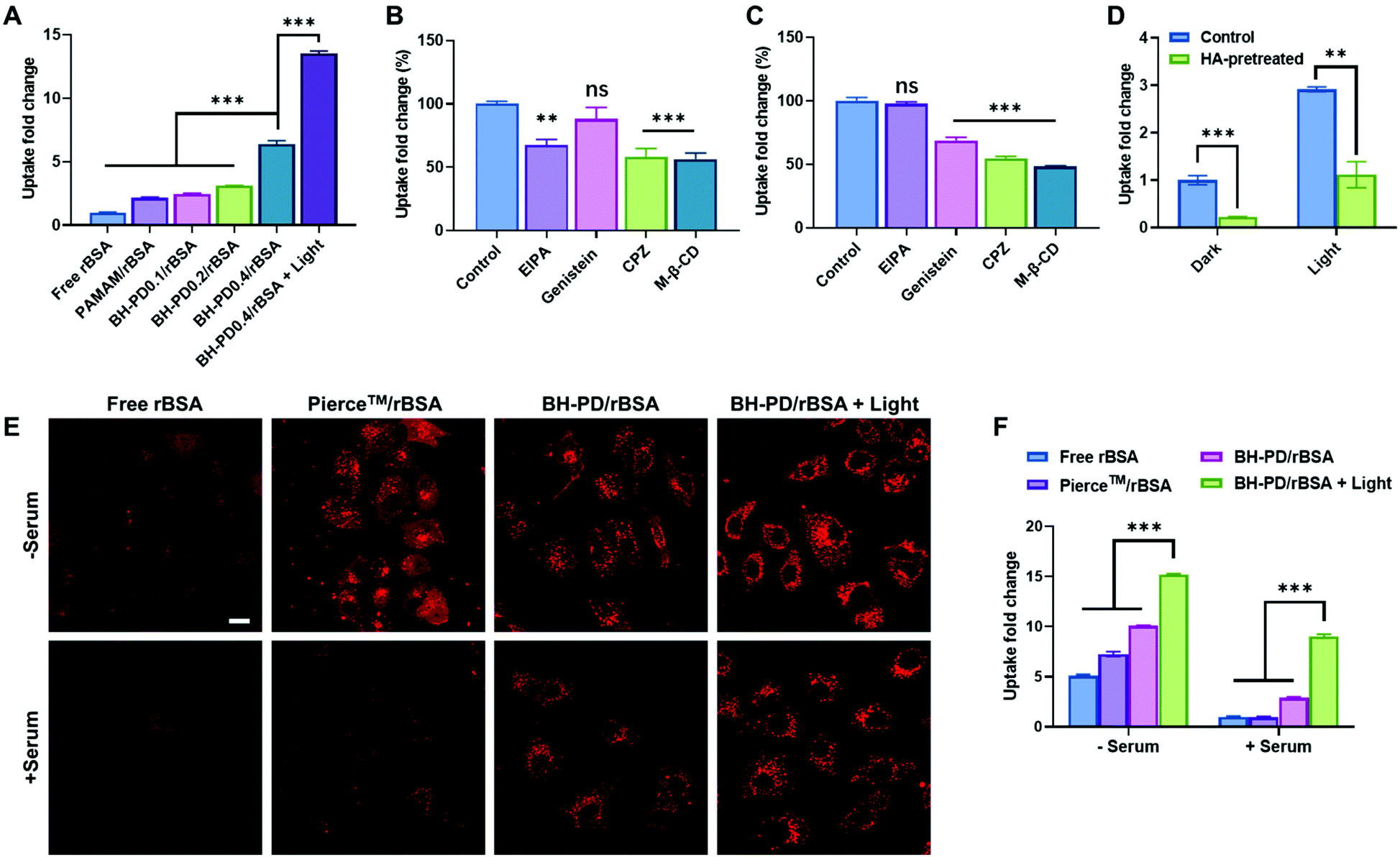 | ||
| Fig. 3 Cytosolic delivery of rBSA. (A) Cellular uptake of free rBSA, PAMAM/rBSA mixture and rBSA nanocomplexes. The free rBSA-treated group was set as the control group. Endocytosis pathway analysis of BH-PD/rBSA nanocomplexes (B) without and (C) with light treatment (EIPA, macropinocytosis inhibitor; Genistein, caveolae-mediated endocytosis inhibitor; CPZ, clathrin-mediated endocytosis inhibitor; M-β-CD, lipid-raft-mediated endocytosis inhibitor). (D) Cellular uptake of BH-PD/rBSA nanocomplexes with or without free HA treatment. (E) Confocal microscopy and (F) flow cytometry analysis of cellular uptake of free rBSA, rBSA with Pierce™ Protein Transfection Reagent Kit, BH-PD/rBSA nanocomplexes, and BH-PD/rBSA nanocomplexes with light irradiation in the medium with or without serum. Scale bar: 20 μm. Irradiation condition: 420 nm, 50 mW cm−2, 2 min. Data analysis: n = 3, ns: not significant, **p < 0.01, ***p < 0.001. | ||
To investigate the cellular uptake routes, cells were pretreated with four endocytosis inhibitors before the incubation with BH-PD/rBSA. In darkness, BH-PD/rBSA uptake was more affected by the macropinocytosis inhibitor (EIPA), clathrin-mediated endocytosis inhibitor (CPZ), and lipid-raft-mediated endocytosis inhibitor (M-β-CD) (Fig. 3B). After light irradiation, the uptake through macropinocytosis was less important, while the caveolae-mediated endocytosis inhibitor (genistein) became more effective to inhibit complex uptake (Fig. 3C). The HA targeting performance was also evaluated by comparing complex uptake with or without pretreatment of free HA (Fig. 3D). In darkness, due to the competition of free HA and HA coatings on the complexes, the HA-pretreated group only showed ∼22% cellular uptake compared with its control group. With light irradiation, the HA-pretreated group showed an elevation of cellular uptake due to the exposed cationic PAMAM. However, the cellular uptake was still lower than that in the group without HA-pretreatment (∼38%). The result indicated the HA layer still contributed to the cellular uptake of rBSA, which also supported the hypothesis that the pieces of dissociated nanocomplexes with HA coating might still encapsulate rBSA temporarily and enhance its intracellular delivery.
The protein uptake before and after light irradiation were then compared with a commercial protein delivery kit, Pierce™ protein transfection reagent (Thermo Fisher), through confocal microscopy (Fig. 3E) and flow cytometry (Fig. 3F). The kit showed a relatively high protein delivery efficiency in serum-free medium. However, its delivery efficiency dramatically decreased in the medium containing 10% (v/v) FBS. In contrast, BH-PD/rBSA nanocomplexes after light irradiation maintained a high protein delivery efficiency regardless of the existence of serum. The excellent serum tolerance of BH-PD/rBSA was presumably attributed to the HA/BSA layer on the surface of the nanocomplexes. BH-PD/rBSA nanocomplexes also presented a time-dependent cellular uptake behavior (Fig. S8†). The dislocation with acidic endosomes after 6 h incubation in serum-containing medium (Fig. S8†) or 1 h incubation in serum-free medium (Fig. S9†) demonstrated an efficient endosomal escape ability of the protein nanocomplexes. We also tested the effect of light irradiation on endosomal escape of the protein (Fig. S10†). After light irradiation, the signal of the lysotracker decreased greatly, presumably due to the “proton sponge” effect of exposed PAMAM. This result indicated that nanocomplex dissociation induced by light irradiation would promote the endosomal escape of proteins.
The nanocomplexes maintained the enzyme activity of the encapsulated proteins
We then examined the binding ability and enzyme activity of the delivery system with a functional enzyme, glucose oxidase (GOx). Through the same nanoprecipitation method, PD0.4 and GOx could form stable nanocomplexes (PD/GOx) of around 118.6 nm in water. We conducted the titration experiment with rhodamine-labelled glucose oxidase (rGOx) (Fig. S11†) and obtained the binding constant Kb of PD0.4 as 1.62 × 106 M−1, which was larger than the Kb of PD0.1 (5.19 × 105 M−1). The result is consistent with the binding ability of rBSA. The PD/GOx nanocomplexes were collected through centrifugation and the unbound/released GOx in the supernatant was analyzed with a standard protocol to indicate GOx binding ability50 (Fig. 4A). In principle, GOx oxidizes glucose to produce H2O2, and horseradish peroxidase (HRP) catalyzes H2O2 to oxidize colorless tetramethylbenzidine (TMB) into a blue product (TMBNH). The absorbance of TMBNH at 370 nm can be used to determine the GOx enzyme activity.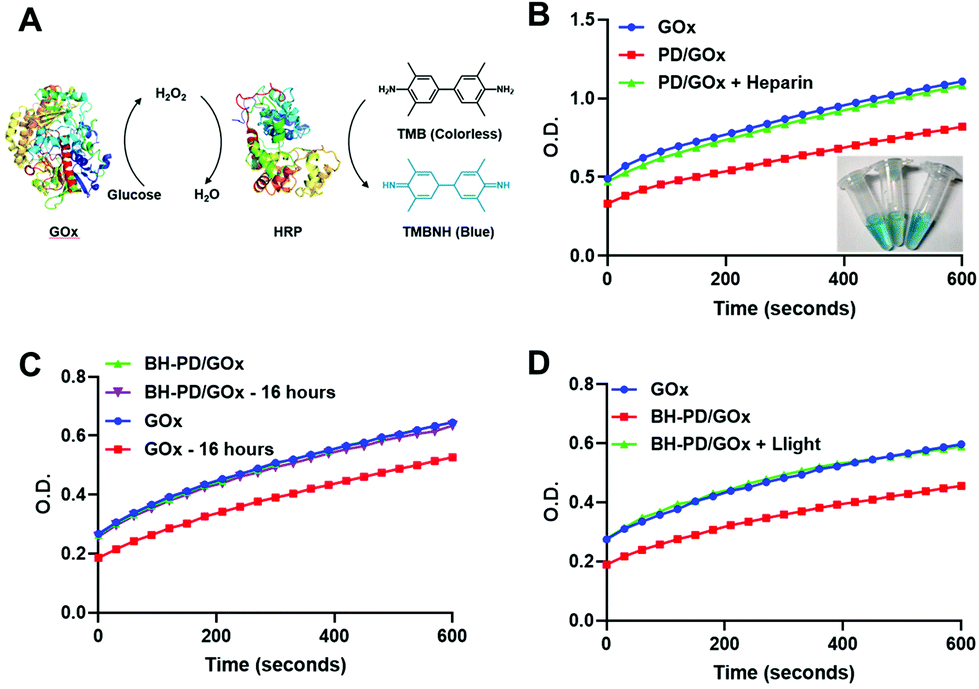 | ||
| Fig. 4 Enzymatic activity analysis of GOx nanocomplexes. (A) Diagram of the TMB protocol. GOx oxidizes glucose to produce H2O2, and HRP catalyzes H2O2 to oxidize colorless TMB into a blue product (TMBNH). The absorbance at 370 nm is used to determine the enzyme activity. (B) GOx enzyme activity determined from the supernatant of free GOx, PD/GOx, and PD/GOx with heparin in water. (C) GOx enzyme activity of GOx and BH-PD/GOx in PBS with 5% (w/v) BSA at 37 °C after 16 h. (D) GOx enzyme activity determined from the supernatant of GOx and BH-PD/GOx before and after light irradiation (420 nm, 50 mW cm−2, 2 min) in PBS containing 5% (w/v) BSA. | ||
With the addition of a high concentration of anionic heparin to compete the binding of PD0.4 against GOx, the release of GOx to the supernatant was observed (Fig. 4B). To prepare stable nanocomplexes in the physiological solution, the coating of HA and BSA was performed (BH-PD/GOx) and the diameter of the nanocomplexes increased to 165.2 nm. The coating did not affect the enzyme activity of GOx. Moreover, BH-PD/GOx showed negligible enzyme activity change in PBS containing 5% (w/v) BSA over 16 h incubation (Fig. 4C), demonstrating the good stability of the system, while free GOx partly lost its enzyme activity in the solution. This result demonstrated that the nanocomplex structure could protect proteins from degradation and inactivation.12
The enzyme activity of the supernatants of BH-PD/GOx nanocomplexes before and after light irradiation was measured to investigate the phototriggered release behavior (Fig. 4D). The enhanced enzyme activity of the light-treated group demonstrated the release of GOx after light irradiation. Thus, we concluded that nanocomplex formation and phototriggered protein release processes would not affect the enzyme activities. The nanocomplex structure could provide an excellent protection for protein delivery.
Cytosolic delivery of cytotoxic proteins
We then prepared GOx nanocomplexes as protein therapeutics. GOx could produce hydrogen peroxide with glucose to kill cancer cells.50 It is a strongly anionic protein at pH 7.4, which is not preferable for cellular uptake. HA-coating endowed the nanocomplexes with binding ability to A549 cells. Moreover, light irradiation significantly enhanced the cellular uptake of GOx, which was confirmed by confocal microscopy (Fig. 5A) and flow cytometry (Fig. 5B). PD0.4 did not show obvious toxicity at the working concentration (<20 μg mL−1, Fig. S12†) against A549 cells regardless of the light irradiation, while BH-PD/GOx showed a high anticancer effect after light irradiation (Fig. 5C), which may result from the enhanced internalization of GOx after light irradiation. These results together demonstrated that PD0.4 was a promising photocontrollable platform for cytosolic protein delivery without interfering with their activity.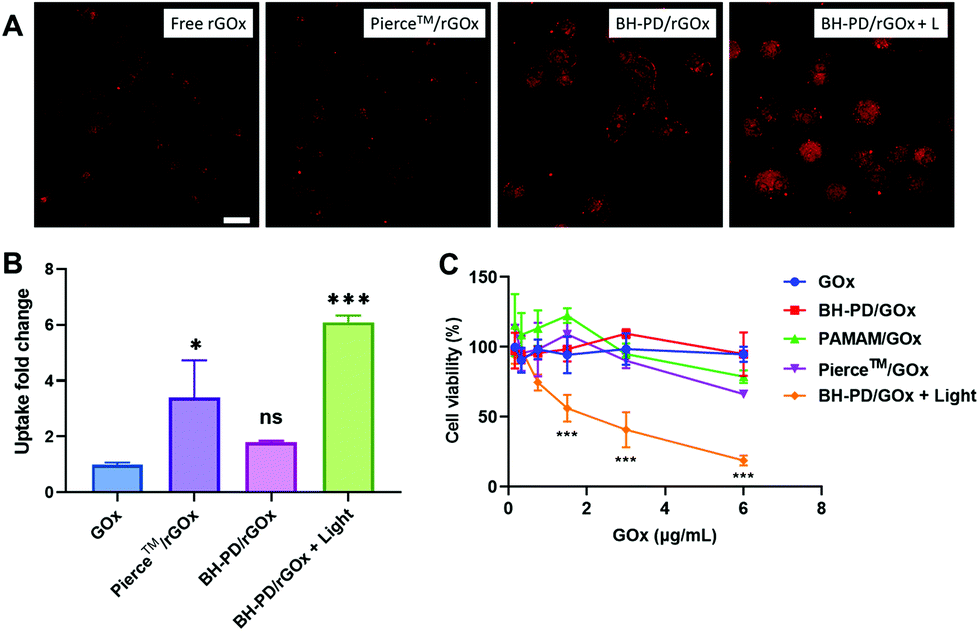 | ||
| Fig. 5 Cytosolic delivery of cytotoxic proteins. (A) Confocal images and (B) flow cytometry analysis of rGOx uptake. (C) Cell viability analysis of GOx treatment. Irradiation condition: 420 nm, 50 mW cm−2, 2 min. Data analysis: n = 3, ns: not significant, *p < 0.05, ***p < 0.001. | ||
Discussion
Many multifunctional stimuli-responsive protein delivery systems have been recently developed to achieve targeted protein delivery and controlled protein release. In these systems, protein modification, complicated organic synthesis, or the addition of extra chemical components are usually involved, which might bring new concerns on the production cost, biocompatibility, and long-term biosafety.51 Here we reported a simple yet efficient photoresponsive protein delivery system with high protein binding ability and delivery efficiency, reducing the need for complex material synthesis and protein modification.In this protein delivery system, the protein binding ability of the photocleavable molecule, DEACM, was reported and utilized for the first time. DEACM was proved to contribute to the photoresponsiveness, protein binding, and also the driving force for the self-assembly of the system. We used one moiety to achieve multiple functions, which reduced the complexity of the synthesis and the usage of additives. Meanwhile, PAMAM supported the easy modification with DEACM and electrostatic interactions with proteins, which provided the foundation for the high protein binding efficiency of this system.
To achieve targeted protein delivery, the high stability of the protein nanocomplexes during circulation and the controlled release should be considered. In our system, the coating of biocompatible HA and BSA achieved high stability of the nanocomplexes in the physiological solution, whereas many other reported co-assembled protein delivery systems did not exhibit stability under physiological conditions. The rigid structure and inactive surface facilitated the high serum tolerance of the system. Under spatiotemporally controlled light irradiation, protein release and enhanced cellular uptake were demonstrated in complete medium, which provided a high possibility for clinical applications. One limitation of this photoresponsive system is that 420 nm light has a short tissue penetration depth, which is limited to treating diseases in superficial tissues or the eye. In the next stage, we would explore longer wavelength light-responsive groups as protein binding moieties, to achieve deeper tissue penetration.
This protein delivery system was demonstrated to be appliable for various proteins like BSA and GOx, which are anionic proteins. Cationic proteins, like trypsin and lysozyme, could not form nanocomplexes with PD0.4 (data not shown). However, fusing with anionic tag-like green fluorescent protein52 or phenylboronic acid53 to provide the negative charge for co-assembly would be an option in future studies.
In summary, a simple and photoresponsive protein delivery system was developed with the help of a photocleavable group, DEACM, which provided photoresponsiveness, protein binding, and the driving force for the self-assembly of the system. The system not only delivered proteins, but also enhanced the cellular uptake of proteins and controlled protein release upon light irradiation. The high delivery efficiency, good serum tolerance, and photocontrollability of the system imply its potential in biomedical applications.
Author contributions
Y. L., Y. Z., and W. W. designed the study. Y. L. and Y. Z. synthesized and characterized the materials. Y. L. did the photolysis study, release study, confocal study, and paper writing. Y. Z. did the cytotoxicity study, data process, and revised the manuscript. K. L., Y. Z. (Yaming Zhang) and T. W. helped with the experiments and revised the manuscript. W. W. supervised the study and revised the manuscript.Conflicts of interest
A US provisional patent application was filed with no. 63/209078.Acknowledgements
We acknowledge the assistance of The University of Hong Kong Li Ka Shing Faculty of Medicine Faculty Core Facility. This work was supported by the National Natural Science Foundation of China (No. 81803469), Research Grants Council of Hong Kong (Early Career Scheme, No. 27115220), and Ming Wai Lau Centre for Reparative Medicine Associate Member Program.References
- M. Ray, Y.-W. Lee, F. Scaletti, R. Yu and V. M. Rotello, Nanomedicine, 2017, 12, 941–952 CrossRef CAS PubMed.
- X. Liu, F. Wu, Y. Ji and L. Yin, Bioconjugate Chem., 2019, 30, 305–324 CrossRef PubMed.
- S. Konermann, M. D. Brigham, A. E. Trevino, J. Joung, O. O. Abudayyeh, C. Barcena, P. D. Hsu, N. Habib, J. S. Gootenberg, H. Nishimasu, O. Nureki and F. Zhang, Nature, 2015, 517, 583–588 CrossRef CAS PubMed.
- T. Nochi, Y. Yuki, H. Takahashi, S. Sawada, M. Mejima, T. Kohda, N. Harada, I. G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS PubMed.
- R. Goswami, T. Jeon, H. Nagaraj, S. Zhai and V. M. Rotello, Trends Pharmacol. Sci., 2020, 41, 743–754 CrossRef CAS PubMed.
- L. Ren, J. Lv, H. Wang and Y. Cheng, Angew. Chem., Int. Ed., 2020, 59, 4711–4719 CrossRef CAS PubMed.
- K. Chen, S. Jiang, Y. Hong, Z. Li, Y.-L. Wu and C. Wu, Prog. Nat. Sci.: Mater. Int., 2019, 29, 617–627 CrossRef CAS.
- M. R. Villegas, A. Baeza and M. Vallet-Regi, Molecules, 2018, 23, 1008 CrossRef PubMed.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- F. M. Veronese, PEGylated protein drugs: basic science and clinical applications, Springer, 2009 Search PubMed.
- H. Murata, J. Futami, M. Kitazoe, T. Yonehara, H. Nakanishi, M. Kosaka, H. Tada, M. Sakaguchi, Y. Yagi, M. Seno, N. H. Huh and H. Yamada, J. Biochem., 2008, 144, 447–455 CrossRef CAS PubMed.
- Y. Jeong, B. Duncan, M. H. Park, C. Kim and V. M. Rotello, Chem. Commun., 2011, 47, 12077–12079 RSC.
- P. Yao, Y. Zhang, H. Meng, H. Sun and Z. Zhong, Biomacromolecules, 2019, 20, 184–191 CrossRef CAS PubMed.
- B. P. Koppolu, S. G. Smith, S. Ravindranathan, S. Jayanthi, T. K. S. Kumar and D. A. Zaharoff, Biomaterials, 2014, 35, 4382–4389 CrossRef CAS PubMed.
- M. Vila-Caballer, G. Codolo, F. Munari, A. Malfanti, M. Fassan, M. Rugge, A. Balasso, M. de Bernard and S. Salmaso, J. Controlled Release, 2016, 238, 31–42 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- H. Omar, J. G. Croissant, K. Alamoudi, S. Alsaiari, I. Alradwan, M. A. Majrashi, D. H. Anjum, P. Martins, R. Laamarti, J. Eppinger, B. Moosa, A. Almalik and N. M. Khashab, J. Controlled Release, 2017, 259, 187–194 CrossRef CAS PubMed.
- M. Yu, Z. Gu, T. Ottewell and C. Yu, J. Mater. Chem. B, 2017, 5, 3241–3252 RSC.
- X. Qin, C. Yu, J. Wei, L. Li, C. Zhang, Q. Wu, J. Liu, S. Q. Yao and W. Huang, Adv. Mater., 2019, 31, e1902791 CrossRef PubMed.
- J. Lv, Q. Fan, H. Wang and Y. Cheng, Biomaterials, 2019, 218, 119358 CrossRef CAS PubMed.
- M. Yu, J. Wu, J. Shi and O. C. Farokhzad, J. Controlled Release, 2016, 240, 24–37 CrossRef CAS PubMed.
- M. Dabkowska, K. Luczkowska, D. Roginska, A. Sobus, M. Wasilewska, Z. Ulanczyk and B. Machalinski, J. Nanobiotechnol., 2020, 18, 120 CrossRef CAS PubMed.
- J. Wu, N. Kamaly, J. Shi, L. Zhao, Z. Xiao, G. Hollett, R. John, S. Ray, X. Xu, X. Zhang, P. W. Kantoff and O. C. Farokhzad, Angew. Chem., Int. Ed., 2014, 53, 8975–8979 CrossRef CAS PubMed.
- K. Okuro, M. Sasaki and T. Aida, J. Am. Chem. Soc., 2016, 138, 5527–5530 CrossRef CAS PubMed.
- H. Chang, J. Lv, X. Gao, X. Wang, H. Wang, H. Chen, X. He, L. Li and Y. Cheng, Nano Lett., 2017, 17, 1678–1684 CrossRef CAS PubMed.
- C. Liu, T. Wan, H. Wang, S. Zhang, Y. Ping and Y. Cheng, Sci. Adv., 2019, 5, eaaw8922 CrossRef CAS PubMed.
- R. Tang, C. S. Kim, D. J. Solfiell, S. Rana, R. Mout, E. M. Velázquez-Delgado, A. Chompoosor, Y. Jeong, B. Yan and Z.-J. Zhu, ACS Nano, 2013, 7, 6667–6673 CrossRef CAS PubMed.
- R. Mout, M. Ray, G. Y. Tonga, Y. W. Lee, T. Tay, K. Sasaki and V. M. Rotello, ACS Nano, 2017, 11, 2452–2458 CrossRef CAS PubMed.
- Z. Zhang, W. Shen, J. Ling, Y. Yan, J. Hu and Y. Cheng, Nat. Commun., 2018, 9, 1377 CrossRef PubMed.
- B. Esteban-Fernandez de Avila, D. E. Ramirez-Herrera, S. Campuzano, P. Angsantikul, L. Zhang and J. Wang, ACS Nano, 2017, 11, 5367–5374 CrossRef CAS PubMed.
- H. He, Y. Chen, Y. Li, Z. Song, Y. Zhong, R. Zhu, J. Cheng and L. Yin, Adv. Funct. Mater., 2018, 28, 1706710 CrossRef.
- N. B. Hentzen, R. Mogaki, S. Otake, K. Okuro and T. Aida, J. Am. Chem. Soc., 2020, 142, 8080–8084 CrossRef CAS PubMed.
- T. Ohtsuki, S. Kanzaki, S. Nishimura, Y. Kunihiro, M. Sisido and K. Watanabe, Nat. Commun., 2016, 7, 12501 CrossRef CAS PubMed.
- Q. Liu, W. Wang, C. Zhan, T. Yang and D. S. Kohane, Nano Lett., 2016, 16, 4516–4520 CrossRef CAS PubMed.
- Y. Li, W. Lv, L. Wang, Y. Zhang, L. Yang, T. Wang, L. Zhu, Y. Wang and W. Wang, Nano Res., 2021, 14, 2630–2636 CrossRef CAS.
- C. Bao, L. Zhu, Q. Lin and H. Tian, Adv. Mater., 2015, 27, 1647–1662 CrossRef CAS PubMed.
- J. Lv, C. Wang, H. Li, Z. Li, Q. Fan, Y. Zhang, Y. Li, H. Wang and Y. Cheng, Nano Lett., 2020, 20, 8600–8607 CrossRef CAS PubMed.
- S. Paul, P. Roy, P. S. Sardar and A. Majhi, ACS Omega, 2019, 4, 7213–7230 CrossRef CAS.
- R. Mogaki, K. Okuro, R. Ueki, S. Sando and T. Aida, J. Am. Chem. Soc., 2019, 141, 8035–8040 CrossRef CAS PubMed.
- N. Akbay, D. Topkaya, Y. Ergün, S. Alp and E. Gök, J. Anal. Chem., 2010, 65, 382–387 CrossRef CAS.
- K. Sharma, P. Yadav, B. Sharma, M. Pandey and S. K. Awasthi, J. Mol. Recognit., 2020, 33, e2834 CrossRef CAS PubMed.
- T. Bayraktutan and Y. Onganer, Spectrochim. Acta, Part A, 2017, 171, 90–96 CrossRef CAS PubMed.
- W. Xu, F.-Q. Luo, Q.-S. Tong, J.-X. Li, W.-M. Miao, Y. Zhang, C.-F. Xu, J.-Z. Du and J. Wang, CCS Chem., 2021, 431–442, DOI:10.31635/ccschem.021.202000696.
- F. Barbero, L. Russo, M. Vitali, J. Piella, I. Salvo, M. L. Borrajo, M. Busquets-Fite, R. Grandori, N. G. Bastus, E. Casals and V. Puntes, Semin. Immunol., 2017, 34, 52–60 CrossRef CAS PubMed.
- C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
- W. Zhang, Q. Cheng, S. Guo, D. Lin, P. Huang, J. Liu, T. Wei, L. Deng, Z. Liang, X. J. Liang and A. Dong, Biomaterials, 2013, 34, 6495–6503 CrossRef CAS PubMed.
- R. Cai and C. Chen, Adv. Mater., 2019, 31, e1805740 CrossRef PubMed.
- L. Palanikumar, S. Al-Hosani, M. Kalmouni, V. P. Nguyen, L. Ali, R. Pasricha, F. N. Barrera and M. Magzoub, Commun. Biol., 2020, 3, 95 CrossRef CAS PubMed.
- W. Wang, Q. Liu, C. Zhan, A. Barhoumi, T. Yang, R. G. Wylie, P. A. Armstrong and D. S. Kohane, Nano Lett., 2015, 15, 6332–6338 CrossRef CAS PubMed.
- J. Li, Y. Li, Y. Wang, W. Ke, W. Chen, W. Wang and Z. Ge, Nano Lett., 2017, 17, 6983–6990 CrossRef CAS PubMed.
- C. Lei, X. R. Liu, Q. B. Chen, Y. Li, J. L. Zhou, L. Y. Zhou and T. Zou, J. Controlled Release, 2021, 331, 416–433 CrossRef CAS PubMed.
- S. Yang, Q. Tang, L. Chen, J. Chang, T. Jiang, J. Zhao, M. Wang and P. R. Chen, Angew. Chem., 2020, 59, 18087–18094 CrossRef CAS PubMed.
- M. Wang, S. Sun, C. I. Neufeld, B. Perez-Ramirez and Q. Xu, Angew. Chem., Int. Ed., 2014, 53, 13444–13448 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04430c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |