
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling plasmonic catalysis and nanocrystal growth through cyclic regeneration of NADH†
Ana
Sánchez-Iglesias
a,
Joscha
Kruse
 b,
Andrey
Chuvilin
cd and
Marek
Grzelczak
*b
b,
Andrey
Chuvilin
cd and
Marek
Grzelczak
*b
aCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), 20014 Donostia – San Sebastián, Spain
bCentro de Física de Materiales (CSIC-UPV/EHU) and Donostia International Physics Center, 20018 Donostia – San Sebastián, Spain. E-mail: marek.g@csic.es
cCIC nanoGUNE BRTA, 20018 Donostia – San Sebastián, Spain
dIkerbasque Basque Foundation for Science, 48013 Bilbao, Spain
First published on 11th August 2021
Abstract
In a typical colloidal synthesis, the molecules of the reducing agent are irreversibly oxidized during nanocrystal growth. Such a scenario is of questionable sustainability when confronted with naturally occurring processes in which reducing agent molecules are cyclically regenerated. Here we show that cofactor molecules once consumed in the nucleation and growth of metallic nanocrystals can be photoregenerated using metallic nanocrystals as photocatalysts and reused in the subsequent nucleation process. Cyclic regeneration of cofactor molecules opens up the possibilities for the sustainable synthesis of inorganic nanoparticles.
In nature, biochemical cycles (e.g., Krebs or futile cycles) are hidden driving force powering processes responsible for self-repair, growth or reproduction. In biocatalytic processes, cyclic reduction/oxidation of intermediate molecules (cofactors) is central in reaching high performance of technologically-relevant chemical transformations, either under batch1 or flow conditions.2 The concept of cycles has only recently attracted the attention of the research community dealing with nanosystems. Thus, theoretical works have shown that periodic light energy delivery can accelerate the rate of templated self-replication of nanoparticles.3,4 Experimental findings evidenced that the cyclic assembly/disassembly of superlattices increases catalytic reaction rates under confinement.5 Thus, switching from a continuous to cyclic variant may favor the sustainability of existing (photo)chemical processes involving inorganic nanosystems by, for example, adapting to the diurnal cycle.
In a light-driven synthesis of plasmonic nanoparticles, the pre-formed nanoparticles act as both catalysts and seeds for the reduction of a metal precursor.6–9 In such a process, photogenerated holes irreversibly oxidize the electron donor molecules (citrate10 or alcohols11), while electrons reduce the metal precursor (gold, silver or palladium) exclusively on the nanocrystal surface.9 As a result, the photocatalytic nanocrystals gain in size maintaining their total number, while the electron donor is irreversibly oxidized. We hypothesized that disentangling metal reduction and the photocatalytic process by using a recyclable reductor can lead to a scenario in which nanoparticles catalyze the formation of other nanoparticles of similar composition. Thus, the total number of nanoparticles can increase. Although light-induced growth of nanoparticles out of the surface of “mother” nanoparticles has been attained on a solid substrate12,13 or in a colloidal phase,14 the processes described suffer from low efficiency because they were carried out with a continuous supply of light energy. Here we propose a cyclic variant of plasmonic catalysis, in which nanocrystals catalyze the photoreduction of intermediate biomolecules that serve as reductors in the nucleation and growth of a new set of nanoparticles under dark conditions (Fig. 1).
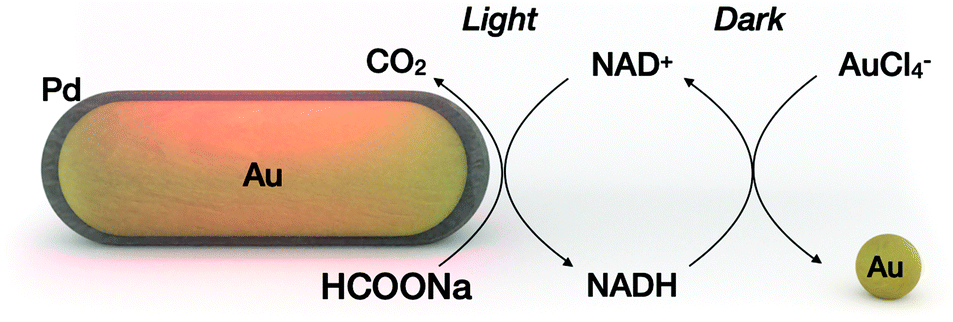 | ||
| Fig. 1 Cyclic photoregeneration of cofactor molecules allows plasmonic catalysis to be coupled to the nucleation and growth of metallic nanoparticles. Under the light conditions, the Au/Pd nanoparticle photocatalyst dehydrogenates sodium formate to NADH and produces CO2 as a byproduct. Under the dark conditions, NADH reduces the metal precursor to gold nanoparticles. The light and dark processes can be performed cyclically. | ||
The evaluation of our central hypothesis should take into account the following constraints: (i) electron shuttle molecules are reversibly reduced by the metal photocatalyst and oxidized by the metal precursor. (ii) The reduced form of the electron shuttle drives the nucleation of new nanoparticles in the dark, whereas other molecules in the medium (electron donors, stabilizer molecules, and byproducts) remain chemically inert. (iii) The molecules involved in the dark reaction (e.g., stabilizer) remain inert in the light reaction. (iv) Dark and light reactions require spatial separation to minimize back reactions (oxidation of the cofactor) and possible poisoning of the photocatalyst surface.
Nicotinamide adenine dinucleotide (NADH), which can be photoregenerated from NAD+ on metallic nanocrystals, was chosen as an electron shuttle.15–19 The standard redox potentials for the NAD+/NADH and AuCl4−/Au pairs are −0.32 V and 1.002 V, respectively, suggesting spontaneous gold reduction by NADH.20,21 In addition, reduction/oxidation of NADH can be easily monitored optically, making it a suitable model electron shuttle. Sodium formate was chosen as an electron donor in the light process. Other groups have shown that sodium formate is a suitable electron donor for molecular hydrogen production on Pd-coated gold nanoparticles.22,23 Our group has shown that dehydrogenation of sodium formate on Pd-coated gold nanoparticles can be coupled with the regeneration of NADH.18 On the other hand, metallic gold (here, the product of the dark reaction) is a poor catalyst for dehydrogenation of sodium formate,24 ensuring dehydrogenation of sodium formate in the light process only (constraints ii and iii). To physically separate the nanoparticle photocatalyst from the nanoparticle product, we chose to perform the light reaction under flow conditions, and the dark reaction in batch.
The entire process is described as follows: Pd-coated gold nanorods catalyze the photoregeneration of cofactor molecules (NAD+ to NADH) using sodium formate as the electron donor and producing CO2 as a byproduct. In the subsequent dark reaction, the NADH molecules promote the nucleation of the metal precursor, forming gold nanocrystals. The oxidized redox mediator is recycled in the subsequent light process by physically separating the nanoparticle product from the mixture containing the oxidized cofactor (NAD+). Throughout the process, light energy, formate ions and the metal precursor are the main ingredients for the synthesis of metallic nanoparticles using other metallic nanoparticles as photocatalysts.
To demonstrate that the nucleation and growth of gold nanoparticles are driven by NADH (constraint ii), we performed time-resolved UV-vis-NIR measurements of the dark reaction by varying the NADH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NAD+ molar ratio ([NAD] = 1 mM, commercially available) in the presence of sodium formate (1 M) at pH 8. No metallic gold was observed at 100% of NAD+, while at 100% of NADH the reduction was completed, forming nanocrystals of 2 nm diameter (Fig. 2a–c). Kinetic studies showed that the increase of the localized surface plasmon resonance (LSPR) band at 510 nm is accompanied by the decrease of the absorbance band at 340 nm, suggesting again that NADH is the driving force in the dark reaction (Fig. S1†). We found that NAD(H) interacts with the newly formed nanoparticles. The Z-potential of gold nanoparticles was −7 mV (Fig. S2†). XPS analysis revealed the presence of N 1s and P 2p (Fig. 2d and e). These measurements suggest that NAD interacts with the gold surface through the nicotinamide moiety, corroborating previous results.25 The NAD–Au interaction is further confirmed by photoluminescence of nanoparticles26 through the emission band at 400 nm (Fig. 2f). It should be noted that NAD(H) retains its chemical integrity during the dark reaction, as demonstrated by NMR analysis of the oxidized and reduced cofactor in the presence of the metallic precursor (Fig. S3†).
:NAD+ molar ratio ([NAD] = 1 mM, commercially available) in the presence of sodium formate (1 M) at pH 8. No metallic gold was observed at 100% of NAD+, while at 100% of NADH the reduction was completed, forming nanocrystals of 2 nm diameter (Fig. 2a–c). Kinetic studies showed that the increase of the localized surface plasmon resonance (LSPR) band at 510 nm is accompanied by the decrease of the absorbance band at 340 nm, suggesting again that NADH is the driving force in the dark reaction (Fig. S1†). We found that NAD(H) interacts with the newly formed nanoparticles. The Z-potential of gold nanoparticles was −7 mV (Fig. S2†). XPS analysis revealed the presence of N 1s and P 2p (Fig. 2d and e). These measurements suggest that NAD interacts with the gold surface through the nicotinamide moiety, corroborating previous results.25 The NAD–Au interaction is further confirmed by photoluminescence of nanoparticles26 through the emission band at 400 nm (Fig. 2f). It should be noted that NAD(H) retains its chemical integrity during the dark reaction, as demonstrated by NMR analysis of the oxidized and reduced cofactor in the presence of the metallic precursor (Fig. S3†).
 | ||
| Fig. 2 Dark reaction. (a) Effect of the NADH:NAD+ molar ratio on the nucleation of gold nanoparticles. Inset in (a): Digital images of nanoparticle solutions obtained at molar ratios ranging from 0 to 100%. (b) Time-dependent spectral change in the solutions at different molar ratios. (c) Size distribution and TEM image of the nanoparticles obtained at 100% NADH. (d and e) XPS analysis showing the presence of nitrogen and phosphorus compounds in the newly formed nanoparticles. (f) Extinction and emission spectra of gold nanoparticles (excitation wavelength = 350 nm). | ||
The use of conventional stabilizers – surfactants and polymers27 – was ruled out because they interfered with the light and dark reactions (vide infra). For example, cationic surfactants (e.g., cetyltrimethylammonium bromide (CTAB)) suppressed the photocatalytic activity of AuPd nanorods through their bromide content (Fig. S4†). In contrast, polyvinylpyrrolidone molecules induced non-selective nanoparticle nucleation even without NADH (Fig. S5†). In order to minimize the interaction of NAD with the gold surface (and address constraint iii), we used sodium triphosphate (STTP), a chemically resistant compound (a food additive) that has been shown to have stabilizing properties on gold nanoparticles.28
We used palladium-coated gold nanorods (length: 45.5 ± 5.0 nm, width: 11.9 ± 1.4 nm) as plasmonic photocatalysts.29–31 Upon Pd reduction on the surface of the gold nanorods, the maximum of LSPR redshifted from 735 to 763 nm (Fig. 3a), indicating the formation of a metallic shell, which was confirmed by HRTEM analysis (Fig. 3e and S6†).
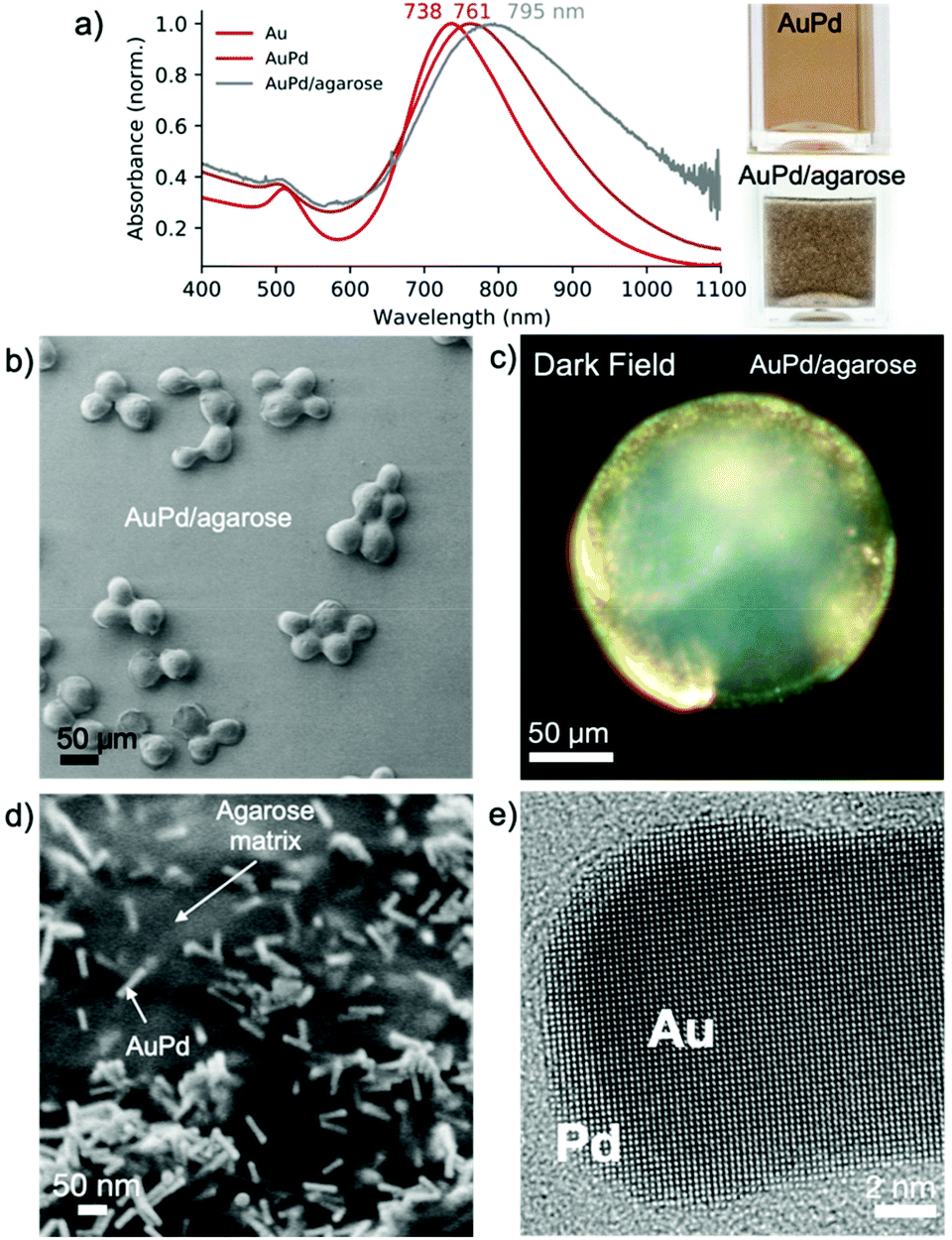 | ||
| Fig. 3 Plasmonic photocatalysts. (a) UV-vis-NIR spectra of bare gold nanorods, Pd-coated gold nanorods and Pd-coated gold nanorods after immobilization in agarose beads. (b) Low-resolution SEM image of AuPd/agarose. (c) Dark field image of individual beads (AuPd/agarose) with visible bright spots confirming the presence of plasmonic nanoparticles. (d) SEM image of a dried agarose bead showing randomly distributed AuPd nanorods. (e) HRTEM image of an AuPd nanorod with the visible palladium shell. | ||
To gain processability of the photocatalyst without compromising the optical properties, we immobilized AuPd nanorods on agarose beads (∼100 μm).32 The chemically rich agarose matrix interacts with the Au/Pd nanoparticles displacing the CTAB bilayer, without changing the optical properties of the nanoparticles even after thorough ethanol washing (Fig. 3a). After immobilization, the LSPR shifted by ∼25 nm due to an increase in the refractive index of the medium, as the refractive index of agarose is 1.34.33 Thermogravimetric analysis showed that the composite contains <1 wt% of photoactive metallic nanoparticles (Fig. S7†). Analysis of individual beads by darkfield microscopy revealed the presence of bright spots, the result of light scattering on the metallic element, which was not observed when using bare beads (Fig. 3c and S8†). SEM analysis showed that the AuPd nanorods are randomly distributed on the agarose beads (Fig. 3d). The small interparticle distance, as shown in Fig. 3d, is due to the shrinkage of highly porous agarose beads under vacuum. A combination of focused ion beam and SEM analysis allowed the visualization of the distribution of the AuPd nanoparticles in the beads. The nanoparticles were found to distribute preferentially close to the surface of the agarose beads (Fig. S9†).
To assess the simultaneous formation of NADH and CO2 under light conditions, a photocatalyst placed at the bottom of the vial was irradiated for two hours (200 mW cm−2, 400–1100 nm) (Fig. 4a, inset). The evolution of NADH was monitored in the liquid phase via UV-vis spectroscopy by measuring the absorbance at 340 nm. The formation of CO2 was tracked in the air phase through GC-MS (Fig. 4a). These measurements confirmed the simultaneous regeneration of NADH and dehydrogenation of formic acid, confirming our previous results.18 To further demonstrate the catalytic role of plasmonic nanoparticles, we determined an action spectrum (7 wavelengths at 5 mW cm−2 for 2 h of irradiation) (Fig. 4b), showing that NADH regeneration follows the AuPd/agarose spectral signature. Since dehydrogenation of formic acid is temperature dependent, we speculate that the plasmonic core favors a photothermal effect, which can be confirmed by the regeneration of NADH (2%) on bare gold nanorods (Fig. S13†).
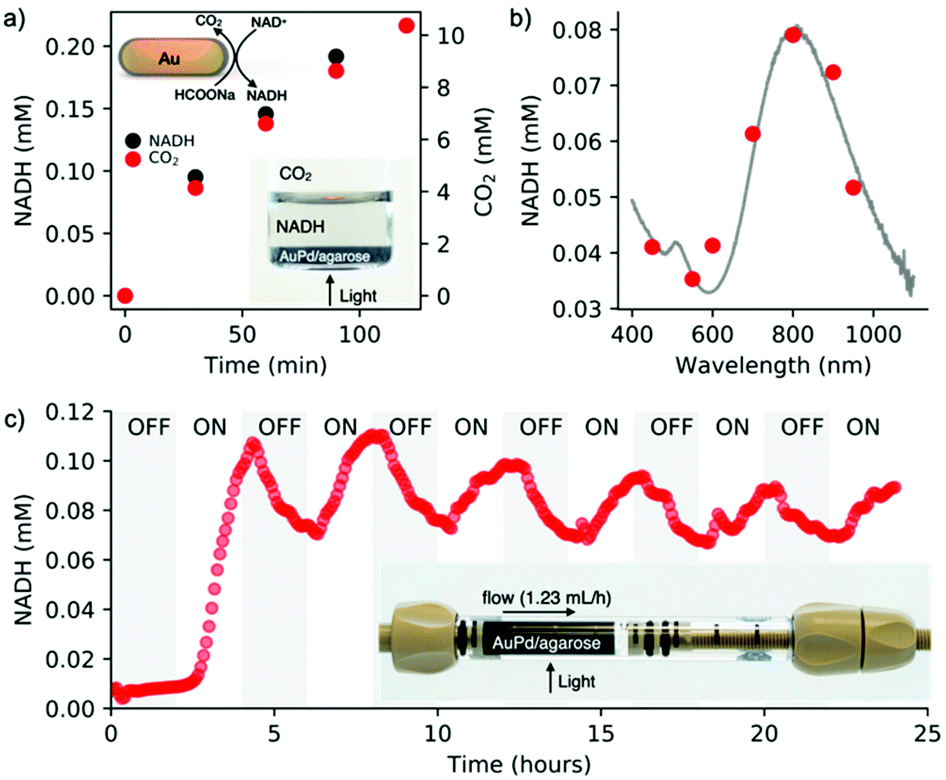 | ||
| Fig. 4 Light reaction. (a) Time-dependent simultaneous evolution of NADH and CO2 during light irradiation. Lower inset: Digital image of a batch photoreactor containing AuPd/agarose in the bottom. (b) Wavelength-dependent regeneration of NADH. (c) Photoregeneration of NADH under flow conditions, showing the change in the NADH concentration at the outlet of the photoreactor under ON–OFF light conditions. Inset: Digital image of a flow photoreactor during operation. | ||
A facile processing of the AuPd/agarose composite allows us to adapt batch photocatalysis to flow conditions. The flow photoreactor consisted of a column coupled at the outlet to an on-line UV-vis spectrophotometer to monitor the amount of regenerated NADH in real time (Scheme S1†). Fig. 4c shows the result of the light reaction run for 24 hours with alternating ON–OFF light states lasting 2 hours each. In the ON state, regeneration of NADH reached ∼10%. The slight decrease in the NADH concentration at longer times was presumably due to catalyst deactivation by evolved CO2. The degradation of photoactive components (AuPd) inside the agarose matrix was excluded, which was confirmed by XPS analyses (Fig. S10†). Note that washing the beads out of the column once the reaction was completed allowed for the recovery of the initial performance of the light process (Fig. S11†). Formic acid dehydrogenation and NADH reduction occur simultaneously on the photocatalyst surface. No NADH regeneration was observed when NAD+ was supplied to the outlet of the photocatalyst column (Fig. S12†).
To evaluate our main hypothesis, we ran three complete cycles of the light/dark reactions by supplying a mixture containing NAD+ (0.001 M), sodium formate (1 M), and STTP (0.05 M) at pH 8 in the photoreactor (Fig. 5a). To increase the efficiency of NADH regeneration, we doubled the light intensity (300 mW cm−2), obtaining ∼55% of the regenerated cofactor (Fig. 5d). The collected mixture was combined with the gold salt and allowed to reduce the gold completely (Fig. 5c). The nanoparticles were separated from the mixture by centrifugal filtration and the collected permeate with the remaining NAD+/NADH was delivered back to the flow photoreactor (Fig. 5b). We observed a decrease in the amount of regenerated NADH with each subsequent light cycle, which is probably due to the interaction of the cofactor with the gold nanoparticles during the dark process or the interaction with the agarose matrix (Fig. 5f, red). We are also aware of (NAD)2 formation,34 as indicated by the remaining band at 340 nm after separation of nanoparticles from the growth solution (Fig. 5b). Consistently, the concentration of metallic gold also decreased with each cycle as the NADH:AuIII molar ratio of 4 remained constant in each run. The diameter of the nanoparticles progressively decreased from 2.5 nm (first cycle) to 1.8 nm (third cycle) (Fig. 5e and S14†). Since the metal nanoparticles catalyze the formation of other metal nanoparticles by recycling the reducing agent, we estimated the turnover number as the ratio of the number of spherical nanoparticle products to the number of AuPd nanorod photocatalysts with each cycle. Given that the photocatalytic bed contained 6 × 1012 nanorods and that after each cycle, the solution contained 6 × 1014, 8 × 1014, and 7 × 1014 spherical gold nanoparticles, we estimated that one nanorod catalyzes the nucleation of 100, 133, and 117 spherical particles in each successive cycle (Fig. 5f, black; ESI – section 12†). These data show that the cyclic recovery of the cofactor allows plasmonic catalysis to be coupled to nucleation and nanoparticle growth, thus satisfying condition I.
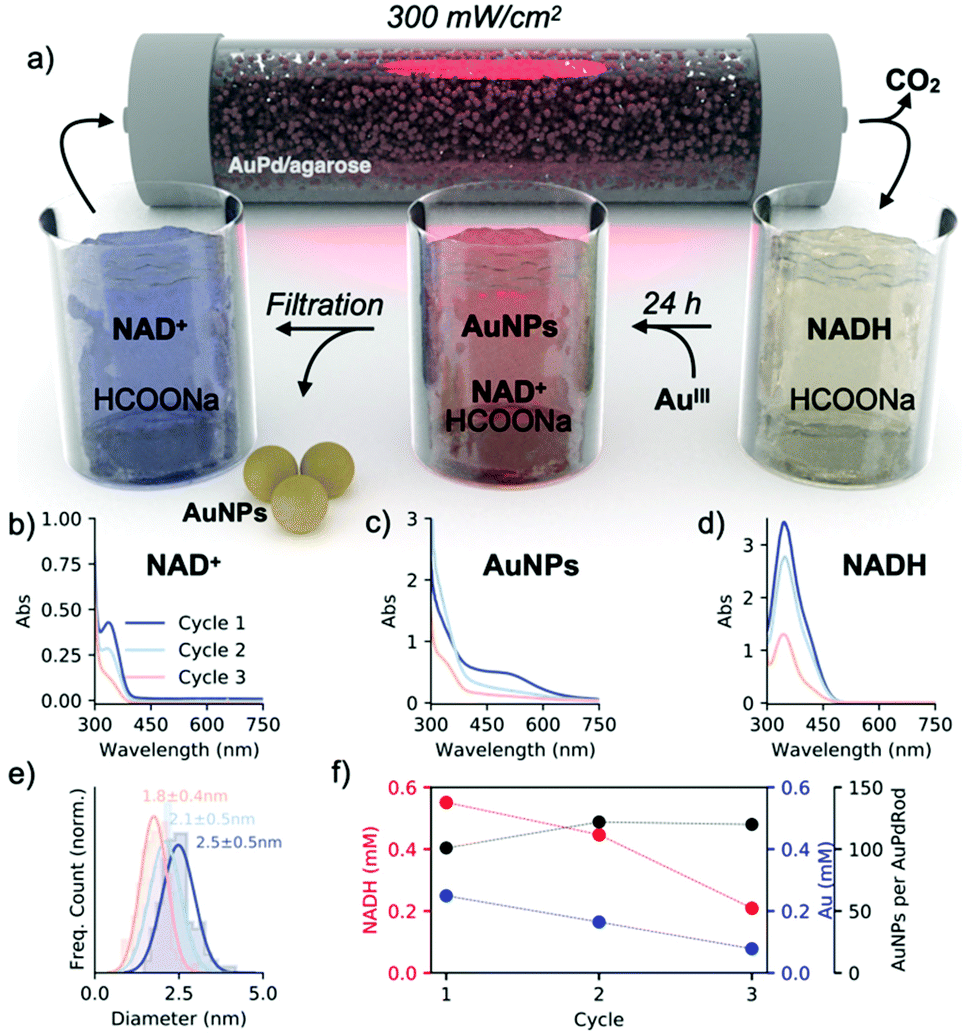 | ||
| Fig. 5 Coupling light and dark reactions through cyclic regeneration of cofactor molecules. (a) Physical and temporal separation of the light reaction in flow and the dark reaction in batch. (b–d) UV-vis spectra of (d) the collected mixtures after the light reaction, (c) the mixture after the dark reaction containing nanoparticles, and (b) permeate solutions without nanoparticles before the subsequent light reaction. (e) Size distributions of the nanoparticles after each dark process. (f) Concentration of NADH (red) and Au (blue) and ratio of new nanoparticles per AuPd nanorod (black) in each cycle. Despite the drop in the NADH concentration, the number of new nanoparticles per photocatalyst nanoparticle remains unchanged. | ||
Conclusions
We provided a proof-of-concept experimental framework showing that cyclic photoregeneration of cofactor molecules can link the process of plasmonic catalysis (light reaction) with the nucleation and growth of metallic nanocrystals (dark reaction). The prime difficulty of coupling the light and dark processes is to minimize the interference of a given component (stabilizer or electron donor) in the antagonistic reaction. Another difficulty is the creation of a positive feedback loop in which the dark reaction product would catalyze the subsequent light reaction, a property that is still missing in the present experimental framework. Note that the presence of the Pd co-catalyst on the gold nanorods was critical for the regeneration of NAD+ in the light reaction, making the new set of Au nanoparticles obtained in the dark reaction unsuitable for the photoregeneration of NADH, that is, metallic gold is unable to catalyze the dehydrogenation of sodium formate. However, replacing the palladium co-catalyst with (macro)molecular co-catalysts may overcome the issue related to the compositional similarity of mother and daughter nanocrystals and thus the conservation of photocatalytic activities over subsequent generations.From a broader perspective, these results raise new promises on sustainable colloidal nanofabrication where nanoparticles by harvesting light energy catalyze the regeneration of their own reducing agent. Thus, future prospects envisage (i) the coupling of the light and dark processes under batch conditions and (ii) the use of water as an electron donor to move closer to the creation of an artificial photosynthetic system.
Author contributions
A. S. I. and J. K. – data curation, investigation, methodology, visualization, and writing – review and editing; A. C. – investigation, funding acquisition, resources, and writing – review and editing; M. G. – conceptualization, funding acquisition, project administration, resources, methodology, validation, supervision, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish MINECO (PID2019-111772RB-I00) and the BBVA Foundation – “Primera convocatoria de ayudas fundacion BBVA a investigadores, innovadores y creadores culturales”. A. C. acknowledges support from the Spanish MINECO under the Maria de Maeztu Units of Excellence Program (MDM-2016-0618). The authors thank Jordi Llop for the help with the measurements of CO2 evolution, Daniel Padro for the help with the NMR analysis and Luis Yate for the assistance with the XPS analysis.References
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- R. Zhang, D. A. Walker, B. A. Grzybowski and M. Olvera de la Cruz, Angew. Chem., Int. Ed., 2014, 53, 173–177 CrossRef CAS.
- F. Lugli and F. Zerbetto, J. Phys. Chem. C, 2019, 123, 825–835 CrossRef CAS.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS.
- A. Henglein and R. Tausch-Treml, J. Colloid Interface Sci., 1981, 80, 84–93 CrossRef CAS.
- R. Jin, Y. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901–1903 CrossRef CAS.
- R. Jin, Y. Charles Cao, E. Hao, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS PubMed.
- M. Grzelczak and L. M. Liz-Marzán, Chem. Soc. Rev., 2014, 43, 2089–2097 RSC.
- X. Wu, E. S. Thrall, H. Liu, M. Steigerwald and L. Brus, J. Phys. Chem. C, 2010, 114, 12896–12899 CrossRef CAS.
- Y. Zhai, J. S. DuChene, Y.-C. Wang, J. Qiu, A. C. Johnston-Peck, B. You, W. Guo, B. DiCiaccio, K. Qian, E. W. Zhao, F. Ooi, D. Hu, D. Su, E. A. Stach, Z. Zhu and W. D. Wei, Nat. Mater., 2016, 15, 889–895 CrossRef CAS PubMed.
- S. J. Lee, B. D. Piorek, C. D. Meinhart and M. Moskovits, Nano Lett., 2010, 10, 1329–1334 CrossRef CAS PubMed.
- E. Kazuma and T. Tatsuma, J. Phys. Chem. C, 2013, 117, 2435–2441 CrossRef CAS.
- Y. Wei, S. Han, D. A. Walker, S. C. Warren and B. A. Grzybowski, Chem. Sci., 2012, 3, 1090–1094 RSC.
- A. Sánchez-Iglesias, A. Chuvilin and M. Grzelczak, Chem. Commun., 2015, 51, 5330–5333 RSC.
- A. Sánchez-Iglesias, J. Barroso, D. Martinez-Solis, J. M. Taboada, F. Obelleiro-Basteiro, V. Pavlov, A. Chuvilin and M. Grzelczak, J. Mater. Chem. A, 2016, 4, 7045–7052 RSC.
- K. Kinastowska, J. Liu, J. M. Tobin, Y. Rakovich, F. Vilela, Z. Xu, W. Bartkowiak and M. Grzelczak, Appl. Catal., B, 2019, 243, 686–692 CrossRef CAS.
- N. Tarnowicz-Staniak, S. Vázquez-Díaz, V. Pavlov, K. Matczyszyn and M. Grzelczak, ACS Appl. Mater. Interfaces, 2020, 12, 19377–19383 CrossRef CAS PubMed.
- S. Roy, V. Jain, R. K. Kashyap, A. Rao and P. P. Pillai, ACS Catal., 2020, 10, 5522–5528 CrossRef CAS.
- Y. Xiao, V. Pavlov, S. Levine, T. Niazov, G. Markovitch and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 4519–4522 CrossRef CAS PubMed.
- L. Zhang, Y. Li, D.-W. Li, C. Jing, X. Chen, M. Lv, Q. Huang, Y.-T. Long and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 6789–6792 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 137, 948–957 CrossRef PubMed.
- B. Wu, J. Lee, S. Mubeen, Y.-S. Jun, G. D. Stucky and M. Moskovits, Adv. Opt. Mater., 2016, 4, 1041–1046 CrossRef CAS.
- X. Wang, Q. Meng, L. Gao, Z. Jin, J. Ge, C. Liu and W. Xing, Int. J. Hydrogen Energy, 2018, 43, 7055–7071 CrossRef CAS.
- A. Damian and S. Omanovic, Langmuir, 2007, 23, 3162–3171 CrossRef CAS PubMed.
- A. Bonanno, I. Pérez-Herráez, E. Zaballos-García and J. Pérez-Prieto, Chem. Commun., 2020, 56, 587–590 RSC.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed.
- J. Xin, L. Miao, S. Chen and A. Wu, Anal. Methods, 2012, 4, 1259–1264 RSC.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- M. Grzelczak, J. Pérez-Juste, B. Rodríguez-González and L. M. Liz-Marzán, J. Mater. Chem., 2006, 16, 3946–3951 RSC.
- X. Ma, Y. Xia, L. Ni, L. Song and Z. Wang, Spectrochim. Acta, Part A, 2014, 121, 657–661 CrossRef CAS.
- E. Fujiwara, T. D. Cabral, M. Sato, H. Oku and C. M. B. Cordeiro, Sci. Rep., 2020, 10, 7035 CrossRef CAS.
- T. Saba, J. Li, J. W. H. Burnett, R. F. Howe, P. N. Kechagiopoulos and X. Wang, ACS Catal., 2021, 11, 283–289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr04400a |
| This journal is © The Royal Society of Chemistry 2021 |