
Facile synthesis of lactoferrin conjugated ultra small large pore silica nanoparticles for the treatment of glioblastoma†
Taskeen Iqbal
Janjua
 a,
Aria
Ahmed-Cox
bcd,
Anand Kumar
Meka
a,
Friederike M.
Mansfeld
bcde,
Helen
Forgham
bcd,
Rosa Mistica C.
Ignacio
bcd,
Yuxue
Cao
a,
Joshua A.
McCarroll
bcd,
Roberta
Mazzieri
*fgh,
Maria
Kavallaris
*bcd and
Amirali
Popat
*ai
a,
Aria
Ahmed-Cox
bcd,
Anand Kumar
Meka
a,
Friederike M.
Mansfeld
bcde,
Helen
Forgham
bcd,
Rosa Mistica C.
Ignacio
bcd,
Yuxue
Cao
a,
Joshua A.
McCarroll
bcd,
Roberta
Mazzieri
*fgh,
Maria
Kavallaris
*bcd and
Amirali
Popat
*ai
aSchool of Pharmacy, The University of Queensland, Brisbane, QLD 4102, Australia. E-mail: a.popat@uq.edu.au
bChildren's Cancer Institute, Lowy Cancer Research Centre, UNSW Sydney, Sydney, NSW 2031, Australia. E-mail: m.kavallaris@ccia.unsw.edu.au
cSchool of Women's and Children's Health, Faculty of Medicine and Health, UNSW Sydney, Sydney, NSW 2052, Australia
dARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Australian Centre for Nanomedicine, UNSW Sydney, Sydney, NSW 2052, Australia
eARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash Institute of Pharmaceutical Sciences, Monash University, Melbourne, VIC 3052, Australia
fDiamantina Institute, Translational Research Institute, The University of Queensland Brisbane QLD, 4102, Australia. E-mail: Roberta.Mazzieri@petermac.org
gPeter MacCallum Cancer Centre, Melbourne, VIC 3000, Australia
hSir Peter MacCallum Department of Oncology, The University of Melbourne, Melbourne, VIC 3010, Australia
iMater Research Institute – The University of Queensland, Translational Research Institute, Woolloongabba QLD 4102, Australia
First published on 6th September 2021
Abstract
The blood brain barrier (BBB) and blood tumour barrier (BTB) remain a major roadblock for delivering therapies to treat brain cancer. Amongst brain cancers, glioblastoma (GBM) is notoriously difficult to treat due to the challenge of delivering chemotherapeutic drugs across the BBB and into the tumour microenvironment. Consequently, GBM has high rates of tumour recurrence. Currently, limited numbers of chemotherapies are available that can cross the BBB to treat GBM. Nanomedicine is an attractive solution for treating GBM as it can augment drug penetration across the BBB and into the heterogeneous tumour site. However, very few nanomedicines exist that can easily overcome both the BBB and BTB owing to difficulty in synthesizing nanoparticles that meet the small size and surface functionality restrictions. In this study, we have developed for the first-time, a room temperature protocol to synthesise ultra-small size with large pore silica nanoparticles (USLP, size ∼30 nm, pore size >7 nm) with the ability to load high concentrations of chemotherapeutic drugs and conjugate a targeting moiety to their surface. The nanoparticles were conjugated with lactoferrin (>80 kDa), whose receptors are overexpressed by both the BBB and GBM, to achieve additional active targeting. Lactoferrin conjugated USLP (USLP-Lf) were loaded with doxorubicin – a chemotherapy agent that is known to be highly effective against GBM in vitro but cannot permeate the BBB. USLP-Lf were able to selectively permeate the BBB in vitro, and were effectively taken up by glioblastoma U87 cells. When compared to the uncoated USLP-NPs, the coating with lactoferrin significantly improved penetration of USLP into U87 tumour spheroids (after 12 hours at 100 μm distance, RFU value 19.58 vs. 49.16 respectively). Moreover, this USLP-Lf based delivery platform improved the efficacy of doxorubicin-mediated apoptosis of GBM cells in both 2D and 3D models. Collectively, our new nano-platform has the potential to overcome both the BBB and BTB to treat GBM more effectively.
 Amirali Popat | Dr Amirali Popat is an Associate Professor and Director of Research at the School of Pharmacy, The University of Queensland, Australia. He obtained his PhD in Nanomedicine from Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Australia in 2012. His group's research focuses on development of stimuli responsive nanomaterials to overcome multiple biological barriers for precision medicine, in particular development of smart drug delivery systems for the treatment of IBD, Diabetes and Cancer. Currently (2020–2021) he serves as a president of Australian Controlled Release Society (AusCRS) and editorial board member of many prestigious journal such as Journal of Controlled Release, Pharmaceutics, DDTR and early career advisory board member of Nanoscale Horizon |
Introduction
Drug delivery to brain tumours is a challenging task and a major obstacle to cure. The presence of biological barriers such as the blood brain barrier (BBB) and brain tumour barrier (BTB) limit the penetration of most chemotherapeutic drugs.1,2 In addition, the complexity of brain tumours and lack of efficient drug delivery techniques to the brain has had significant ramifications on patient survival. Among the different types of brain tumours, glioblastoma (GBM) is a very invasive, aggressive, and malignant tumour. The survival rate with existing therapy is poor, with less than 5% of patients surviving more than five years post diagnosis.3 Moreover, the median time to develop tumour recurrence for GBM is seven months post-diagnosis and typically, these patients survive only up to 14 months.4,5 Challenges for treating GBM stem from the high levels of local immunosuppression which limit immune response against the tumour, the significant inter- and intra-patient tumour heterogeneity that promotes niche populations of treatment resistant cancer cells, and above all, the presence of the BBB and BTB which impede drug delivery to GBM.2,6–8 Therefore, there is an urgent need to improve GBM prognosis with the development of advanced and effective drug delivery strategies.Within the last two decades, many nanotechnology-based drug delivery platforms have been developed and some are already being used in the clinic to treat various cancers.9,10 Examples of these nanomedicine include liposomal formulation of doxorubicin (100 nm Doxil ® or 180 nm Myocet®) used to treat sarcoma or breast cancer.2 Inorganic nanoparticles such as hafninum oxide nanoparticles (50 nm NBTXR3) have also gained approval as radio enhancers for soft tissue sarcomas.2,11 Nanotherm – an iron oxide nanoparticle (15 nm) has been approved by European Medicines Agency for magnetic hyperthermic treatment of GBM.12 To the best of our knowledge, no nanoparticle based formulations have so far been approved for delivery of chemotherapy drugs to treat brain tumours. The lack of effective nanoparticles which meet the size and functionality required to permeate the BBB and BTB have slowed clinical progression in this space. Ideally, smaller nanoparticles are more likely to permeate across the BBB, especially if the size is below the 40 nm size threshold as demonstrated by numerous studies.13–16 Moreover, active targeting across the BBB and into the tumour is an important factor that needs to be considered when developing nano-based therapy for cancers of the brain such as GBM. Active targeting of nanomedicines to specific cell surface proteins can enhance uptake by the BBB and tumour cells in order to limit drugs concentrating in non-cancerous tissues. Transporters for proteins such as transferrin, lactoferrin or albumin are abundantly expressed on the surface of the BBB and can therefore be utilised for efficient and active delivery of nanomedicines across the BBB and to the tumour.2,6,16,17 Among many different targeting agents, lactoferrin is the most attractive for GBM.2 Firstly, it has been reported that lactoferrin receptors are highly expressed by both the BBB and GBM cells which makes it an ideal cascade targeting ligand.18 Secondly, lactoferrin based targeting has less endogenous competition owing to low levels of circulating lactoferrin in the blood stream.19,20 Lastly, lactoferrin unidirectional transport from the blood to brain reduces the possibility of a lactoferrin conjugated compounds effluxing back into the blood circulation, thus minimising off-target effects.18
Doxorubicin (DOX) is an anthracycline that has been shown to be a highly effective chemotherapy drug which is currently used in clinic for the treatment of numerous cancers such as bladder carcinoma, breast cancer, lymphomas, sarcomas and neuroblastoma.21In vitro studies suggest that DOX can efficiently kill GBM cells by reducing the activity of topoisomerase II and thereby prevent DNA synthesis and cancer cell replication.14 Moreover, intracranial delivery of DOX directly into the tumour in various animals models such as mice15,22,23 and dogs24 have shown significant reduction in GBM tumour volume and improvement in clinical symptoms. However, in the clinical setting, DOX is not delivered intracranially for GBM as it is a invasive and risky procedure. Owing to the inability of DOX to permeate the BBB and reach therapeutic doses at the tumour site, it must be delivered at high doses systemically with known systemic side-effects, particularly cardiotoxicity.25,26 Therefore, drug delivery systems that can increase DOX intracerebral concentrations is expected to not only show efficacy against GBM but also minimise off-target, adverse effects.
In recent years, mesoporous silica nanoparticles (MSN) have gained significant traction owing to their excellent properties such as good biocompatibility, large surface area with capacity for high drug loading, versatility to control porosity and size, and ability to attach targeting moieties to specifically target cancerous tissues.27–29 Recent studies have showed that nanoparticles less than 50 nm are more efficient for drug delivery to the brain.13,14,30 For instance, Mo et al. developed 20, 40 and 80 nm MSN and demonstrated that smaller (20 and 40 nm) MSN were more efficient in delivering DOX to cancer cells across in vitro BBB models.14 However, these MSN had small pore sizes (2 nm) and therefore had comparatively low drug loading capacity (20 nm particles had 15 μg DOX per 1 mg MSN).14
Increased porosity of MSN would increase drug and macromolecule loading onto the nanoparticle carrier.31 So far, synthesis of large pore MSNs has been limited to 50 nm with complex synthesis conditions requiring high temperatures which can increase the cost of synthesis.32–34 Herein, we report facile synthesis of ultra-small, large pore silica nanoparticles (USLP) at room temperature using a biphasic sol–gel method. The USLP have small size (30 nm) and large pore size (>7 nm) which can not only load DOX but also encapsulate large proteins such as lactoferrin (80 kDa) as a targeting moiety to efficiently cross the BBB and penetrate GBM cells. This USLP based delivery system was tested for its ability to efficiently penetrate the BBB in vitro and into GBM cells using 2D and 3D tumour spheroid models. DOX loaded USLP systems were also compared with free DOX in their efficacy against GBM in vitro in 2D cell culture as well as 3D U87 spheroid models.
Experimental
Materials
Tetraethyl orthosilicate (TEOS, 99%), cetyltrimethyl ammonium chloride (CTAC, 25 wt% in water), triethanolamine (TEA, ≥99%), 3-aminopropyl-triethoxy silane (ATPES, 99%), 3-trihydroxysilyl-propyl methyl-phosphonate (THMP, 50 wt% in H2O), % low-melt agarose, phenol red free DMEM, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil) were obtained from Sigma-Aldrich Australia. Dimethyl sulfoxide (Chem-Supply, Gillman, SA, Australia), phosphate buffered saline tablets (MP Biomedicals, Santa Ana, CA, USA), sodium hydroxide (Chem-Supply, Gillman, SA, Australia). Doxorubicin Hydrochloride (Thermofisher Scientific, Australia). Bovine lactoferrin (NatraFerrin) from MG Nutritionals®, Burnswick, Australia. Pacific blue Annexin 5 binding buffer (An5B, Biolegend Australia cat 640918), 7-AAD Viability Staining Solution (7AAD, Biolegend Australia cat 420404). Flat-bottom 96-well plates (In Vitro Technologies). Adenosine Tri Phosphate (ATP) (Life Technologies). CellTiter Glo reagent (Promega), Cyanine 5 NHS ester (Cy5 Dye) (cat#53020) was sourced from lumiprobe.Room temperature synthesis of USLP
USLP were synthesized by addition of 0.6 g TEA into 18 mL deionised water and 1 mL CTAC in 100 mL conical flask. The solution was stirred for 1 hour at 1000 rpm in ambient room temperature. 2 mL TEOS and 18 mL n-hexane were then added and this solution was then stirred continuously for 24 hours. USLP were obtained by centrifugation (Jouan Centrifuge KR22i in the rotor AK50-22) at 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700g for 10 minutes. The supernatant was discarded, and pellet was washed with ethanol three times to remove any unreacted species. After washing, the obtained pellet was dried for 12 hours in 60 °C oven. This dried pellet was crushed into fine white powder with use of mortar and pestle. To study the influence of temperature, the synthesis was also conducted at 4 °C and 50 °C with all other parameters kept constant.
700g for 10 minutes. The supernatant was discarded, and pellet was washed with ethanol three times to remove any unreacted species. After washing, the obtained pellet was dried for 12 hours in 60 °C oven. This dried pellet was crushed into fine white powder with use of mortar and pestle. To study the influence of temperature, the synthesis was also conducted at 4 °C and 50 °C with all other parameters kept constant.
To remove any residual surfactants from USLP, the dried nanoparticles were placed in a muffle furnace (Thermo scientific, Australia) at 550 °C for 5 hours.
Surface modification of USLP
Surface modification was conducted as per previous reports with slight changes as follows.35,36 Amine surface functionalisation (USLP-NH2) was conducted by taking 300 mg USLP and placing it into a vacuum dryer (Salvis, Switzerland) for 12 hours at 150 mbar 60 °C to remove moisture.35 USLP was added into 40 mL toluene and stirred at 50 °C in a sealed environment to prevent any moisture entering the flask. After 30 minutes of stirring the temperature was increased to 115 °C and 300 μL of 3-aminopropyl-triethoxy silane was added and the mixture was refluxed for 18 hours. USLP-NH2 were collected as pellets by centrifugation (24700g for 10 minutes) and washed three times with 70% ethanol. Pellet obtained was dried in 60 °C oven for 12 hours and then crushed into a fine powder.
Phosphate surface functionalisation (USLP-PO3) was carried out by preparing a solution of 0.3 mL of 3-trihydroxysilyl-propyl methyl-phosphonate(THMP) dissolved in 30 mL Milli-Q water.35 To minimise the silanol group hydroxylation and dissolution, the pH of this solution was reduced to pH 5.5 with dilute 0.1% hydrochloric acid. 300 mg USLP was added to this solution and bath sonicated for 10 minutes. Lower pH is also important to catalyse condensation reaction between the silanol group present on THMP and USLP.16,35,36 The mixture was refluxed for 18 hours at 100 °C with continuous stirring at 1000 RPM. USLP-PO3 were collected as pellets by centrifugation (24700g for 10 minutes) and washed three times with 70% ethanol. Pellets obtained were dried in 60 °C oven for 12 hours and then crushed into a fine powder.
Cy5 dye conjugated silica (USLP-NH2-Cy5) were prepared by sonicating 30 mg of USLP-NH2 in 3 mL of DMSO. Cy5 dye 0.6 mL (1 mg mL−1) was added to this mixture and stirred at room temperature for 2 hours. Particles were obtained with centrifuge 24700g for 5 minutes, washed five times with 70% ethanol until a clear supernatant was observed. The particles were vacuum dried at 150 mbar for 24 hours at room temperature.
Drug loading on USLP
50 mg USLP-PO3 or USLP-NH2 was added to solution of DOX (12.5 mg in 4 mL deionised water) and stirred for 500 RPM for 2 hours, particles were collected by centrifugation (24700g, 10 minutes) and pellet freeze dried to remove remaining solvent. The samples USLP-PO3-Dox or USLP-NH2-DOX were stored in −20 °C for future use.
Lactoferrin conjugation with USLP
Lactoferrin coating (USLP-NH2-Cy5-Lf) was carried out by sonicating 10 mg USLP-NH2 Cy5 in 1.7 mL deionised water and adding to 2.5 mL lactoferrin (1.5 mg mL−1 water solution) and mixed via a rotating wheel for 2 hours. This was then centrifuged (24700g for 10 minutes) to obtain pellet and washed once with 1 mL of water. The USLP-NH2-Cy5-Lf were obtained by freeze drying for 24 hours.
For lactoferrin coating of drug loaded USLP, 20 mg of USLP-PO3-DOX or USLP-NH2-DOX were stirred in 2 mL lactoferrin coating solution (10 mg lactoferrin in 2 mL water) for 2 hours, particles were collected by centrifugation (24700g for 10 minutes) and pellet freeze dried to remove any remaining solvent. The samples USLP-PO3-DOX-Lf or USLP-NH2-DOX-Lf were stored in −20 °C for future use.
Physiochemical characterization
The USLP prepared were characterized for their shape, particle size, zeta potential, functional group grafting, pore size and surface area as per the minimum information reporting of bio-nanomaterials.37 Particle size data was obtained by both Transmission Electron Microscopy (TEM) and Dynamic Light Scattering (DLS). For TEM images HITACHI HT7700 (Tokyo, Japan) was used. The machine was operated at 80 kV with condenser. Samples were prepared by suspending USLP in 1 mg mL−1 ethanol and sonicating for 10 minutes. This suspension was then added dropwise to carbon-coated copper grid which was air-dried and viewed under the microscope. DLS Nano series ZS instrument (Malvern Panalytical, United Kingdom) was used with measurements recorded in both back scatter and non-invasive backscatter (NIBS). All sample were run in triplicates. Each run had 100 sub runs of 10 seconds each and measurements were performed at 25 °C. Refractive index of silica was used as 1.42 and measurements for both size and zeta potential were taken by diluting 100 times the 1 mg mL−1 USLP in water at pH 7.4 in disposable folded capillary zeta cell (Malvern Instruments, DTS1060).Pore size and surface area information was obtained using Micromeritics Tristar II 3020 system (Micrometrics Tristar II, United Kingdom). Between 70–100 mg of USLP powder was taken and degassed (60 °C) for 24 hours under vacuum pipeline. Measurements were carried out at −196 °C using liquid nitrogen. Barrett–Joyner–Halenda (BJH) adsorption branch was used to measure the pore size distribution.
Quantitative analysis of functional group grafting and/or drug mass loading was performed using Mettler-Toledo Thermogravimetric analysis instrument (StarE, Switzerland). Both Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) information was obtained. About 5–10 mg of particles were accurately weighed using a microbalance and placed in an alumina crucible (70 μL). Temperature range of 50 °C to 900 °C was selected with a temperature ramp rate of 10 °C per minute. Further qualitative analysis of different functional group on USLP was conducted by Fourier Transformed Infrared Spectroscopy (FTIR) using the PerkinElmer FTIR-ATR spectrometer.
In vitro release of DOX
A calibration curve of DOX was prepared for concentrations ranging from 0.078 to 10 μM in (R2 ∼ 0.99) in PBS pH 7.4 and pH 5.5 using a fluorescence plate reader with emission λ 470 nm and excitation λ 560 nm. For in vitro release 1000 μg DOX, USLP-PO3-DOX, USLP-PO3-DOX-Lf containing equivalent 300 μg DOX were suspended in either 5 mL PBS pH 7.4 or 5.5 and at predetermined time, 1 mL sample was taken and replaced with 1 mL PBS. Samples were centrifuged (24700g for 10 minutes). The supernatant was diluted according to respective calibration curve and analysed by fluorescence spectroscopy.
In vitro USLP cytotoxicity and cell uptake
Preparation of nanoparticle solutions
Nanoparticles were suspended in DMEM at 0.6 mg mL−1, kept on ice and sonicated with a probe sonicator set to 10 W for 1 minute in several short bursts. Samples were diluted to the required concentrations with DMEM (0.1 to 10 μg mL−1).Cellular uptake
Cells were plated into 24-well glass bottom plates pre-treated with Collagen I and incubated for 48 hours to allow cell attachment. At time points 1, 4 and 24 hours, freshly prepared nanoparticle solutions were added to give a final nanoparticle concentration of 10 μg mL−1. Immediately prior to imaging cells were stained with H33342 nuclear stain, washed and fresh cell culture medium was added. Cells were imaged on a Zeiss LSM 780 equipped with an environmental chamber which controls temperature for live-cell imaging using a 20× 0.8 NA air objective. Acquired images were processed using the Fiji software package.In vitro cell apoptotic studies of USLP-DOX vs. USLP-DOX-Lf against U87 Cells
U87 cells were plated in 6-well plates at a cell density of 300000 cells per well. After 24 hours incubation, cells were washed with PBS and treatment was added. 1 μg mL−1 equivalent free DOX or USLP-PO3-DOX, USLP-PO3-DOX-Lf were added as treatments. Controls with no treatment or equivalent USLP-PO3 or USLP-PO3-Lf were also used. Cell were incubated for 72 hours and collected using trypsin (0.5 mL) followed by neutralization with 2 mL fresh media. Floating cells present in the culture media and PBS washes before trypsinisation were also collected and pooled with the detached cells. Cells were washed twice with sterile PBS to remove any remaining culture media and resuspended in 100 μL cold Annexin V buffer solution (An5B) containing 5 μL Annexin V (An5, 40 μg mL−1) and 5 μL 7AAD (50 μg mL−1). Samples were incubated in the dark for 15 minutes before analysis. For each sample, a minimum of 10000 events were recorded using LSR Fortessa X20 machine. Florescent minus one controls single stains were used for compensation and gating purposes. Data were analysed using FlowJo software.
In vitro 2D blood brain barrier permeation
A blood–brain barrier (BBB)-GBM cell in vitro model was established using a previously described protocol.38–40 In brief, 2 × 105 human HBEC-5i cells were seeded onto the apical side of individual transwell inserts. To encourage the formation of a tight barrier, cells were cultured in human astrocyte conditioned medium as previously described.38–40 After 15 days the ability of the cell monolayer to block the permeability of macromolecules was measured using TRITC-dextran (155 kDa) as previously described.39,40 Expression of the tight junction protein, Zonula occluden 1 (ZO-1) on the surface of HBEC-5i cell monolayer was confirmed by immunofluorescence and confocal microscopy. U87 cells were seeded at 5 × 103 cells per well onto collagen on the basolateral side of individual transwell inserts. The next day, Cy 5-labelled USLP with or without lactoferrin coating were added (10 μg mL−1) to the apical side of the transwell inserts. Twenty-four hours later, U87 cells were collected, fixed and stained with actin as previously described.40 Cy5 nanoparticle cell uptake was assessed using confocal microscopy.Tumour spheroid preparation
For live imaging of spheroids, glioblastoma (U87) cells were stained with DiI (1 μM, 15 minutes before being spun down (1200 RMP, 3 minutes) and resuspended in fresh media DMEM + 10% FBS. These cells were seeded at 2000 cells per 200 μL per well in ultra-low attachment U-bottom 96-well plates (Lonza) for 60 hours. Spheroids were then carefully transferred in 1% low-melt agarose (sigma) to glass-bottom 24-well plates. Phenol red free DMEM + 10% FBS was added to all wells for imaging. For nanoparticle uptake, nanoparticles were added to wells with spheroids to a final concentration of 10 μg mL−1. Sterile 1 X PBS was added to outer wells in the plate to reduce evaporation and drift during imaging.Uptake of nanoparticles into U87 3D tumour spheroids and efficacy of USLPs loaded with DOX against U87 3D tumour spheroids
Efficacy of USLP with or without DOX was investigated in 3D GBM (U87) tumour spheroids and measured using the CellTiter Glo assay (Promega). Briefly, U87 cells were seeded (2000 cells per 100 μL DMEM + 10% FBS per well) in ultra-low adherent round-bottom 96-well plates (Lonza) for a total of three days to enable spheroid formation. Spheroids were then treated in triplicates with varying concentrations of either free DOX, or USLP with DOX loading, at concentrations indicated (normalised to DOX concentration). USLP-PO3 and USLP-NH2 without DOX were also tested at the maximum concentrations to ensure any impact on spheroid viability was related to the doxorubicin loading of the nanoparticle, and not the particle itself. After three and six hours, spheroids were gently transferred to wells with 200 μL fresh media which removed any particles or doxorubicin that had not already been taken up in the spheroids. Spheroids were then left for a total of five days post-treatment. Representative brightfield images of U87 spheroids five days post-treatment were acquired on an Olympus CKX41 inverted microscope with UPlanFL N 4×/0.13na objective Following, spheroids were transferred in 50 μL total volume to white flat-bottom 96-well plates. Media controls were used as blanks for each condition. An ATP standard curve was pipetted in duplicate using 10 mM ATP diluted in DMEM + 10% FBS to a range of 0.3125 μM–10 μM. CellTiter Glo reagent was added at 1:1 ratio and plates transferred to an Orbit 4 Benchtop shaker for spheroid dissociation (60 RPM, 30 minutes). ATP concentrations were then measured using 1.0 second luminescence exposure on a Wallac3 Victor plate reader (PerkinElmer) and exported as Excel files for analysis.
Image acquisition of nanoparticle uptake into tumour spheroids
Nanoparticle uptake was imaged on a Zeiss LSM 880 inverted laser scanning confocal microscope with incubation (37 °C) and 5% CO2 using a Plan-Apochromat 10×/0.45 M27 objective. Fluorescence was captured at 1.4 times zoom, minimum 1024 by 1024 pixels using frame-fast Airy with two tracks (Track 1: 561 nm, 7% laser; Track 2: 633 nm, 20% laser), 488/561/633 beam splitter and BP 570–620/LP 645 filters. Acquisition was adjusted to acquire z-stacks from the core of the spheroids through to the circumference (∼250 μm) every 30 minutes for a total of 24 acquisitions (12 hours). Raw data was exported to Zen (2.1 SP3, Zeiss) for Airy processing (3D, automated settings at 6.0 strength) before post-processing for maximum intensity projections, ortholog images and subsequent analysis.Analysis of nanoparticle uptake into tumour spheroids
Mid-plane z-stacks of spheroids were imported as virtual image stacks into Fiji. A linear gradient (50 pixel width) through the diameter was defined and quantified using PlotProfile. Raw intensities were exported and graphed in PRISM (V7.04).Results and discussion
Rational design and synthesis of USLP delivery system
To achieve effective and targeted drug delivery to brain tumours, parameters such as the size, surface functionality and porosity of nanoparticles need to be considered.14,30,41 The size and porosity of the nanoparticle are important for the optimum drug loading and to ensure penetration to the cancerous tissue. In addition, for drug delivery purposes, nanoparticles need to be uniform in size and shape to ensure consistent dosage of drug is released at the target site. In this study, ultra-small size particles were explored for their ability to penetrate the BBB and the BTB.14 Large pores were considered because it was expected to provide improved drug loading especially when considering multiple cargos in the future. Finally, silica nanoparticles were chosen because of their excellent biocompatibility and relative ease to control parameters such as size and surface morphology.Herein, we report the for the first time synthesis of silica nanoparticles which are; (1) synthesised at room temperature, (2) ultra small size of 30 nm and, (3) large porous structure (>7 nm). USLP were synthesised using one-pot, biphasic sol–gel method with all synthesis steps conducted at room temperature. The first step involved mixing of water phase consisting of cationic surfactant cetyltrimethylammonium chloride (CTAC) and basic catalyst triethanolamine (TEA) under aqueous conditions. TEA was chosen as a catalyst as it has been reported to control particle size by preventing particle size growth and agglomeration42–45 As the concentration of surfactant was above the critical micelle concentration, the surfactant self-assembles into micelle with hydrophobic core. Once micelles were formed, silica precursor tetraethyl orthosilicate (TEOS) was added in the same synthesis pot to initiate hydrolysis and condensation. The anionic TEOS reacted with cationic micelle to generate USLP structure with the help of known pore expander n-hexane46,47 (Fig. S1†). The combination of room temperature synthesis, TEA, mixing speed and n-hexane have contributed to the formation of USLP.
The reaction temperature needs to be above the surfactant critical micellar temperature or the cloud point (temperature at which oil/water phase separate). Typically, large pore silica nanoparticle synthesis requires high temperature. High temperature (>60 °C) is required for specific surfactants and swelling agents to synthesise large pore silica nanoparticles (>7 nm) such as Santa Barbara Amorphous type material (SBA-15) but generally these are large size nanoparticles (>100 nm). However, silica nanoparticles have also been synthesised in room temperature with use of n-hexane but they had large size (100–200 nm).47 The critical challenge for ultra-small, large pore silica nanoparticles (USLP) was to reproduce synthesis with both small size (<40 nm) and large pore characteristics (>7 nm). Previously in our lab, we have synthesised large pore silica nanoparticles (∼10 nm) but the size of these nanoparticles was too large for brain drug delivery (∼200 nm).48,49 At reduced temperature, it has been reported that the rate of nucleation decreases which reduces the particle size.50,51 The room temperature synthesis of USLP is therefore an important factor for USLP's small particle size. The role of temperature was confirmed by keeping all other parameters unchanged and synthesizing at 50 °C. It was found that the size of particle grow with increase in temperature (>100 nm) and when the temperature is reduced to 4 °C the particles tend to aggregate (Fig. S3†). This phenomena has also been reported by Wang et al., as they developed large pore dendritic (>20 nm) silica nanoparticles but with diameter larger than USLP (i.e. 50 nm) in room temperature conditions.50 To generate ultra-small particles with large pore sizes, iterative tests were conducted to optimise stirring speed, temperature, synthesis reaction time and use of different oil phases (summarised in Fig. S2 and S3†). When dichlormethane or toulene were used as oil phase the size of particles increased to 100 nm and 70 nm respectively (Fig. S2†). The rate of TEOS hydrolysis is slowest in n-hexane as it the most hydrophobic (logP 3.90) when compared to cyclohexane (logP 3.44), toulene (logP 2.73) and dichlormethane (logP 1.25). Due to n-hexane relatively higher hydrophobicity the rate of TEOS diffusion from the oil phase into water phase was slowest which decreased the rate of TEOS hydrolysis.47 These results confirmed that n-hexane is very important in controlling both the size and porosity features. Stirring speed is another important factor when synthesising these nanoparticles. Stiring speed controls the rate in which oil phase interacts with water in the biphasic synthesis mixture and thereby influences the speed of silica hydroxylation and condensation. It was observed that higher stirring speed yielded more uniform particle size. From these sets of experiments, we could identify the parameters that needed to be controlled for facile synthesis of uniform USLP i.e. high stirring speed, n-hexane and room temperature synthesis. Optimised synthesis conditions yielded ultra-small mesoporous silica nanoparticles (30.74 ± 4.00 nm) with large pore sizes (>7 nm) and negative surface charge (zeta potential −11.7 ± 0.6 mV). The particle synthesis protocol was replicated several times to ensure the protocol was robust and yields similar particle shape and size with every batch (Fig. S4†).
To remove any residual surfactant from silica nanoparticle, calcination or solvent extraction techniques are used.52 For USLP, calcination was used for complete removal of surfactant because solvent extraction has been reported to be inadequate.52 Surfactant (CTAC) removal is important to ensure porosity feature of USLP are obtained and no residual surfactant remains in the formulation to prevent any side effect. Evidence of surfactant removal can be identified by both a lack of weight loss of organic mass post calcination between 100 °C to 600 °C using TGA (Fig. S9†) and disappearance of surfactant peak at 2980 and 2930 cm−1 FTIR spectra post-calcination (Fig. S5†).
Surface charge is an important parameter as it can influence nanoparticle penetration into desired cells53,54 and effect drug release rates.2,35 One key advantage of silica nanoparticle is that the surface charge can be easily modified. USLP surface charge was modified, using a post-synthesis functionalisation method. Surface functionalisation was conducted with organosilane (i.e. APTES for cationic amine groups and THMP for anionic phosphonate group). The surface modified with an amine (–NH2) shifted zeta-potential to 19 ± 0.6 mV and the phosphate (–PO3) functionalisation shifted to −32.3 ± 0.4 mV (Fig. S6†). Post-functionalisation, the surface area and pore volume of USLP decreased. This effect was more pronounced in USLP-NH2 compared to USLP-PO3 (Fig. S7† and Table 1). Subsequent drug loading with doxorubicin (DOX) and coating with the lactoferrin was conducted to achieve cascade targeting across the BBB and into GBM. From TEM images, it was confirmed that particles are spherical shaped with uniform size and large porous structures visible (Fig. 1). The average diameter of USLP was determined to be 30 nm (TEM size) with and without surface functionalisation (Table 1). However, the size of USLP increased after conjugation with lactoferrin i.e. 30 nm vs. 35 nm as measured by TEM (Table 1). These results did not corroborate with DLS data showing slightly larger size of nanoparticles due to the hydrodynamic layer (Fig. S8†). This phenomenon is well established and has also been reported by many other researchers.35,55–57
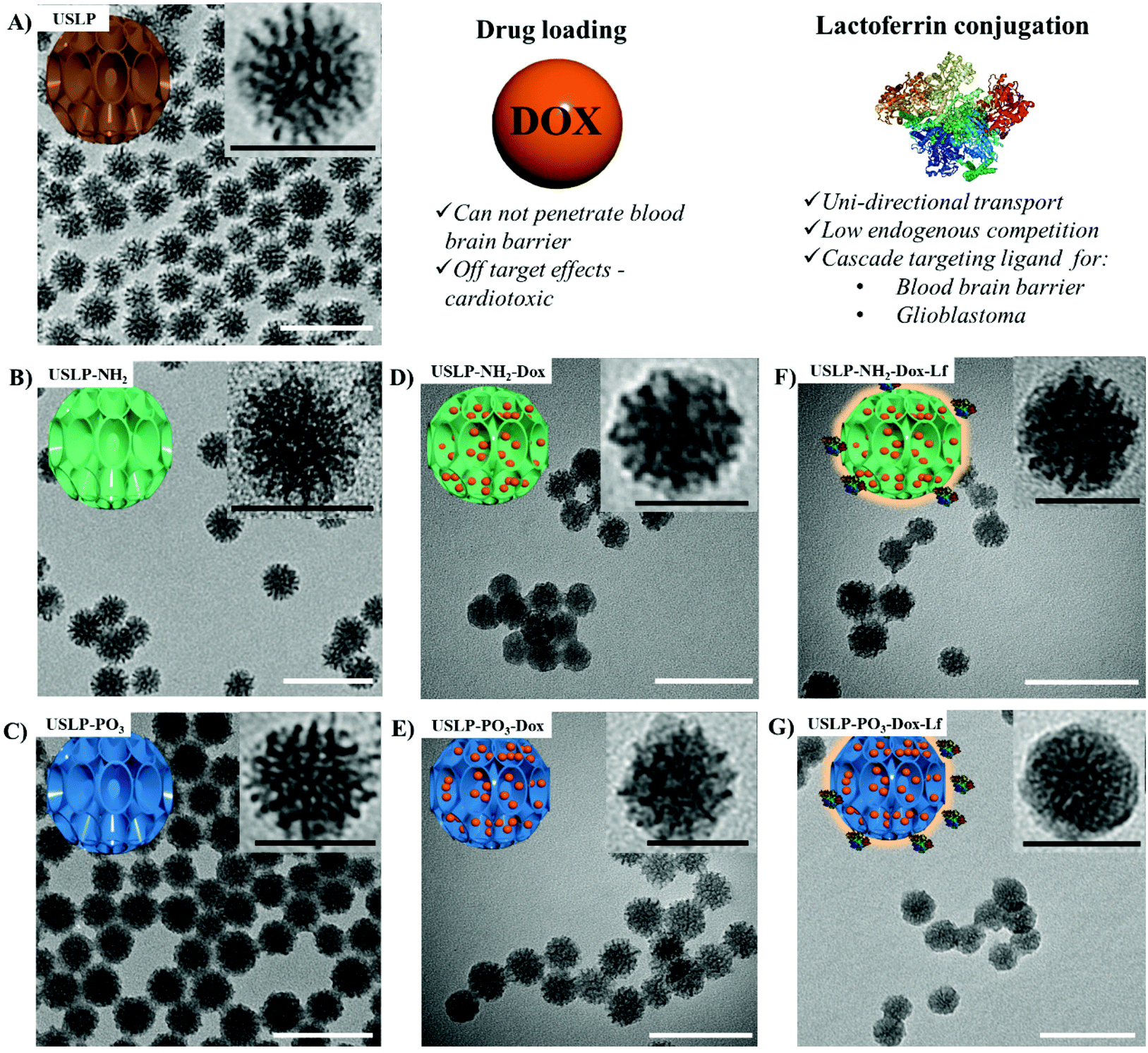 | ||
| Fig. 1 Schematic summary for the synthesis of ultra-small size (∼30 nm) with large pore (>7 nm) silica nanoparticles (USLP), surface functionalisation, and surface lactoferrin coating with lactoferrin. Characterisation of USLP by transmission electron microscopic images of nanoparticles (A) USLP, (B) USLP-NH2, (C) USLP-PO3, (D) USLP-NH2-Dox, (E) USLP-PO3-Dox, (F) USLP-NH2-Dox-Lf, (G) USLP-PO3-Dox-Lf. Images were prepared by suspending 1 mg of nanoparticles in ethanol and sonicated for 10 min before placing on TEM copper grids. White scale bar 100 nm, inset black scale bar 30 nm. | ||
| Sample | Average size nm (TEM) | BET surface area (m2 g−1) | BJH pore size (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| USLP | 30.74 ± 4.00 | 1144.63 | 7.70 | 1.97 |
| USLP-NH2 | 32.54 ± 4.06 | 293.32 | 7.35 | 0.99 |
| USLP-PO3 | 33.82 ± 4.93 | 583.86 | 7.98 | 1.10 |
| USLP-NH2-Lf | 34.95 ± 5.78 | — | — | — |
| USLP-PO3-Lf | 36.32 ± 4.96 | — | — | — |
USLP based delivery system efficiently penetrate in vitro BBB and internalise into GBM cells
After successful synthesis of nanoparticle with desired size and porosity, we then investigated these USLP for their superior penetration across the BBB and BTB. To examine whether silica nanoparticles coated with lactoferrin could traverse an in vitro BBB model and internalize into GBM cells, a BBB-tumour cell model was established as previously described38,40 (Fig. 2A). To simulate the BBB we used the human brain endothelial cell line HBEC-5i, on the apical surface of transwell inserts. Addition of human astrocyte conditioned media simulated the HBEC-5i to grow as a tight monolayer. To ensure the integrity of the BBB was maintained, the expression of the tight junction protein, Zonula occluden 1 (ZO-1), on the surface of HBEC-5i cell monolayer was confirmed by immunofluorescence and the penetration of macromolecules was blocked as confirmed by using TRITC-dextran (155 kDa) (results not shown) as previously described.39,40Fig. 2B, shows that silica nanoparticles coated with lactoferrin were able to traverse the HBEC5i cell monolayer and internalize into U87 cells. These results suggest that the lactoferrin coating on the surface of the USLP nanoparticles plays an important role in promoting their attachment to the surface of endothelial cells. Our results are in agreement with literature, as other types of nanoparticles when conjugated with lactoferrin have also improved nanoparticle cellular uptake.58,59 It was also expected that uncoated USLP would be able to traverse BBB owing to their small size. However, the small (∼30 nm) uncoated nanoparticles were not detected in U87 cells (Fig. 2C) at the dose of 10 μg mL−1. This could be due either to their inability to cross the BBB or poor cellular uptake by the U87 cells due to low dose which was then investigated further.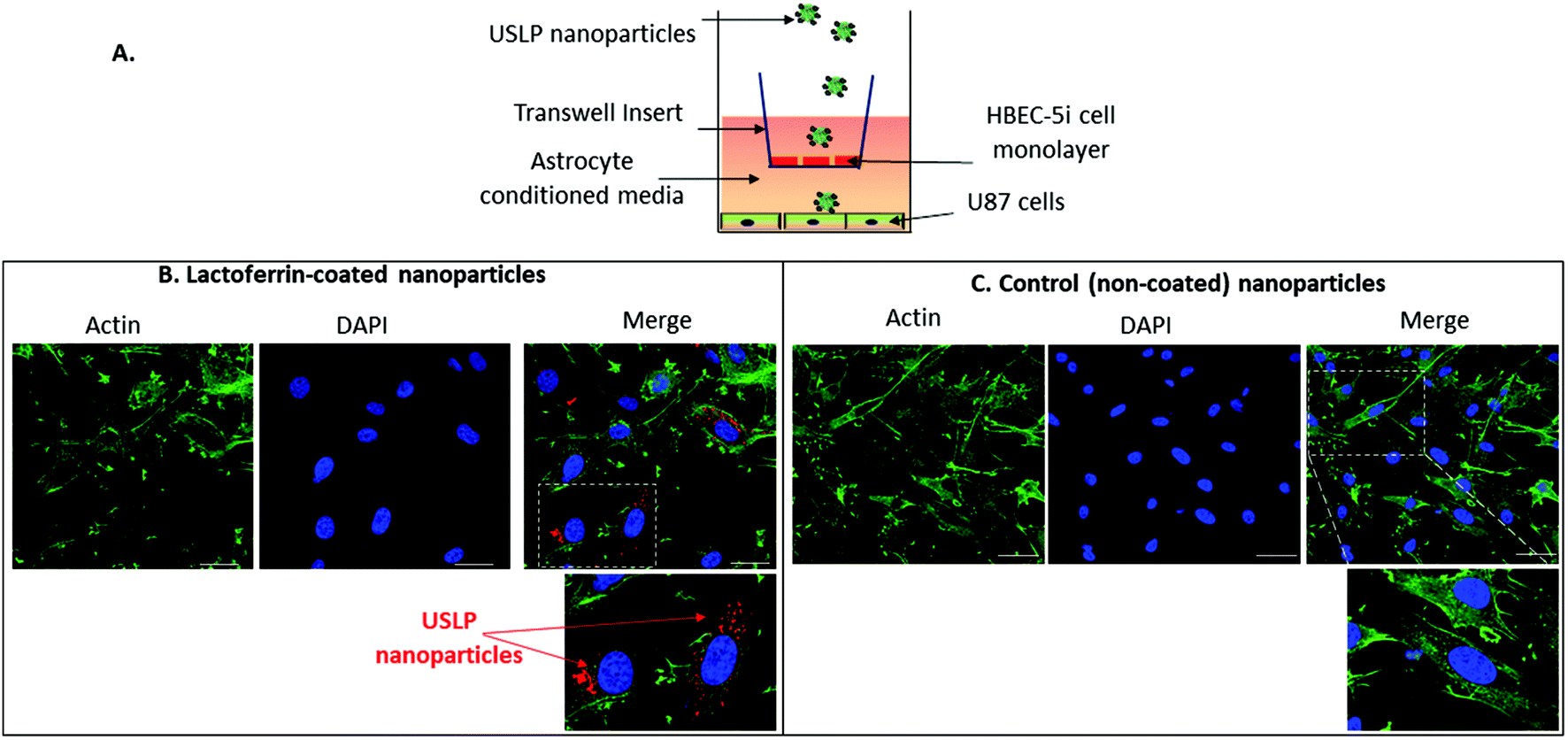 | ||
| Fig. 2 Blood–brain barrier permeability in vitro of USLP. (A) Schematic diagram of the blood–brain barrier (BBB)-tumour cell model. (B) Representative images showing that Cy5-labelled silica nanoparticles coated with lactoferrin can traverse an in vitro BBB and internalize into U87 cells 24 h post-treatment compared to non-coated silica nanoparticles (control) (C). Green = Actin, Blue = Nucleus, Red = Cy5-labelled nanoparticles, scale bars = 30 μm, n = 3 independent experiments. White dotted box indicates area of the zoomed image. | ||
After establishing the feasibility of USLP to penetrate an in vitro BBB, we further studied USLP penetration into U87-GBM cells. Effective cellular uptake of USLP by the GBM cells is an important determinant for effective drug delivery. It is known that the majority of nanomedicine uptake in cells occurs via cellular endocytosis.60 As such, conjugation with specific targeting ligands like lactoferrin could help nanoparticles identify and bind specifically to GBM cells which are known to overexpress the lactoferrin receptor. This would augment the receptor mediated USLP internalization and ultimately achieve targeted drug delivery. Owing to their small size, USLP showed good uptake in the U87 GBM cell line (Fig. 3). Results from 2D imaging studies show that both USLP-NH2-Cy5 and USLP-NH2-Cy5-Lf can be taken up by the U87 cells (Fig. 3). However, based on the qualitative data available, there is no significant difference between uncoated USLP and lactoferrin conjugated USLP in terms of nanoparticle uptake in the U87 GBM cells (Fig. 3). The camouflage of USLP surface with lactoferrin could be enabling uptake of nanoparticles but perhaps the small size of USLP also plays an important role in the uptake into U87 cells. From this study, it is clear that USLP are taken up by U87 cells, however it cannot be established if lactoferrin has significant influence on nanoparticle uptake by U87 cells in 2D cultures.
 | ||
| Fig. 3 Uptake of uncoated and lactoferrin-coated nanoparticles into U87 glioblastoma cells over time visualised by confocal microscopy. Scale bar, 20 μm. Blue H33342 nuclear stain, red Cy5 labelled nanoparticles. | ||
3D tumour spheroid models are better predictive models for successful delivery system when compared to 2D cell culture models.61 The key benefit of 3D tumour spheroids is that they can efficiently simulate the multicellular framework and heterogeneous nature of GBM.62 Consequently, we can more reliably predict nano-formulations penetration and chemo-resistance in 3D GBM models. The peripheral cells present within the tumour spheroid have been reported to simulate the actively growing tumour cells in proximity to blood vessels but the cells in the core of the tumour often become dormant and are responsible for tumour recurrence.63 From our results in Fig. 4, the USLP labelled with Cy5 dye are able to penetrate into the tumour spheroid within 12 hours. The result from tumour spheroid penetration shows that both coated and uncoated USLP can penetrate the tumour spheroid within 12 hours (Fig. 4A and C). This data is in agreement with 2D confocal data suggesting that the small size of USLP facilitate uptake by U87 cells. However, from the 3D model, it was observed that lactoferrin coating has substantial influence in increasing the USLP penetration (Fig. 4D) compared to uncoated USLP (Fig. 4C and ESI video†). After one hour, at the distance of 100 μm from the tumour spheroid periphery, the lactoferrin coated USLP have significantly higher fluorescence intensity compared to uncoated USLP (1.26 RFU vs. 3.20 RFU). After 12 hours, both types of USLP penetrate into the tumour spheroid but the fluorescence intensity of lactoferrin coated is significantly higher compared to USLP without lactoferrin coating, within the tumour spheroids (at 100 μm distance RFU value 19.58 vs. 49.16) (Fig. 4B and D). This data indicates that firstly, the small size USLP can penetrate tumour spheroids, however penetration is augmented when using lactoferrin coated nanoparticles. Thus, the lactoferrin mediated transport of nanoparticles plays a significant role in permeation of nanoparticles into the tumour. Taken together, the USLP nanoparticles can efficiently penetrate across the BBB and accumulate in GBM.
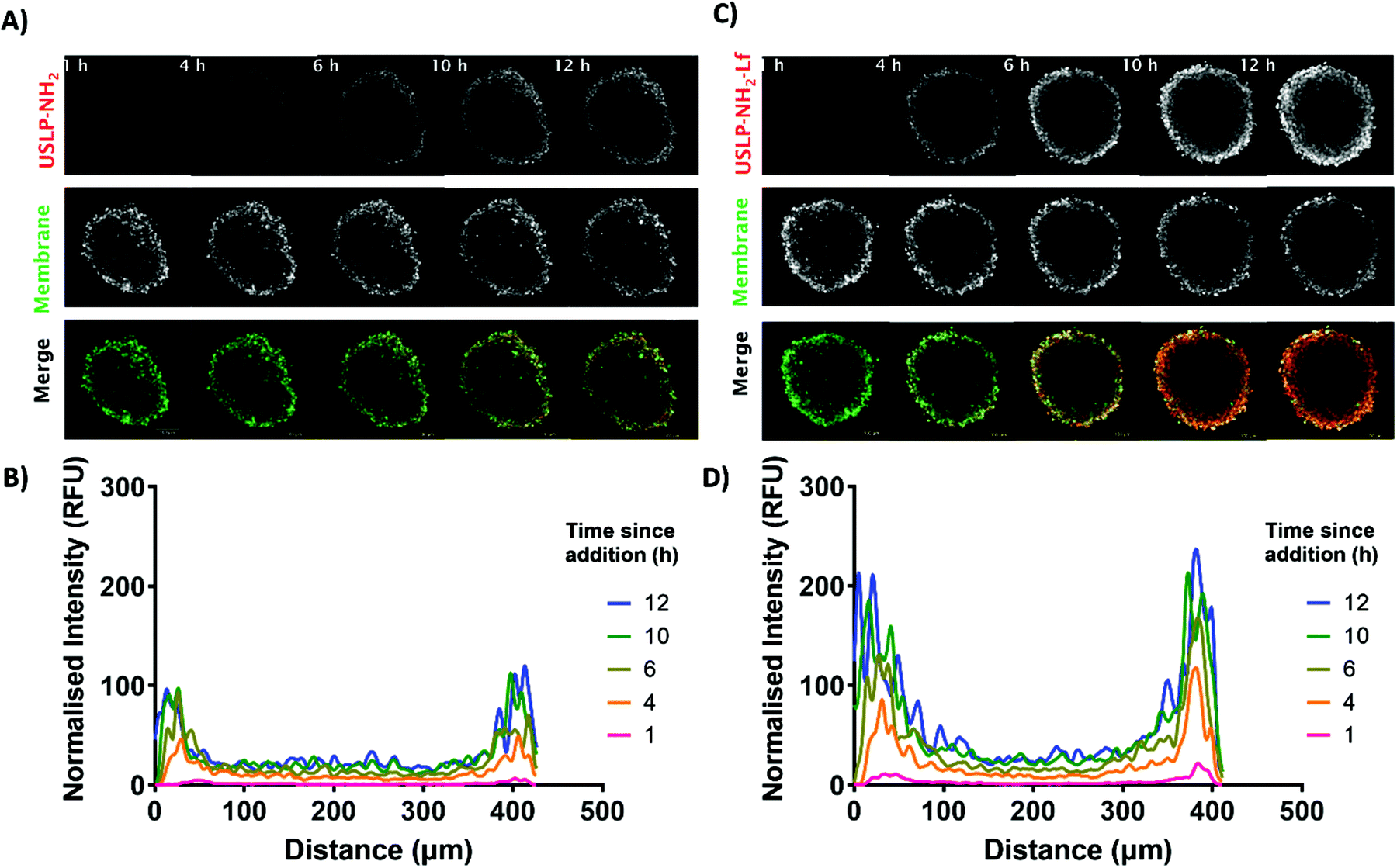 | ||
| Fig. 4 Cy5 labelled USLP uptake in glioblastoma (U87) 3D tumour spheroids over 12 hours. (A) Representative z-stack of uncoated USLP, membrane dye (DiI) and merge at 1, 4, 6, 10, 12 hours post addition (Red, USLP; Green, Membrane). (B) Quantified changes in fluorescence intensity of USLP over time using linear correlation for time points above. (C) Representative z-stack of lactoferrin coated (USLP-NH2-Lf), membrane dye (DiI) and merge at 1, 4, 6, 10, and 12 hours post addition (Red, USLP-NH2-Lf; Green, Membrane). (D) Quantified changes in fluorescence intensity of USLP-NH2-Lf over time using linear correlation for time points above (Fiji). Images acquired using a Zeiss LSM 880 confocal microscope (Fast airy, sequential frame-fast laser excitation at 561 nm and 633 nm, 10× objective). Analysis conducted in (Fiji). Scale bar in (A) and (C), 100 μm. | ||
USLP-mediated delivery of Dox in 2D and 3D in vitro cultures of GBM cells
Chemotherapeutic drugs such as DOX, often have maximum tolerated doses which are dictated by systemic toxicities as a result of unintended, “off-target” interactions with normal tissues when administrated systemically as free compound; for instance, DOX is known for its acute cardiotoxicity.21 On the contrary, a sustained release system can prevent the premature release of DOX into the blood circulation, ensuring DOX is released at the tumour site with minimal exposure to critical tissues or organs. For example, modified formulations of DOX available in the market, such as liposomal coated DOX (Doxil®), have been shown to have reduced cardiotoxicity.64,65 To investigate whether USLP allow for a controlled release profile of DOX, we analysed their release rate over a period of time (48 hours) in different pH systems (Fig. 5). USLP were loaded with DOX were quantified using TGA (Fig. S9†). All USLP tested slowed the release of DOX when compared with free DOX. Moreover, the release rate was slower at pH 7.4 which simulated the plasma environment, whereas a faster release rate was observed at pH 5.5, mimicking both the lyso-endosome conditions and acidic micro-environment of GBM.66 The faster release in lower pH is desirable as it can be expected that after injection of nanoparticles in the blood stream, less drug will be released systemically with less affects on non-tumour cells. DOX has a pKa of 7.3–8.4 and at the lower pH of 5.5, it becomes protonated due to the presence of primary amine in its structure.67 This cationic DOX at pH 5.5 has increased solubility which explains faster release at lower pH. Other researchers such as Kamba et al. have observed similar phenomena.68 Moreover, conjugation with lactoferrin further slowed the release in both pH systems. This was expected as lactoferrin surface coating acts as physical barrier slowing the release of DOX from the pores within the nanoparticles. This, together with the slower release at the plasma pH, could be advantageous since it may avoid drug release until particles cross the BBB. This in turn will prevent excessive release of DOX into the circulation, minimise off target side effects, and at the same time, achieve higher drug delivery at the tumour site. Finally, we found that release was faster in phosphate functionalised USLP compared to amino functionalised USLP. Phosphate functionalised USLP have a larger pore size and pore volume which could explain the faster release rate (Table 1).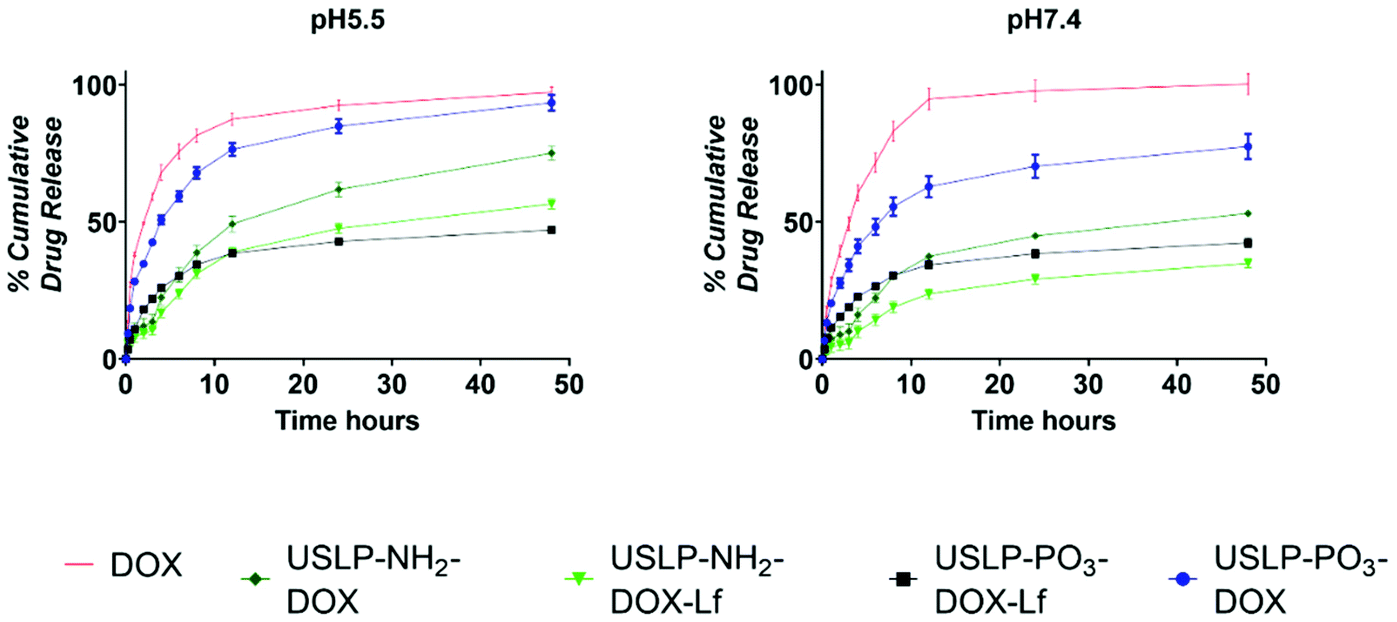 | ||
| Fig. 5 In vitro drug release study. To study the in vitro release of USLP-PO3-DOX, USLP-PO3-DOX-Lf, USLP-NH2-DOX, USLP-NH2-DOX-Lf and DOX were added into phosphate buffer solution at pH 5.5 and pH 7.4. Release of DOX was measured by fluorescence spectroscopy emission λ 470 nm and excitation λ 560 nm. Data is presented as mean ± SEM, n = 3. | ||
As USLP were shown to have efficient uptake by GBM cells, next we treated U87 with DOX loaded USLP or free DOX and investigated its ability to induce apoptosis in a growing 2D cell culture. DOX is known to induce cytotoxicity in tumours by stimulating apoptosis via initiating intercalation with DNA and inhibiting DNA replication by acting on the topoisomerase enzyme.21 We observed a significant increase in the percentage of total, late, and early apoptotic cells in all the DOX treated group when compared to the cells in the non-treated group or nanoparticles without DOX (P < 0.001) (Fig. 6). Importantly, the results show that USLP-PO3 and USLP-PO3-Lf without DOX have no effect on cell viability in both 2D and 3D culture (Fig. 6 and 7). This suggests that on their own the nanoparticles are not toxic to the cells. Moreover, USLP-PO3-Lf had the highest percentages of early and late apoptotic cells, and consequently the lowest proportion of live cells. This suggests that the presence of lactoferrin on USLP could help deliver DOX to the U87 cells and increase the level of apoptosis. Numerous studies have shown that lactoferrin receptor is highly expressed by GBM and aids in drug delivery to the tumour.58,59,69
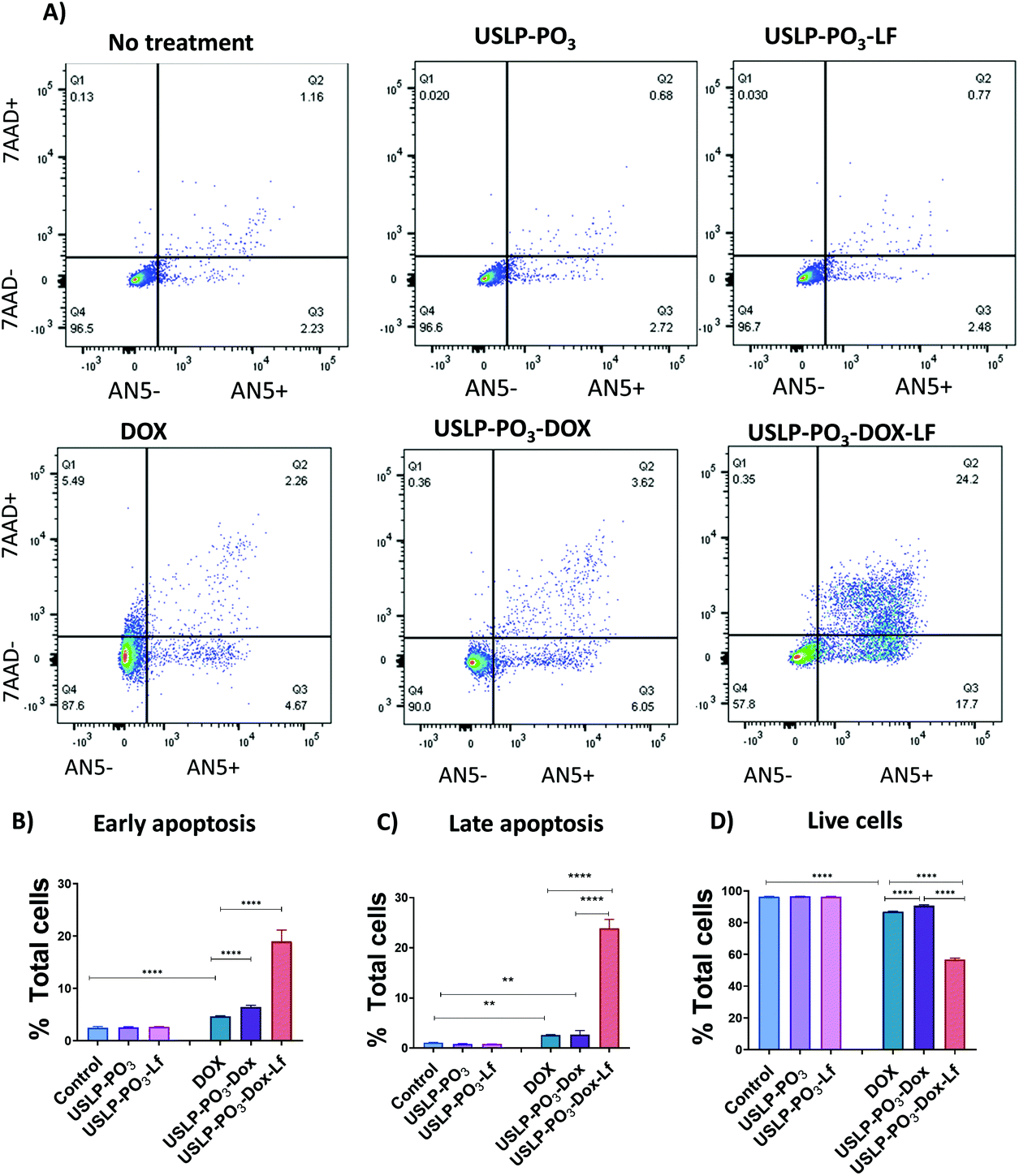 | ||
| Fig. 6 Higher levels of GBM cell apoptosis were achieved when DOX was loaded into USLP-Lf nanoparticles. In vitro cell apoptosis studies were performed by incubating human U87 GBM cells for 3 days with 1 μg mL−1 equivalent of free Dox, USLP-PO3-Dox or USLP-PO3-Dox-Lf. (A) Representative dot plots showing the gating of early apoptotic (Q3: AN5 + 7AAD−), late apoptotic (Q2: AN5 + 7AAD+), necrotic (Q1: AN5 − 7AAD+) and live cells (Q4: AN5 − 7AAD−), as quantified by FACS upon Annexin V (AN5) and 7AAD staining. Cells are gated on single cells. (B–D) Percentage of cells in (B) early apoptosis, (C) late apoptosis and (D) live cells. Data presented as means ± SEM. Results were analysed by unpaired one-way ANOVA. * p < 0.05, ** p < 0.01, *** p < 0.001. | ||
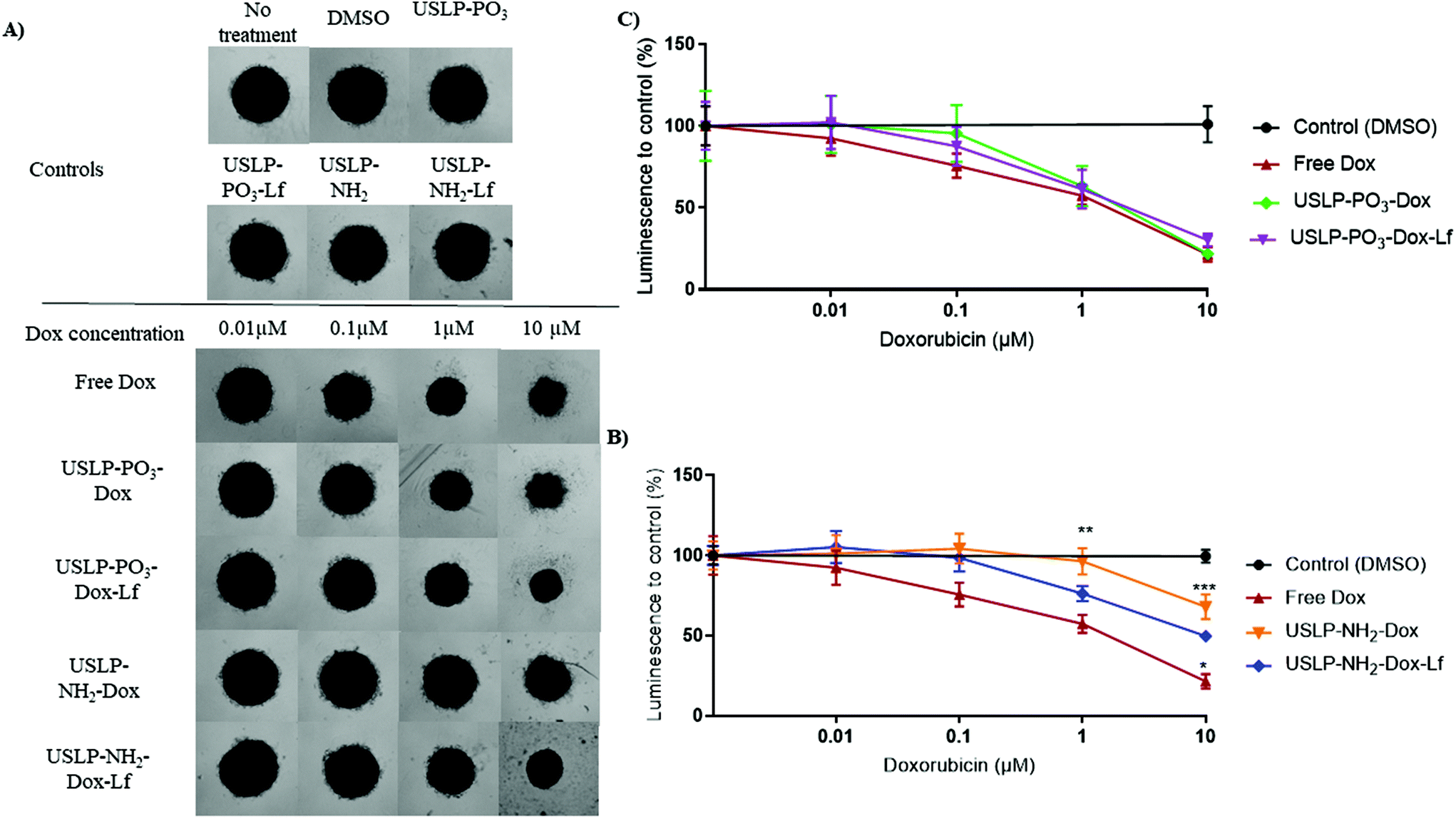 | ||
| Fig. 7 Doxorubicin loaded mesoporous silica nanoparticle (USLP) 3D cytotoxicity data in glioblastoma (U87) tumour spheroids. Spheroids were cultured for three days, treated for 6 hours and then media replaced for five days of growth post-treatment. (A) Representative Brightfield images of U87 spheroids five days post-treatment with Free DOX, USLP-PO3-DOX, USLP-PO3-DOX-Lf, USLP-NH2-DOX and USLP-NH2-DOX-Lf. DMSO was used as a vehicle control for free DOX. Unloaded USLP used as a control for each treatment group. Images acquired on an Olympus CKX41 inverted microscope with UPlanFL N 4×/0.13na objective. Scale bar, 500 μm. (B and C) Quantified changes in luminescence intensity compared to controls with various doxorubicin concentrations after 6-hour treatment. Points, mean of n = 3 independent experiments. Error, SEM. Significance to free Dox at concentration indicated, two-way ANOVA, * p < 0.05, ** p < 0.01, *** p < 0.001. | ||
As our data showed USLP and USLP-Lf were readily taken up by 3D U87 tumour spheroids, we further explored the efficacy of doxorubicin loaded USLP in reducing tumour spheriod size. The literature suggests that 3D tumour spheroids are a superior model for understanding the localized spatio-temporal effects of DOX on GBM cells.70 Although we didn't see significant difference between free drug and nanoparticles, the negatively charged USLP-PO3 (with and without lactoferrin) formulations had better efficacy in reducing tumour spheroid size compared to positively charged USLP-NH2 in both 3 hours and 6 hours treatments (Fig. 7 and Fig. S8†). The presence of lactoferrin coating in the case of amino-functionalised USLP, both enhanced the penetration of nanoparticle in tumour spheroids and decreased tumour growth (Fig. 4 and 7). However, in the case of phosphate functionalised USLP, both USLP-PO3-Dox and USLP-PO3-Dox-Lf had similar efficacy as free DOX. Similar trends were observed in both 3 h and 6 h treatments (Fig. 7 and Fig. S11†). Differences in the performance of phosphate and amino functionalised USLP against U87 tumour spheroids can be explained by both their difference in their interaction with the tumour spheroids, and their different drug release profiles. In addition, when compared to the cationic USLP-NH2, it could be expected that the anionic USLP-PO3 surface charge can have stronger interaction with cationic DOX leading to improved physiological stability. Our findings are in agreement with literature suggesting that compared to cationic nanoparticles, the anionic nanoparticles have better interaction and efficient uptake by brain cancer cells such as U87 GBM cells.54,71,72 This needs further evaluation in vivo to conclusively determine if the anionic USLP are better at traversing BBB and delivering chemotherapeutics to decrease tumour size. It is important to note that although in the U87 3D tumour spheroid model the efficacy of DOX was not significantly enhanced relative to free DOX, the utility of free DOX for brain tumour therapy is hindered by many shortcomings. For instance, (1) DOX cannot cross the BBB (2) has poor permeability across brain tumours and (3) has off target effects. By using the USLP based delivery system, we can ensure that DOX is able to both penetrate the BBB and into tumour parenchyma to efficiently exert its effects against GBM.
Conclusions
In this study, we have developed for the first time a facile room temperature synthesis protocol for ultra-small silica nanoparticles with large pores (30 nm, USLP). These USLP are capable of loading doxorubicin and the large protein lactoferrin (80 kDa) as a tumour targeting moiety. We have shown that this formulation is highly efficient in permeating the BBB (in vitro), has significantly better tumour permeation (3D U87 tumour spheroid model) and has potential to therefore increase the efficacy of doxorubicin for brain tumour therapy. With this platform we can significantly improve the utility of many chemotherapeutic drugs such as DOX to cross the BBB and improve penetration of our nanoparticles deep into the tumour with the aim to improve survival and quality of life of patients with brain cancer. Next, we will assess this novel platform's biodistribution and efficacy in pre-clinical models of glioblastoma.Conflicts of interest
There are no conflicts to declare.Author contributions
TIJ, AA-C, RM, MK, and AP supported the conception and design of the project. TIJ, AKM and AP contributed toward development and synthesis optimisation of USLP. TIJ and AA-C completed majority of experiments. FMM contributed towards 2D cellular uptake studies. HF, RMI and JAC helped with BBB permeation study. TIJ and YC contributed towards visualiation of data presented. TIJ prepared the original draft of the manuscript. All authors contributed towards the review, editing of manuscript and approved the submitted version.Acknowledgements
We acknowledge the School of Pharmacy, University of Queensland for the research facilities. We thank the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. TIJ and Y.C are thankful for the financial support from the University of Queensland Graduate School Scholarship (UQGSS). AA-C acknowledges support from the Scientia PhD Scholarship Scheme (RSRE9000, UNSW Sydney), the Josee Hilton Excellence Award and Children's Cancer Institute Postgraduate Top-up Scholarship. A.P. acknowledges financial support from National Health and Medical Research Council for the Early Career and Career Development Fellowships. R. M. is thankful to the Cure Brain Cancer Foundation for the Innovation Grant (21471) and to the Children Hospital Foundation for the Accelerator grant. MK acknowledges financial support from National Health and Medical Research (Program Grant APP1091261 and Principal Research Fellowship APP1119152), Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036), and Cancer Institute New South Wales (2019/TPG2037). JM acknowledges financial support from Cancer Australia/Kids Cancer Project (APP1184840) and the Mark Hughes Foundation for the innovation grant (MHF1461). MK, JM, HF, FMM and AA-C would like to thank the Biomedical Imaging Facility at the Mark Wainwright Analytical Centre, UNSW Sydney, for their support and resources involved in this work.References
- C. D. Arvanitis, G. B. Ferraro and R. K. Jain, Nat. Rev. Cancer, 2020, 20, 26–41 CrossRef CAS PubMed.
- T. I. Janjua, P. Rewatkar, A. Ahmed-Cox, I. Saeed, F. M. Mansfeld, R. Kulshreshtha, T. Kumeria, D. S. Ziegler, M. Kavallaris, R. Mazzieri and A. Popat, Adv. Drug Delivery Rev., 2021, 171, 108–138 CrossRef CAS.
- Q. T. Ostrom, H. Gittleman, P. Liao, C. Rouse, Y. Chen, J. Dowling, Y. Wolinsky, C. Kruchko and J. Barnholtz-Sloan, Neuro. Oncol., 2014, 16, iv1–iv63 CrossRef PubMed.
- C. Kelly, P. Majewska, S. Ioannidis, M. H. Raza and M. Williams, J. Neurooncol., 2017, 135, 621–627 CrossRef.
- M. Rapp, J. Baernreuther, B. Turowski, H.-J. Steiger, M. Sabel and M. A. Kamp, World Neurosurg., 2017, 103, 733–740 CrossRef.
- R. P. Moura, C. Martins, S. Pinto, F. Sousa and B. Sarmento, Expert Opin. Drug Delivery, 2019, 16, 271–285 CrossRef CAS.
- A. I. Oliveira, C. Pinho, B. Sarmento and A. C. P. Dias, Drug Deliv. Transl. Res., 2021, 1–15 Search PubMed.
- B. Sánchez-Dengra, I. González-Álvarez, F. Sousa, M. Bermejo, M. González-Álvarez and B. Sarmento, Eur. J. Pharm. Biopharm., 2021, 163, 120–126 CrossRef.
- S. Rawal and M. M. Patel, J. Controlled Release, 2019, 301, 76–109 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, 51–67 CrossRef.
- S. Bonvalot, P. L. Rutkowski, J. Thariat, S. Carrère, A. Ducassou, M.-P. Sunyach, P. Agoston, A. Hong, A. Mervoyer, M. Rastrelli, V. Moreno, R. K. Li, B. Tiangco, A. C. Herraez, A. Gronchi, L. Mangel, T. Sy-Ortin, P. Hohenberger, T. de Baère, A. Le Cesne, S. Helfre, E. Saada-Bouzid, A. Borkowska, R. Anghel, A. Co, M. Gebhart, G. Kantor, A. Montero, H. H. Loong, R. Vergés, L. Lapeire, S. Dema, G. Kacso, L. Austen, L. Moureau-Zabotto, V. Servois, E. Wardelmann, P. Terrier, A. J. Lazar, J. V. M. G. Bovée, C. Le Péchoux and Z. Papai, Lancet Oncol., 2019, 20, 1148–1159 CrossRef CAS.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC.
- J. H. Kang, J. Cho and Y. T. Ko, J. Drug Target., 2019, 27, 103–110 CrossRef CAS PubMed.
- J. Mo, L. He, B. Ma and T. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 6811–6825 CrossRef CAS PubMed.
- J. Kuang, W. Song, J. Yin, X. Zeng, S. Han, Y.-P. Zhao, J. Tao, C.-J. Liu, X.-H. He and X.-Z. Zhang, Adv. Funct. Mater., 2018, 28, 1800025 CrossRef.
- R. Guillet-Nicolas, A. Popat, J.-L. Bridot, G. Monteith, S. Z. Qiao and F. Kleitz, Angew. Chem., Int. Ed., 2013, 52, 2318–2322 CrossRef CAS PubMed.
- E. Neumann, E. Frei, D. Funk, M. D. Becker, H.-H. Schrenk, U. Müller-Ladner and C. Fiehn, Expert Opin. Drug Delivery, 2010, 7, 915–925 CrossRef CAS.
- C. Fillebeen, L. Descamps, M.-P. Dehouck, L. Fenart, M. Benaïssa, G. Spik, R. Cecchelli and A. Pierce, J. Biol. Chem., 1999, 274, 7011–7017 CrossRef CAS PubMed.
- P. F. Levay and M. Viljoen, Haematologica, 1995, 80, 252–267 CAS.
- R. Qiao, Q. Jia, S. Hüwel, R. Xia, T. Liu, F. Gao, H.-J. Galla and M. Gao, ACS Nano, 2012, 6, 3304–3310 CrossRef CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440 CrossRef CAS PubMed.
- O. Turan, P. A. Bielecki, V. Perera, M. Lorkowski, G. Covarrubias, K. Tong, A. Yun, G. Loutrianakis, S. Raghunathan, Y. Park, T. Moon, S. Cooley, D. Dixit, M. A. Griswold, K. B. Ghaghada, P. M. Peiris, J. N. Rich and E. Karathanasis, Adv. Ther., 2019, 2, 1900118 CrossRef CAS.
- A. Iorio, M. Da Ros, C. Pisano, M. de Martino, L. Genitori and I. Sardi, J. Clin. Med., 2019, 8, 331 CrossRef CAS PubMed.
- J. A. MacDiarmid, V. Langova, D. Bailey, S. T. Pattison, S. L. Pattison, N. Christensen, L. R. Armstrong, V. N. Brahmbhatt, K. Smolarczyk, M. T. Harrison, M. Costa, N. B. Mugridge, I. Sedliarou, N. A. Grimes, D. L. Kiss, B. Stillman, C. L. Hann, G. L. Gallia, R. M. Graham and H. Brahmbhatt, PLoS One, 2016, 11, e0151832 CrossRef.
- I. Sardi, O. Fantappiè, G. la Marca, M. G. Giovannini, A. L. Iorio, M. da Ros, S. Malvagia, S. Cardellicchio, L. Giunti, M. de Martino and R. Mazzanti, Cancer Lett., 2014, 353, 242–247 CrossRef CAS PubMed.
- Y. Octavia, C. G. Tocchetti, K. L. Gabrielson, S. Janssens, H. J. Crijns and A. L. Moens, J. Mol. Cell. Cardiol., 2012, 52, 1213–1225 CrossRef CAS PubMed.
- R. Mohammadpour, M. Yazdimamaghani, D. L. Cheney, J. Jedrzkiewicz and H. Ghandehari, J. Controlled Release, 2019, 304, 216–232 CrossRef CAS.
- R. Mohammadpour, D. L. Cheney, J. W. Grunberger, M. Yazdimamaghani, J. Jedrzkiewicz, K. J. Isaacson, M. A. Dobrovolskaia and H. Ghandehari, J. Controlled Release, 2020, 324, 471–481 CrossRef CAS PubMed.
- M. Manzano, M. Vallet-Regí and M. Vallet-Regí, Adv. Funct. Mater., 2019, 30, 1902634 CrossRef.
- T. D. Brown, N. Habibi, D. Wu, J. Lahann and S. Mitragotri, ACS Biomater. Sci. Eng., 2020, 6, 4916–4928 CrossRef CAS.
- M. M. Abeer, P. Rewatkar, Z. Qu, M. Talekar, F. Kleitz, R. Schmid, M. Lindén, T. Kumeria and A. Popat, J. Controlled Release, 2020, 326, 544–555 CrossRef CAS PubMed.
- E. Juère, R. Caillard, D. Marko, G. Del Favero and F. Kleitz, Chem. – Eur. J., 2020, 26, 5195–5199 CrossRef.
- C. Xu, M. Yu, O. Noonan, J. Zhang, H. Song, H. Zhang, C. Lei, Y. Niu, X. Huang, Y. Yang and C. Yu, Small, 2015, 11, 5949–5955 CrossRef CAS PubMed.
- J. Feng, Y. Liu, C. Liu, W. Hu, C. Zhang, S. Li, Y. Song and C. Yu, Microporous Mesoporous Mater., 2020, 307, 110504 CrossRef CAS.
- Z. Chaudhary, S. Subramaniam, G. M. Khan, M. M. Abeer, Z. Qu, T. Janjua, T. Kumeria, J. Batra and A. Popat, Front. Bioeng. Biotechnol., 2019, 7, 225 CrossRef.
- Z. Chaudhary, G. M. Khan, M. M. Abeer, N. Pujara, B. Wan-Chi Tse, M. A. McGuckin, A. Popat and T. Kumeria, Biomater. Sci., 2019, 7, 5002–5015 RSC.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS.
- A. Al-Shehri, M. E. Favretto, P. V. Ioannou, I. A. Romero, P.-O. Couraud, B. B. Weksler, T. L. Parker and P. Kallinteri, Pharm. Res., 2015, 32, 1072–1083 CrossRef CAS PubMed.
- C. Puech, S. Hodin, V. Forest, Z. He, P. Mismetti, X. Delavenne and N. Perek, Int. J. Pharm., 2018, 551, 281–289 CrossRef CAS PubMed.
- K. C. Tjandra, N. McCarthy, L. Yang, A. J. Laos, G. Sharbeen, P. A. Phillips, H. Forgham, S. M. Sagnella, R. M. Whan, M. Kavallaris, P. Thordarson and J. A. McCarroll, J. Med. Chem., 2020, 63, 2181–2193 CrossRef CAS PubMed.
- M. Shilo, A. Sharon, K. Baranes, M. Motiei, J.-P. M. Lellouche and R. Popovtzer, J. Nanobiotechnol., 2015, 13, 19 CrossRef PubMed.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef.
- C. Urata, Y. Aoyama, A. Tonegawa, Y. Yamauchi and K. Kuroda, Chem. Commun., 2009, 5094 RSC.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862 RSC.
- K.-C. Kao and C.-Y. Mou, Microporous Mesoporous Mater., 2013, 169, 7–15 CrossRef CAS.
- S. Ma, Y. Wang and Y. Zhu, J. Porous Mater., 2011, 18, 233–239 CrossRef CAS.
- M. M. Abeer, A. K. Meka, N. Pujara, T. Kumeria, E. Strounina, R. Nunes, A. Costa, B. Sarmento, S. Z. Hasnain, B. P. Ross and A. Popat, Pharmaceutics, 2019, 11, 418 CrossRef CAS.
- A. K. Meka, P. L. Abbaraju, H. Song, C. Xu, J. Zhang, H. Zhang, M. Yu and C. Yu, Small, 2016, 12, 5169–5177 CrossRef CAS PubMed.
- Y. Wang, H. Song, M. Yu, C. Xu, Y. Liu, J. Tang, Y. Yang and C. Yu, J. Mater. Chem. B, 2018, 6, 4089–4095 RSC.
- M. Yu, L. Zhou, J. Zhang, P. Yuan, P. Thorn, W. Gu and C. Yu, J. Colloid Interface Sci., 2012, 376, 67–75 CrossRef CAS.
- M. Barczak, New J. Chem., 2018, 42, 4182–4191 RSC.
- S. Saha, V. Yakati, G. Shankar, M. M. C. S. Jaggarapu, G. Moku, K. Madhusudana, R. Banerjee, S. Ramkrishna, R. Srinivas and A. Chaudhuri, J. Mater. Chem. B, 2020, 8, 4318–4330 RSC.
- D. Gonzalez-Carter, A. E. Goode, D. Kiryushko, S. Masuda, S. Hu, R. Lopes-Rodrigues, D. T. Dexter, M. S. P. Shaffer and A. E. Porter, Nanoscale, 2019, 11, 22054–22069 RSC.
- T. G. F. Souza, V. S. T. Ciminelli and N. D. S. Mohallem, J. Phys. Conf. Ser., 2016, 733, 012039 CrossRef.
- R. Guillet-Nicolas, A. Popat, J.-L. Bridot, G. Monteith, S. Z. Qiao and F. Kleitz, Angew. Chem., Int. Ed., 2013, 52, 2318–2322 CrossRef CAS PubMed.
- M. Kaasalainen, V. Aseyev, E. von Haartman, D. Ş. Karaman, E. Mäkilä, H. Tenhu, J. Rosenholm and J. Salonen, Nanoscale Res. Lett., 2017, 12, 74 CrossRef PubMed.
- Y.-C. Kuo and Y.-C. Chen, Int. J. Pharm., 2015, 479, 138–149 CrossRef CAS PubMed.
- S. Kumari, S. M. Ahsan, J. M. Kumar, A. K. Kondapi and N. M. Rao, Sci. Rep., 2017, 7, 6602 CrossRef.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- J. Hoarau-Véchot, A. Rafii, C. Touboul and J. Pasquier, Int. J. Mol. Sci., 2018, 19, 181 CrossRef PubMed.
- J. Guyon, L. Andrique, N. Pujol, G. V. Røsland, G. Recher, A. Bikfalvi and T. Daubon, J. Visualized Exp., 2020, 158 DOI:10.3791/60998.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS PubMed.
- D. Jain, J. Nucl. Cardiol., 2000, 7, 53–62 CrossRef CAS PubMed.
- T. Safra, F. Muggia, S. Jeffers, D. D. Tsao-Wei, S. Groshen, O. Lyass, R. Henderson, G. Berry and A. Gabizon, Ann. Oncol., 2000, 11, 1029–1034 CrossRef CAS PubMed.
- P. W. Halcrow, N. Khan, G. Datta, J. E. Ohm, X. Chen and J. D. Geiger, Cancer Rep., 2019, 2, e1193 CAS.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 31703, Doxorubicin, https://pubchem.ncbi.nlm.nih.gov/compound/Doxorubicin, (accessed 26 August 2021).
- S. A. Kamba, M. Ismail, S. H. Hussein-Al-Ali, T. A. T. Ibrahim and Z. A. B. Zakaria, Molecules, 2013, 18, 10580–10598 CrossRef CAS PubMed.
- G. Bu, E. A. Maksymovitch, H. Geuze and A. L. Schwartz, J. Biol. Chem., 1994, 269, 29874–29882 CrossRef CAS.
- M. V. Sheraton, G. G. Y. Y. Chiew, V. Melnikov, E. Y. Tan, K. Q. Luo, N. Verma and P. M. A. A. Sloot, BMC Cancer, 2020, 20, 1201 CrossRef CAS.
- N. Qi, Y. Zhang, X. Tang and A. Li, Drug Des., Dev. Ther., 2020, 14, 1825–1836 CrossRef CAS PubMed.
- B. Kim, G. Han, B. J. Toley, C. Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465–472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03553c |
| This journal is © The Royal Society of Chemistry 2021 |