
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo step promotion of a hot tumor immune environment by gold decorated iron oxide nanoflowers and light-triggered mild hyperthermia†
Alba
Nicolas-Boluda
ab,
Gautier
Laurent
c,
Rana
Bazzi
c,
Stéphane
Roux
c,
Emmanuel
Donnadieu
 b and
Florence
Gazeau
*a
b and
Florence
Gazeau
*a
aUniversité de Paris, MSC, CNRS-UMR 7057, Paris, France. E-mail: florence.gazeau@u-paris.fr
bUniversité de Paris, Institut Cochin, INSERM U1016/CNRS UMR 8104, Paris, France
cInstitut UTINAM, CNRS UMR 6213, Université Bourgogne Franche-Comté, Besançon, France
First published on 14th October 2021
Abstract
Nanoparticle-mediated photothermal therapy (PTT) is an emerging modality to treat tumors with both spatial and temporal control provided by light activation. Gold decorated iron oxide nanoflowers (GIONF) are good candidates for PTT due to their biocompatibility, biodegradability and light-to-heat conversion. Profound changes in the tumor immune environment might be early induced by the gold and iron oxide metallic agents in addition to the photothermal effects. This study aims to elucidate the outcome of GIONF on their own, and of GIONF-induced mild hyperthermia in the tumor immune infiltrate in a murine model of triple negative breast cancer. First we explored the effects of 24 h GIONF exposure on bone-marrow derived macrophages (BMDM), revealing significant effects on the BMDM phenotype and secretion, 6 days post-incubation, with important downregulation of several cytokines and MHCII expression, predominantly towards a pro-inflammatory response. Intratumoral administration of GIONF promoted an increase in monocyte recruitment at day 1 post-administration, shifting towards a pro-inflammatory anti-tumor microenvironment with lower Treg population and a 4 fold lower CD4/CD8 ratio compared to the control at day 12. On top of the GIONF effects, mild hyperthermia (43 °C for 15 min), although it does not induce significant changes in tumor growth, resulted in an additional increase of CD8+ T lymphocytes and pro-inflammatory cytokines. The combination of a timely controlled immune response to GIONF and to mild hyperthermia could be used as a remotely triggered adjuvant treatment to immunotherapy approaches at the best favorable time-window.
Introduction
Nanoparticle-mediated physical therapies based on the use of electromagnetic or acoustic waves (such as high-intensity focused ultrasound, magnetic hyperthermia or photothermal therapy (PTT)) have been intensely studied in the last decade. They represent an alternative or combinatorial strategy to be applied in chemoresistant tumors. The main principle of these therapies is the use of nanoparticles that can remotely be activated by an external stimulus such as a laser or an alternating magnetic field, generating a controlled nanoparticle-induced hyperthermia in the target region. In particular, PTT is a minimally invasive hyperthermia treatment based on the conversion of optical energy into thermal energy by nanoparticles upon near-infrared laser irradiation. Remotely activated nanoparticles offer high specificity, minimal invasiveness and spatiotemporal selectivity. Hence it dramatically improves the spatial and temporal precision of the hyperthermic treatment that can be combined or alternated with the administration of chemotherapy or immunotherapy at the best favorable “time window”. In a clinical setup, PTT is making possible by the endoscopic intratumoral (IT) administration of nanoparticles (considered as a medical device) which is the best option to maximize the tumor concentration of nanoheaters and spare the surrounding healthy tissue. PTT is nowadays regarded as a potent precision nanomedicine approach to augment the efficacy of standard of care chemotherapy and/or radiotherapy by modulating the tumor microenvironment (TME) in solid tumors, reaching the stage of clinical trials.1,2 However the multifactorial effect of nanoparticle-mediated PTT is not fully understood and might strongly depend on the nanoparticles used as well as on the complexity of the TME.3 Particularly immune cells and their interplay with tumor and stromal cells play a decisive role in the different stages of tumor development, up to the point that the reinvigoration of the immune response against cancer using immunotherapies can lead to complete regression of tumors in certain cases. The immune status of solid tumors may vary from a “hot” inflammatory environment comprising inflammatory macrophages and cytotoxic T cells able to attack the tumor cells to an immune-excluded TME or immune desert in which T cells cannot reach the tumor cells or cannot penetrate the tumor.4 The concept of T-cell driven immunosurveillance against cancer has led to the development of immunotherapies based either on the reinvigoration of the T cell function in situ, mainly via antibodies targeting immune checkpoint receptors, or on the transfer of genetically modified autologous T cells with enhanced antitumoral activity, mainly chimeric antigen receptor (CAR)-expressing T cells.5,6 Both strategies have provided an unprecedented level of long-term antitumor activity in patients with several metastatic cancers. However, the majority of patients with advanced cancers still do not experience sustained clinical benefits from immunotherapy highlighting the presence of barriers that one needs to identify in order to overcome them and propose more powerful strategies. Recently, PTT has been shown to transform a “cold” immunosuppressive TME into a “hot” inflammatory TME in preclinical syngenic tumor models.7–10 However, there is a need for better understanding the multifactorial effects of local heating in the TME depending on the tumor type, which should give the opportunity to finely control their biological outcome and synergy with other treatment modalities such as immunotherapies. Moreover, nanoparticles by themselves can play the role of an immunostimulating adjuvant by modulating the activation of immune cells in various contexts. In particular, metallic nanoparticles, especially gold and iron based, have the potential to be used as vaccine adjuvants.11 Their high surface for molecular interactions and the direct activation or modulation of intracellular signaling pathways allow them to trigger the recruitment of myeloid and lymphoid cells.12 Nanoparticles do not always induce a pro-inflammatory immune response, but can also exert an anti-inflammatory reaction. This depends greatly on the physico-chemical properties such as size and shape,13 coating,14 protein corona or metal nature.15,16 For this reason, the effect of metallic nanoparticles on the immune system should be considered prior to their use for therapy. Previous studies have already demonstrated unexpected effects induced by nanoparticles in the tumor immune infiltrate resulting in positive therapeutic effects. In particular, the induction of a phenotypic switch in macrophages from an M2-like anti-inflammatory phenotype to a pro-inflammatory M1-like phenotype by FDA approved iron oxide nanoparticles has been reported. This switch resulted in an inhibition of tumor growth caused solely by the exposure of iron oxide nanoparticles.17 Hence nanoparticle-mediated PTT can result in the reprogrammation of the immune response against tumors in at least two successive or parallel stages resulting from interactions with nanoparticles and from generated local hyperthermia and subsequent perturbation of tissue homeostasis.In this study, we evaluate the effect of gold decorated iron oxide nanoflowers (GIONF) and of GIONF-mediated PTT on the tumor immune infiltrate in a mouse tumor model. The choice of nanoparticles for this study was based mainly on their multimodal applications: iron oxide nanoflowers (IONF) are interesting due to their multifunctional properties ranging from imaging and diagnosis to therapeutics.18 Their outstanding cooperative magnetic properties19 make them excellent nanoheaters under alternating magnetic field and MRI contrast agents coupled with a controlled biodegradability in the cellular environment.20 On the other hand, ultrasmall gold nanoparticles have been proposed as radiosensitizing nanoagents to enhance the efficacy and local control of radiotherapy, while allowing image-guided therapy and fast renal clearance.21 The grafting of ultrasmall radiosensitizing nanoparticles onto large IONF results in a hybrid multimodal nanoparticle suitable for combinatory therapies such as PTT, radiosensitization and magnetic hyperthermia. Previous studies performed by our group have already demonstrated the efficacy of these nanoparticles as PTT agents22 for softening and shrinking of cholangiocarcinoma human tumor xenograft. GIONF were shown to have particular behavior in the context of desmoplastic tumors, targeting the abundant population of cancer associated fibroblasts. However, many of the preclinical studies are based on experiments in which human tumor cell lines are grafted into immune-deficient mice. In such xenograft settings, the effect of nanoparticles on the tumor immune landscape is somewhat overlooked. Interestingly several studies have shown that the pharmacokinetics of nanoparticles and the tumor uptake were different in immune-deficient mice compared to the immune-competent mice.23
In the present study we focus on the immunomodulation properties of GIONF and GIONF-induced mild hyperthermia (43 °C for 15 min) in the context of triple negative breast cancer. For this purpose, we used the preclinical model of MET-1 murine breast tumor that was derived from the genetic engineered mouse model MMTV-PyMT (mouse mammary tumor virus-polyoma middle tumor-antigen) mouse mammary gland carcinoma model of breast cancer.24 We first evaluate the effect of GIONF alone on bone-marrow derived macrophages. Then we study the influence of intratumoral administration of GIONF on the immune infiltrate of this breast tumor model. Finally, the differential effect of GIONF-mediated mild hyperthermia is investigated and we shed light on its potential application as a therapeutic vaccine.
Results and discussion
GIONFs change the phenotype of bone-marrow derived macrophages and their cytokine secretion, predominantly towards a pro-inflammatory response
Gold-decorated iron oxide nanoflowers (GIONF) used in this study are isotropic clusters of nanocrystalline iron oxide nanoparticles of approximately 11 nm that are assembled in a flower-like monocrystalline structure with a diameter of 25–30 nm coated with dithiolated diethylenetriaminepentaacetic acid gold NPs (Au@DTDTPA) of 2–3 nm, the number of gold nanoparticles per IONF being approximatively 28. They have been fully characterized in a previous study.22 These hybrid nanostructures are designed to maximize the tumor retention of gold NPs and prolong their radiosensitization effects which is otherwise compromised by premature renal clearance of the tiny gold NPs.25 Gold decoration also improves the efficiency of iron oxide nanoflowers as photothermal agents for PTT under NIR light irradiation. Our previous study has shown that cell viability was not affected by GIONF incubated for 24 h up to 500 μg mL−1 iron concentration in murine RAW264.7 macrophages.22 In order to investigate the immune modulatory properties of these hybrid metallic nanostructures, we first examined the response of bone-marrow derived macrophages to the exposure to GIONF at a concentration of [Fe] = 122 μg mL−1.Bone-marrow derived macrophages (BMDM) were derived from BALB/C mouse tibia bone marrow following a protocol published elsewhere.26 BMDM were incubated with GIONF overnight, rinsed and left in culture up to 6 days without GIONF. The supernatant was analyzed using a multiplex cytokine kit, in order to determine the levels of different cytokines released by BMDM that have been incubated or not with GIONF (Fig. 1A). Cells, on the other hand, were analyzed by flow cytometry in order to detect changes in their phenotype (Fig. 1A). Macrophages were defined as CD11b+ Ly6C− Ly6G− F4/80+ as depicted in Fig. S1A.†
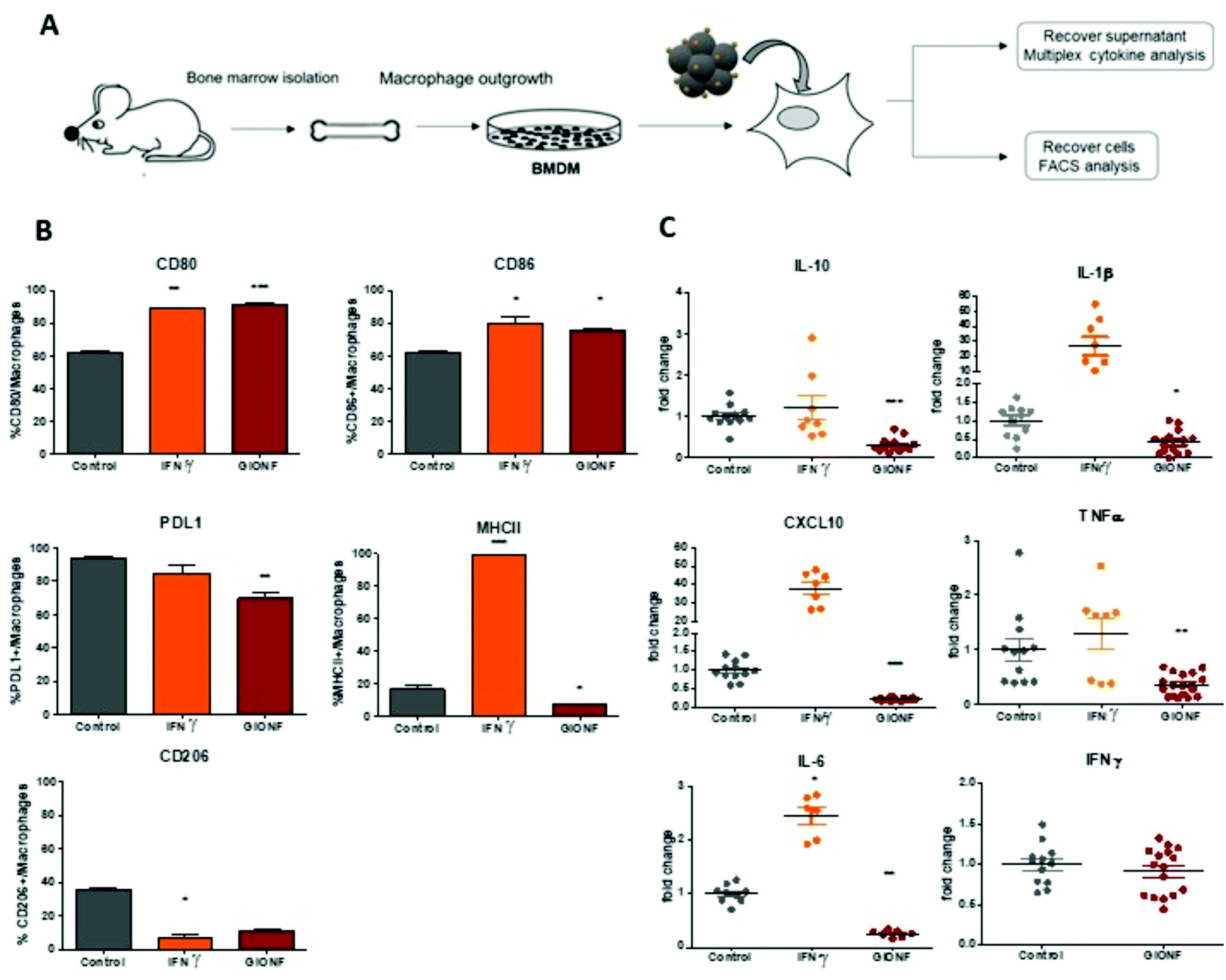 | ||
| Fig. 1 (A) Schema of the experimental setup for the analysis of the effect of GIONF exposition on BMDMs. (B) Percentage of CD80+, CD86+, PDL1+, MHCII+ and CD206+ macrophages analyzed by flow cytometry after 6 days under control conditions or after IFNγ (2 ng mL−1) or GIONF (122 μg mL−1) exposition for 24 h. (C) Multiplex dosage of cytokines in BMDM culture of the supernatant 6 days after treatment with IFNγ (2 ng mL−1) or GIONF (122 μg mL−1) for 24 h. Results are shown as mean ± SEM of n = 3–6 per condition of three independent experiments. One-way ANOVA. Kruskal–Wallis *** p-value <0.001 ** p-value <0.01 *p-value <0.05. | ||
Macrophages undergo specific differentiation depending on the local tissue environment. They respond to environmental cues within tissues such as damaged cells, activated lymphocytes, or microbial products and nanoparticles to differentiate into distinct functional phenotypes.17 The M1 macrophage phenotype is characterized by the production of high levels of pro-inflammatory cytokines, an ability to mediate resistance to pathogens, strong microbicidal properties and promotion of Th1 responses. M1 macrophages are implicated in initiating and sustaining inflammation, which in the context of cancer can activate the immune response against cancer cells. In contrast, M2 macrophages are characterized by their involvement in tissue remodeling, efficient phagocytic activity, immune regulation and tumor promotion via tumor immunosuppression. In the tumoral context, the reprogramming of protumoral M2 macrophages into inflammatory M1 subtypes is part of antitumoral strategies that can be mediated by iron oxide nanoparticles.17
M1-like markers such as the co-stimulatory molecules CD80 and CD86 and the major histocompatibility complex class II molecules (MHCII) as well as the M2-like marker CD206 were quantified by flow cytometry in BMDMs exposed or not to GIONF following the gating strategy described in ESI Fig. S1† (macrophages being first gated as CD45+ CD11b+ Ly6C− Ly6G− F4/80+ CD64+ cells). As a positive control for classical M1 macrophage activation, BMDMs were challenged with IFNγ.
BMDM exposure to GIONF induced an increase in the expression of several M1-like related markers notably CD86 and CD80. Around 80% of cells exposed to GIONF were CD86+ and CD80+ in comparison with about 60% for untreated conditions, reaching the level of the positive control challenged with IFNγ. However, GIONF also induced a significant downregulation of MHCII. The main function of major histocompatibility complex (MHC) class II molecules is to present processed antigens, which are derived primarily from exogenous sources, to CD4+ T-lymphocytes. MHC class II molecules thereby are critical for the initiation of the antigen-specific immune response. The fraction of MHCII+ macrophages was reduced by a factor of two, counting 10% of GIONF-treated macrophages (Fig. 1B). Regarding the M2 polarization, the number of CD206+ cells – a marker of M2 mouse macrophage also called mannose receptor C type 1 – was strongly reduced in GIONF-treated cells to the level of cells treated with IFNγ, a positive control for M1 polarization.
Programmed cell death protein 1 (PD-1) and its ligand PD-L1 and PD-L2 are coinhibitory molecules on the surface of T lymphocytes, suppressing T cell activation and allowing tumor immune escape. PD-L1 can be located in hematopoietic cells, including T cells, B cells, dendritic cells, and macrophages. We thus checked the expression of PD-L1 in BMDM following exposition to GIONF. Interestingly the immune suppressive marker PD-L1 was downregulated in GIONF-exposed macrophages, resulting in a 15% decrease of PD-L1+ macrophages (Fig. 1B).
Additionally, we observed that the uptake of GIONF by macrophages could be detected using the side scattering in flow cytometry, which generally provides information about the internal complexity (i.e. granularity) of a cell. In this case the granularity of the cell changes when loaded with GIONF (Fig. S1C†).
To complement this analysis of the cell phenotype, the cytokines released by the macrophages were analyzed using a multiplex assay. The analysis revealed that GIONF provokes a decrease in the secretion of the pro-inflammatory cytokines TNFα, IL-1β, IL-6 and CXCL10 normally abundantly secreted by M1-like macrophages (as we can see in the IFNγ positive control) (Fig. 1C). However, the secretion of the immune suppressive cytokine (IL-10), normally secreted by M2-like macrophages, was also decreased by GIONF exposure (Fig. 1C). IFNγ secretion was not increased due to GIONF exposure (Fig. 1C).
Overall our results indicate that GIONF exposure for 24 h has significant effects on the BMDM phenotype and secretion, 6 days post-incubation. Particularly this reveals important downregulation of several cytokines and MHCII expression. We cannot simply classify these effects as a polarization of macrophages towards an M1 or an M2-like phenotype. A possible explanation would be that the treated macrophages are in an intermediate stage of differentiation at this given time-point. The M1/M2 classification of macrophages is now considered an oversimplified approach that does not adequately fit the spectrum of macrophage populations. Particularly in the tumoral context the identification of tumor associated macrophages (TAMs) is not adequately described by M1/M2 classification.27 The cytokine balance in the TME can reflect a variety of TAM subpopulations, expressing a continuum of M1 and M2 polarization hallmarks.28 It is however generally admitted that switching TAMs to a predominantly M1 phenotype might be beneficial for anti-cancer immunotherapeutic treatment.29 For this reason, we thought to characterize the effects of GIONF directly in the TME of the MET-1 syngeneic mouse model of triple-negative breast cancer.
GIONF induces a shift towards an antitumor immune environment 12 days after intratumoral administration
In the following we examined how tumor immunogenicity can be influenced by the intratumoral injection of GIONFs. BALB/C mice were inoculated with MET-1 cells to induce the formation of subcutaneous tumors. When the tumor reached 5–6 mm3 size, GIONF were injected intratumorally (420 μg iron). Intratumoral injection was chosen because associated with PTT, it is one of the clinically relevant options to treat unresectable tumors that must be shrunk before surgery. It allows controlling and maximizing nanoparticle concentration in the tumor (which is important for PTT efficiency) while minimizing release in the systemic circulation and accumulation in organs of the reticulo-endothelial system. Regarding the investigation of the immune response to GIONF, intratumoral administration allows getting rid of the systemic effect (e.g. opsonization, capture of nanoparticles by the RES system) downstream to nanoparticle infiltration in the tumor. The mice were sacrificed at two different time points after injection in order to analyze the early and late effects of GIONF in the immune infiltrate. The two chosen time points were 1 day and 12 days after GIONF injection (Fig. S2A†). To confirm the diffusion of GIONF in the TME, histology analysis of the tumors 24 hours after injection was performed. MET-1 tumors have a structure that resembles that of certain breast tumors with stromal and tumor compartments that are intertwined in contrast to the clear compartmentalization in desmoplastic tumors. Results confirm the diffusion inside the tumor and their uptake by cells in the tumor as well as localization in the extracellular matrix (Fig. S3†). Tumors were analyzed by flow cytometry and the different myeloid and lymphoid populations were analyzed following the gating strategies shown in Fig. S4.† In parallel, the tumor growth was monitored.GIONF injection into MET-1 tumors did not affect tumor growth over the 12 days post-treatment (Fig. S2B†). However, 24 hours after GIONF injection, there was a significant recruitment of monocytes in the tumor compared to the control (Fig. 2A) whereas all the other cellular subsets do not seem to be significantly influenced by GIONF at this early time point. Such monocyte recruitment can be due to the need to process the GIONF present in the tumor and would be shortly followed by macrophage differentiation and uptake of nanoparticles. Indeed, we observe a non-significant tendency towards the increase of F4/80+ macrophage (CD45+ CD11b+ Ly6C− Ly6G− F4/80+ CD64+) infiltration at 24 hours. In contrast, 12 days after GIONF injection, the situation is inverted. The monocyte population is significantly decreased compared to the control at day 12. On the other hand, dendritic cells, very rare cells within tumors, show a tendency for a decrease at 24 hours and no changes at 12 days.
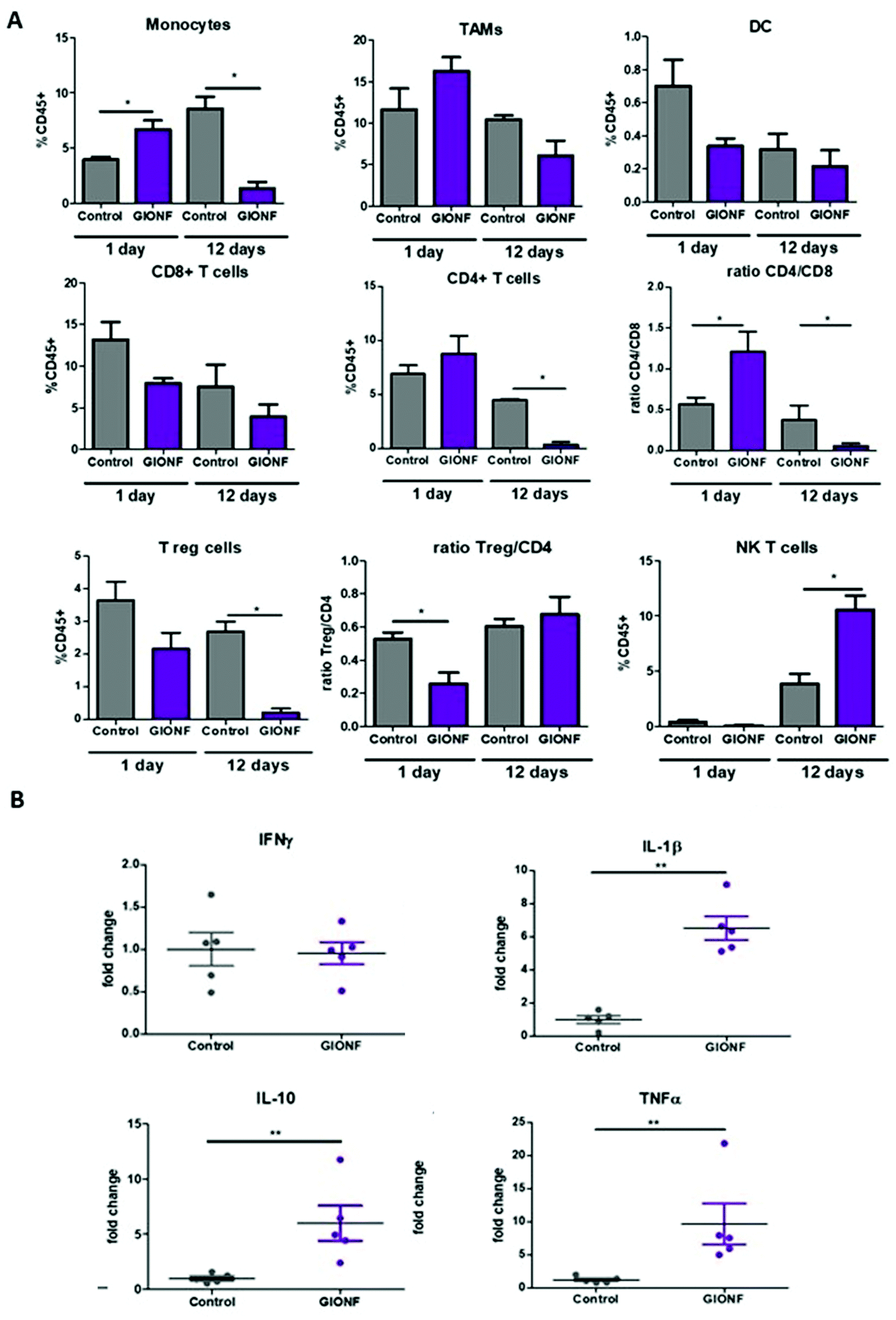 | ||
| Fig. 2 Effect of GIONF in the MET-1 immune infiltrate at early and late time points. (A) Percentage of the different myeloid (monocytes, tumor-associated macrophages (TAMs), dendritic cells (DCs)) and lymphoid (CD8+ T cells, CD4+ T cells, T regulatory cells (CD4+/FoxP3+, Treg, NK T cells (Nkp46+) in control (PBS) and GIONF treated tumors at two different time points 1 and 12 days after GIONF injection. Markers and gating strategies are indicated in Fig. S4.† (B) Multiple dosage of cytokines in MET-1 tumor slice supernatants at 12 days after GIONF injection. Results are shown as mean ± SEM of n = 5–6 per condition. T-Student. Mann Whitney (control vs. GIONF in each point), **p-value <0.01 *p-value <0.05. | ||
The tumor infiltrating lymphocytes play a major role in cancer surveillance. Both CD4 and CD8 T cell responses and their interplay are part of the cancer immune cycle and both populations significantly influence the clinical outcome and control of tumor growth. Cytotoxic CD8+ T cells are the key effector cell population mediating effective anti-tumor immunity resulting in better clinical outcomes. CD4+ T cells are critical for priming of tumor-specific CD8+ T cells. However, intra-tumoral CD4+ T cells can have negative prognostic effects on breast cancer patient outcomes, mediated by the immunosuppressive Tregs subpopulation, which suppress or downregulate induction and proliferation of effector T cells. Tregs prevents effective anti-tumor immunity representing a major obstacle to the success of immunotherapy. Here we observed a significant decrease of CD4+ T cells 12 days after GIONF injection compared to the control tumor at the same time point as well as to GIONF-treated tumors at day 1 (Fig. 2A). This drastic decrease of CD4+ T cells at day 12 also impacts the T reg population (that represents approximately 50–60% of CD4+ T cells in this tumor model). In fact, the Treg/CD4 ratio is significantly decreased at the early time point (1 day after injection). This long lasting decrease in CD4 and in particular Tregs might contribute to a pro-inflammatory environment, potentiating anti-tumoral actions. CD8+ T cells tend to decrease in GIONF injected tumors both at day 1 and day 12. However, we observe a switch in the CD4/CD8 ratio between the two time points (Fig. 2A) which dramatically reduces at day 12 in comparison with day 1, and more particularly for GIONF-treated tumors.
GIONFs induce first an increase of CD4/CD8 ratio at day 1 which turns to diminish drastically at day 12 in comparison with the control. A high CD4/CD8 ratio has been correlated with tumor progression in breast cancer tumor patients,30 hence this switch towards a lower CD4/CD8 contributes to the establishment of an anti-tumoral microenvironment. Consistently among regulating T cells, we also observed a significant increase in the NK T cell infiltration 12 days after GIONF injection (Fig. 2A).
To go deeper into the immune status of the MET-1 TME after administration of GIONF, the tumor cytokine network was quantified at day 12 post injection by collecting the supernatant of slices made from MET-1 tumors (Fig. 2B). Cytokine analysis at 12 days reveals an increase of pro-inflammatory cytokines such as TNF-α and IL-1β, but also of IL-10 known to have mainly anti-inflammatory functions. The increase of IL-10 secretion by monocytes is consistent with the depletion of CD4+ T cells.31
Even when nanoparticles have been injected into the tumor (intratumoral injection), it has been reported that they are capable of extravasation into the blood vessels and biodistribution among other organs32,33 particularly in liver and spleen resulting in off-target effects34–36 such as an alteration of lymphocyte subpopulation in the spleen due to exposure to copper nanoparticles.36 Based on this we decided to examine whether the lymphoid cells in the spleen were affected by i.t. injection of GIONF. Results show no significant effect on CD8+, CD4+ and NK T subsets at short term exposure (day 1) of GIONF. However, 12 days after GIONF i.t. injection there is a significant increase of NK T cells in the spleen (Fig. 3), which correlates with the increase of NK T cells seen in the tumor at the same time point (Fig. 2A).
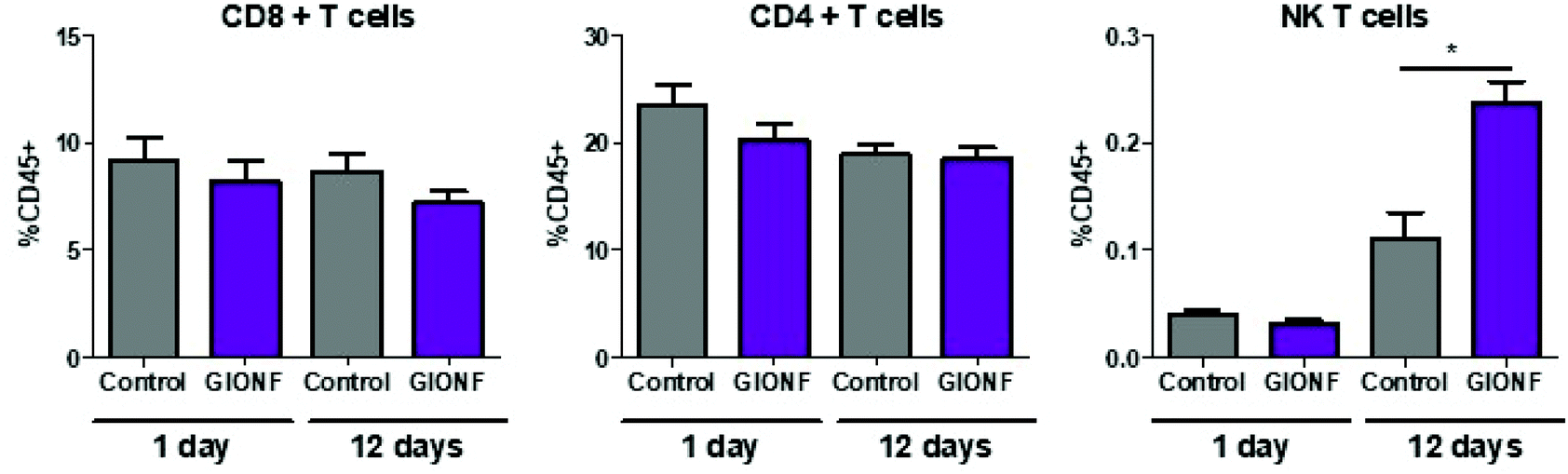 | ||
| Fig. 3 Changes in the lymphoid subset in the spleen 1 day and 12 days after GIONF exposure. Results are shown as mean ± SEM of n = 5–6 per condition. T-Student. Mann–Whitney (control vs. GIONF in each time point) * p-value <0.05. | ||
Overall, GIONF nanoparticles seem to induce a mild and transient inflammatory response in the tumor at early-time points with the increase in monocyte recruitment and macrophage differentiation presumably to phagocytose the injected nanoparticles. At longer time points, a dynamic shift towards a more anti-tumor microenvironment 12 days after injection was revealed by a 15 fold and 10 fold lower proportion of CD4+ T and Treg cells, respectively, a 4 fold lower CD4/CD8 ratio and a 3 fold increase in NK T cells in comparison with the control at the same time-point accompanied by pro-inflammatory cytokines such as TNF-α and IL-1β.
GIONF-mediated mild photo-hyperthermia in the MET-1 model triggers a delayed infiltration of cytotoxic CD8+ T cells
Once the effect of GIONF in the tumor immune infiltrate was evaluated, we moved on to determine the additional outcome of GIONF-mediated PTT in the tumor immune infiltrate. We recently demonstrated the use of GIONF as PTT agents in a model of xenografted cholangiocarcinoma.22 Here, we first confirmed GIONF internalization in MET-1 tumors cells and their heating capacities once internalized in cells. MET-1 cells were incubated with GIONF for 24 hours at [Fe] = 112, 56 and 11 μg mL−1. Quantification of iron by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES) shows that GIONF are taken up by MET-1 cell in a dose-dependent manner (Fig. 4A) without inducing cytotoxicity after 24 h incubation as shown by Alamar Blue assay (Fig. S5†). Upon 24 hours internalization in cells, GIONF are localized and confined in endo-lysosomal compartments (Fig. 4D). To check the heating capability of GIONF in the MET-1 tumor cell, cells were incubated for 24 hours at a concentration of [Fe] = 122 μg mL−1 (Fig. 4A) and washed to discard the excess of nanoparticles not internalized by cells. 1 × 106 GIONF-loaded MET-1 cells were pelleted and resuspended in a small volume (100 μL) for PTT experiments. The cell pellet was then irradiated with an 808 nm laser at a power of 2.2 W cm−2 for 5 minutes (Fig. 4A). GIONF internalized in MET-1 cells resulted in moderate heating efficiencies, reaching an increase of temperature of around 2 °C (Fig. 4B).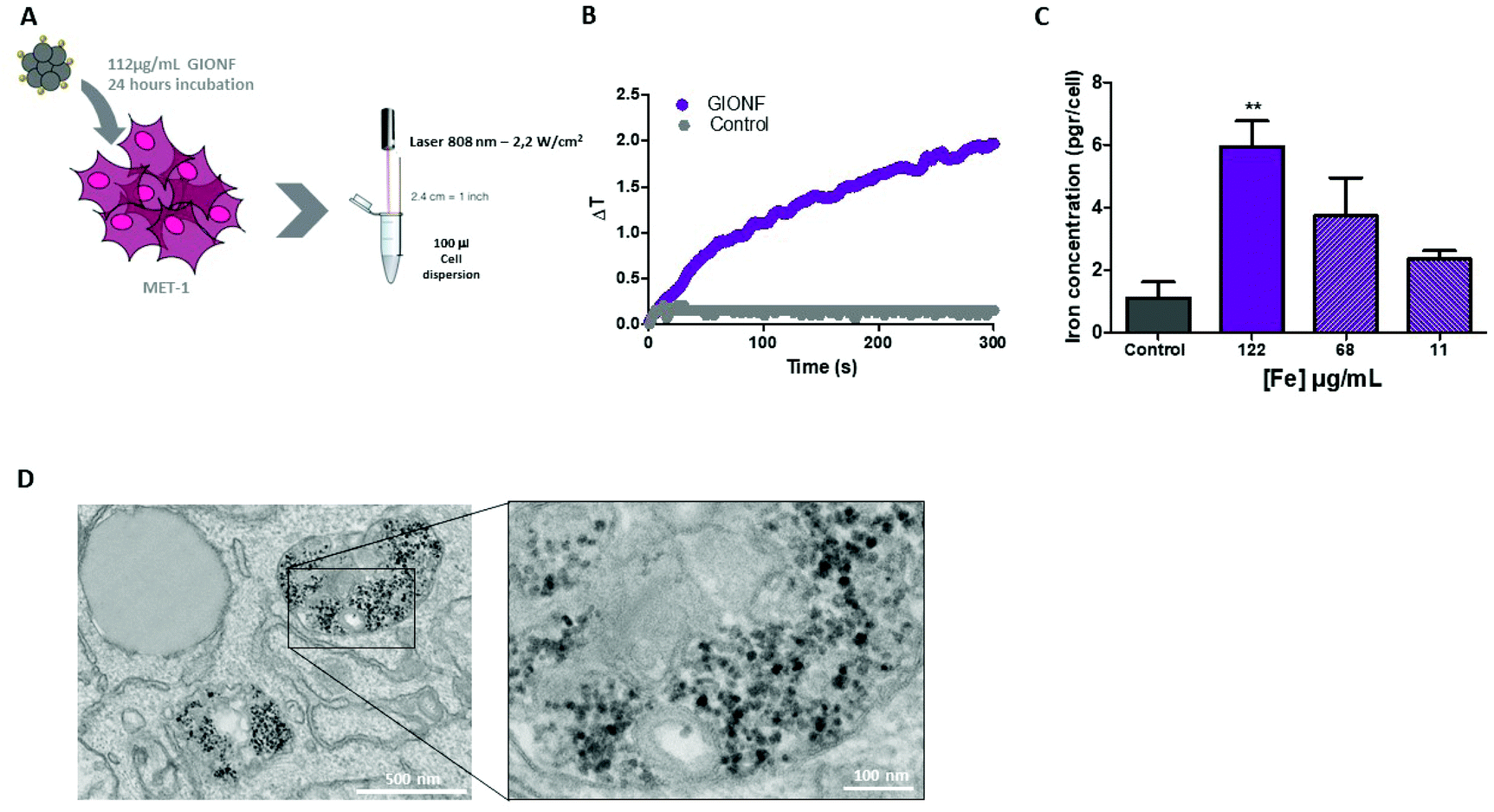 | ||
| Fig. 4 GIONF heating capacity in MET-1 cells. (A) Illustrative schema of the experimental setup. (B) Temperature increase of GIONF internalized in MET-1 cells upon irradiation with an 808 nm laser (2.2 W cm−2). (C) Quantification of iron in MET-1 cells when incubated with GIONF at different concentrations: 112, 56 and 11 μg mL−1. (D) TEM images of MET-1 cells after 24-hour incubation with GIONF. | ||
Once we confirmed the heating capacity of GIONF in MET-1 cells, we performed PTT in vivo in the MET-1 tumor model. 24 hours after the intratumoral administration of GIONF, the tumors were irradiated with an 808 nm laser for 15 minutes at an average power laser of 1 W cm−2. The goal was to assess the effect of nanoparticle-mediated mild hyperthermia in the immune infiltrate; therefore the laser power was modulated in order to obtain an average surface temperature of 43 °C monitored using an infrared camera (Fig. 5C and D). The calculation of the cumulative equivalent minutes at 43 °C (CEM43 °C) is used to determine the thermal dose of a hyperthermia protocol and to compare different approaches.37 In this experiment the CEM43 °C was of 15. The mice were sacrificed at day 2 and day 13 days after GIONF administration, i.e. day 1 and day 12 after PTT, in order to analyze the early and late effects of GIONF-mediated PTT in the tumor immune infiltrate (Fig. 5A).
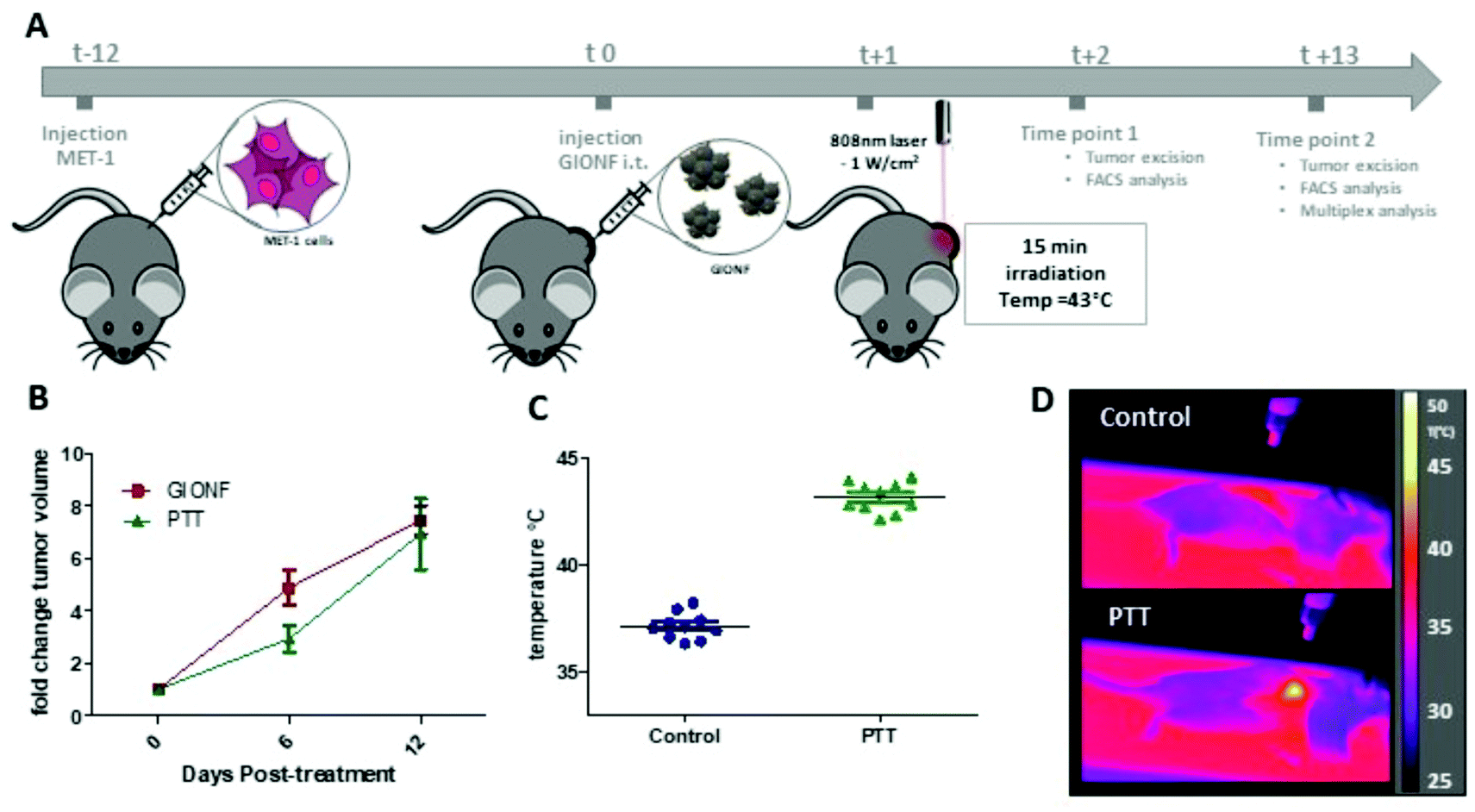 | ||
| Fig. 5 GIONF-mediated PTT in MET-1 tumors (A) schema of the experimental procedure. (B) MET-1 tumor volume fold change of GIONF-injected tumors and GIONF-mediated PTT tumors (n = 6 per group). (C) Average surface temperature of tumors during 15 min of 808 nm laser irradiation (1 W cm−2). (D) Representative infrared images illustrating the localized tumor hyperthermia. | ||
GIONF-mediated mild hyperthermia (one PTT session for 15 min, CEM43 °C = 15) did not have any significant impact on tumor growth (Fig. 5B). This sub-therapeutic condition was chosen deliberately to focus on the effect of intracellular local heating on the immune response independently of a massive cell death that would lead to tumor shrinkage. To isolate the effect of intracellular hyperthermia, we have compared the immune infiltrate after the PTT session to that of tumors injected with GIONF without laser activation considered as the control in this experiment. No significant difference could be observed in any of the analyzed immune cell populations at day 1 post-PTT (Fig. 6A). A slight decrease of the monocyte and macrophage populations as well as CD4+ cells was noted after PTT in GIONF-injected tumors. As we observed an early increase of monocytes/macrophages in GIONF-injected tumors that would presumably process the particles (Fig. 2A), these populations would have most probably been depleted by hyperthermia at short time post-irradiation. TEM pictures of tumors actually feature large aggregates of GIONF within endo-lyososomal compartments of dying cells (Fig. S6†).
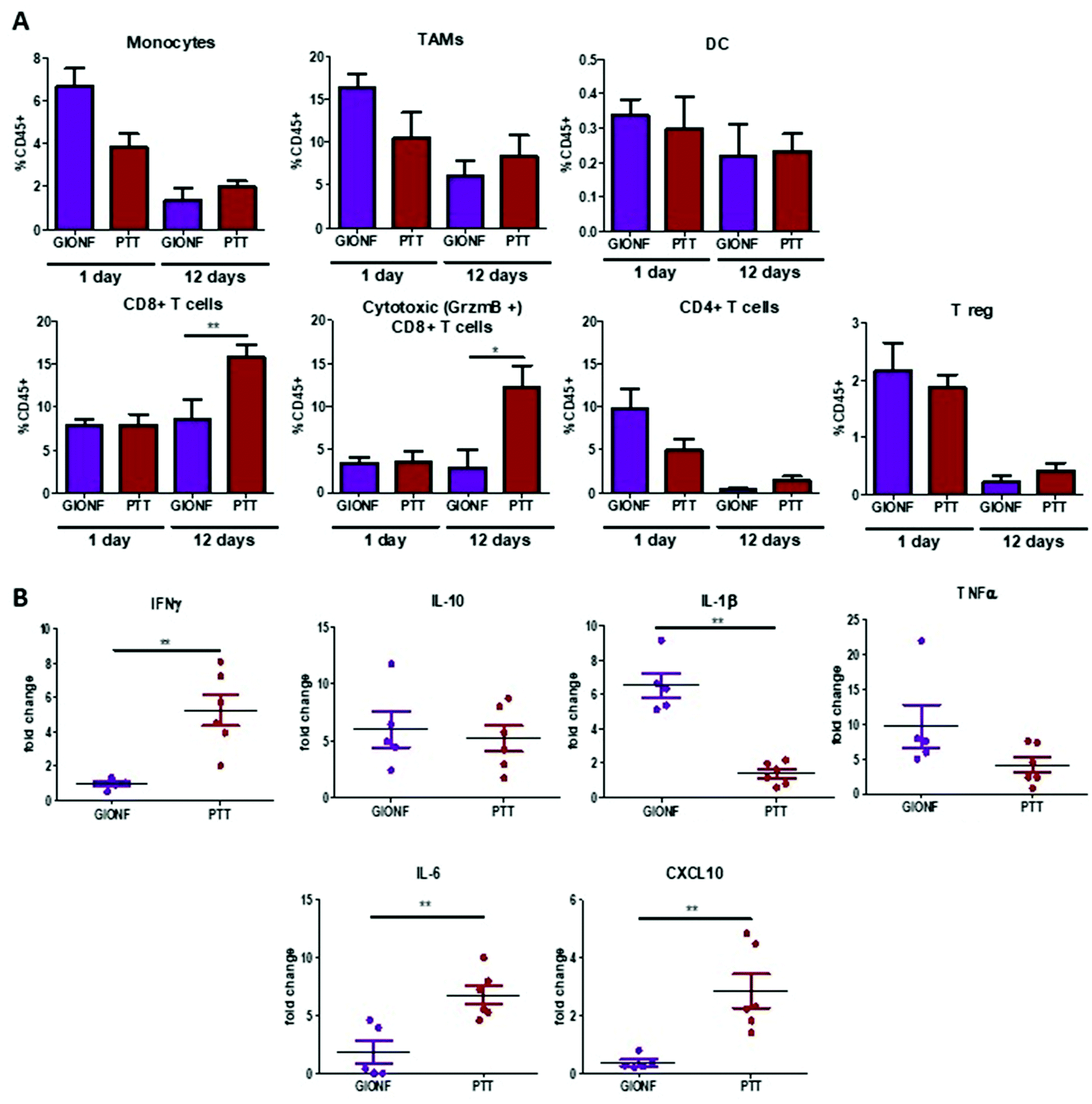 | ||
| Fig. 6 Effect of GIONF-mediated PTT in MET-1 immune infiltrate at early and late time points. (A) Percentage of different myeloid (monocytes, tumor associated macrophages (TAM), dendritic cells (DC) and lymphoid (CD8+ T cells, cytotoxic T cells (CD8+/GrzmB+), CD4+ T cells, T regulatory cells (CD4+/FoxP3+) population in control (injected with GIONF) and in GIONF-mediated PTT treated tumors at two different time points 1 and 12 days after GIONF-mediated PTT. (B) Multiplex dosage of cytokines in MET-1 tumor slice supernatants 12 days after GIONF-mediated PTT. Results are shown as mean ± SEM of n = 5–6 per condition. T-Student. Mann–Whitney (GIONF vs. PTT in each time point), **p-value <0.01 * p-value <0.05. | ||
When looking at the long term response to GIONF-mediated PTT at day 13, the most striking difference with non-irradiated GIONF injected tumors is a significant increase of CD8+ T cells and more particularly of the cytotoxic CD8+/GrzmB+ T cells (Fig. 6A). In contrast, the decrease in CD4+ T cells with respect to the earlier time points is observed for both GIONF-injected tumors, irradiated or not (Fig. 2A). The analysis of cytokine release by MET-1 tumor slices reveals a significant increase in IFNγ, IL-6 and CXCL10, and a significant decrease of IL-1β secretion (Fig. 6B) in comparison with the tumor injected with GIONF. The increase in IFNγ is correlated with the increase in cytotoxic CD8+ T cell infiltration since they are one of the main producers of this cytokine.38 IFNγ is a pro-inflammatory cytokine that activates macrophages and induces their differentiation into the M1-like phenotype. The increase in CXCL10 secretion is correlated with the levels of IFNγ, since different cell types such as monocytes, endothelial cells and fibroblasts secrete CXCL10 in response to IFNγ.39 CXCL10 contributes to the attraction of monocytes and T cells to the tumor.40
Overall, a single session of GIONF-mediated mild hyperthermia (CEM43 = 15) can elicit a progressive recruitment of CD8+ T cells to the tumors, together with inflammatory cytokines observed at day 12 post-treatment. A plausible hypothesis is that intracellular hyperthermia can trigger the release of antigens, intracellular organelles and damage associated molecular patterns (DAMPs) such as heat shock proteins. All of this can induce dendritic cell activation, through activation of the nuclear factor kappa-light-chain enhancer of activated B cells (NK-κβ) pathway, that in turns promotes the expression of co-stimulatory molecules CD80/CD86.41 Activated dendritic cells migrate to the tumor draining lymph node where they will prime CD8+ effector T cells. This would explain the increase in tumor CD8+ T cell infiltration observed here. Similar results have already been reported by others using different nanoparticles for PTT including aluminum oxide nanoparticles,42 Prussian blue nanoparticles,43 carbon nanotubes,44 and gold nanoshells.45 However, this increase in cytotoxic CD8+ T cells did not induce a significant change in tumor growth (Fig. 5B); this is probably due to the existence of other mechanisms that inhibit CD8+ T cells such as check point proteins. Although combination therapy is out of the scope of this study, our results suggest that the combination of PTT with immune check point inhibitors may be of interest for this setting. Several studies have already demonstrated the success of the combination of nanoparticle-mediated PTT with anti CTLA-4,43,44 anti PD-L1,46 and anti-PD1 therapy.47 GIONF-mediated PTT could therefore be used as an adjuvant therapy in combination with immunotherapies for the treatment of solid tumors. Our study also sheds light on the importance of looking at the immune response over time after treatment with both nanoparticles and mild hyperthermia in order to be able to select an appropriate time window for combination with immune check-point inhibitors.
Conclusion
Prior to using metallic nanoparticles for nanomedicine applications, it is important to precisely assess their effects on immune cells. As previously mentioned, until now most biocompatibility tests have been mainly based on checking the induction of apoptosis or cell proliferation; however immunogenicity is not always verified. This is of importance based on the results presented in this paper. GIONF, that proved to be biocompatible, have shown to trigger phenotypic changes in bone marrow derived macrophage in vitro and alter the tumor immune microenvironment in vivo. These changes did not prove to have a significant effect on tumor growth; however it is important to consider them since they may affect the outcome of a therapeutically combinatory strategy. In this particular case, the decrease of T regs and the increase in NKT potentially contributes to a favorable pro-inflammatory, anti-tumoral microenvironment induced by GIONF alone.Nanoparticle-mediated PTT is an effective anti-tumor therapy that has largely been developed in the last decades. Most studies focus on the targeting of tumor cells and the reduction of tumor burden. However, the effect on the tumor immune infiltrate is seldom tackled. GIONF-mediated mild hyperthermia (CEM43 of 15) resulted in a later increase of CD8+ T lymphocyte tumor infiltration and an increase in the levels of pro-inflammatory cytokines. The combination of this with the decrease of Tregs induced by GIONF themselves generates an overall pro-inflammatory immune microenvironment. Nevertheless, it is not enough to induce a significant effect on tumor growth. This may be due to the emergence of other pro-tumoral effectors. For example, we have not specifically measured the presence of myeloid derived suppressor cells (MDSC) that may impede T cells from exerting their antitumoral functions. Other studies have reported that PTT can increase the frequency of MDSC systemically.45 Despite the effective T cell response induced by GIONF-mediated PTT, this response is not robust enough to overcome the suppressive effects from MDSC. For this reason, the combination of nanoparticle-mediated PTT and immunotherapies thus could provide promising results.
This “vaccine” like effect that we have described produced by PTT was demonstrated in a few studies using different nanoparticles.43–45,48 Traditional cancer vaccines are usually used to trigger specific immune responses to complement immune checkpoint-based cancer therapy. However, the variability of neoantigens between different tumors can drive the vaccine to fail the triggering of specific anti-cancer immune response.49 PTT can potentially be a solution to this issue. The production of immunogenic cancer cell debris that consequently activates DC and antigen presentation could be considered as a “whole cancer cell vaccine”. It can potentially cover the shortages of immune checkpoint therapies by turning on the immunogenicity of tumors and therefore favoring the transition of a “cold” tumor devoid of T cells, to a “hot” tumors well infiltrated in CD8+ T cells. In contrast to previous studies that performed thermal ablation, meaning that the average temperature reached was over 50 °C and CEM43 was much larger,43,44,48 the “vaccine”-like effect described here was consecutive to a single mild hyperthermia treatment. Mild hyperthermia has been seen to promote apoptosis rather than necrosis50 mostly found after thermal ablation. This could explain why the increase in effector T cells is mainly seen at late-time points. Overall these results suggest that immune check point inhibitors could be advantageously used once the GIONF and mild hyperthermia priming of the TME elicited massive T cell infiltration.
Materials and methods
GIONF synthesis
Iron oxide nanoflowers (IONFs) were first synthesized following the procedure published by Hugounenq et al.19 FeCl3·6H2O (2.164 g; 8 mmol) and FeCl2·4H2O (0.795 g; 4 mmol) were completely dissolved in DEG (75 mL). The solution was stirred for one hour. The black-colored solution was poured with NMDEA (75 mL) and stirred again for one hour. Separately, NaOH pellets (1.42 g; 35.6 mmol) were dissolved in a mixture of polyols (40 mL DEG and 40 mL NMDEA). This solution was added to the solution of iron chlorides and the resulting mixture was stirred for another three hours. Then, the temperature was elevated to 220 °C using a regular heating (2 °C min−1). Once the temperature is set to 220 °C, the solution is stirred for 4 hours, and then cooled down slowly to room temperature by removing the heating plate. The black sediment was separated magnetically and washed with mixture of ethanol and ethyl acetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) several times to eliminate organic and inorganic impurities. Possible iron hydroxides were removed by treatment with 10% nitric acid. Iron(III) nitrate (Fe(NO3)3·9H2O) (2 g, 4.951 × 10−3 mol) is then dissolved in water (20 mL) and added to the nanoparticles. The resulting mixture is heated to 80 °C for 45 min to achieve a complete oxidation of the nanoparticles. After another treatment with 10% nitric acid, the particles were washed twice with acetone and diethyl ether and redispersed in water. At this stage, an aqueous dispersion of IONF is obtained that is stable under acid or basic conditions with a point of zero charge near pH 7.3.
:1, v/v) several times to eliminate organic and inorganic impurities. Possible iron hydroxides were removed by treatment with 10% nitric acid. Iron(III) nitrate (Fe(NO3)3·9H2O) (2 g, 4.951 × 10−3 mol) is then dissolved in water (20 mL) and added to the nanoparticles. The resulting mixture is heated to 80 °C for 45 min to achieve a complete oxidation of the nanoparticles. After another treatment with 10% nitric acid, the particles were washed twice with acetone and diethyl ether and redispersed in water. At this stage, an aqueous dispersion of IONF is obtained that is stable under acid or basic conditions with a point of zero charge near pH 7.3.
To obtain chelator-coated gold nanoparticles functionalized with dopamine (Au@DTDTPAd), an aqueous solution (3 mL) containing EDC (0.207 g; 108 × 10−3 mol) and NHS (0.247 g; 2150 × 10−3 mol) was added to a suspension of Au@DTDTPA gold nanoparticles (6 mL, 10 g Au L−1). The synthesis and the complete characterization of Au@DTDTPA were described elsewhere.25 These gold nanoparticles are composed of a gold core (2–3 nm) which is coated with a dithiolated derivative of diethylenetriaminepentaacetic acid (DTDTPA). The suspension was stirred at pH 6 for 90 minutes. Afterwards, an aqueous solution (4 mL) containing dopamine (9.45 × 10−3 g; 2.25 × 10−5 mol) was added to the suspension under stirring at pH 7.5. The mixture is stirred overnight. The purification of the suspension of gold nanoparticles was performed by dialysis against water (MWCO: 6–8 kDa) for 12 h. The water bath was changed three times every 3 h.
To assemble the Au@DTDTPAd with IONFs, the suspension of Au@DTDTPA modified by dopamine (Au@DTDTPAd) was mixed under stirring with the suspensions of IONFs (6 mL; 35 g Fe per L). The mixture with a pH value of 5.5 was heated at 50 °C for 24 h. Successive washings were performed with ultrapure water, acetone, and diethyl ether until a clear supernatant was obtained. After purification the gold decorated IONFs (GIONFs) were introduced into ultrapure water (6 mL) in order to provide an aqueous suspension with an iron concentration of 35 g L−1.
Cell culture
The MET-1 cell line (obtained from Robert Cardiff – University of California, Davis) and L929 (ATCC CCL-1) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, L-glucose and sodium pyruvate (Gibco).To produce bone-marrow derived macrophages (BMDM), L929 cell supernatants need to be previously prepared. To do so, L929 cells were grown to confluence in complete DMEM at 37 °C, 5% CO2. At confluence, the media were replaced by fresh complete DMEM and cells were cultured for 10 days without replacing the media. The supernatant was then collected and centrifuged at 2200 rpm for 10 minutes, and cell pellets were discarded. The supernatants were stored at −20 °C until they were used.
To obtain the bone marrow of the mouse, the mouse was euthanized by cervical dislocation. The hind legs were cut off and skin and flesh were removed leaving the bare bones. The bones were washed in ice cold sterile 1× PBS. The two extremities of the bone were cut and using an 25G needle complete DMEM is flushed inside the bone in order to recover the bone marrow. This step was repeated three times in order to ensure that all the bone marrow cells are collected. The supernatant was filtered through 70 μm Nylon cell strainers to remove solid fragments and then centrifuged at 15000 rpm for 10 min at 4 °C. The supernatant was discarded and the cell pellet was dissociated gently in 10 mL of complete DMEM containing 15% L929 cell supernatant. The cells were then filtered through a 40 μm Nylon cell strainer. The filtrate was recovered and added to complete DMEM supplemented with 15% L929 cell media in a Petri dish. The cells were grown and left to differentiate for 7 days during which the cell media were replaced every 2–3 days.
Flow-cytometry analysis of BMDM
BMDM were harvested by flushing with ice cold PBS-EDTA solution. The solution was then centrifuged at 1500 rpm for 5 minutes, the supernatant was discarded, and cells were counted and resuspended in fresh complete DMEM and seeded in 6-well plates at a seeding density of 500000 cells per well. Three different treatments were made: (i) incubation with GIONF, (ii) IFNγ treatment and (iii) control. BMDMs were incubated overnight with GIONF ([Fe] = 122 μg mL−1), and cells are then washed twice with PBS to remove any GIONF leftovers and replaced with fresh medium leaving cells in culture up to 6 days, changing the media after 3 days. As a positive control for M1 classical activation, one group of BMDM were treated with IFNγ (Peprotech, 2 ng mL−1). The control group was just incubated with complete DMEM.
To harvest the GIONF-containing BMDM, the BMDM were washed twice with complete DMEM and incubated for 5 minutes with ice cold PBS-EDTA solution. After flushing several times, the cells were detached and recovered. To recover the cells, the suspension was centrifuged at 1500 rpm for 5 minutes, the supernatant was discarded and the cells were filtered through a 70 μm cell strainer and centrifuged. Red blood cell lysis with ACK buffer was performed on the remaining pellet and subsequently filtered on a 40 μm cell strainer, and the cell suspension was rinsed in PBS and stained in 96-well round-bottom plates with a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) for 20 min at 4 °C. Cells were then washed and stained with antibodies for surface markers for 20 min at 4 °C. The anti-mouse antibodies used were the following:
| Antigen | Fluorophore | Clone | Company |
|---|---|---|---|
| CD11b | BV421 | M1/70 | BD biosystems |
| CD45 | AF700 | 30-F11 | Biolegend |
| Ly-6C | APC-Cy7 | HK1.4 | Biolegend |
| Ly-6G | BV510 | 1A8 | Biolegend |
| MHC II | BV785 | M5/114.15.2 | Biolegend |
| CD80 | FITC | 16-10A1 | Biolegend |
| CD64 | PE | X54-5/7.1 | Biolegend |
| CD206 | BV605 | CO68C2 | Biolegend |
| F4/80 | BV650 | A3-1 | Biolegend |
| CD86 | AF647 | GL-1 | Biolegend |
| CD274 (PDL1) | BV711 | 10F.9G2 | Biolegend |
After surface staining, cells were fixed with BD Fixation and Permeabilization Solution for 20 min at 4 °C. After washing in PBS, cells were resuspended in PBS 2% FBS and analyzed with a BDFortessa flow cytometer (BD Bioscience). Data were analyzed using FlowJo software.
Cytokine detection of the BMDM supernatant
BMDM were harvested by flushing with ice cold PBS-EDTA solution. The solution was then centrifuged at 1500 rpm for 5 minutes, the supernatant was discarded, and cells were counted and resuspended in fresh complete DMEM and seeded in 96-well plates at a seeding density of 20000 cells per well. Three different treatments were made: (i) incubation with GIONF, (ii) IFNγ treatment and (iii) control. BMDMs were incubated overnight with GIONF ([Fe] = 122 μg mL−1), and cells are then washed twice with PBS to remove any GIONF leftovers and replaced with fresh medium leaving cells in culture up to 6 days, changing the media after 3 days. As a positive control for M1 classical activation, one group of BMDM was treated with IFN γ (Peprotech, 2 ng mL−1). The control group was just incubated with complete DMEM.
The supernatant of each well was collected and centrifuged at 1500 rpm to eliminate suspension cells. Cell-free supernatants were frozen and stored at 80 °C. IL-10, IL-1β, TNF α, CXCL10, IL-6 and IFNγ release was assayed by Multiplex technology (U-PLEX, Meso Scale Discovery multiplex assay).
Quantification of GIONF uptake by cells in vitro
In order to study the uptake of GIONF, MET-1 cells were incubated at different concentrations ([Fe] = 112, 56 and 11.2 μg mL−1) for 24 hours. The concentration of iron in MET-1 cells was performed using an ICP atomic emission spectrometer (model iCAP6200 duo. Thermo Fischer Scientific, analysis performed in the PARI platform, IPGP, Paris, France). For each iron measurement, cell pellets were digested in 289 μL of 69% nitric acid (trace metal basic grade; Sigma) at room temperature overnight. The solutions were then diluted in ultrapure water for analysis. The calibration standards and the quality controls were provided by SCP SCIENCE and ChemLab. The analyzer drift is regularly corrected by quality controls measurements to ensure a drift lower than 5%. An ASX-520 autosampler was used. Argon 4.5 Linde (min, 99.995%) was used as plasma and purge gas. All samples were analyzed in 2% nitric acid.Cellular photothermal measurements
MET-1 cells were incubated with [Fe] = 112 μg ml−1 GIONF for 24 h in 6-well cell culture plates. Cells were then trypsinized, washed with PBS1× and counted. 1 × 106 cells were resuspended in 100 μl PBS 1× (500 μl tube). The cell suspension was then subjected to an 808 nm beam laser for 5 min (2.2 W cm−2, height 4 cm from the liquid/air interface, Laser Diode Drivers, BWT), and temperature was recorded with an infrared camera SC7000 from FLIR Systems.In vivo photothermal therapy experiments
All animal experiments were performed in agreement with institutional animal use and care regulations after approval by the local Ethics Committee from Université de Paris. 106 MET-1 cells suspended in 50 μL of PBS were injected subcutaneously into the flank of a 6-week-old female FVB mice (Janvier France). Tumor growth was followed with a caliper, and the tumor volume (V) was calculated as follows: xenograft volume = (xy2)/2 where x is the longest and y, the shortest of two perpendicular diameters.Prior to treatment, animals were randomized into three groups as follows: (i) control group: PBS intratumoral (i.t.) injection (50 μL), (ii) GIONF control group: [Fe] 420 μg per tumor GIONF injected i.t. (50 μL, iron concentration of 8.4 μg μL−1) and (iii) [Fe] 420 μg per tumor GIONF i.t injection combined with one photothermal therapy sessions (808 nm laser irradiation at 1 W cm−2 for 15 min (height from tumor skin 4 cm, Laser Diode Drivers, BWT)). Surface temperature was recorded with an infrared camera SC7000 from FLIR Systems during the laser treatment and analyzed using ResearchIR Software (FLIR). Mice were sacrificed 24 hours and 12 days after the photothermal therapy session. All animal experiments were performed in agreement with institutional animal use and care regulations after approval by the local Ethics Committee. The cumulative number of equivalent minutes at 43 °C (CEM43 °C) was calculated in each of the experiments:
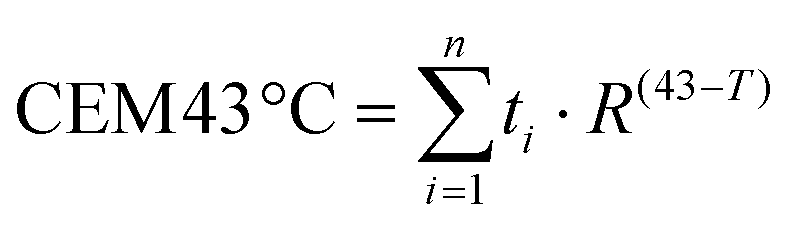
Flow cytometry of tumors
Tumors were mechanically dissociated and digested for 45 min at 37 °C in RPMI 1640 with 37.5 μg mL−1 Liberase TM (Roche) and 8000 U mL−1 DNase I, bovine pancreas (Merck Millipore). The resulting digestion was filtered through a 70 μm cell strainer and centrifuged. Red blood cell lysis with ACK buffer was performed on the remaining pellet and subsequently filtered on a 40 μm cell strainer, and the cell suspension was rinsed in PBS and stained in 96-well round-bottom plates with a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) for 20 min at 4 °C. Cells were then washed and stained with antibodies for surface markers for 20 min at 4 °C. The anti-mouse antibodies used were the following:| Antigen | Fluorophore | Clone | Company |
|---|---|---|---|
| CD11b | PE-Cy7 | M1/70 | Biolegend |
| CD11b | BV421 | M1/70 | BD Biosystems |
| CD11c | BV605 | N418 | Biolegend |
| CD45 | AF700 | 30-F11 | Biolegend |
| Ly-6C | APC-Cy7 | HK1.4 | Biolegend |
| Ly-6G | BV510 | 1A8 | Biolegend |
| CD4 | BV 711 | GK1.5 | BD Biosystems |
| CD8a | PerCP-e710 | 53-6.7 | eBioscience |
| TCRβ | BV605 | H57-597 | BD Biosystems |
| Nkp46 | eFluor660 | 29A1.4 | eBioscience |
| MHC II | BV785 | M5/114.15.2 | Biolegend |
| CD64 | PE | X54-5/7.1 | Biolegend |
| CD24 | PerCP-Cy5.5 | M1/69 | Biolegend |
| F4/80 | BV650 | A3-1 | Biolegend |
After surface staining, cells were fixed with BD Fixation and Permeabilization Solution for 20 min at 4 °C. In the case of intracellular staining, cells were incubated overnight with the following anti-mouse antibodies:
| Antigen | Fluorophore | Clone | Company |
|---|---|---|---|
| FoxP3 | FITC | MF-14 | Biolegend |
After washing in PBS, cells were resuspended in PBS 2% FBS and analyzed with a BDFortessa flow cytometer (BD Bioscience). Data were analyzed using FlowJo software.
Cytokine detection in the tumor slice supernatant
Fresh MET-1 tumors were embedded in 5% low-gelling-temperature agarose (type VII-A; Sigma-Aldrich) prepared in PBS. Slices (100 μm) were cut with a vibratome (VT 1000S; Leica) in a bath of ice-cold PBS. Tumor slices were kept at 37 °C in 24-well plates with 0.5 mL RPMI per well. Four to five slices were put in culture for each mouse. Eighteen hours later supernatants were collected and centrifuged at 1500 rpm to eliminate suspension cells. Cell-free supernatants were frozen and stored at 80 °C. IL-10, IL-23, IL-17, IL-1β, TNF α and IFNγ release was assayed by Multiplex technology (U-PLEX, Meso Scale Discovery multiplex assay).Histology
Tumors were fixed overnight at 4 °C in a periodate–lysine–paraformaldehyde solution [0.05 M phosphate buffer containing 0.1 M L-lysine (pH 7.4), 2 mg mL−1 NaIO4, and 10 mg mL−1 paraformaldehyde]. The tumors were dehydrated in graded solutions of ethanol and embedded in paraffin. For histology, five micrometer tissue sections were stained with Trichrome Masson and Perls staining.Transmission electron microscopy (TEM)
Fresh tumor sections (<1 mm) were washed with PBS. They were then resuspended in fixing buffer (0.1 M sodium cacodylate, 2.5% glutaraldehyde) and mixed gently for 24 h at 4 °C. After washing twice, tissues were resuspended in 0.1 M sodium cacodylate buffer until the TEM procedure. In case of cells, MET-1 cells were incubated with GIONF at an iron concentration of [Fe] 112 μg mL−1 for 24 hours at 37 °C. Following incubation cells MET-1 were detached by trypsinization, washed twice with PBS and fixed with 2% glutaraldehyde in 0.1 M Na cacodylate buffer pH 7.2, for 1 hour at room temperature.Samples were then contrasted with Oolong Tea Extract (OTE) 0.5% in cacodylate buffer, post-fixed with 1% osmium tetroxide containing 1.5% potassium cyanoferrate, gradually dehydrated in ethanol (30% to 100%) and substituted gradually in a mixture of ethanol-Epon and embedded in Epon (Delta microscopie – Labège France). Thin sections (70 nm) were collected onto 200 mesh cooper grids and counterstained with lead citrate. Grids were examined with a Hitachi HT7700 electron microscope operated at 80 kV (Elexience – France), and images were acquired with a charge-coupled device camera (AMT). MIMA2 MET – GABI, INRAE, Agroparistech, 78352 Jouy-en-Josas, France.
Statistical analysis
Results were analyzed using the GraphPad Prism 5.0 statistical software. Data are shown as means ± standard error of the mean (SEM). For comparisons between two groups, parametric Student's t-test or non-parametric Mann–Whitney test was used. For comparisons between more than two groups, the parametric One-Way analysis of variance (ANOVA) test followed by a posteriori Kruskal–Wallis test was performed.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We acknowledge Christine Péchoux (INRAE, UMR 1313, Plateforme MIMA2, Jouy en Josas, France) for electron microscopy preparation and observation, Maryline Favier, Fabiola Ely-Marius and Rachel Onifarasoaniania from HistIM Plate-Form (Institut Cochin, Paris France).This project was funded by ANR CARGOLD (ANR-16-CE09-0026) (FG, RB, SR, LG), European Union's Horizon 2020 Research and Innovation Program under grant agreement no. 685795 and no. 801305 (FG), IdEx Université de Paris, ANR-18-IDEX-0001 (FG), Region Ile de France under the Convention SESAME 2019 – IVETh (no. EX047011) (FG), French Ligue Nationale Contre le Cancer (Equipes Labellisées) (ED), Plan Cancer (Tumor Heterogeneity and Ecosystem Program) (ED), CARPEM (Cancer Research for Personalized Medicine) (ED). ANB received a PD fellowship by the Institute Thematique Multi-Organismes (ITMO) Cancer, the Doctoral School Frontières du Vivant (FdV) – Programme Bettencourt and the Fondation ARC pour la recherche sur le cancer.
References
- A. R. Rastinehad, et al., Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(37), 18590 CrossRef CAS PubMed.
- J. M. Stern, et al., Initial Evaluation of the Safety of Nanoshell-Directed Photothermal Therapy in the Treatment of Prostate Disease, Int. J. Toxicol., 2016, 35(1), 38–46 CrossRef CAS PubMed.
- F. R. Balkwill, M. Capasso and T. Hagemann, The tumor microenvironment at a glance, J. Cell Sci., 2012, 125(23), 5591 CrossRef CAS PubMed.
- J. Galon and D. Bruni, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies, Nat. Rev. Drug Discovery, 2019, 18(3), 197–218 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Elements of cancer immunity and the cancer-immune set point, Nature, 2017, 541(7637), 321–330 CrossRef CAS PubMed.
- W. A. Lim and C. H. June, The Principles of Engineering Immune Cells to Treat Cancer, Cell, 2017, 168(4), 724–740 CrossRef CAS PubMed.
- L. Huang, et al., Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy, Nat. Commun., 2019, 10(1), 4871 CrossRef PubMed.
- N. Zhang, et al., Photothermal therapy mediated by phase-transformation nanoparticles facilitates delivery of anti-PD1 antibody and synergizes with antitumor immunotherapy for melanoma, J. Controlled Release, 2019, 306, 15–28 CrossRef CAS PubMed.
- X. Liu, et al., Combination of MAPK inhibition with photothermal therapy synergistically augments the anti-tumor efficacy of immune checkpoint blockade, J. Controlled Release, 2021, 332, 194–209 CrossRef CAS PubMed.
- M. Zhou, et al., Combining Photothermal Therapy-Induced Immunogenic Cell Death and Hypoxia Relief-Benefited M1-Phenotype Macrophage Polarization for Cancer Immunotherapy, Adv. Ther., 2021, 4(2), 2000191 CrossRef CAS.
- L. M. Marques Neto, A. Kipnis and A. P. Junqueira-Kipnis, Role of Metallic Nanoparticles in Vaccinology: Implications for Infectious Disease Vaccine Development, Front. Immunol., 2017, 8, 239–239 Search PubMed.
- S. S. Bawage, et al., Gold nanorods inhibit respiratory syncytial virus by stimulating the innate immune response, Nanomedicine, 2016, 12(8), 2299–2310 CrossRef CAS PubMed.
- K. Niikura, et al., Gold nanoparticles as a vaccine platform: influence of size and shape on immunological responses in vitro and in vivo, ACS Nano, 2013, 7(5), 3926–3938 CrossRef CAS PubMed.
- M. S. Boyles, et al., Chitosan functionalisation of gold nanoparticles encourages particle uptake and induces cytotoxicity and pro-inflammatory conditions in phagocytic cells, as well as enhancing particle interactions with serum components, J. Nanobiotechnol., 2015, 13, 84 CrossRef PubMed.
- L. A. Dykman, et al., Gold nanoparticles as an adjuvant: Influence of size, shape, and technique of combination with CpG on antibody production, Int. Immunopharmacol., 2018, 54, 163–168 CrossRef CAS PubMed.
- S. Fogli, et al., Inorganic nanoparticles as potential regulators of immune response in dendritic cells, Nanomedicine, 2017, 12(14), 1647–1660 CrossRef CAS PubMed.
- S. Zanganeh, et al., Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues, Nat. Nanotechnol., 2016, 11(11), 986–994 CrossRef CAS PubMed.
- A. Curcio, et al., Iron Oxide Nanoflowers @ CuS Hybrids for Cancer Tri-Therapy: Interplay of Photothermal Therapy, Magnetic Hyperthermia and Photodynamic Therapy, Theranostics, 2019, 9(5), 1288–1302 CrossRef CAS PubMed.
- P. Hugounenq, et al., Iron Oxide Monocrystalline Nanoflowers for Highly Efficient Magnetic Hyperthermia, J. Phys. Chem. C, 2012, 116(29), 15702–15712 CrossRef CAS.
- Y. Javed, et al., Biodegradation mechanisms of iron oxide monocrystalline nanoflowers and tunable shield effect of gold coating, Small, 2014, 10(16), 3325–3337 CrossRef CAS PubMed.
- I. Miladi, et al., The in vivo radiosensitizing effect of gold nanoparticles based MRI contrast agents, Small, 2014, 10(6), 1116–1124 CrossRef CAS PubMed.
- A. Nicolás-Boluda, et al., Photothermal Depletion of Cancer-Associated Fibroblasts Normalizes Tumor Stiffness in Desmoplastic Cholangiocarcinoma, ACS Nano, 2020, 14(5), 5738–5753 CrossRef PubMed.
- P. Korangath, et al., Nanoparticle interactions with immune cells dominate tumor retention and induce T cell-mediated tumor suppression in models of breast cancer, Sci. Adv., 2020, 6(13), eaay1601 CrossRef CAS PubMed.
- E. Y. Lin, et al., Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases, Am. J. Pathol., 2003, 163(5), 2113–2126 CrossRef.
- C. Alric, et al., The biodistribution of gold nanoparticles designed for renal clearance, Nanoscale, 2013, 5(13), 5930–5939 RSC.
- V. Trouplin, et al., Bone marrow-derived macrophage production, J. Visualized Exp., 2013,(81), e50966 Search PubMed.
- B.-Z. Qian and J. W. Pollard, Macrophage diversity enhances tumor progression and metastasis, Cell, 2010, 141(1), 39–51 CrossRef CAS PubMed.
- A. Sica and A. Mantovani, Macrophage plasticity and polarization: in vivo veritas, J. Clin. Invest., 2012, 122(3), 787–795 CrossRef CAS PubMed.
- C. D. Mills, L. L. Lenz and R. A. Harris, A Breakthrough: Macrophage-Directed Cancer Immunotherapy, Cancer Res., 2016, 76(3), 513–516 CrossRef CAS PubMed.
- X. Yang, et al., Prognostic signficance of CD4/CD8 ratio in patients with breast cancer, Int. J. Clin. Exp. Pathol., 2017, 10(4), 4787–4793 CAS.
- D. G. Brooks, et al., IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(7), 3018–3023 CrossRef CAS PubMed.
- T. Lammers, et al., Effect of intratumoral injection on the biodistribution and the therapeutic potential of HPMA copolymer-based drug delivery systems, Neoplasia, 2006, 8(10), 788–795 CrossRef CAS PubMed.
- H. Xie, et al., Effect of intratumoral administration on biodistribution of 64Cu-labeled nanoshells, Int. J. Nanomed., 2012, 7, 2227–2238 CrossRef CAS PubMed.
- Z. Chen, et al., Acute toxicological effects of copper nanoparticles in vivo, Toxicol. Lett., 2006, 163(2), 109–120 CrossRef CAS PubMed.
- X. Sang, et al., The chronic spleen injury of mice following long-term exposure to titanium dioxide nanoparticles, J. Biomed. Mater. Res., Part A, 2012, 100(4), 894–902 CrossRef PubMed.
- X. Zhou, et al., The Toxic Effects and Mechanisms of Nano-Cu on the Spleen of Rats, Int. J. Mol. Sci., 2019, 20(6), 1469 CrossRef CAS PubMed.
- S. A. Sapareto and W. C. Dewey, Thermal dose determination in cancer therapy, Int. J. Radiat. Oncol., Biol., Phys., 1984, 10(6), 787–800 CrossRef CAS.
- J. R. Schoenborn and C. B. Wilson, Regulation of interferon-gamma during innate and adaptive immune responses, Adv. Immunol., 2007, 96, 41–101 CAS.
- A. D. Luster, J. C. Unkeless and J. V. Ravetch, Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins, Nature, 1985, 315(6021), 672–676 CrossRef CAS PubMed.
- J. H. Dufour, et al., IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking, J. Immunol., 2002, 168(7), 3195–3204 CrossRef CAS PubMed.
- S. Basu, et al., Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway, Int. Immunol., 2000, 12(11), 1539–1546 CrossRef CAS PubMed.
- W. Chen, et al., Combining photothermal therapy and immunotherapy against melanoma by polydopamine-coated Al(2)O(3) nanoparticles, Theranostics, 2018, 8(8), 2229–2241 CrossRef CAS PubMed.
- J. Cano-Mejia, et al., Prussian blue nanoparticle-based photothermal therapy combined with checkpoint inhibition for photothermal immunotherapy of neuroblastoma, Nanomedicine, 2017, 13(2), 771–781 CrossRef CAS PubMed.
- C. Wang, et al., Immunological responses triggered by photothermal therapy with carbon nanotubes in combination with anti-CTLA-4 therapy to inhibit cancer metastasis, Adv. Mater., 2014, 26(48), 8154–8162 CrossRef CAS PubMed.
- A. S. Bear, et al., Elimination of metastatic melanoma using gold nanoshell-enabled photothermal therapy and adoptive T cell transfer, PLoS One, 2013, 8(7), e69073 CrossRef CAS PubMed.
- Q. Lu, et al., Photothermally activatable PDA immune nanomedicine combined with PD-L1 checkpoint blockade for antimetastatic cancer photoimmunotherapy, J. Mater. Chem. B, 2019, 7(15), 2499–2511 RSC.
- L. Luo, et al., Sustained release of anti-PD-1 peptide for perdurable immunotherapy together with photothermal ablation against primary and distant tumors, J. Controlled Release, 2018, 278, 87–99 CrossRef CAS PubMed.
- Q. Chen, et al., Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy, Nat. Commun., 2016, 7, 13193 CrossRef CAS PubMed.
- T. N. Schumacher and R. D. Schreiber, Neoantigens in cancer immunotherapy, Science, 2015, 348(6230), 69–74 CrossRef CAS PubMed.
- Y. Zhang, et al., Temperature-dependent cell death patterns induced by functionalized gold nanoparticle photothermal therapy in melanoma cells, Sci. Rep., 2018, 8(1), 8720 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03201a |
| This journal is © The Royal Society of Chemistry 2021 |