
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-induced in situ chemical activation of a fluorescent probe for monitoring intracellular G-quadruplex structures†
Marco
Deiana‡
 *a,
Maëlle
Mosser‡
b,
Tangui
Le Bahers
b,
Elise
Dumont
b,
Marta
Dudek
c,
Sandrine
Denis-Quanquin
b,
Nasim
Sabouri
a,
Chantal
Andraud
b,
Katarzyna
Matczyszyn
c,
Cyrille
Monnereau
*b and
Laure
Guy
*b
*a,
Maëlle
Mosser‡
b,
Tangui
Le Bahers
b,
Elise
Dumont
b,
Marta
Dudek
c,
Sandrine
Denis-Quanquin
b,
Nasim
Sabouri
a,
Chantal
Andraud
b,
Katarzyna
Matczyszyn
c,
Cyrille
Monnereau
*b and
Laure
Guy
*b
aDepartment of Medical Biochemistry and Biophysics, Umeå University, 90187 Umeå, Sweden. E-mail: marco.deiana@umu.se
bUniv Lyon, ENS de Lyon, CNRS UMR 5182, Université Claude Bernard Lyon 1, Laboratoire de Chimie, F69342, Lyon, France. E-mail: cyrille.monnereau@ens-lyon.fr; laure.guy@ens-lyon.fr
cAdvanced Materials Engineering and Modelling Group, Faculty of Chemistry, Wroclaw University of Science and Technology, Wyb. Wyspianskiego 27, 50-370 Wroclaw, Poland
First published on 7th July 2021
Abstract
Light-activated functional materials capable of remote control over duplex and G-quadruplex (G4) nucleic acids formation at the cellular level are still very rare. Herein, we report on the photoinduced macrocyclisation of a helicenoid quinoline derivative of binaphthol that selectively provides easy access to an unprecedented class of extended heteroaromatic structures with remarkable photophysical and DNA/RNA binding properties. Thus, while the native bisquinoline precursor shows no DNA binding activity, the new in situ photochemically generated probe features high association constants to DNA and RNA G4s. The latter inhibits DNA synthesis by selectively stabilizing G4 structures associated with oncogenic promoters and telomere repeat units. Finally, the light sensitive compound is capable of in cellulo photoconversion, localizes primarily in the G4-rich sites of cancer cells, competes with a well-known G4 binder and shows a clear nuclear co-localization with the quadruplex specific antibody BG4. This work provides a benchmark for the future design and development of a brand-new generation of light-activated target-selective G4-binders.
Introduction
Besides the fundamental role of genomic double-stranded (ds-) DNA in genetic information storage, expression, replication and transmission, there is growing evidence that other non-canonical forms of DNA structures also play major regulatory roles in the cell life cycle and metabolism. In particular, G-quadruplex (G4) structures, consisting of short single-stranded sequences self-organized into folded guanine-rich tetrameric units,1,2 are increasingly seen as components of prime importance in the regulation of senescent cells’ replication.3–5 G4s are particularly prominent in oncogene promoters as well as in telomeres, and these regions have been identified as attractive targets for anticancer therapeutics. While a large number of organic and coordination compounds have been reported to target duplex and G4 structures with different levels of selectivity, there are very few reports of small molecules that can bind to DNA only after activation by an external stimulus.6–12 Prominent recent examples of such active and dynamic G4-binders include a metal salphen complex that is activated in the presence of bio-reductants11 and a metal-mediated conformational switching of a G4-binder based on a pyridodicarboxamide unit.6 Among the various possible triggers, photon irradiation offers distinct advantages because of its higher spatiotemporal precision, tunable energetic resolution, remote action and absence of waste chemical products. Yet, the development of photoresponsive G4-ligands has received scant attention to date: a molecular photoswitch based on an azobenzene unit,7 stilbene- and dithienylethene-derived ligands12,13 and (photo)caged G4-binders9,10 constitute rare examples of such molecules and translation of their activity into relevant physiological media has been rarely reported.10,12In a recent series of papers, we have described a low-cost and high-yielding, gram-scale synthetic procedure that afforded enantiopure binaphthol-like compounds featuring extended quinoline moieties, a motif quite commonly found in bioactive molecules.14–16 In the present paper, we describe how, upon UV light irradiation of 1, selective conversion to a new emissive photoproduct 2 is achieved where the intramolecular macrocyclization of the individual quinoline moieties occurs with significant extension of the aromatic skeleton of the molecule (Scheme 1 and Fig. 1A). This π-extended dicationic chromophore that remains soluble in physiological media structurally belongs to the class of compounds sometimes referred to as “azonia”,17 a class of molecules with remarkable photophysical properties18,19 that we study in detail through a combined spectroscopic study and computational modeling.
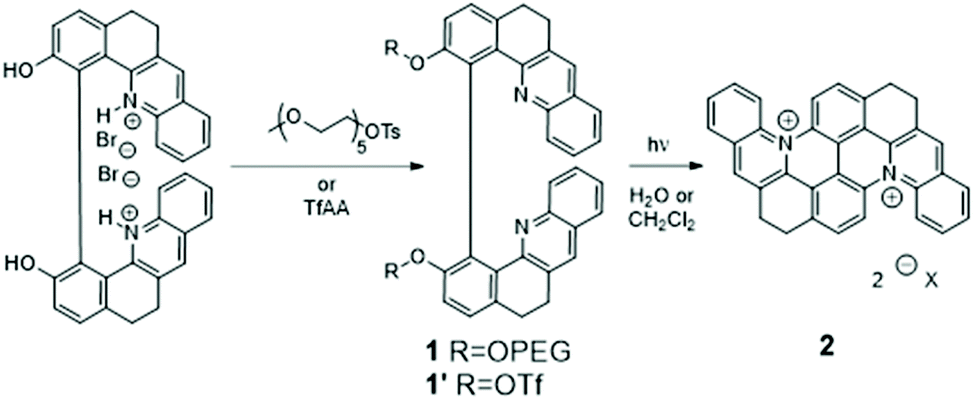 | ||
| Scheme 1 Synthetic pathway of the precursors and photogenerated products. Chemical structures and synthetic procedures of 1, 1′ and 2. | ||
 | ||
| Fig. 1 Light-driven generation of 2 and the corresponding theoretical kinetic model. (A) UV/Vis spectral evolution of 1 (in deionized H2O) upon irradiation at 355 nm. The absorption spectra were recorded at 5 min intervals. The black arrows show the emergence of isosbestic points. (B) Evolution of the fluorescence recorded at 480 nm during irradiation at 355 nm. | ||
Given the well-known affinity of flat, extended poly(hetero)aromatic structures for DNA in general and the reported specific affinity of some, i.e., extended quinolizinium derivatives or other crescent shaped analogues for G4 in particular,20–23 we then evaluate the properties of compound 2 as a light-activated G4 reporter/stabilizer.
We show that, while the native compound 1 displays no or very weak affinity towards duplex and G4 DNAs, the photochemically synthesized probe 2 binds both duplex and quadruplex structures, with a preference for G4s. The polymerase stop assay shows that 2 inhibits the progression of the Taq-DNA polymerase by selectively stabilizing the G4 in the DNA template. Finally, we demonstrate that photoconversion from 1 to 2 can operate under mild irradiation in cancer cells.
Results and discussion
Molecular design, structural characterization and spectroscopy
The water-soluble bisquinoline 1 is obtained in one step by O-pegylation of its previously described phenol precursor14 according to Scheme 1 (see the ESI for the full synthetic procedure, and NMR spectra in Fig. S1–S3†). The two PEG chains confer good water solubility to the synthesized compounds (7.6 × 10−4 M in deionized water). The absorption spectrum of 1 in water features a classical π–π* band, centered at ≈340 nm (Fig. 1A, blue curve). TD-DFT calculations confirm the localized nature of the intense band measured at 340 nm and predict a large oscillator strength, while the tail measured in the visible part is due to a series of charge transfer transitions characterized by very low oscillator strengths <0.04 a.u. (Fig. S18†). Only weak fluorescence is observed, with an apparent QY < 0.001 in agreement with the TD-DFT computed existence of weakly emissive first singlet excited states (Table S2 and Fig. S18 and S19†).Upon prolonged irradiation in the π–π* band (355 nm), compound 1 is progressively converted into an extended π-conjugated compound 2 as shown in Scheme 1. Fig. 1A shows the accompanying evolution of the UV/Vis spectrum of 1. It undergoes a progressive marked change, mainly characterized by the arising of a 420 nm centered broad band, and the simultaneous disappearance of the initial 340 nm centered transition. Throughout the whole transformation process, three isosbestic points are observed at 273, 318 and 361 nm, characteristic of a clean conversion process from the starting material to a single photo-generated product.
This conversion occurs with a very marked enhancement of the photoluminescence efficiency, which reaches 0.12 for the product. Meanwhile, the position and shape of the luminescence band are left unmodified by the transformation, strongly suggesting that the newly formed product was already present as traces in the initial sample (Fig. S23†), which was confirmed by the measurement of the excitation spectrum of the latter (not shown). In order to assess the kinetics of this transformation, changes in the luminescence intensity (480 nm) were monitored upon prolonged irradiation of the sample as shown in Fig. 1B. The irradiation wavelength was set near the isosbestic point of the reactant/product system (ca. 355 nm), in order to maintain a constant absorbance value throughout the whole experiment.
A sigmoidal curve was obtained, with an initial onset stage characterized by a slow increase in luminescence, followed by a much faster growth to reach a quasi-plateau value (Fig. 1B).
This evolution indicates that the reaction may occur either through an autocatalyzed photoinduced process24 (where the photogenerated product catalyzes the reaction by which it is generated) or through several successive reactions.
According to the literature,25 we were able to discriminate between both hypotheses by studying the slope of the tangent at the origin (t = 0) for the conversion of 1 in different initial system compositions (Fig. S24†) which enabled us to conclude in favor of a mechanism based on successive reactions.
The structure of 2, as photogenerated in a spectrometer cell as a low concentration solution in D2O, was elucidated in situ using 1 GHz NMR (structure of 2 is featured in Scheme 1, for complete NMR study, attribution, and printouts of the NMR spectra, see Fig. S6–S12†). Additional 2D NMR experiments and DOSY measurements confirmed the removal of the PEG chains in the course of the macrocyclization step. Meanwhile, HRMS analyses supported the existence of compound 2, which was found at m/z = 229.0896 (z = 2, M2+).
The DFT computed NMR signature for molecule 2 matched the experimental data (Table S1†), with a correlation factor above 0.99 (Fig. S16 and S17†). Moreover, absorption and emission spectra modelized by TD-DFT evidenced a good match between the positions (with a systematic error <0.1 eV) and relative intensities of the computed and experimental spectra for both the reactant and photogenerated product (Tables S2, S3 and Fig. S18–S22†).
While the aforementioned synthetic strategy is remarkably efficient in achieving almost quantitative in situ photoconversion of 1 to 2 in a physiological environment, we were not able to isolate the target compound 2 with scales and analytical purity compatible with a detailed and reliable evaluation of its binding properties with DNA/RNA molecules. In order to tackle this issue, we developed a new procedure starting from an alternative precursor (1′, see Scheme 1 and the ESI† for the detailed synthetic procedure). Taking advantage of the difference in the solubility of precursor 1′ in water, we managed to isolate 2 as its chloride salt in water by simple extraction of the dichloromethane reaction medium with a NaCl (30 μM) solution. The purity (as confirmed by 1H NMR, Fig. S13, 13C NMR, Fig. S14, and HPLC, Fig. S15†) and the scale are compatible with the subsequent use of 2 in biological tests.
Evaluation of the duplex/G4-interactive binding properties of 1 and 2
1 and 2 were then separately involved in affinity measurements with ds-DNA and G4s (the oligo sequences used in this study are provided in Table S4† and the morphological characterization of the studied G4 sequences in Fig. S29–S32†) after having ascertained a clean photoconversion of 1 to 2 in the buffered conditions used for the DNA association measurements (Fig. S33†). While significant spectral changes were observed in both cases for 2 (Fig. 2 and S34–S39†), no significant changes in the absorption band of 1 were observed under similar titration conditions, indicating no or very weak and unspecific binding interactions of 1 for DNA molecules, both in double-stranded and G4 conformations (Fig. 2A and S34, S36 and S38†).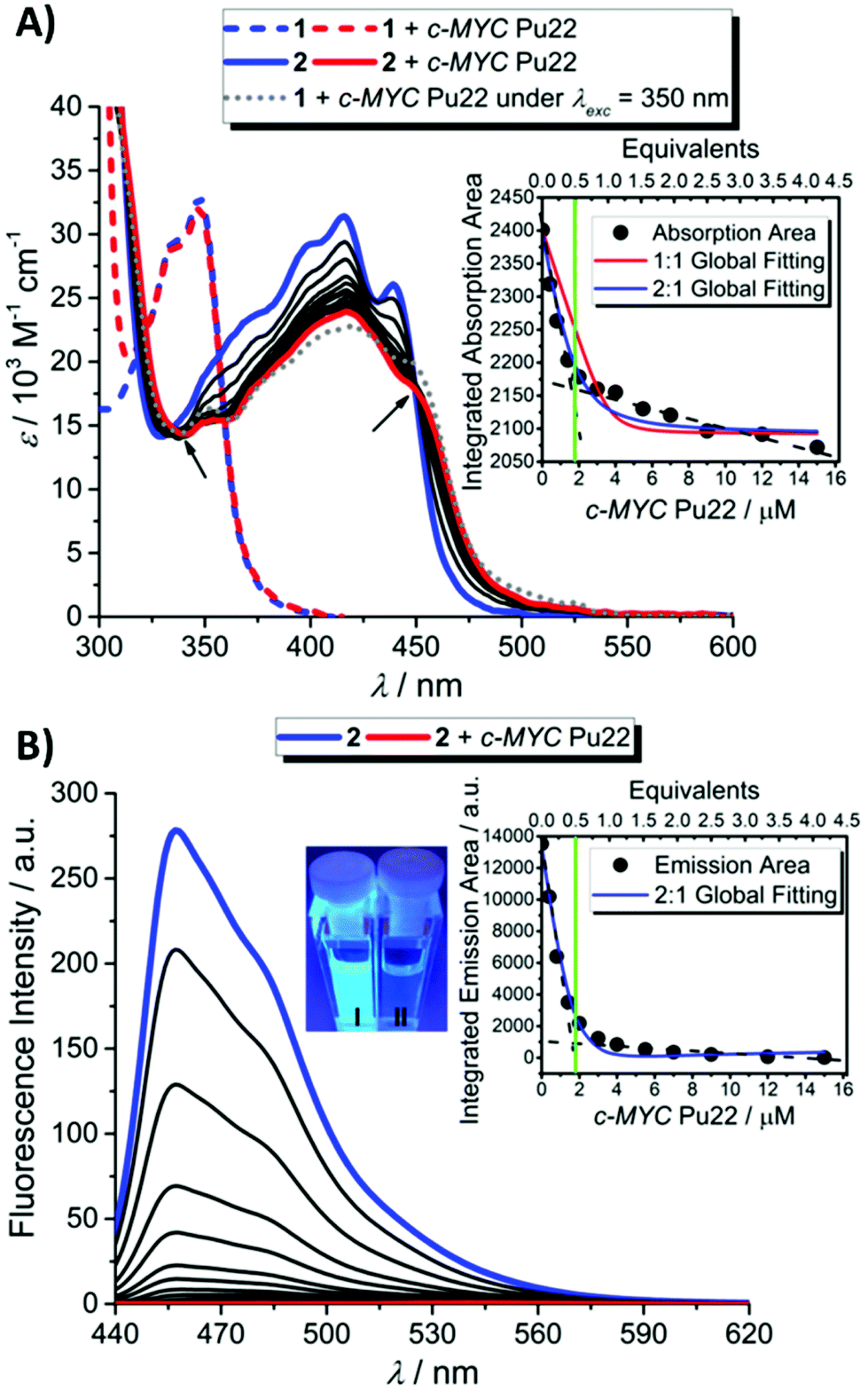 | ||
Fig. 2 Photoactivated G4 binding process. (A) UV/Vis absorption spectra of 1 (dashed lines) and 2 (solid lines) in the presence of c-MYC Pu22 (C1 = 3.8 μM, C2 = 3.6 μM, Cc-MYC Pu22 = 0 to 15 μM, CKCl = 100 mM, and CTris = 50 mM pH 7.4). The gray dotted line shows the photoconversion of 1 into 2 in the presence of c-MYC Pu22 under λexc = 350 nm for 60 min. The arrows indicate the isosbestic points. Inset: nonlinear fitting procedure obtained by using a global 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and noncooperative 2:1 binding model. The dashed black and the vertical green lines aim to show the inflection point supporting a 2:1 binding stoichiometry. (B) Emission spectra of 2 in the presence of c-MYC Pu22 (C2 = 3.6 μM, Cc-MYC Pu22 = 0 to 15 μM, CKCl = 100 mM, and CTris = 50 mM pH 7.4, λexc = 328 nm) and associated naked-eye detection of 2 in the absence (I: bluish cuvette) and presence of the G4 structure (II: colorless cuvette) under 312 nm UV light exposure. Inset: nonlinear fitting procedure obtained by using a global noncooperative 2:1 binding model. The dashed black and the vertical green lines indicate the inflection point supporting a 2:1 binding stoichiometry. :1 and noncooperative 2:1 binding model. The dashed black and the vertical green lines aim to show the inflection point supporting a 2:1 binding stoichiometry. (B) Emission spectra of 2 in the presence of c-MYC Pu22 (C2 = 3.6 μM, Cc-MYC Pu22 = 0 to 15 μM, CKCl = 100 mM, and CTris = 50 mM pH 7.4, λexc = 328 nm) and associated naked-eye detection of 2 in the absence (I: bluish cuvette) and presence of the G4 structure (II: colorless cuvette) under 312 nm UV light exposure. Inset: nonlinear fitting procedure obtained by using a global noncooperative 2:1 binding model. The dashed black and the vertical green lines indicate the inflection point supporting a 2:1 binding stoichiometry. | ||
In particular, upon addition of c-MYC Pu22, a well-characterized parallel G4 structure which constitutes a major reference for G4-interactive binding studies,26–29 the absorption bands of 2 underwent a marked hypochromic shift along with a bathochromic shift (Fig. 2A). The presence of isosbestic points characterized a well-defined 2-c-MYC Pu22 complex.21 A qualitatively similar evolution pattern was observed with other G4 structures (Fig. S34 and S36†), suggesting that 2 is not a sequence or topology-specific G4 binder. Interestingly, titration of 2 in the presence of the complementary c-MYC Pu22 single-stranded (ss-) C-rich sequence, under neutral pH conditions, led to very weak optical changes indicating a good binding selectivity for G4 structures (Fig. S40†). Under acidic pH conditions (pH 5), this C-rich sequence is also capable of forming an intercalated motif (i-motif) structure as probed by electronic circular dichroism (ECD) spectroscopy (Fig. S41†).30 The luminescence properties of 2 were not modified upon the addition of the i-motif structure, further supporting the binding selectivity of 2 for G4s (Fig. 3).
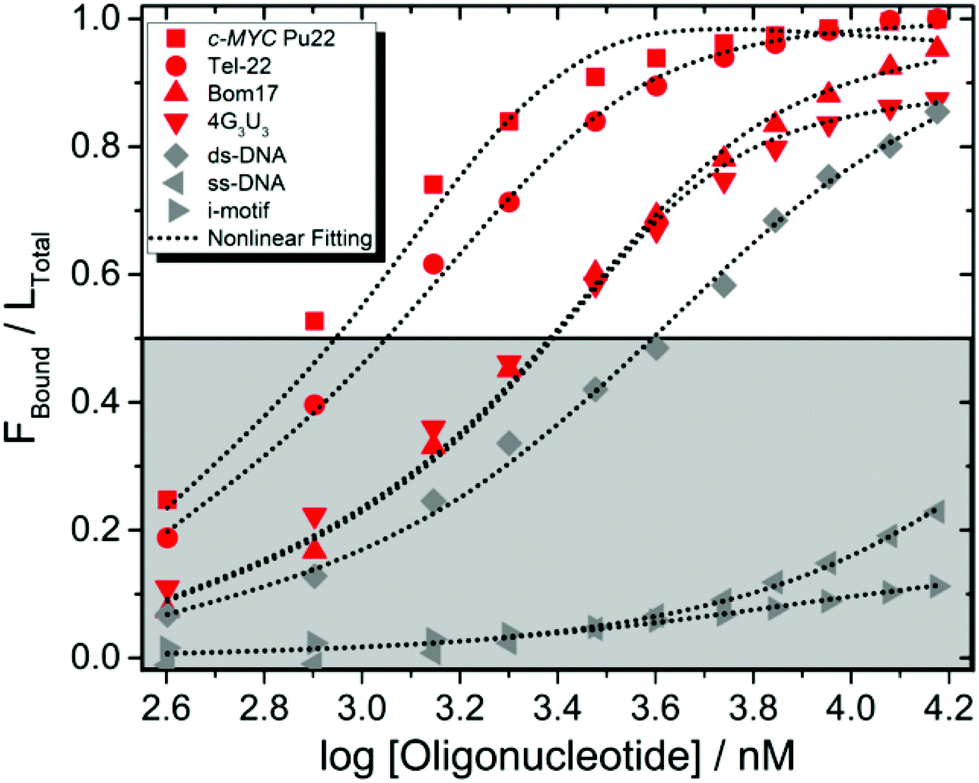 | ||
| Fig. 3 Comparison of the binding profiles of 2 complexed with G4 and non-G4 structures derived by fluorimetric titrations. The ligand's fraction bound (Fbound, i.e. Ltotal − Lfree) over the total ligand concentration (Ltotal) is plotted as a function of oligonucleotide concentration. Titrations involving G4s, ds-DNA and ss-DNA were performed in 50 mM Tris buffer (pH 7.4) and 100 mM KCl. Titration involving i-motifs was done in 50 mM cacodylate buffer (pH 5.0) and 100 mM KCl to maintain the same ionic strength condition. | ||
Next, we investigated the possibility of inducing in situ the photoswitching G4-binding properties of 1 in the presence of c-MYC Pu22. As depicted in Fig. 2A (dotted gray line), photoconversion of 1 into 2 in the presence of the G4 structure eventually led to a spectral signature of the mixture similar to that obtained upon complexation of 2 with the same G4, thereby confirming the ability of this probe to behave as a “smart” photo-activated G4-binder.
Steady-state fluorescence titrations were conducted in buffered aqueous solutions. In order to avoid excitation artifacts resulting from the decrease in molar extinction coefficient at the absorption maximum in the complexed systems, all samples were excited at 328 nm, close to one of the isosbestic points of the system. Evolution of the fluorescence spectrum of 2 upon the addition of G4s (Fig. 2B and S35 and S37†) or ds-DNA (Fig. S39†) resulted in a steady decrease in the photo-luminescence with an almost complete quenching upon full complexation with parallel and hybrid DNA G4s. This evolution pattern brings evidence of the effective interaction between the fluorophores and the different DNA motifs. Furthermore, the very efficient quenching of the emission intensity of molecule 2 that accompanies G4 binding greatly facilitates visual monitoring of the complexation process (inset Fig. 2B). The qualitatively similar optical changes exhibited by 2 in the presence of duplex or quadruplex structures can be expected by considering the di-cationic nature of the ligand which may facilitate long-range interactions with the negatively charged phosphate groups distributed along the DNA duplex grooves.31–33 Off-target binding interactions are also reported for the benchmark cationic G4-stabilizer Phen-DC3.34,35
In order to gain further insights into the binding mode and quantitatively compare the affinity of 2 with different DNA architectures, a global nonlinear curve fitting procedure was carried out on both spectrophotometric and fluorimetric titrations.36,37
The plot of the relative absorption changes or luminescence intensity variations vs. duplex/G4 concentration resulted in classical hyperbolic binding curves (Fig. 2 and S34–S39†). The binding constants determined from the nonlinear global fitting analysis showed that 2 displayed higher binding affinities towards noncanonical DNA structures (c-MYC Pu22 and Tel-22) than DNA duplex (note that the molecular size of the templates is comparable) (Table 1). Indeed, the output data derived from the 2-c-MYC Pu22 and 2-Tel-22 systems could be only fitted with great accuracy exclusively by using a 2:1 binding model. In this model, two molecules of 2 coordinate with a single G4 molecule, and a global association constant (Kglobal) with units in M−2 is provided by multiplying K11 (M−1) × K21 (M−1).38 Since the physical units describing a 1:1 and a 2:1 binding model are different, no direct quantitative comparison of the data can be made. However, with two molecules of compound 2 complexed to a single G4 template, the resulting association constants derived for 2-c-MYC Pu22 or 2-Tel-22 systems are likely to be much higher compared to the K value calculated for the 2-ds-DNA system. Altogether, all collected data point out a very efficient binding process of 2 to G4 structures (c-MYC Pu22 and Tel-22), with good selectivity over ds-DNA binding, thus confirming the relevance of this newly described class of compounds as specific G4 binders. The fluorimetric data supporting the interactive binding properties of 2 toward DNA G4s and non-G4 structures are shown in Fig. 3.
:1 and noncooperative 2:1 binding models
| System | 1:1 fitting |
2:1 fitting |
||||||
|---|---|---|---|---|---|---|---|---|
| K (M−1) | Cov(Fit) | Error (%) |
K
11b (M−1) |
K
21b (M−1) |
K
Globalc (M−2) |
Cov(Fit) | Error (%) | |
|
a
K = association constant derived by using a global 1:1 fitting model obtained with BindFit by using the Nelder–Mead method.
b
K
11 = association constant derived by using a global 2:1 noncooperative fitting model through two identical binding sites (i.e. K11 = 4 K21) obtained with BindFit by using the Nelder–Mead method.
c
K
Global = global association constant defined as KGlobal = K11 × K21.
d N.D. = not determined, very poor fit with uncertainties >100%.
|
||||||||
| 2-c-MYC Pu22 (UV/Vis) | 2.3 × 107 | 0.0500 | 47.7 | 2.5 × 106 | 6.2 × 105 | 1.5 × 1012 | 0.0071 | 2.9 |
| 2-c-MYC Pu22 (emission) | N.D.d | — | — | 3.8 × 106 | 9.5 × 105 | 3.6 × 1012 | 0.0044 | 2.2 |
| 2-Tel-22 (UV/Vis) | 1.3 × 107 | 0.0242 | 22.9 | 1.4 × 106 | 3.5 × 105 | 4.9 × 1011 | 0.0027 | 1.4 |
| 2-Tel-22 (emission) | N.D.d | — | — | 2.1 × 106 | 5.2 × 105 | 1.1 × 1012 | 0.0009 | 0.8 |
| 2-Bom17 (UV/Vis) | 2.9 × 106 | 0.0080 | 5.2 | — | — | — | — | — |
| 2-Bom17 (emission) | 1.7 × 106 | 0.0014 | 1.2 | — | — | — | — | — |
| 2-ds-DNA (UV/Vis) | 9.0 × 105 | 0.0057 | 2.1 | — | — | — | — | — |
| 2-ds-DNA (emission) | 4.8 × 105 | 0.0018 | 0.7 | — | — | — | — | — |
The recognition mechanism exerted by 2 toward G4 structures was explored by unbiased all-atom molecular dynamic (MD) simulations. MD simulations corroborated a stable end-stacking interaction mode of 2 with c-MYC Pu22 (Fig. 4, Table S5 and additional ESI† for details) with π-stacking above and below the plane of 2 (with G-quads and flanking nucleobases A, T or G) and proximity to the phosphate groups providing a stabilizing environment by electrostatic complementarity. MD simulations performed using telomeric G4s (hybrid I, hybrid II, hybrid III and antiparallel) having different folding patterns showed heterogeneous ligand binding models depending on the G4 structure considered with mainly 2 either end-stacked or lying along the G4's grooves (Fig. S64–S67†).
 | ||
| Fig. 4 Molecular dynamics simulations. Representative structures of 2 in the presence of c-MYC Pu22 in (A) side view and (B) top view, obtained after 200 ns of all-atom molecular dynamics simulations. Two molecules of 2 coordinate with the c-MYC Pu22 template at the terminal G-tetrad ends. | ||
In order to attempt to identify the specific site localization of our probe on the c-MYC Pu22 template, we performed 1D 1H NMR titration experiments. As shown in Fig. S42,†c-MYC Pu22 formed a highly defined G4 conformation as indicated by the well-resolved imino proton peaks appearing between 10 and 12 ppm. Upon addition of 2 to the G4 template, a line broadening of all the signals in the imino-region of c-MYC Pu22 was observed. On comparing the chemical shift of all imino protons, we found that those corresponding to the G-tetrad ends, especially at the 5′-end, were the most perturbed. The line broadening of the signals is indicative of fast on–off rates between free and bound ligand states which confirms that 2 strongly binds to the c-MYC Pu22 G4 structure. Similar findings were recently reported for a number of G4-ligands.39,40 In order to confirm that the observed line broadening was an effect of exchange processes caused by a strong binding rather than a compound-induced unfolding of the G4 structure, we performed ECD spectroscopy. ECD was used to assess the morphological changes occurring on the c-MYC Pu22 and telomeric G4 structures in the presence of 2 (Fig. S43 and S44†). ECD spectra showed that the secondary structures of both c-MYC Pu22 and Tel-22 were fully conserved upon the addition of 2. Therefore, in spite of their high association constants, complexation with 2 did not induce any marked topological changes on the G4 scaffolds, a highly important feature in the design of ideal in vivo G4 fluorescent probes. The lack of any well-resolved induced circular dichroism (ICD) signals further corroborates the hypothesis of a π-stacking binding mode.41–43 The latter was also confirmed by fluorescence displacement assay using the G4 end-stacker, Phen-DC3,44 which was able to replace 2 from the external G-tetrads of c-MYC Pu22 indicating competition of the two molecules for the same binding site (Fig. S45†).
Interestingly, MD simulations using DNA duplex as a template showed 2 lying along the groove (Fig. 5A and B). These findings can be expected by considering the large π-conjugated distorted structure of 2 which can prevent the molecule from intercalating among the duplex nucleobases.
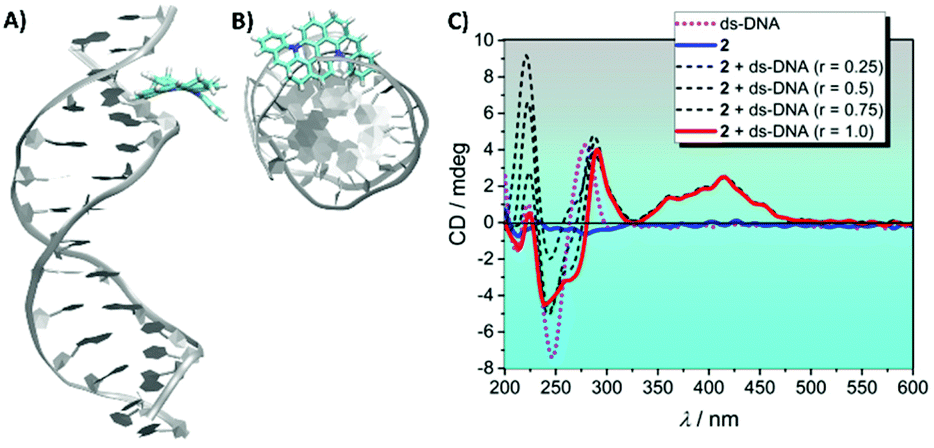 | ||
| Fig. 5 (A) Molecular dynamics simulations for 2 bound to the DNA duplex's groove. (B) Top-view image of 2 lying along the DNA groove. (C) ECD spectra of 2 complexed with ds-DNA. The emergence of a positive ICD signal in the 2-ds-DNA complex supports the groove binding mode of 2. (C2 = 10.0 μM, Cc-MYC Pu22 = 0, 2.5, 5.0, 7.5 and 10.0 μM, and CTris = 10 mM pH 7.4). | ||
Experimental support for this hypothesis was provided by ECD experiments performed by titrating ds-DNA into a fixed concentration of 2. As depicted in Fig. 5C, a positive ICD signal that matches the linear absorption of the ligand is observed upon duplex addition. The ICD band results from the coupling of the electric transition moments of 2 and the DNA bases.45 Positive ICD signals are usually observed for DNA groove binders and the absence of exciton coupling with the characteristic bi-signate shape ruled out the formation of dimer or higher order complexes.
G4-selective stabilization effects
In order to further confirm the ability of compound 2 to selectively stabilize G4s over duplex structures, we performed ECD-based thermal assays. A high level of thermal stabilization (ΔTm) on the G4 templates upon complexation with 2 was observed (Table 2, Fig. 6 and S46†). Depending on the G4 structure considered, this stabilization reached up to ∼30 °C (large enough to exclude unfolding of the G4 structure) after the addition of 4 equivalents of 2. Conversely, under the same experimental conditions used to probe G4 stabilization, very weak or no stabilizing effects on the duplex templates were observed (Fig. 6). Overall, these data indicate that 2 can specifically stabilize G4 structures over duplex conformations. | ||
| Fig. 6 Results of the ECD-based thermal assays performed on a panel of DNA/RNA G4s or duplexes. (A) The radar graph shows the extent of thermal stabilization induced by 2 on G4 and duplex templates. (B and C) 2 exerts negligible thermal stabilization effects on the RNA duplex but largely stabilized the RNA G4 structure. (C2 = 8.0 μM, Cds-RNA/4G3U3 = 2.0 μM, CKCl = 5 mM, and CTris = 10 mM pH 7.4). | ||
| System | T m (G4/duplex) | T m (2-G4/duplex) | ΔTma |
|---|---|---|---|
| a ΔTm values are calculated as: Tm (2-G4/duplex) − Tm (G4/duplex). b These values are an estimation because of the high thermal stability of the 2-c-MYC Pu22 complex. | |||
| 2-c-MYC Pu22 | 68 | >80b | >12b |
| 2-Tel-22 | 47 | 72 | 25 |
| 2-Bom17 | 36 | 61 | 25 |
| 2-4G3U3 | 59 | 89 | 30 |
| 2-ds-DNA | 57 | 59 | 2 |
| 2-scr ds-DNA | 48 | 49 | 1 |
| 2-GC-rich ds-DNA | 47 | 51 | 4 |
| 2-ds-RNA | 57 | 55 | −2 |
As G4 formation can stall DNA replication causing genomic instability, stabilization of these noncanonical DNA structures could play important roles in view of cancer therapeutic applications. Therefore, we evaluated 1 and 2 for their ability to affect the progression of the Taq DNA polymerase on two different G4 DNA templates, c-MYC and telomeric G4 DNA, and loaded these reactions on denaturing polyacrylamide gels. We systematically compared these results with those obtained using a non-G4 control sequence to determine the specificity of the molecules (Fig. 7A).46 Selective stabilization of the G4 structure by 1 and/or 2 should enhance the pausing/arrest of the DNA polymerase on the G4 DNA template, resulting in lower amounts of full-length products (Fig. S47†). Similarly, unspecific stabilization of the non-G4 control template by 1 and/or 2 would also result in reduced amounts of replicated full-length products.
 | ||
| Fig. 7 G4-stabilization through 1-to-2 photoconversion resolved by Taq-DNA polymerase stop assays. (A) Sequences of non-G4 DNA, c-MYC Pu24T and Tel-22 templates used in this assay. The region for the primer annealed to the complementary template DNA is marked in bold. The Gs forming the G4 structures are indicated in red. Asterisks represent the pausing sites detected in (B) and (D). (B) Taq-DNA polymerase stop assay with 1. (C) Relative quantification of full-length products of the gels in (B) normalized to the control sample (C1 = 0 μM). (D) Taq-DNA polymerase stop assay with 2. (E) Relative quantification of full-length products of the gels in (D) normalized to the control sample (C2 = 0 μM). Non-G4 DNA, c-MYC Pu24T and Tel-22 templates were tested for 1 and photoconverted 2. Concentrations of compounds used were (C1/2 = 0.0, 0.05, 0.15, 0.25, 0.5, 1.5, 2.5, 3.5 and 5 μM). Black arrows indicate the start of the reaction (non-extended primer); red arrows represent full-length products; asterisks represent pausing sites. Error bars represent standard deviations for 3 independent experiments. | ||
The conditions used in these experiments allowed multiple cycles/attempts to replicate each template by the Taq DNA polymerase, therefore only highly stable pausing sites are visible in the gels. The Taq polymerase stop assays performed with different concentrations of 2 (obtained after photoconversion of 1) showed a clear concentration-dependent G4 stabilization effect (Fig. 7D and E). 2 displayed a clear-cut binding preference for the G4 DNA sequences than for the non-G4 template, as no changes of full-length products were detected on this control template (Fig. 7D and E). In agreement with the spectroscopic data, 1 did not show any stabilization effects on either G4 or the non-G4 templates, an essential feature for a photoactivable prodrug (Fig. 7B and C). Next, the stabilization efficiency of 2 was compared with that of CX-5461, a very potent G4 stabilizer that is now tested in clinical trials for breast cancer patients.47 We found that 2 and CX-5461 exerted similar levels of G4 stabilization effects on both G4 templates (Fig. S48†). These results clearly shed light on the possible therapeutic potential of this molecule and may open novel (photo)therapeutic strategies that harness photophysical properties that have long been known but seldom combined in a single molecular scaffold.
Remote-light controlled intracellular duplex/G4 recognition
Molecules capable of binding nucleic acids with high affinity are known to efficiently inhibit most DNA/RNA-associated processes (replication, transcription, translation, and repair) with sometimes disastrous adverse effects,48 but also with many potential applications in medicine, and most predominantly in cancer therapy.49 In this context, molecules whose binding affinities specifically can be triggered in situ from a “passive” to an “active” form using light present many advantages in view of designing target-specific photochemotherapeutic agents.50Recently, the intracellular localization of duplex/G4 turn-off probes51,52 including the benchmark G4-interactive compound Phen-DC3 was reported.53 For instance, Phen-DC3 is a weak fluorophore and its associated emission intensity was further quenched in the presence of DNA/RNA G4s (turn-off). Conversely, in cells, Phen-DC3 showed a bright and well-defined fluorescence signal that was used to track DNA/RNA G4 formation. Even more recently, Di Antonio and co-workers reported on a G4-specific fluorescent probe that enabled single-molecule detection of DNA G4 structures in cells.54 The authors were able to provide an estimation of the total DNA G4 nuclear concentration on a single U2OS cell of ∼500 nM. Since the concentration of the fluorophores commonly used to detect G4 structures in cells by confocal laser scanning microscopy (CLSM) is within the micromolar range, it turns out that an excess of ligand compared to the total number of G4s is used. Under these conditions, the emissive properties of the fluorophore are only slightly modified (turned-off) by the intracellular G4 sites. Indeed, the large excess of the ligand may lead to the formation of multiple and heterogeneous ligand:G4 adducts having different stoichiometries/affinities and fluorescence quantum yields. This aspect dramatically differs from the in vitro data, in which a well-defined complex is usually formed at equimolar concentration of G4 and ligands.
These findings prompted us to evaluate the cellular emission fingerprint of 1 and 2 in fixed HeLa cells by CLSM. Firstly, we tested the in cellulo photoconversion of 1 into 2 (Fig. 8 and S49†). The cells were incubated with 1 and then either left in the dark (no UV light) or irradiated for 1, 2, 5 and 10 minutes at 312 nm before imaging. 1′-Treated cells in the absence of UV light showed no detectable fluorescence signal (Fig. 8 and S49†). However, upon 1 minute irradiation, mostly cytoplasmic staining was observed (Fig. 8 and S49†). After 2 minutes of irradiation, the staining pattern was dramatically changed and a punctate emission confined mostly within the nucleolar sites was observed (Fig. 8 and S49†). Increasing the time of exposure to UV light (5 and 10 minutes) resulted in an even brighter and well-defined nuclear signal (Fig. 8 and S49†). Based on these data, we can conclude that the fluorescence signal observed in the cells arose from the photoconversion of 1 into 2 along with subsequent re-localisation of the photoproduct from the cytoplasm to the nucleus and its binding to DNA/RNA structures.
 | ||
| Fig. 8 In cellulo photoconversion of compound 1. Confocal fluorescence images of MeOH-fixed HeLa cells stained with 1 (10 μM) along with the corresponding bright field (BF) and overlay images. The cells were either nonirradiated or irradiated with a 312 nm UV lamp for 1, 2, 5 and 10 minutes. Scale bar = 5 μm. Experimental settings: a 405 nm diode laser was used to image 1λexc = 405 nm, λem 430–630 nm. | ||
In order to confirm that the staining pattern of the photogenerated compound was independent of the synthetic protocol used (i.e.2 photogenerated in cellulo from 1 or separately isolated as a salt from 1′), we performed CLSM with the isolated pure form of 2.
The cells treated with 2 revealed an intense fluorescence signal in the nuclear and G4-rich nucleolar compartments (Fig. 9A, S50 and S51†) in strong agreement with the aforementioned results using 1in cellulo as a precursor.51,53,55–58 Therefore, we decided to use the isolated form of 2 in subsequent cell studies.
 | ||
| Fig. 9 G4 recognition in cancer cells. (A) Confocal fluorescence images of MeOH-fixed HeLa cells stained with 2 (10 μM) or TMPyP4 (10 μM) along with the corresponding bright field (BF) and overlay images. (B) Fluorescence images of fixed HeLa cells stained with 2 (10 μM) before and after treatment with RNase (upper panel). Fluorescence images of ThT (5 μM) before and after treatment with RNase (lower panel). (C) Fluorescence images of fixed HeLa cells stained with 2 (10 μM) before and after treatment with DNase (upper panel). Fluorescence images of Hoechst 33342 (5 μM) before and after treatment with DNase (lower panel). (D) Fluorescence displacement assay between 2 (10 μM) and BRACO-19 (10 μM). (E) Confocal fluorescence images of PFA-fixed HeLa cells stained with 2 (10 μM) and BG4. Merged fluorescence images demonstrate the nuclear co-localization between 2 and BG4. No fluorescence signal was observed for the control cells that were not treated with BG4 but treated with the secondary antibodies. Scale bar = 5 μm. Experimental settings (A–D): a 405 nm diode laser was used for 2 and Hoechst 33342 λexc = 405 nm, λem 430–630 nm for 2 and λem 430–530 nm for Hoechst 33342; an argon laser was used for TMPyP4 visualization λexc = 514 nm, λem = 550–700 nm; the argon laser was used for ThT visualization λexc = 458 nm, λem = 480–680 nm. Experimental settings (E): a 405 nm diode laser was used for 2λexc = 405 nm, λem 430–480 nm; a white light laser (WLL) was used for BG4 λexc = 598 nm, λem 620–750 nm. | ||
The localization of 2 in the nucleoli (dense multifunctional domains in which ribosome biogenesis begins) is relevant for future target applications, as the nucleoli are enriched with both DNA and RNA G4 structures.59
To get a first hint about the possible interaction of 2 with DNA in the cells, we compared its staining pattern with that of TMPyP4, a porphyrin-based fluorescent duplex/G4 binder. The TMPyP4-treated cell showed, similar to 2, an intense fluorescence signal localized mainly in the nucleus and nucleoli, suggesting a high level of co-localization between the two probes (Fig. 9A and S50†). In order to confirm the nature of the main binding targets of 2, we then treated the MeOH-fixed cells with either RNase or DNase before incubating with 2. RNase pretreatment largely modified the nucleolar staining pattern of 2 indicating the possible binding of 2 to RNA molecules (Fig. 9B and S52A†). The almost complete disappearance of the 2-associated fluorescence signal in the nucleoli upon RNase treatment prevents the possibility to perform quantitative data analysis. The interactive binding interactions of 2 toward RNA G4 were investigated by in vitro fluorimetric titrations and indicated a relatively good binding affinity of 2 to the RNA G4 (K = 2.6 × 106 M−1) (Fig. S53†).
To further examine the RNA binding properties of 2, we used Thioflavin-T (ThT) as a control. ThT is a fluorescent G4 probe with high affinity to RNA G4s. Thus, we expected that RNase pretreatment before incubating the cells with ThT would reduce the levels of the detected RNA G4 structures, similar to what was seen for 2.60 Indeed, we observed that the nucleolar ThT signal is reduced after RNase treatment (Fig. 9B, S52B and S54†). Together, these data suggest that part of the nucleoli-signal detected by 2 and ThT are from RNA G4 structures.
Treatment of the fixed HeLa cells by DNase before incubating the cells with 2 showed a different staining pattern (Fig. 9C and S55A†). The DNase treatment strongly reduced the nuclear and nucleolar fluorescence signal associated with the presence of 2 (Fig. 9C, S55A and S56†). These data suggest that the fluorescence signal of 2 in the nucleus stems from duplex/G4 DNA binding. These conclusions were confirmed by the minor groove binder Hoechst 33342, showing that pre-treatment with DNase does indeed affect the nuclear signal (Fig. 9C and S55B†). Together, these last sets of experiments indicate that 2 mainly binds to DNA in the nucleus and to DNA/RNA in the nucleoli of HeLa cells. Finally, we performed a competition displacement assay using the G4-binder BRACO-19 to determine if BRACO-19 and 2 compete for the same binding sites in the cells.61In vitro experiments showed that BRACO-19 could efficiently replace 2 from the c-MYC Pu22 template (Fig. S57†). In cells, BRACO-19 almost completely displaced 2 from the nuclear and nucleoli regions, showing that the compounds share the same binding sites (Fig. 9D, S58 and S59†). Replacement of 2 by BRACO-19 may be further explained by the ability of BRACO-19 to bind to both duplex and G4 structures as observed by photometric titrations (Fig. S60–S62†). These results are expected, considering the poly-cationic nature of BRACO-19 which can easily establish electrostatic interactions with the phosphate groups of the DNA duplex.
Finally, we used the highly specific G4 antibody BG4 in immunofluorescence experiments to compare the localization of BG4 with 2 (Fig. 9E and S63†).62–64 As expected, 2 mainly stained the nuclei of HeLa cells. On the other hand, BG4 showed punctate nuclear and cytoplasmic staining in agreement with its ability to target both DNA and RNA G4s, respectively. The punctate fluorescence foci associated with BG4 staining usually differ from the less localized optical output observed for G4 sensors. This is expected by considering the different molecular sizes between BG4 and small optical reporters. Fluorogenic dyes can easily diffuse and accumulate within different cellular compartments providing spatially heterogeneous distributions. By merging both the individual fluorescent channels of 2 and BG4, we could observe that our probe fairly well co-localized with the G4-specific antibody mostly at the nuclear level. Importantly, in the absence of the primary BG4 antibody, no detectable fluorescent signal was observed supporting the targeting and visualization of DNA/RNA G4s in HeLa cells by BG4 (Fig. 9E). Collectively, the results described above support the potential of 2 to bind and detect G4 structures in cells.
Conclusions
In this paper, we described the synthesis and detailed study of a (diazonia)tetrabenzonaphtacene compound 2 that combines relevant spectroscopic properties with compelling photo-triggerable binding properties to G4s.The photochemical transformation associated with the conversion of 1 in water (or its closely related derivative 1′ in organic solvent) takes place with high efficiency and selectivity, making it a very interesting new protocol in the toolbox of chemical synthesis of extended poly(hetero)aromatic compounds.65–67
Spectroscopic features were fully addressed and rationalized on the basis of DFT and TD-DFT calculations.
In striking contrast with its parent helicoidal compound 1, polycyclic compound 2 undergoes efficient association with both duplex and quadruplex structures. In all cases, association results in marked changes in the absorption spectrum and in an almost quantitative quenching of the 460 nm centered luminescence enabling G4 naked eye detection. 1H NMR titration studies, displacement assays and MD simulations supported the binding of 2 to c-MYC Pu22 through the end-stacking mode. ECD-based thermal measurements and Taq polymerase stop assays clearly showed the ability of 2 to impart a strong G4 stabilization effect to an extent that is comparable to one of the most potent G4 binders reported so far. Finally, 2 preferentially targeted both DNA in the nuclei and DNA/RNA in the nucleoli of cancer cells.
This last feature, combined with the possible in cellulo photoconversion of 1 to 2 that was clearly evidenced in this paper, make this new class of molecules very appealing candidates in view of G4 targeted cancer therapeutic approaches, based on the in cellulo photoactivation of a prodrug.
Experimental
Materials
Solvents, reagents, chemicals and biological templates were purchased from commercial suppliers (Sigma-Aldrich and Eurofins Genomics) and used without further modifications unless otherwise stated. Oligonucleotides were diluted with ultrapure water (DNase and RNase free) to a concentration of 1 mM and stored at 5 °C. The exact oligonucleotide concentration was determined by UV/Vis spectroscopy using the molar extinction coefficients (ε260) provided in Table S4† and calculated by using an oligo analyzer on the IDT website. An aqueous stock solution (1 M) of TRIS buffer was prepared by dissolving tris(hydroxymethyl)aminomethane in water and the pH was then adjusted to 7.4.Oligonucleotide annealing and morphological characterization
Oligonucleotides were heated at 95 °C for 5 min in the presence of 100 mM KCl and then slowly allowed to reach room temperature overnight. I-motif formation was induced by using 50 mM cacodylate buffer at pH 5.0. The list of the oligonucleotides used in this study is provided in Table S4.† Evidence for most of the G4 folding sequences used in this study was provided by electronic circular dichroism (ECD) measurements and isothermal difference spectra (IDS), and the latter is defined as the difference between unfolded and prefolded normalized UV/Vis spectra. ECD spectra were normalized to Δε (M−1 cm−1) = θ/(32980 × c × l) based on G-quadruplex strand concentration, where θ is the ECD ellipticity in millidegrees, c is the DNA concentration in mol L−1, and l is the path length in cm.General procedures
The absorption spectra were recorded on a Hitachi U-2900 spectrophotometer (at WUST), on a T90+ UV/Vis spectrometer (PG Instruments Ltd, at Umeå University) or on a Jasco V-650 spectrophotometer (at ENS de Lyon). Steady-state emission spectra were recorded with a Hitachi F-7000 fluorescence spectrophotometer (at WUST), with a Jasco FP-6500 spectrofluorometer equipped with a JascoPeltier-type temperature controller (ETC2736, at Umeå University) or with a Horiba Jobin-Ivon Fluorolog-3 fluorimeter (at ENS de Lyon). 1.0 cm path length quartz cells were used throughout these measurements.Spectra of 2 (procedure A) recorded at 1 GHz for the structure elucidation
NMR spectra (1H, 13C) of 2 were recorded at 298 K on a Bruker Avance III 1000 spectrometer at CRMN operating at 1000.3 MHz and 251.53 MHz for 1H and 13C, respectively, at CRMN, Villeurbanne. A 5 mm TCI H-C/N-D cryoprobe equipped with a z-gradient coil was used. The temperature was calibrated with a tube containing 40 mm of 99.8% MeOH-d4. Data were listed in parts per million (ppm) and were reported relative to tetramethylsilane. Residual solvent peaks were used as internal standards. 1H NMR spectra were recorded with a single pulse sequence with a 30° pulse angle, and 16 scans were collected (relaxation delay, 1 s). A 0.3 Hz line broadening was applied before Fourier transformation and a baseline correction was performed on spectra before integration with Bruker software. 1H chemical shifts are referenced to D2O (4.7 ppm). The gCOSY experiments were performed with 8192 data points with 64 scans for each of 256 t1 increments being acquired with spectral widths of 13369 Hz in F2 and F1. The required acquisition time was 306 ms. A recycle delay of 2 s was used. The gHSQC experiments were performed with 8192 data points with 16 scans for each of 256 t1 increments being acquired with spectral widths of 16026 Hz in F2 and 41505 Hz in F1. The required acquisition time was 256 ms. A recycle delay of 5 s was used. The gHMBC experiments were performed with 8192 data points with 128 scans for each of 256 t1 increments being acquired with spectral widths of 13021 Hz in F2 and 55868 Hz in F1. The required acquisition time was 315 ms. A recycle delay of 2 s was used. The gNOESY experiments were performed with 6144 data points with 64 scans for each of 256 t1 increments being acquired with spectral widths of 14098 Hz in F2 and F1. The required acquisition time was 218 ms. A mixing time of 2 s and a recycle delay of 2 s were used. 1H DOSY 2D experiments were acquired using a LED experiment. Gradients were linearly sampled in 32 to 40 points. 16 to 128 scans were acquired on 16k data points, with a recycle time of 3 s. The gradient pulse length was d/2 = 1.2 ms and the D diffusion delay was 50 ms. A Fourier transform was applied in F2 with 1 Hz exponential broadening. The DOSY spectra were obtained by fitting independently all columns of the F2 transformed spectrum, using a Levenberg–Marquardt algorithm embedded into the commercial software BRUKER TopSpin™.
Proton and carbon chemical shift assignments are provided in Table S1.†
Spectra of 2 (procedure B)
NMR spectra (1H and 13C) of 2 following procedure B via1′ (Fig. S4 and S5† for 1H and 13C NMR spectra) are reported in the ESI.†HPLC of 1′ to 2 conversion
HPLC/MS were recorded on an Agilent Poroshell SB-C18 column Øi.3 × L.100 mm using CH3CN/H2O (10 mM ammonium formate), flow rate 1 mL min−1, detection UV at 254 nm, and 10 μL injected. Chromatograms are provided in Fig. S15.†Theoretical studies: NMR chemical shift calculations
DFT/GIAO (Gauge independent atomic orbitals) calculations have been performed at the PBE0/6-311G(d,p)//PBE0/6-31+G(d) level of theory, in order to corroborate the assignments (Table S1†) of the experimental peaks for 1H and 13C NMR spectra. An implicit solvation scheme was used, the integral equation formalism polarizable continuum model (IEFPCM) to account for the solvation in water. The agreement between experimental vs. calculated 1H and 13C NMR chemical shifts, using tetramethylsilane as a reference, is shown in Fig. S16 and S17.†Kinetic experiments and fitting models
Spectra associated with the study of the photochemical reaction along with the applied kinetic equations and models are provided in the ESI (Fig. S23–S28†).Spectrophotometric and fluorimetric titrations
A solution of 1 (3.8 μM) or 2 (3.6 μM) was prepared by diluting the stock solution in a suitable amount of buffered water (CKCl = 100 mM, TRIS buffer 50.0 mM, pH = 7.4). The freshly prepared solutions were then titrated with the annealed oligonucleotide buffered solutions and left to equilibrate for several minutes before recording the UV/Vis or emission spectra.Nonlinear analysis of the saturation curves
Quantification of the association constants (K, K11 and K21) between 2 and canonical and noncanonical DNA structures was carried out by using a global nonlinear curve fitting procedure. The variation in the absorption and fluorescence signals of 2 was monitored upon the addition of incremental duplex and quadruplex concentrations and provides typical saturation binding curves. Binding constants were obtained with Bindfit by using multiple global fitting methods (Nelder–Mead method) on both the UV/Vis data in the range of 365–475 nm and fluorescence data in the range 445–545 nm. Dilution corrections were included in the fitting option.Fluorescence displacement assays
Fluorescence displacement assays were performed by titrating the 2-c-MYC Pu22 binary mixture by increasing the concentration of the well-known G4-binders Phen-DC3 or BRACO-19 until no change in the 2′ fluorescence intensity was observed.Electronic circular dichroism (ECD) spectra and ECD-based thermal melting assays
ECD spectra were recorded with a Jasco J-720 spectropolarimeter equipped with a JascoPeltier-type temperature controller (PTC-423L) and are presented as the sum of 4 accumulations by using a dual path length cuvette. ECD spectra were recorded in the range of 225–575 nm. Before use, the optical chamber of the CD spectrometer was deoxygenated with dry nitrogen and was held under a nitrogen atmosphere during the measurements. Appropriate references were subtracted from the obtained ECD spectra. CD melting data were acquired by increasing the temperature from 20 to 95 °C and monitoring the characteristic CD peaks of each sequence used. The G4 folding of these sequences, under the experimental conditions used for thermal assays (i.e. 5 mM KCl for c-MYC Pu22, Tel-22, and 4G3U3 and 15 mM KCl for Bom17), is reported in Fig. S29–S32.† Melting values were estimated by fitting the normalized melting curves with a dose response function using OriginPro 2015 software.1H NMR G4 titration studies
c-MYC Pu22 and 2 were dissolved in 10 mM potassium phosphate buffer (K2HPO4/KH2PO4, pH = 7.4) and 35 mM KCl. 10% D2O was added to the solutions. All spectra were recorded at 298 K on a Bruker 850 MHz Avance III HD spectrometer equipped with a 5 mm TCI cryoprobe. Excitation sculpting was used in the 1H NMR titration experiments, and 256 scans were recorded. Processing was performed in Topspin 3.6 (Bruker Biospin, Germany).Molecular dynamics simulations of 2 interacting with ds-DNA and G4s
All-atom explicit solvent molecular dynamics simulations were performed to probe the interaction mode of 2 with a ds-DNA sequence, telomeric G4s, and c-MYC Pu22 and details are provided in the ESI.†DNA polymerase stop assays
Reactions containing 40 nM template DNA (G4 or non-G4) annealed to a TET-labeled primer were incubated with different concentrations of 1, photoconverted 2 and CX-5461 in the presence of 50 mM KCl. The control reaction contained no ligands. 0.625 U μL−1 Taq-DNA polymerase was used in the reactions. Reactions with Taq-DNA polymerase were incubated for 30 minutes at 50 °C. The following oligonucleotide sequences were used:Primer 5′-3′:
TET-TGAAAACATTATTAATGGCGTCGAGCGTCCG
c-MYC Pu24T 5′-3′:
ATATATATATTGAGGGTGGTGAGGGTGGGGAAGGATATATATATCGGACGCTCGACGCCATTAATAATGTTTTCA
Tel-22 5′-3′:
ATATATATATAGGGTTAGGGTTAGGGTTAGGGATATATATATCGGACGCTCGACGCCATTAATAATGTTTTCA
Non-G4 5′-3′:
GAGACCATTCAAAAGGATAATGTTTGTCATTTAGTATATGCCCCTGCTCGTCTTCCCTTCTCCGGACGCTCGACGCCATTAATAATGTTTTCA
Fluorescence microscopy
HeLa cells (cervical cancer cell line) were cultured at 37 °C (5% CO2) in a DMEM high glucose medium (Gibco) supplemented with 10% fetal bovine serum and penicillin–streptomycin. For fixed cell imaging, the cells were seeded on glass-bottomed microwell dishes (MaTek Corp.), fixed with cold MeOH for 10 min at −20 °C and permeabilized with PBST (phosphate buffered saline supplemented with 0.1% Triton X-100). For the in cellulo photoconversion (Fig. 8 and S49†), cells treated with 1 (10 μM) for 30 minutes were either kept in the dark or irradiated by using a 312 nm UV light for 1, 2, 5 and 10 minutes before recording the fluorescence images. In the cellular experiments reported in Fig. 9 and S50, S52, S55, S58 and S63,† the cells were treated with the isolated form of 2 (10 μM) for 30 min at room temperature. For RNA degradation, the MeOH fixed cells were treated with 1 mg ml−1 RNase A (Thermo Fisher) and incubated for 2 hours at 37 °C. For DNA degradation, the MeOH fixed cells were treated with 50 U μL−1 DNase I (Thermo Fisher) and incubated for 2 hours at 37 °C. For the fluorescence competition assay 2 (10 μM) was incubated with BRACO-19 (10 μM) for 30 min at room temperature. Images were acquired using a Leica SP8 FALCON (FAst Life time CONtrast) confocal microscope using an HC PL APO 63×/1.20 Water motCORR CS2 objective. Maximum intensity projection of Z-stack images was used for data presentation. Quantitative data analysis was performed by selecting the regions of interest and measuring the average fluorescence signal from the selected areas. All data were processed by using ImageJ software available at https://imagej.nih.gov/ij/.Immunofluorescence experiments
BG4 with a FLAG-tag (Ab00174-30.126) was purchased from Absolute Antibody Ltd (Oxford, UK). 50000 cells were seeded the day before treatment on glass-bottomed microwell dishes (MatTek Corp.). The cells were fixed with 4% PFA for 15 min and then permeabilized in 0.1% Triton X-100 for 20 min at room temperature. The cells were blocked with 2% nonfat milk dissolved in 1× PBS buffer and incubated for 1 h at 37 °C. BG4 (dilution: 1:1000) was dissolved in 1% tween-20/1× PBS supplemented with 5% goat serum (Sigma Merck, G9023) and the cells treated with BG4 were incubated for 1 h at 37 °C. The cells were washed with 1× PBS and incubated with Rabbit anti-Flag (Cell Signaling, cat. num 14793) (dilution: 1:800) dissolved in 1% tween-20/1× PBS supplemented with 5% goat serum for 1 h at 37 °C. Then, the cells were washed with 1× PBS and incubated with goat anti-rabbit IgG (H + L) Alexa Fluor 594 (Life Technologies, cat. num. A11012) (dilution 1:1000) dissolved in 1% tween-20/1× PBS supplemented with 5% goat serum for 1 h at 37 °C. Finally, the cells were washed multiple times with 1× PBS and incubated with 2 (10 μM) for 30 min at room temperature. Control experiments were also performed in the absence of the primary BG4 antibody. Images were acquired using a Leica SP8 FALCON confocal microscope. Maximum intensity projection of Z-stack images was used for data presentation. All data were processed by using ImageJ software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Centre de RMN à Haut Champs, CRMN, FR CNRS 3050, for the access to their 1 GHz NMR facilities. MM, TLB, ED, SDQ, CA, CM and LG thank the SYSPROD project and AXELERA Pôle de Compétitivité for financial support (PSMN Data Center). NS and MDe thank the Biochemical Imaging Center at Umeå University, the National Microscopy Infrastructure (VR-RFI 2016-00968) and NMR for Life (http://www.nmrforlife.se), for NMR spectroscopy support. MM, TLB, ED, SDQ, CA, CM and LG thank the “fond recherche” of the Ecole Normale Supérieure de Lyon for funding, in the framework of the DiazBioLic project. Work in Sabouri's lab was supported by the Knut and Alice Wallenberg Foundation (KAW2015-0189), Cancerfonden (2019/126) and the Swedish Research Council (VR-MH 2018-02651). MDe was supported by a fellowship from the MIMS Excellence by Choice Postdoctoral Programme. Work in Matczyszyn's lab was supported by the NCN Opus grant (UMO-2019/35/B/ST4/03280).Notes and references
- J. R. Williamson, Annu. Rev. Biophys. Biomol. Stuct., 1994, 23, 703–730 CrossRef CAS PubMed
.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770 CrossRef CAS PubMed
- J.-L. Mergny, J.-F. Riou, P. Mailliet, M.-P. Teulade-Fichou and E. Gilson, Nucleic Acids Res., 2002, 30, 839–865 CrossRef CAS PubMed
- J. W. Shay, Cancer Discovery, 2016, 6, 584–593 CrossRef CAS PubMed
- D. Monchaud, P. Yang, L. Lacroix, M.-P. Teulade-Fichou and J.-L. Mergny, Angew. Chem., Int. Ed., 2008, 47, 4858–4861 CrossRef CAS PubMed
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5305–5309 CrossRef CAS PubMed
- M. L. Di Pietro, F. Puntoriero, F. Tuyéras, P. Ochsenbein, P. P. Lainé and S. Campagna, Chem. Commun., 2010, 46, 5169–5171 RSC
- T. Nakamura, K. Iida, M. Tera, K. Shin-ya, H. Seimiya and K. Nagasawa, ChemBioChem, 2012, 13, 774–777 CrossRef CAS PubMed
- P. Murat, M. V. Gormally, D. Sanders, M. D. Antonio and S. Balasubramanian, Chem. Commun., 2013, 49, 8453–8455 RSC
- S. Bandeira, J. Gonzalez-Garcia, E. Pensa, T. Albrecht and R. Vilar, Angew. Chem., Int. Ed., 2018, 57, 310–313 CrossRef CAS PubMed
- M. P. O'Hagan, J. Ramos-Soriano, S. Haldar, S. Sheikh, J. C. Morales, A. J. Mulholland and M. C. Galan, Chem. Commun., 2020, 56, 5186–5189 RSC
- M. P. O'Hagan, S. Haldar, M. Duchi, T. A. A. Oliver, A. J. Mulholland, J. C. Morales and M. C. Galan, Angew. Chem., Int. Ed., 2019, 58, 4334–4338 CrossRef PubMed
- A. Bensalah-Ledoux, D. Pitrat, T. Reynaldo, M. Srebro-Hooper, B. Moore, J. Autschbach, J. Crassous, S. Guy and L. Guy, Chem. – Eur. J., 2016, 22, 3333–3346 CrossRef CAS PubMed
- L. Jierry, S. Harthong, C. Aronica, J.-C. Mulatier, L. Guy and S. Guy, Org. Lett., 2012, 14, 288–291 CrossRef CAS PubMed
- S. Speed, F. Pointillart, J.-C. Mulatier, L. Guy, S. Golhen, O. Cador, B. Le Guennic, F. Riobé, O. Maury and L. Ouahab, Eur. J. Inorg. Chem., 2017, 2017, 2100–2111 CrossRef CAS
- D. Sucunza, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, J. Org. Chem., 2016, 81, 10126–10135 CrossRef CAS PubMed
- X. Liu, M. Li, T. Han, B. Cao, Z. Qiu, Y. Li, Q. Li, Y. Hu, Z. Liu, J. W. Y. Lam, X. Hu and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 11259–11268 CrossRef CAS PubMed
- Y. Chen, X.-R. Wei, R. Sun, Y.-J. Xu and J.-J. Ge, Anal. Methods, 2019, 11, 3523–3531 RSC
- H. Bertrand, A. Granzhan, D. Monchaud, N. Saettel, R. Guillot, S. Clifford, A. Guédin, J.-L. Mergny and M.-P. Teulade-Fichou, Chem. – Eur. J., 2011, 17, 4529–4539 CrossRef CAS PubMed
- A. Granzhan and H. Ihmels, Synlett, 2016, 1775–1793 CAS
- A. Granzhan, H. Ihmels and K. Jäger, Chem. Commun., 2009, 1249–1251 RSC
- A. Marchand, A. Granzhan, K. Iida, Y. Tsushima, Y. Ma, K. Nagasawa, M.-P. Teulade-Fichou and V. Gabelica, J. Am. Chem. Soc., 2015, 137, 750–756 CrossRef CAS PubMed
- W. Luo, C. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. Fan, ACS Nano, 2010, 4, 7451–7458 CrossRef CAS PubMed
-
R. Schaal, in Chemical Kinetics of Homogeneous Systems, ed. R. Schaal, Springer, Dordrecht, Holland, 1974 Search PubMed
- A. T. Phan, Y. S. Modi and D. J. Patel, J. Am. Chem. Soc., 2004, 126, 8710–8716 CrossRef CAS PubMed
- E. Hatzakis, K. Okamoto and D. Yang, Biochemistry, 2010, 49, 9152–9160 CrossRef CAS PubMed
- R. D. Gray, J. O. Trent, S. Arumugam and J. B. Chaires, J. Phys. Chem. Lett., 2019, 10, 1146–1151 CrossRef CAS
- K.-B. Wang, M. S. A. Elsayed, G. Wu, N. Deng, M. Cushman and D. Yang, J. Am. Chem. Soc., 2019, 141, 11059–11070 CrossRef CAS
- Z. Dvoráková, D. Renciuk, I. Kejnovská, P. Školáková, K. Bednárová, J. Sagi and M. Vorlícková, Nucleic Acids Res., 2018, 46, 1624–1634 CrossRef PubMed
- M. Deiana, B. Mettra, K. Matczyszyn, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Biomacromolecules, 2016, 17, 3609–3618 CrossRef CAS PubMed
- M. Dudek, M. Deiana, Z. Pokladek, P. Mlynarz, M. Samoc and K. Matczyszyn, Nanoscale, 2018, 10, 11302–11306 RSC
- M. Deiana, B. Mettra, L. Martinez-Fernandez, L. M. Mazur, K. Pawlik, C. Andraud, M. Samoc, R. Improta, C. Monnereau and K. Matczyszyn, J. Phys. Chem. Lett., 2017, 8, 5915–5920 CrossRef CAS
- E. Largy, N. Saettel, F. Hamon, S. Dubruille and M. P. Teulade-Fichou, Curr. Pharm. Des., 2012, 18, 1992–2001 CrossRef CAS PubMed
- E. Largy, F. Hamon and M.-P. Teulade-Fichou, Anal. Bioanal. Chem., 2011, 400, 3419–3427 CrossRef CAS PubMed
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC
- A. Marchand, D. Strzelecka and V. Gabelica, Chem. – Eur. J., 2016, 22, 9551–9555 CrossRef CAS
- W. Liu, Y.-F. Zhong, L.-Y. Liu, C.-T. Shen, W. Zeng, F. Wang, D. Yang and Z.-W. Mao, Nat. Commun., 2018, 9, 3496 CrossRef
- M. P. O'Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Chem. – Eur. J., 2020, 26, 6224–6233 CrossRef PubMed
- V. Grande, F. Doria, M. Freccero and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7520–7524 CrossRef CAS PubMed
- M. Deiana, K. Chand, J. Jamroskovic, I. Obi, E. Chorell and N. Sabouri, Angew. Chem., Int. Ed., 2020, 59, 896–902 CrossRef CAS PubMed
- V. Grande, C.-A. Shen, M. Deiana, M. Dudek, J. Olesiak-Banska, K. Matczyszyn and F. Würthner, Chem. Sci., 2018, 9, 8375–8381 RSC
- W. J. Chung, B. Heddi, F. Hamon, M.-P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS PubMed
- N. C. Garbett, P. A. Ragazzon and J. B. Chaires, Nat. Protoc., 2007, 2, 3166–3172 CrossRef CAS PubMed
- H. Han, L. H. Hurley and M. Salazar, Nucleic Acids Res., 1999, 27, 537–542 CrossRef CAS
- H. Xu, M. Di Antonio, S. McKinney, V. Mathew, B. Ho, N. J. O'Neil, N. D. Santos, J. Silvester, V. Wei, J. Garcia, F. Kabeer, D. Lai, P. Soriano, J. Banáth, D. S. Chiu, D. Yap, D. D. Le, F. B. Ye, A. Zhang, K. Thu, J. Soong, S.-C. Lin, A. H. C. Tsai, T. Osako, T. Algara, D. N. Saunders, J. Wong, J. Xian, M. B. Bally, J. D. Brenton, G. W. Brown, S. P. Shah, D. Cescon, T. W. Mak, C. Caldas, P. C. Stirling, P. Hieter, S. Balasubramanian and S. Aparicio, Nat. Commun., 2017, 8, 14432 CrossRef CAS PubMed
- L. R. Ferguson and W. A. Denny, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2007, 623, 14–23 CrossRef CAS PubMed
- J. W. Nial, R. B. Craig, J. G. Collins, K. Sharon and R. A.-W. Janice, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef PubMed
- Y. Yang, J. Mu and B. Xing, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1408 Search PubMed
- O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Angew. Chem., Int. Ed., 2019, 58, 8007–8012 CrossRef CAS PubMed
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. MacNeil and J. A. Thomas, Chem. Sci., 2019, 10, 3502–3513 RSC
- M. Deiana, J. Jamroskovic, I. Obi and N. Sabouri, Chem. Commun., 2020, 56, 14251–14254 RSC
- M. Di Antonio, A. Ponjavic, A. Radzevičius, R. T. Ranasinghe, M. Catalano, X. Zhang, J. Shen, L. M. Needham, S. F. Lee, D. Klenerman and S. Balasubramanian, Nat. Chem., 2020, 12, 832–837 CrossRef CAS PubMed
- J. Li, X. Yin, B. Li, X. Li, Y. Pan, J. Li and Y. Guo, Anal. Chem., 2019, 91, 5354–5361 CrossRef CAS
- F. Doria, M. Nadai, M. Zuffo, R. Perrone, M. Freccero and S. N. Richter, Chem. Commun., 2017, 53, 2268–2271 RSC
- J. Jamroskovic, M. Doimo, K. Chand, I. Obi, R. Kumar, K. Brännström, M. Hedenström, R. Nath Das, A. Akhunzianov, M. Deiana, K. Kasho, S. Sulis Sato, P. L. Pourbozorgi, J. E. Mason, P. Medini, D. Öhlund, S. Wanrooij, E. Chorell and N. Sabouri, J. Am. Chem. Soc., 2020, 142, 2876–2888 CrossRef CAS PubMed
- M. Deiana, K. Chand, J. Jamroskovic, R. N. Das, I. Obi, E. Chorell and N. Sabouri, Nanoscale, 2020, 12, 12950–12957 RSC
- J. Lefebvre, C. Guetta, F. Poyer, F. Mahuteau-Betzer and M.-P. Teulade-Fichou, Angew. Chem., Int. Ed., 2017, 56, 11365–11369 CrossRef CAS PubMed
- S. Zhang, H. Sun, H. Chen, Q. Li, A. Guan, L. Wang, Y. Shi, S. Xu, M. Liu and Y. Tang, Biochim. Biophys. Acta, 2018, 1862, 1101–1106 CrossRef CAS
- E. Ruggiero and S. N. Richter, Nucleic Acids Res., 2018, 46, 3270–3283 CrossRef CAS PubMed
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS
- G. Biffi, M. Di Antonio, D. Tannahill and S. Balasubramanian, Nat. Chem., 2014, 6, 75–80 CrossRef CAS PubMed
- S. Zhang, H. Sun, L. Wang, Y. Liu, H. Chen, Q. Li, A. Guan, M. Liu and Y. Tang, Nucleic Acids Res., 2018, 46, 7522–7532 CrossRef CAS PubMed
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2016, 45, 1542–1556 RSC
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR spectra, HPLC, theoretical studies, spectra associated with the study of the photochemical reactions, G4 binding studies, cell imaging and quantification. See DOI: 10.1039/d1nr02855c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |