
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Drug delivery, biodistribution and anti-EGFR activity: theragnostic nanoparticles for simultaneous in vivo delivery of tyrosine kinase inhibitors and kinase activity biosensors†
Robin
Bofinger‡§
a,
Gregory
Weitsman‡
 b,
Rachel
Evans
bc,
Matthias
Glaser
ad,
Kerstin
Sander
ad,
Helen
Allan
a,
Daniel
Hochhauser
c,
Tammy L.
Kalber
e,
Erik
Årstad
ad,
Helen C.
Hailes
a,
Tony
Ng
*bc and
Alethea B.
Tabor
*a
b,
Rachel
Evans
bc,
Matthias
Glaser
ad,
Kerstin
Sander
ad,
Helen
Allan
a,
Daniel
Hochhauser
c,
Tammy L.
Kalber
e,
Erik
Årstad
ad,
Helen C.
Hailes
a,
Tony
Ng
*bc and
Alethea B.
Tabor
*a
aDepartment of Chemistry, University College London, 20, Gordon Street, London WC1H 0AJ, UK. E-mail: a.b.tabor@ucl.ac.uk
bSchool of Cancer and Pharmaceutical Sciences, King's College London, London, SE1 1UL, UK. E-mail: tony.ng@kcl.ac.uk
cUCL Cancer Institute, Paul O'Gorman Building, University College London, London WC1E 6DD, UK
dCentre for Radiopharmaceutical Chemistry, Kathleen Lonsdale Building, 5 Gower Place, London WC1E 6BS, UK
eCentre for Advanced Biomedical Imaging, Paul O'Gorman Building, University College London, London WC1E 6DD, UK
First published on 3rd November 2021
Abstract
In vivo delivery of small molecule therapeutics to cancer cells, assessment of the selectivity of administration, and measuring the efficacity of the drug in question at the molecule level, are important ongoing challenges in developing new classes of cancer chemotherapeutics. One approach that has the potential to provide targeted delivery, tracking of biodistribution and readout of efficacy, is to use multimodal theragnostic nanoparticles to deliver the small molecule therapeutic. In this paper, we report the development of targeted theragnostic lipid/peptide/DNA lipopolyplexes. These simultaneously deliver an inhibitor of the EGFR tyrosine kinase, and plasmid DNA coding for a Crk-based biosensor, Picchu-X, which when expressed in the target cells can be used to quantify the inhibition of EGFR in vivo in a mouse colorectal cancer xenograft model. Reversible bioconjugation of a known analogue of the tyrosine kinase inhibitor Mo-IPQA to a cationic peptide, and co-formulation with peptides containing both EGFR-binding and cationic sequences, allowed for good levels of inhibitor encapsulation with targeted delivery to LIM1215 colon cancer cells. Furthermore, high levels of expression of the Picchu-X biosensor in the LIM1215 cells in vivo allowed us to demonstrate, using fluorescence lifetime microscopy (FLIM)-based biosensing, that EGFR activity can be successfully suppressed by the tyrosine kinase inhibitor, released from the lipopolyplexes. Finally, we measured the biodistribution of lipopolyplexes containing 125I-labelled inhibitors and were able to demonstrate that the lipopolyplexes gave significantly higher drug delivery to the tumors compared with free drug.
Introduction
Current challenges in next-generation medicine have stimulated the rapid development of theragnostic agents. These are of increasing importance for diseases such as cancer, where no two patients will have exactly the same biomarkers and oncogenic mutations, and where most currently available therapeutic agents have limited target selectivity, poor localization, and undesirable side-effects. Theragnostic agents can be based on small molecules conjugated to a targeting moiety, such as an antibody; on engineered mammalian cells; and on various types of nanoparticles (e.g. iron oxide, gold, polymeric or liposomal).1 Whilst all of these have limitations,2 liposome-based nanoparticles have several key advantages. These include the ability to design multimodal nanoparticles with several functionalities contained in, or attached to, a single liposome: rapid cellular uptake; a wide range of cellular compatibilities and low toxicity; and long circulating half-life combined with eventual biodegradability.3Many theragnostic agents have now been developed which are capable of diagnosing, through imaging, the presence of cancerous cells and then delivering a precisely targeted therapeutic intervention.4 Whilst these approaches can be used to measure biodistribution and accumulation of small molecule drugs at the tumor, none of the theragnostic agents reported to date can quantify the effectiveness of the therapeutic at the molecular level in vivo. This is particularly important in targeted cancer therapy. Although diagnostic assays are available to determine protein expression status for some types of cancer, for many tumor types there are either no validated assays, or the expression status of the protein cannot in practice be correlated with the clinical response to the drug.5 The development of theragnostic nanoparticles which would improve drug delivery, whilst also allowing both the biodistribution and response to the drug to be measured, would be a major step towards the development of more effective cancer therapies, tailored towards the driving molecular phenotype of an individual tumor.
Epidermal growth factor receptor (EGFR) is found to be overexpressed and/or constitutively activated in a variety of tumors including breast, lung and colon6 and is a validated target for drug development. The EGFR family7 consists of four members (EGFR, HER2, HER3 and HER4) with more than 10 different ligands8 able to activate downstream signaling, leading to growth proliferation and inhibition of apoptosis, angiogenesis and metastasis. Several therapeutic strategies targeting EGFR have been devised. In particular, small molecule inhibitors of the EGFR intracellular tyrosine kinase activity (TKI), and antibodies blocking ligand interaction have been developed and tested in preclinical and clinical studies.9,10 Unfortunately, the factors that predict clinical response to anti-EGFR agents remain unclear, and the success rate for these reagents in the clinic is low. Expression of EGFR does not correlate with efficacy of the EGFR antibody cetuximab11 and the use of the antibody is limited by toxicity.12 In recent clinical studies of EGFR-expressing breast13 and colorectal14 cancer patients, response rates to EGFR-targeted therapies were low and varied between cancer types, with the majority of patients eventually developing resistance to these reagents. It is clear that in order for these strategies to be successful, much more research is needed to understand how EGFR inhibitors and antibodies mechanistically function in vivo at both the cellular and molecular level.15
A strategy for overcoming the poor pharmacokinetic profiles and low target selectivity of anticancer drugs is to use liposomes, liposome-based nanoparticles and polymersomes as delivery vehicles.16 The polyethylene glycol (PEG)-shielded liposomal doxorubicin (DOXIL®) is clinically approved for the treatment of ovarian and breast cancer, with significantly longer circulation times and lower cardiotoxicity compared to the free drug, and several other liposomal drug formulations are in Phase II trials for other cancers.17 At the preclinical level, radiolabeling18 of liposome-encapsulated dasatinib with 18F and liposome-encapsulated Mo-IPQA labeled19 with 124I have enabled quantification, demonstrating some of the highest tumor uptakes in vivo of TKI so far observed. Non-targeted liposomal drug formulations were believed to rely on the passive accumulation of these nanoparticles (100–200 nm) in tumors through the Enhanced Permeability and Retention (EPR) effect. However, many clinical studies have failed to show either increased nanoparticle accumulation in tumors or increased efficacy, and it is now clear that this effect is, at best, heterogeneous in humans.20,21 Ligand-mediated cell-selective targeting offers additional advantages in terms of target cell specificity and cellular uptake. For example, immunoliposomes have been prepared using anti-EGFR antibodies as targeting moieties and were used to deliver drugs such as 5-fluorouracil,22 cisplatin23 and doxorubicin24 to cancer cells in vivo, with moderate to significant improvements in cytotoxicity and cell selectivity compared to non-targeted liposomes. Likewise, polymersomes encapsulating plitidepsin and targeted with an anti-EGFR antibody showed greatly increased cytotoxicity and cellular uptake in colorectal cancer cell lines compared to an untargeted polymersome.25 Liposomes targeted with EGFR-binding peptides have also been used to deliver doxorubicin26 and cisplatin27 effectively to cancer cells. Finally, lipoplexes targeted with anti-EGFR antibodies have been formulated to deliver siRNA to non-small cell lung cancer cell lines28 and into EGFR-overexpressing hepatocellular carcinoma.29 Surface modification was necessary for specific uptake of the siRNA into the cells and resulted in enhanced tumor accumulation when compared to untargeted lipoplexes.
We have previously reported30 the development of EGFR-targeted lipopolyplexes for the delivery of pDNA encoding an EGFR biosensor.31 This allowed us to monitor EGFR activity in a breast cancer model in vivo, before and after separate treatment with a TKI, Mo-IPQA.30 As well as successfully demonstrating the inhibition of tyrosine phosphorylation by EGFR in vivo, and showing a significant degree of intratumoral heterogeneity in EGFR activity, our results showed that small molecule TKIs have a very poor uptake in cancer cells, and that the uptake is unselective. In this paper, we report the development of the first targeted, lipopolyplex-based theragnostic nanoparticle that improves drug delivery to cancer cells, allows quantification of the biodistribution, and also provides a readout of TKI mediated enzyme inhibition. As the reasons for the heterogeneity of response of colorectal cancer cells to EGFR-targeted therapies, and the rapid emergence of chemotherapy-resistant strains, are still poorly understood15 these nanoparticles have been used for preliminary investigation of biodistribution and uptake in a murine xenograft colorectal cancer model.
Results and discussion
Design of lipopolyplex-based theragnostic nanoparticles
In our previous work32–38 we have developed lipopolyplexes formulated from a ternary mixture of lipids, peptides and DNA for the targeted delivery of pDNA to tumor cells. The peptide component is a trifunctional sequence comprising: a linear K16 domain to condense pDNA; a cell-targeting sequence that is displayed at the surface of the nanoparticle; and a linker sequence, RVRR, that is a substrate for the endosomal enzyme furin. Mixtures of cationic and neutral lipids are used and are co-formulated with the neutral lipid dioleoylphosphatidylethanolamine (DOPE). The cationic lipids include both DOTMA39 and the analogue DODEG432 which has a short ethylene glycol oligomer headgroup. These give shielded nanocomplexes that have better stability within the systemic circulation but do not impede cellular uptake.The targeting peptide sequence at the surface of these lipopolyplexes binds to the cell of interest and mediates internalisation via receptor-mediated endocytosis: cleavage of the peptide at the RVRR sequence allows partial disassembly of the nanoparticle after internalisation,33 and the neutral lipid DOPE promotes fusion with the endosomal membrane and mediates escape of the pDNA/peptide complex from the endosome.40 These nanoparticles are able to transfect tumor cells in vivo with transfection efficiencies comparable to viral methods,34 and lipopolyplexes generally have lower cytotoxicity than commercially available transfection reagents such as Lipofectamine2000.37,38,41,42 We have previously carried out biophysical characterisation of these lipopolyplexes by a variety of techniques including TEM,37 freeze-fracture EM35 and cryo-EM.36 These EM studies suggest that these lipopolyplexes form spherical particles with a dense internal core and an irregular outer layer.
To report the levels of EGFR activity in cells using these ternary lipopolyplexes, we used a genetically encoded CrkII-based biosensor (Picchu-X) which undergoes conformational changes upon phosphorylation of tyrosine-221 by EGFR.31,43 These changes are quantified by fluorescence resonance energy transfer (FRET) as monitored by fluorescence lifetime imaging microscopy (FLIM). We transfected this biosensor into EGFR-positive tumor cells using targeted ternary liposome-base nanoparticles bearing EGFR-binding peptides at the surface, and measured the response of these tumor cells to a TKI, Mo-IPQA, administered separately.30
In order to achieve our goals of selectively delivering a TKI to colorectal cancer cells, imaging the inhibition of EGFR within the tumor and evaluating the biodistribution of the TKI, we designed enhanced multifunctional lipopolyplex-based nanoparticles (Fig. 1). These lipopolyplex formulations had to include: mixtures of lipids that would enhance the particle stability and provide steric shielding; peptide targeting sequences that would promote uptake from colorectal cancer cells; and a cationic peptide–TKI bioconjugate that would enable incorporation of high levels of both the TKI and the pDNA within the lipopolyplex. Additionally, we needed to develop synthetic strategies to ensure that the cationic peptide–TKI bioconjugate could be radiolabelled with 125I, thus allowing the biodistribution of both the TKI and the lipopolyplex to be assessed.
 | ||
| Fig. 1 (a) Structures of the previously reported 18F PET radiotracer F-[PEG6-IPQA]52 and the EGFR TKI Mo-IPQA;51 (b) peptide, lipid and plasmid DNA components of the lipopolyplex nanoparticles used in this work. | ||
With this next generation of theragnostic lipopolyplexes, in addition to DOPE, DOTMA and DODEG-4 we included cholesterol to further increase bilayer stability,44 and Cy-5-DOTMA30 to enable the uptake of the nanoparticles to the tumor cells to be easily monitored. In order to target the nanoparticles efficiently to cell lines over-expressing EGFR, we synthesized trifunctional peptides with DNA-binding linear K16, RVRR and cell-targeting sequences, as before. We have evaluated three peptide sequences for their ability to target colorectal cancer cells. Peptides P1 and P4 use the LARLLT (D4) sequence,45P2 and P5 the AEYLR46 and P3 and P6 the YHWYGYTPQNVI (GE11) sequence.47,48 These sequences had been previously reported to bind tightly to EGFR and, when displayed at the surface of nanoparticles, mediate uptake to EGFR+ tumor cells. Indeed, we have previously demonstrated that lipopolyplexes formulated with P1 effectively deliver pDNA encoding for a biosensor to HCC1954 and MDA-MB-231 breast cancer cell lines in vivo,30 and that lipopolyplexes formulated with P1, P2 or P3 effectively transfect HCC1954 cells in vitro.49
The major challenge to the design of these self-assembling nanoparticles was the co-encapsulation of the small molecule TKI with pDNA. Whilst several groups have developed multifunctional nanoparticles for combination therapy with small molecules and nucleic acids,50–54 most of these approaches have focused on the co-delivery of siRNA, and many of the reported approaches are based on dendrimers, which have associated toxicities and result in the formation of heterogeneous mixtures.55 In some cases where liposomal co-formulation of both nucleic acid and small molecule was attempted, aggregation of the liposome and leakage of the drug was seen.56 In order to ensure that the TKI was successfully encapsulated in the self-assembling nanoparticle, and was retained within the nanoparticle after formulation, we sought to reversibly bioconjugate the TKI to one of the components of the nanoparticle. We therefore designed a cationic peptide–TKI bioconjugate 1 (Fig. 1) with a disulfide linkage between the two moieties. We based our design on the known EGFR TKI, F-[PEG6-IPQA], a modified form of Mo-IPQA19,51 that had been developed previously as an 18F PET radiotracer (Fig. 1a).57 As the PEG moiety of this TKI is predicted to protrude from the active site of the enzyme58 we reasoned that minimal modification to give a thiol-terminated PEG6 would not adversely affect the potency or selectivity of this inhibitor, but would allow bioconjugation to K16Cys to give K16Cys-S-S-[PEG6-IPQA] 1. It was also envisaged that the K16 cationic sequence of this bioconjugate would bind to pDNA with the same efficiency as the trifunctional K16-RVRR-[targeting sequence] or [targeting sequence]-RVRR-K16 peptides P1–P6, and that we could therefore prepare lipopolyplexes using a mixture of 1 and peptides P1–P6. After internalization of the lipopolyplex, we predicted that the disulfide linkage would be cleaved in the reducing environment of the endosome,59,60 releasing PEG6-IPQA into the cytoplasm.
Synthesis of K16Cys-S-S-[PEG6-IPQA] 1 and MeO-[PEG6-IPQA] 9
In order to prepare K16Cys-S-S-[PEG6-IPQA] 1, we required the key intermediate NPys-S-PEG6-IPQA 2, with the SH group activated as the NPys derivative61 for coupling to the peptide (Scheme 1). The previously reported 17-((4-((3-iodophenyl)amino)-6-nitroquinazolin-7-yl)oxy)-3,6,9,12,15-pentaoxaheptadecyl methanesulfonate (3) was synthesized in 52% yield from 7-fluoro-6-nitro-3H-quinazolin-4-one in 3 steps using modified literature procedures (see ESI† for details). | ||
| Scheme 1 Reagents and conditions: (i) Ph3CSH, NaH, THF, rt, 2 h (82%); (ii) SnCl2·H2O, THF, reflux, 3 h (61%); (iii) isobutyl chloroformate, acrylic acid, Et3N, −40 °C, 35 min (57%); (iv) DTNP, CH2Cl2, CF3COOH, rt, 2 h (48%); (v) CysLys167, MeOH, rt, 1 h (59%). | ||
This was subsequently reacted with the sodium salt of triphenylmethanethiol to yield the trityl protected thiol compound 4. Reduction of the nitro group was carried out using tin(II)chloride which gave the desired amino intermediate 5 in 61% yield. Using a previously reported procedure58 compound 5 was reacted with acryloyl isobutyl carbonate, prepared in situ from acrylic acid and isobutyl chloroformate, to afford compound 6 in 57% yield. At this point, the trityl protecting group was replaced by NPys in a one pot reaction under acidic conditions using 2,2′-dithiobis(5-nitropyridine)62 to afford 2 in 48% yield. In order to prevent competing Michael reaction between the thiol group and the unsaturated amide, it was necessary to maintain the thiol protection throughout the synthetic sequence, and to carry out the replacement of Trt by NPys at acidic pH. Finally, reaction63 with the cationic peptide CysLys167 in MeOH followed by HPLC gave 1 in 59% yield.
In vitro optimization of nanoparticle formulation
To test the ability of lipopolyplexes to deliver pDNA and inhibitor we used LIM1215 colon cancer cells. These are known to have a high level of EGFR expression and response to the TKI inhibitor,64,65 and we had previously demonstrated that a reliable xenograft model could be established.30 As a control, to check the response of the colorectal cancer cell lines to this class of TKI, we prepared an analogue of 1, with the disulfide-linked peptide replaced with an –OMe group. TKI analogue MeO-[PEG6-IPQA] 8 was synthesized from intermediate 3via a similar route to that of 1 (ESI†) and demonstrated to act as a potent inhibitor of EGFR activation in SW48 cells (Fig. S1†).Initially, the sequence of the trifunctional peptides was investigated to see whether placing the targeting sequence at the N or the C terminal had any effect on lipopolyplex uptake and expression in colorectal cancer cells. Preliminary experiments (ESI, Fig. S2†) suggested that the expression levels were lower for all peptides with the AEYLR sequence (P2, P5), and higher for LARLLT (P1, P4) and GE11 (P3, P6) in DLD1 colorectal cancer cells. In this cell line, the C-terminal LARLLT (P1) and N-terminal GE11 (P3) peptides yielded the highest expression levels. The best performing formulation (CLA1, using the P1 sequence) was tested in DLD1 and LIM1215 cells (Fig. S3†). Expression levels of Picchu-X biosensor were far higher in LIM1215 cells, which were selected for further optimisation of the lipopolyplex formulations. Subsequently, the impact of plasmid DNA concentration and targeting peptide (P1–P6) on the transfection efficiency in LIM1215 cells were assessed (Fig. S4 and S5†). Although formulations with the C-terminal LARLLT peptide (P1) still showed the best transfection efficiency, formulations with N-terminal AEYLR (P5) and N-terminal GE11 (P6) also performed well in this cell line. In order to optimize the receptor binding and transfection properties of these lipopolyplexes, we then investigated formulations using different targeting peptides P1, P5, P6 and differing lipid compositions (Table 1), but initially without K16Cys-S-S-[PEG6-IPQA] 1. While we observed that all lipopolyplex formulations were equally internalized by the target cells, changes in the lipid composition and in the concentrations of bifunctional peptides P1, P5, P6, had an impact on the transfection of the Picchu biosensor.
| DODEG4 | DOPE | DOTMA | Cholesterol | Cy5 | Targeting peptide | K16Cys-S-S-[PEG6-IPQA] 1 | |
|---|---|---|---|---|---|---|---|
| CLA1 | 39 | 21 | 15 | 23 | 2 | P1 | — |
| CLA2 | 39 | 44 | 15 | 0 | 2 | P1 | — |
| CLA3 | 39 | 37 | 23 | 0 | 2 | P1 | — |
| CLA4 | 30 | 37 | 23 | 8 | 2 | P1 | — |
| NAE1 | 39 | 21 | 15 | 23 | 2 | P5 | — |
| NAE2 | 39 | 44 | 15 | 0 | 2 | P5 | — |
| NAE3 | 39 | 37 | 23 | 0 | 2 | P5 | — |
| NAE4 | 30 | 37 | 23 | 8 | 2 | P5 | — |
| NGE1 | 39 | 21 | 15 | 23 | 2 | P5 | — |
| NGE2 | 39 | 44 | 15 | 0 | 2 | P6 | — |
| NGE3 | 39 | 37 | 23 | 0 | 2 | P6 | — |
| NGE4 | 30 | 37 | 23 | 8 | 2 | P6 | — |
| F1LA1 | 39 | 21 | 15 | 23 | 2 | P1 | 8 μM |
| F1LA2 | 39 | 21 | 15 | 23 | 2 | P1 | 16 μM |
| F1LA3 | 39 | 21 | 15 | 23 | 2 | P1 | 32 μM |
| F2LA1 | 39 | 44 | 15 | 0 | 2 | P1 | 8 μM |
| F2LA2 | 39 | 44 | 15 | 0 | 2 | P1 | 16 μM |
| F2LA3 | 39 | 44 | 15 | 0 | 2 | P1 | 32 μM |
In contrast to our initial screen, these studies demonstrated that the CLA1 formulation, formulated with P1 (Table 1) gave the maximum transfection efficiency (Fig. 2A, S3 and S4†), and the optimal final concentration of lipopolyplexes in the media was found to be 10 μM (based on the concentration of the lipids) (Fig. S6†). We then prepared variant lipopolyplex formulations, based on CLA1 and including the EGFR inhibitor K16Cys-S-S-[PEG6-IPQA] 1 (Table 1). These lipopolyplex formulations (with increasing amount of inhibitor per particle) gave slightly decreased transfection efficiency (Fig. 2B) compared to particles without the drug (Fig. 2A), however the overall number of transfected tumor cells was not affected. For efficient perfusion through the leaky endothelial cells of the tumor, lipopolyplexes must be <150 nm in diameter. The lipopolyplex formulations were characterised using dynamic light scattering (DLS) and zeta potential measurements (ESI Table S1†) and were found to have particle sizes within the ideal diameter for tumor delivery, reasonable polydispersities, and positive zeta potentials which would minimize particle aggregation and afford a reasonable degree of serum stability. We also carried out in vitro studies to check that the disulfide linker between K16Cys and PEG-IPQA was labile under pseudo-intracellular conditions (GSH/GSSG, HEPES, pH 7.2).66 These indicated that under reducing conditions the linkage would be cleaved within 5 min (ESI Fig. S7†). Next, we assessed the effectiveness of K16Cys-S-S-[PEG6-IPQA] 1 as an inhibitor of EGFR activity as a free therapeutic or within lipopolyplexes by utilizing FRET-FLIM and validating our results by western blot (WB) techniques. Activation of EGFR in cells treated with EGF (natural ligand for EGFR) was evident by decreasing lifetime of green fluorescent protein (GFP) in Picchu-X biosensor (Fig. 2c) and by appearance of phosphorylated EGFR band in WB image (Fig. 2d). In both cases pre-treatment with MeO-[PEG6-IPQA] 8 for 1 h abolished these changes: lifetime of GFP remained at baseline levels (Fig. 2c) and the level of EGFR phosphorylation was significantly lower than in the sample without inhibitor (Fig. 2d, line 8 vs. line 2).
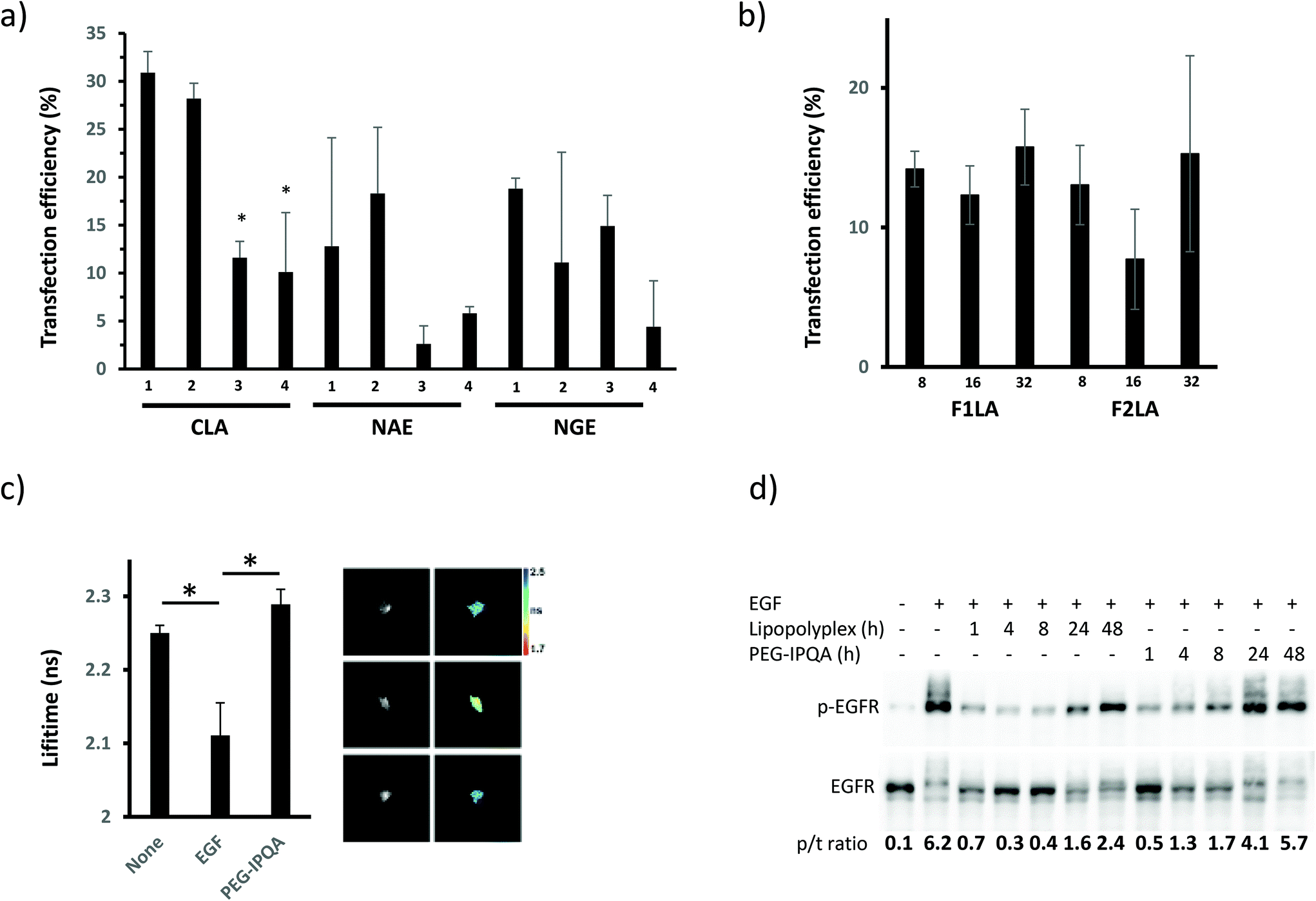 | ||
| Fig. 2 Effect of different lipopolyplex formulations and drug loading on the expression of EGFR in targeted cells and inhibition of its activity upon stimulation with EGF. LIM1215 cells transfected with different lipopolyplexes without inhibitor (CLA1–4, NAE1–4, NGE1–4) (a) or with increasing amount of inhibitor per particle (F1LA1–3, F2LA1–3) (b) 24 h after seeding. High resolution images of 2 × 2 mm area used to quantify transfection efficiency (number of cells expressing biosensor, GFP channel, over total number of cells in field of view, UV channel. (c) FRET-FLIM analysis of the inhibition of EGF-induced EGFR activity in cells by free K16Cys-S-S-[PEG6-IPQA] 1 (10 mM). *P = 0.03, the difference is statistically significant between the groups. (d) Time course of the inhibitory effect of K16Cys-S-S-[PEG6-IPQA] 1 delivered to the cells by lipopolyplex or as a free agent. | ||
Furthermore, incorporation of K16Cys-S-S-[PEG6-IPQA] 1 within lipopolyplexes not only decreased EGFR activity to equally low levels as the free drug but was effective for longer duration (Fig. 2d, lines 3–7 vs. line 2). We observed EGFR inhibition when 1 was delivered within lipopolyplexes for up to 48 h, whereas free MeO-[PEG6-IPQA] 8 was effective only for 24 h (Fig. 2d). These results clearly reveal F1LA1 to be the optimal formulation for lipopolyplexes to achieve maximum transfection efficiency and prolong inhibition of EGFR in cells stimulated with ligand.
Radiolabeling and biodistribution
In order to assess the in vivo biodistribution of these liposome-based theragnostic nanoparticles, we required a [125I]-labelled analogue of 1. As we particularly wished to measure the biodistribution of the IPQA–peptide bioconjugate, we elected to attach the radiolabelled PEG-[125I]IPQA moiety to K16Cys via a maleimide linkage, which would not be subject to reduction in vivo. The analogue K16Cys-SMal-[PEG3-[125I]IPQA] 9 was therefore designed with a similar linker length between the K16 DNA-binding peptide and IPQA analogue 1. PEG3-IPQA precursor 10 was prepared using a modification of the literature procedure58 reported for PEG6-IPQA (ESI†). Similar methodology to that developed for the synthesis of 6 was used to access the key stannylated intermediate 16. Thus, the free PEG terminus of 10 was protected to give TBDPS (tbutyldiphenylsilyl) ether 11, followed by reduction to give amine 12. Stannylation to give 13 and amide formation to afford 14 were followed by TBDPS deprotection to give 15. N-Maleoyl-β-alanine was then coupled to the PEG chain to give the key intermediate 16. Conjugation of 16 to K16Cys (7) gave 17 as a precursor for radiolabeling. Incubation of 17 with sodium [125I]iodide in an iodogen coated tube (30 min, ambient temperature), followed by purification with semi-preparative radio-HPLC, provided the desired K16Cys-SMal-[PEG3-[125I]-IPQA] 9 in 28% radiochemical yield (Scheme 2). | ||
| Scheme 2 Reagents and conditions: (i) TBDPSCl, imidazole, CH2Cl2, 0 °C to rt, 3 h (98%); (ii) SnCl2·2H2O, THF, 60 °C, 3.5 h, (65%); (iii) Pd(PPh3)4, Sn2Me6, dioxane, reflux, 2.5 h, (95%); (iv) isobutyl chloroformate, acrylic acid, Et3N, −40 °C, 30 min (53%); (v) TBAF, THF, 0 °C to rt, 1 h (95%); (vi) N-maleoyl-β-alanine, DCC, DMAP, CH2Cl2, rt, 2 h, (89%); (vii) CysLys167, NaHCO3, rt, 1 h (55%); (viii) sodium [125I]iodide, iodogen, 30 min rt, 28% radiochemical yield. | ||
Radiolabeled [125I]-9 was subsequently formulated as the F1LA1 K16-Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex, which was obtained in 64% radiochemical yield. Radiolabeled (MeO-[PEG6-[125I]-IPQA]) 8 (Scheme 1) was prepared using the same protocol as for [125I]-9, and was obtained in 38% radiochemical yield. The two radiolabeled tracers, [125I]-8 and F1LA1 K16-Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex, were synthesized in order to determine the biodistributions of the small molecule TKI inhibitor and the nanoparticle construct, respectively.
In vivo results
LIM1215 xenografts were established in standard immunocompromised CD1 nude mice (Materials and methods section). The biodistribution data (Fig. 3) showed that the free drug, radiolabelled MeO-[PEG6-[125I]-IPQA] 8 was rapidly cleared via the hepatobiliary route with high uptake in gallbladder and intestines at 3 and 6 h. | ||
| Fig. 3 In vivo radioactive organ biodistribution data presented in % Injected Dose per gram (%ID g−1) for MeO-[PEG6-[125I]-IPQA] 8 (black columns) and F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA] 9 lipopolyplex (grey columns) 3, 6 and 24 h after injection. The tumor %ID g−1 at 3, 6 and 24 h for both tracers is represented on a separate graph. | ||
Whereas F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex showed uptake within the liver and spleen as well as high uptake in the gallbladder and intestines consistent with lipopolyplex uptake and degradation by the Kupffer cells of the reticuloendothelial system. Tumor uptake was highest for both MeO-[PEG6-[125I]-IPQA] 8 and F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex at 3 h with gradual reduction over the 24 h time course. However, the uptake of F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex within tumors was higher than free MeO-[PEG6-[125I]-IPQA] 8 at all time points.
After gamma counting, the tumors were snap frozen, allowing simultaneous expression of biosensor and inhibition of EGFR activity by the liposome-encapsulated K16Cys-SMal-[PEG3-[125I]-IPQA] 9. Fig. 4 shows representative images of tissue from mice injected with CLA1 lipopolyplexes, which only contain the pDNA encoding for the Picchu-X biosensor (top images) and from mice injected with F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplexes, which contain both Picchu-X biosensor pDNA and PEG-IPQA (bottom images). Lifetime measurements of GFP revealed that EGFR activity can be successfully suppressed (Fig. 4) by PEG-IPQA released from the F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplexes. Tissues treated with inhibitor had significantly higher GFP lifetime than control tissues (Fig. 4), which can also be seen on the images (shift from red-yellow to blue colours in pseudocolor map).
 | ||
| Fig. 4 The effect of the inhibitor on EGFR activity measured by Picchu-X biosensor delivered by lipopolyplexes to tumor cells in vivo. Fresh frozen LIM1215 xenografts from animals injected with CLA1 lipopolyplex with pDNA encoding for Picchu-X biosensor alone, or F1LA1 K16Cys-SMal-[PEG3-[125I]-IPQA]-lipopolyplex (additionally loaded with K16Cys-SMal-[PEG3-[125I]-IPQA]) were cut and lifetime images were taken (see Material and methods section). Tumors from control group (injected with lipopolyplex without inhibitor) exhibited low lifetime of GFP in the biosensor (yellow-red colors in pseudocolor map) indicating high level of EGFR activation in the cells. In tumors from animals injected with lipopolyplex loaded with EGFR inhibitor we found high lifetime of GFP (blue color on pseudocolor map) indicating inhibition of EGFR activity in cells. *P = 0.004 (N = 8 images per group), the difference is statistically significant between the groups. | ||
Conclusions
Liposome-based theragnostic nanoparticles hold considerable promise for the delivery of small molecule therapeutic drugs to tumours, and for determining their biodistribution and efficacy in vivo. A major barrier to the success of such approaches is the difficulty encountered in loading sufficient quantities of the drug into the liposome-based formulation, particularly where co-delivery of DNA or RNA is required.67 In this work, we present a novel solution to this problem. The reversible bioconjugation of the small molecule therapeutic to a cationic peptide allows the co-formulation of both a TKI EGFR inhibitor and pDNA coding for a biosensor into a targeted lipopolyplex. The lipopolyplex is designed to disassemble on internalisation into the target cells. Our results indicate that release of the components from the endosome is accompanied by release of the TKI from the bioconjugate and inhibition of EGFR.This approach also allowed us to further investigate the basis for the enhanced circulation and better tumour selectivity afforded by nanoparticle delivery of small molecule therapeutics. The biodistribution data in Fig. 3 suggests different kinetics for either uptake of free drug vs. liposomes, and/or elimination of the free drug vs. drug encapsulated/shielded in liposome in the cells. This phenomenon will need further investigation of potential differences in trafficking of the drug within the cell post-uptake, both in terms of the subcellular compartments involved and kinetics of intracellular trafficking. However, we believe that the in vivo trafficking/clearance differences may be more significant and therefore relevant for the therapeutic effectiveness. The clearance in vivo of radiolabelled Mo-IPQA has recently been studied,30,57 and has shown that this highly lipophilic inhibitor has limited tumour selectivity and undergoes significant hepatobiliary clearance. More hydrophilic PEGylated analogues, such as [18F]F-PEG6-IPQA,68 which formed the basis for the design of our own bioconjugate, show improved pharmacokinetics and quicker in vivo clearance, but still have little selectivity for tumour cells.69,70 In contrast, liposomal formulations of the structurally-related, radiolabelled quinazoline TKI inhibitor, SKI-212243 had a significantly longer circulation time, increased overall tumour uptake and selectivity compared to the free drug.19 Moreover, a different pattern of biodistribution was observed, suggesting that the liposomal formulation is cleared through a different route to the free drug.
Many potent TKI have been developed as anti-EGFR agents, and have shown excellent inhibitory properties in vitro. However, translation to clinically viable agents, particularly for the treatment of colorectal cancer, has generally been unsuccessful, and the reasons for the poor clinical efficacy of these treatments remains elusive. Knowing how these therapies actually act at a molecular level when administered to patients would be a crucial step towards improving the success rate of such inhibitors. Our results demonstrate that lipopolyplexes are a successful delivery system for both an EGFR biosensor and a TKI inhibitor, which will allow surveillance of EGFR activity while testing the potency of such drugs in vivo. This approach in turn opens a new possibility for manipulating and monitoring the activity of EGFR in situ, which in turn will allow a deeper understanding of the dynamics of EGFR activation/inhibition and sensitivity of target cells to EGFR inhibition. Whilst we have previously demonstrated that it is possible to image EGFR activity in vivo utilizing an endoscopy-based fluorescence lifetime imaging methodology,71 this was based on an endoscope requiring physical contact with the area of interest to be imaged, and is a highly invasive procedure. Currently, we envisage that the most useful application of our methodology will be to investigate human derived xenografts to test the responsiveness of the tumour to different treatment regimes.
Finally, we have demonstrated that the reversible bioconjugation of TKIs to cationic peptides and their formulation into targeted nanoparticles results in greatly improved biodistribution relative to the free drug. This in turn will minimize toxicity due to off-target effects. Ultimately, we believe that this approach can be generalised. It will allow for the targeted co-delivery of small molecule/toxic gene, small molecule/gene therapy, and small molecule/siRNA combinations,72 opening up exciting possibilities for combined therapeutic delivery and sensing.
Experimental section
Chemical synthesis
General methods for chemical synthesis and solid phase peptide synthesis, sources of chemicals, purification and analytical methods are in the ESI.† Peptides P1,49P2,49P3,49P4,30 and P630 were synthesized according to the published literature procedures. Experimental procedures for the synthesis of 3, 8, 10, S6, P5 and 7; HPLC/HRMS for peptides 1, 17, P5, 7 and 1H/13C NMR spectra for novel compounds can be found in the ESI.†Synthesis of K16Cys-S-S-[PEG6-IPQA] (1)
Synthesis of K16-Cys-SMal-[PEG3-N-(4-((3-trimethylstannyl)amino)quinazolin-6-yl)acrylamide] (17)
Liposome and lipopolyplex formulation
DODEG4: 50 mol% → 500 μl of 1 mM lipid stock for 1 mL of liposome solution;
DOTMA: 20 mol% → 200 μl of 1 mM lipid Stock for 1 mL of liposome solution;
DOPE: 30 mol% → 300 μl of 1 mM lipid Stock for 1 mL of liposome solution;
CHOL: 30 mol% → 300 μl of 1 mM lipid Stock for 1 mL of liposome solution.
This gave a total concentration of the lipids in the liposomes of 1.3 mM. The solvent in the lipid mixture was slowly removed via evaporation in vacuo to form a lipid thin film and dried for at least 1 h on a high vacuum line. The appropriate amount of distilled water was added to reach a concentration of 1.3 mM total lipid in water. The sample was sonicated for 5 min in a bath sonicator and subsequently on a probe tip sonicator over 10 min using a cycle of 10 s sonication at level 3 and 3 s cooling time. The lipid solution was kept on ice during the sonication process to avoid degradation of the lipid components and overheating of the solution. This procedure gives liposomes of <100 nm and surface charge around +20 meV or above.
The liposomes were transferred into a Falcon tube and to this solution was added targeting peptide P1 (K16-RVRR-LARLLT (1 mM stock)) to a final concentration of 170 μM followed by the drug peptide bioconjugate (K16Cys-S-S-[PEG6-IPQA] 1 (0.7 mM stock)) to a final concentration of 100 μM. The liposome peptide solution was then diluted to 3.5 mL. 680 μg Picchu-X plasmid DNA (1.68 μg μL−1 stock, 404 μL) was added to a Falcon tube and diluted to 3.5 mL. The DNA solution was then slowly (dropwise but steadily) added to the liposome–peptide solution while constantly swirling the solution, in order to avoid the concentration of DNA and peptide gets locally too high and precipitates. Once the whole solution has been added the now freshly formed lipopolyplexes have been transferred to a spin-column (MWCO above 5000 Da) and concentrated to the initial volume of 1 mL.
Radiolabeling
General methods for radioiodination work and HPLC traces can be found in the ESI.†In vivo and in vitro testing
Author contributions
Robin Bofinger, Gregory Weitsman, Rachel Evans, Matthias Glaser, Kerstin Sander and Tammy Kalber performed the experiments (chemical and peptide synthesis, cancer cell biology, FRET/FLIM, radiochemistry and biodistribution). Helen Allan, Robin Bofinger, Gregory Weitsman and Alethea Tabor wrote the initial draft, and all authors contributed to writing and reviewing the final draft. Erik Arstad, Daniel Hochhauser, Tammy Kalber, Helen Hailes, Tony Ng and Alethea Tabor supervised the research groups, and Tammy Kalber, Erik Arstad, Tony Ng and Alethea Tabor acquired the financial support for the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted within the King's College London-UCL Comprehensive Cancer Imaging Centre (CCIC) supported by Cancer Research UK and the EPSRC, in association with MRC and DoH (UK). The authors would like to thank Cancer Research UK and the EPSRC (C5255/A15935 (RB, GW, MG, KS)) for financial support. We also thank the EPSRC for the award of an Early Career Fellowship EP/L006472/1 (TK). The radiochemistry work (MG, KS, EA) was undertaken at the UCL Centre for Radiopharmaceutical Chemistry, which is funded in part by the Department of Health's NIHR Biomedical Research Centres funding scheme. We thank Dr Kersti Karu and Dr Abil Aliev for assistance with mass spectrometry and NMR, and also acknowledge Dr Stephen McCarthy and Aamina Murtza for additional HPLC and NMR characterization.Notes and references
- R. Kojima, D. Aubel and M. Fussenegger, Curr. Opin. Chem. Biol., 2015, 28, 29–38 CrossRef CAS PubMed.
- S. Svenson, Mol. Pharmaceutics, 2013, 10, 848–856 CrossRef CAS PubMed.
- M. R. Chowdhury, C. Schumann, D. Bhakta-Guha and G. Guha, Biomed. Pharmacother., 2016, 84, 291–304 CrossRef PubMed.
- C. Zavaleta, D. Ho and E. J. Chung, SLAS Technol., 2018, 23, 281–293 CAS.
- P. Nuciforo, N. Radosevic-Robin, T. Ng and M. Scaltriti, Breast Cancer Res., 2015, 17, 53 CrossRef PubMed.
- D. S. Salomon, R. Brandt, F. Ciardiello and N. Normanno, Crit. Rev. Oncol. Hematol., 1995, 19, 183–232 CrossRef CAS PubMed.
- Y. Yarden and M. X. Sliwkowski, Nat. Rev. Mol. Cell Biol., 2001, 2, 127–137 CrossRef CAS PubMed.
- J. T. Jones, R. W. Akita and M. X. Sliwkowski, FEBS Lett., 1999, 447, 227–231 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, Clin. Cancer Res., 2001, 7, 2958–2970 CAS.
- W. J. Slichenmyer and D. W. Fry, Semin. Oncol., 2001, 28, 67–79 CrossRef CAS PubMed.
- K. Y. Chung, J. Shia, N. E. Kemeny, M. Shah, G. K. Schwartz, A. Tse, A. Hamilton, D. Pan, D. Schrag, L. Schwartz, D. S. Klimstra, D. Fridman, D. P. Kelsen and L. B. Saltz, J. Clin. Oncol., 2005, 23, 1803–1810 CrossRef CAS PubMed.
- M. E. Lacouture, M. Anadkat, A. Jatoi, T. Garawin, C. Bohac and E. Mitchell, Clin. Colorectal Cancer, 2018, 17, 85–96 CrossRef PubMed.
- L. A. Carey, H. S. Rugo, P. K. Marcom, E. L. Mayer, F. J. Esteva, C. X. Ma, M. C. Liu, A. M. Storniolo, M. F. Rimawi, A. Forero-Torres, A. C. Wolff, T. J. Hobday, A. Ivanova, W.-K. Chiu, M. Ferraro, E. Burrows, P. S. Bernard, K. A. Hoadley, C. M. Perou and E. P. Winer, J. Clin. Oncol., 2012, 30, 2615–2623 CrossRef CAS PubMed.
- L. B. Saltz, N. J. Meropol, P. J. Loehrer, M. N. Needle, J. Kopit and R. J. Mayer, J. Clin. Oncol., 2004, 22, 1201–1208 CrossRef CAS PubMed.
- K. Khan, N. Valeri, C. Dearman, S. Rao, D. Watkins, N. Starling, I. Chau and D. Cunningham, Crit. Rev. Oncol. Haematol., 2019, 143, 153–163 CrossRef PubMed.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975–999 CrossRef CAS PubMed.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef CAS PubMed.
- M. Benezra, D. Hambardzumyan, O. Penate-Medina, D. R. Veach, N. Pillarsetty, P. Smith-Jones, E. Phillips, T. Ozawa, P. B. Zanzonico, V. Longo, E. C. Holand, S. M. Larson and M. S. Bradbury, Neoplasia, 2012, 14, 1132–1143 CrossRef CAS PubMed.
- O. P. Medina, N. Pillarsetty, A. Glekas, B. Punzalan, V. Longo, M. Gönen, P. Zanzonico, P. Smith-Jones and S. M. Larson, J. Controlled Release, 2011, 149, 292–298 CrossRef CAS PubMed.
- F. Danhier, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS PubMed.
- A. A. Natfji, D. Ravishankar, H. M. I. Osborn and F. Greco, J. Pharm. Sci., 2017, 106, 3179–3187 CrossRef CAS PubMed.
- R. Petrilli, J. O. Eloy, F. P. Saggioro, D. L. Chesca, M. C. de Souza, M. V. S. Dias, L. L. P. daSilva, R. J. Lee and R. F. V. Lopez, J. Controlled Release, 2018, 283, 151–162 CrossRef CAS PubMed.
- J. Jung, S. Y. Jeong, S. S. Park, S. H. Shin, E. J. Ju, J. Choi, J. Park, J. H. Lee, I. Kim, Y. A. Suh, J. J. Hwang, S. Kuroda, J. S. Lee, S. Y. Song and E. K. Choi, Int. J. Oncol., 2015, 46, 1268–1274 CrossRef CAS PubMed.
- J. Lehtinen, M. Raki, K. A. Bergstrom, P. Uutela, K. Lehtinen, A. Hiltunen, J. Pikkarainen, H. M. Liang, S. Pitkanen, A. M. Maatta, R. A. Ketola, M. Yliperttula, T. Wirth and A. Urtti, PLoS One, 2012, 7, e41410 CrossRef CAS PubMed.
- F. Goni-de-Cerio, J. Thevenot, H. Oliveira, E. Perez-Andres, E. Berra, M. Masa, B. Suarez-Merino, S. Lecommandoux and P. Heredia, J. Biomed. Nanotechnol., 2015, 11, 2034–2049 CrossRef CAS PubMed.
- H. L. Tang, X. J. Chen, M. J. Rui, W. Q. Sun, J. Chen, J. L. Peng and Y. H. Xu, Mol. Pharmaceutics, 2014, 11, 3242–3250 CrossRef CAS PubMed.
- Y. F. Wang, J. H. Zhou, L. H. Qiu, X. R. Wang, L. L. Chen, T. Liu and W. Di, Biomaterials, 2014, 35, 4297–4309 CrossRef CAS PubMed.
- A. A. Mokhtarieh, S. Cheong, S. Kim, B. H. Chung and M. K. Lee, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1633–1641 CrossRef CAS PubMed.
- J. Gao, Y. S. Yu, Y. Y. Zhang, J. J. Song, H. W. Chen, W. Li, W. Z. Qian, L. Deng, G. Kou, J. M. Chen and Y. Guo, Biomaterials, 2012, 33, 270–282 CrossRef CAS PubMed.
- G. Weitsman, N. J. Mitchell, R. Evans, A. Cheung, T. L. Kalber, R. Bofinger, G. O. Fruhwirth, M. Keppler, Z. V. F. Wright, P. R. Barber, P. Gordon, T. de Koning, W. Wulaningsih, K. Sander, B. Vojnovic, S. Ameer-Beg, M. Lythgoe, J. N. Arnold, E. Årstad, F. Festy, H. C. Hailes, A. B. Tabor and T. Ng, Oncogene, 2017, 36, 3618–3628 CrossRef CAS PubMed.
- K. Kurokawa, N. Mochizuki, Y. Ohba, H. Mizuno, A. Miyawaki and M. Matsuda, J. Biol. Chem., 2001, 276, 31305–31310 CrossRef CAS PubMed.
- C. A. Hurley, J. B. Wong, J. Ho, M. Writer, S. A. Irvine, M. J. Lawrence, S. L. Hart, A. B. Tabor and H. C. Hailes, Org. Biomol. Chem., 2008, 6, 2554–2559 RSC.
- M. F. Mohd Mustapa, S. M. Grosse, L. Kudsiova, M. Elbs, E.-A. Raiber, J. B. Wong, A. P. R. Brain, H. E. Armer, A. Warley, M. Keppler, T. Ng, M. J. Lawrence, S. L. Hart, H. C. Hailes and A. B. Tabor, Bioconjugate Chem., 2009, 20, 518–532 CrossRef PubMed.
- S. M. Grosse, A. D. Tagalakis, M. F. Mohd Mustapa, M. Elbs, Q.-H. Meng, A. Mohammadi, A. B. Tabor, H. C. Hailes and S. L. Hart, FASEB J., 2010, 24, 2301–2313 CrossRef CAS PubMed.
- M. F. Mustapa, P. C. Bell, C. A. Hurley, A. Nicol, E. Guénin, S. Sarkar, M. J. Writer, S. E. Barker, J. B. Wong, M. A. Pilkington-Miksa, B. Papahadjopoulos-Sternberg, P. A. Shamlou, H. C. Hailes, S. L. Hart, D. Zicha and A. B. Tabor, Biochemistry, 2007, 46, 12930–12944 CrossRef CAS PubMed.
- A. Mohammadi, L. Kudsiova, M. F. Mohd Mustapa, F. Campbell, D. Vlaho, K. Welser, H. Story, A. D. Tagalakis, S. L. Hart, D. J. Barlow, A. B. Tabor, M. J. Lawrence and H. C. Hailes, Org. Biomol. Chem., 2019, 17, 945–957 RSC.
- K. Welser, F. Campbell, L. Kudsiova, A. Mohammadi, N. Dawson, S. L. Hart, D. J. Barlow, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Pharm., 2013, 10, 127–141 CrossRef CAS PubMed.
- L. Kudsiova, K. Welser, F. Campbell, A. Mohammadi, N. Dawson, L. Cui, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Biosyst., 2016, 12, 934–951 RSC.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS PubMed.
- Z. ur Rehman, I. S. Zuhorn and D. Hoekstra, J. Controlled Release, 2013, 166, 46–56 CrossRef CAS PubMed.
- K. Van der Jeught, S. De Koker, L. Bialkowski, C. Heirman, P. T. Joe, F. Perche, S. Maenhout, S. Bevers, K. Broos, K. Deswarte, V. Malard, H. Hammad, P. Baril, T. Benvegnu, P.-A. Jaffrès, S. A. A. Kooijmans, R. Schiffelers, S. Lienenklaus, P. Midoux, C. Pichon, K. Breckpot and K. Thielemans, ACS Nano, 2018, 12, 9815–9829 CrossRef CAS PubMed.
- X. Lai, X. Xu, Z. Zhu and Z. Hua, Int. J. Nanomed., 2018, 13, 6603–6623 CrossRef PubMed.
- L. M. Carlin, K. Makrogianneli, M. Keppler, G. O. Fruhwirth and T. Ng, Methods Mol. Biol., 2010, 616, 97–113 CrossRef CAS PubMed.
- R. A. Demel, K. R. Bruckdorfer and L. L. M. van Deenen, Biochim. Biophys. Acta, 1972, 255, 321–330 CrossRef CAS.
- S. Song, D. Liu, J. Peng, H. Deng, Y. Guo, L. X. Xu, A. D. Miller and Y. Xu, FASEB J., 2009, 23, 1396–1404 CrossRef CAS PubMed.
- C. Y. Han, L. L. Yue, L. Y. Tai, L. Zhou, X. Y. Li, G. H. Xing, X. G. Yang, M. S. Sun and W. S. Pan, Int. J. Nanomed., 2013, 8, 1541–1549 Search PubMed.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978–1985 CrossRef CAS PubMed.
- I. Genta, E. Chiesa, B. Colzani, T. Modena, B. Conti and R. Dorati, Pharmaceutics, 2018, 10, 2 CrossRef PubMed.
- R. Bofinger, M. Zaw-Thin, N. J. Mitchell, P. S. Patrick, C. Stowe, A. Gomez-Ramirez, H. C. Hailes, T. L. Kalber and A. B. Tabor, J. Pept. Sci., 2018, 24, e3131 CrossRef PubMed.
- J. Li, Y. Wang, Y. Zhu and D. Oupicky, J. Controlled Release, 2013, 172, 589–600 CrossRef CAS PubMed.
- W. W. Xu, D. Y. Liu, Y. C. Cao and X. Y. Wang, Int. J. Nanomed., 2017, 12, 6461–6470 CrossRef CAS PubMed.
- B. Steinborn, I. Truebenbach, S. Morys, U. Lächelt, E. Wagner and W. Zhang, J. Gene Med., 2018, 20, e3041 CrossRef PubMed.
- R. Bahadur, B. Thapa, J. Valencia-Serna, H. M. Aliabadi and H. Uludag, J. Controlled Release, 2017, 256, 153–169 CrossRef PubMed.
- S. Dong, X. Zhou and J. Yang, Biomed. Pharmacother., 2016, 84, 954–961 CrossRef CAS PubMed.
- S. Svenson, Chem. Soc. Rev., 2015, 44, 4131–4144 RSC.
- J. Jiang, S. J. Yang, J. C. Wang, L. J. Yang, Z. Z. Xu, T. Yang, X. X. Liu and Q. Zhang, Eur. J. Pharm. Biopharm., 2010, 76, 170–178 CrossRef CAS PubMed.
- A. Pal, A. Glekas, M. Doubrovin, J. Balatoni, T. Beresten, D. Maxwell, S. Soghomonyan, A. Shavrin, L. Ageyeva, R. Finn, S. M. Larson, W. Bornmann and J. G. Gelovani, Mol. Imaging Biol., 2006, 8, 262–277 CrossRef CAS PubMed.
- A. Pal, J. A. Balatoni, U. Mukhopadhyay, K. Ogawa, C. Gonzalez-Lepera, A. Shavrin, A. Volgin, W. Tong, M. M. Alauddin and J. G. Gelovani, Mol. Imaging Biol., 2011, 13, 853–861 CrossRef PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- K. M. Wang, N. Liu, P. Zhang, Y. Y. Guo, Y. C. Zhang, Z. X. Zhao, Y. X. Luan, S. Z. Li, J. H. Cai and J. M. Cao, Curr. Org. Chem., 2016, 20, 1477–1489 CrossRef CAS.
- M. S. Bernatowicz, R. Matsueda and G. R. Matsueda, Int. J. Pept. Protein Res., 1986, 28, 107–112 CrossRef CAS PubMed.
- A. L. Schroll, R. J. Hondal and S. Flemer, J. Pept. Sci., 2012, 18, 1–9 CrossRef CAS PubMed.
- E. Vivès and B. Lebleu, Tetrahedron Lett., 1997, 38, 1183–1186 CrossRef.
- S. Fanayan, J. T. Smith, L. Y. Lee, F. F. Yan, M. Snyder, W. S. Hancock and E. Nice, J. Proteome Res., 2013, 12, 1732–1742 CrossRef CAS PubMed.
- Y. Miyamoto, N. Muguruma, S. Fujimoto, Y. Okada, Y. Kida, F. Nakamura, K. Tanaka, T. Nakagawa, S. Kitamura, K. Okamoto, H. Miyamoto, Y. Sato and T. Takayama, Cancer Sci., 2019, 110, 1921–1930 CAS.
- C. Borrás, J. M. Esteve, J. R. Viña, J. Sastre, J. Viña and F. V. Pallardó, J. Biol. Chem., 2004, 33, 34332–34335 CrossRef PubMed.
- B. Almeida, O. K. Nag, K. E. Rogers and J. B. Delehanty, Molecules, 2020, 25, 5672 CrossRef CAS PubMed.
- H. H. Yeh, K. Ogawa, J. Balatoni, U. Mukhapadhyay, A. Pal, C. Gonzalez-Lepera, A. Shavrin, S. Soghomonyan, L. Flores, D. Young, A. Y. Volgin, A. M. Najjar, V. Krasnykh, W. Tong, M. M. Alauddin and J. G. Gelovani, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1603–1608 CrossRef CAS PubMed.
- M. A. Pantaleo, E. Mishani, C. Nanni, L. Landuzzi, S. Boschi, G. Nicoletti, S. Dissoki, P. Paterini, P. P. Piccaluga, F. Lodi, P.-L. Lollini, S. Fanti and G. Biasco, Mol. Imaging Biol., 2010, 12, 616–625 CrossRef PubMed.
- E. B. Corcoran and R. N. Hanson, Med. Res. Rev., 2014, 34, 596–643 CrossRef PubMed.
- G. O. Fruhwirth, A. Ameer-Beg, R. Cook, T. Watson, T. Ng and F. Festy, Opt. Express, 2010, 18, 11148–11158 CrossRef CAS PubMed.
- Y. Li, X. Tan, X. Liu, L. Liu, Y. Fang, R. Rao, Y. Ren, Y. Yang and W. Liu, Asian J. Pharm. Sci., 2020, 15, 646–660 CrossRef PubMed.
- P. R. Barber, I. D. C. Tullis, G. P. Pierce, R. G. Newman, J. Prentice, M. I. Rowley, D. R. Matthews, S. M. Ameer-Beg and B. Vojnovic, J. Microsc., 2013, 251, 154–167 CrossRef CAS PubMed.
- P. R. Barber, S. M. Ameer-Beg, J. Gilbey, R. J. Edens, I. Ezike and B. Vojnovic, Proc. SPIE, 2005, 5700, 171–181 CrossRef CAS.
- M. I. Rowley, P. R. Barber, A. C. C. Coolen and B. Vojnovic, Proc. SPIE, 2011, 7903, 790325 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S7, general methods for chemical synthesis, solid phase peptide synthesis and radiolabelling experiments, additional experimental procedures, 1H/13C NMR/HPLC spectra for novel compounds, DLS and zeta potential data. See DOI: 10.1039/d1nr02770k |
| ‡ These authors contributed equally to this work. |
| § Present address: ANSES, French Agency for Food, Environmental and Occupational Health & Safety, Fougères Laboratory, Residues and Contaminants Analysis Unit, 10B rue Claude Bourgelat, Bioagropolis, Javené, F-35306 Fougères, France (E-mail: E-mail: robin.bofinger@anses.fr). |
| This journal is © The Royal Society of Chemistry 2021 |