
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredictions of attainable compositions of layered quaternary i-MAB phases and solid solution MAB phases†
Martin
Dahlqvist
 * and
Johanna
Rosen
*
* and
Johanna
Rosen
*
Materials Design, Department of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83 Linköping, Sweden. E-mail: martin.dahlqvist@liu.se; johanna.rosen@liu.se
First published on 22nd October 2021
Abstract
MAB phases are layered materials combining metallic and ceramic attributes. Their ternary compositions, however, have been limited to a few elemental combinations which makes controlled and tailored properties challenging. Inspired by the recent discovery of Mo4/3Y2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases, i.e., quaternary layered MAB phases with in-plane chemical order, we perform an extensive first-principles study to explore formation of chemical order and solid-solutions upon metal alloying of M2AB2 phases of 1092 compositions (M from group 3 to 9 and A = Al, Ga, In, Si, Ge, Sn). This large dataset provides 39 chemically ordered (i-MAB) and 52 solid solution (MAB) phases that are predicted to be thermodynamically stable at typical synthesis temperatures, of which a majority have not yet been experimentally reported. The possibility for realizing both i-MAB and solid solution MAB phases, combined with the multiple elemental combinations previously not observed in these boride-based materials, allows for an increased potential for property tuning and potential chemical exfoliation into 2D derivatives.
Introduction
The spurred interest in layered ternary transition metal borides like M2AlB2 (M = Cr, Mn, Fe in space group Cmmm),1–6M3AlB4 (M = Cr in space group Immm),3,7,8M4AlB6 (M = Cr in space group Cmmm),5,7MAlB (M = Mo, W in space group Cmcm),9–11 and M4AlB4 (M = Cr in space group Immm)12 comes from the discovery of, e.g., magnetocaloric properties,4 high-temperature oxidation resistance,13 and electrocatalytic properties.14,15 Another potential area is to use these materials as precursors for being exfoliated into two-dimensional (2D) counterparts, MBenes, however, realization of individual single-layer sheets remains a quest.14,16–20In 2015 these phases were collectively coined MAB phases by Ade and Hillebrecht due to their resemblance with layered carbide and nitride MAX phases, where M is an early transition metal, A is mostly a group 13 or 14 element (e.g. Al, Ga, and Si), and X is C and/or N.5 MAB phases typically have an orthorhombic crystal structure, but with the recent addition of Ti2InB2 in space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2, hexagonal symmetry was introduced to the family.21 In a follow-up theoretical work, five M2AB2 and two M3AB4, all with a hexagonal crystal structure, were predicted to be stable.22
m2, hexagonal symmetry was introduced to the family.21 In a follow-up theoretical work, five M2AB2 and two M3AB4, all with a hexagonal crystal structure, were predicted to be stable.22
The attainable chemistries in synthesized MAB phases have for a long time been limited to A = Al and M = Cr, Mo, W, Mn, and Fe, apart from the recently discovered Ti2InB2. A route for expanding attainable compositions of MAB phases is through alloying by adding a fourth element on M, A, or B sites. This allows for property tuning and has been realized for disordered solid-solution MAB phases (Mo1−xWx)AlB and (Mo1−xCrx)AlB with Cmcm symmetry,23,24 (Fe1−xMx)2AlB2 (M = Fe, Co) and (Mn1−xCrx)2AlB2 with Cmmm symmetry.25–28 and (Mn1−xCrx)3AlB4 with Immm symmetry.25 Potential out-of-plane ordering with alternating M-layers based on one M-element only have been investigated theoretically in the Cr3AlB4 crystal structure (with Immm symmetry) although experimental verification is awaiting.29 Alloying on the A-site has been less explored with limited solubility of Si (x ≥ 0.03) in MoAl1−xSixB.30
Alloying has been demonstrated as an attainable path towards expanding chemistries in materials resembling the MAB phases (MAX phases) resulting in either a solid solution or a chemically ordered (o-MAX with two metals ordered out-of-plane and i-MAX with two metals ordered in-plane) distribution of the alloying elements.31–33 In a similar way, an ordered distribution of alloying elements is possible in MAB phases other than disordered solid solutions. Recently, we considered metal alloying as a path towards expanding the attainable chemistries in orthorhombic and hexagonal M2AlB2 phases, and discovered two in-plane ordered MAB phases, Mo4/3Y2/3AlB2 and Mo4/3Sc2/3AlB2,34 coined i-MAB in analogy to i-MAX.35 This finding was accompanied with theoretical stability predictions of an additional 13 chemically ordered i-MAB phases and 16 solid solution MAB phases. This exemplifies alloying as a prospective path towards expanding the MAB phase chemistries, which combined with the various MAB structures would allow for a range of possibilities when it comes to novel elemental combinations, bond modification, and property tuning. Bond modification in a MAB phase could for example allow for potential conversion into its 2D derivative, in the same way as conversion of i-MAX to i-MXene.36
In this work we use a systematic theoretical approach which includes high-throughput stability predictions to demonstrate an expansion of the possible chemistries for quaternary M2AB2 phases, (M′1−xM′′x)2AB2, beyond A = Al, by exploiting metal alloying. We consider both chemically ordered (i-MAB) and a solid solution distribution of M′ and M′′, the latter by considering both the Cmmm and Pm2 space group symmetries. We considered a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of M′ and M′′ motivated by the in-depth investigation in ref. 34 where further details can be found. In short, a 2:1 ratio allows in-plane chemical ordering, as confirmed in experiments. A ratio diverging significantly from 2:1, such as 3:1 or 1:1, will likely give a solid solution, if such composition is stable. In a previous report, the related in-plane ordered i-MAX phase (Mo2/3Sc1/3)2AlC were investigated and it was demonstrated that small compositional deviations from the ideal 2:1 ratio is possible while still maintaining the i-MAX structure, however, diverging composition is detrimental for MXene formation.37
:1 ratio of M′ and M′′ motivated by the in-depth investigation in ref. 34 where further details can be found. In short, a 2:1 ratio allows in-plane chemical ordering, as confirmed in experiments. A ratio diverging significantly from 2:1, such as 3:1 or 1:1, will likely give a solid solution, if such composition is stable. In a previous report, the related in-plane ordered i-MAX phase (Mo2/3Sc1/3)2AlC were investigated and it was demonstrated that small compositional deviations from the ideal 2:1 ratio is possible while still maintaining the i-MAX structure, however, diverging composition is detrimental for MXene formation.37
Guided by thermodynamic stability calculations, we confirm the stability for experimentally known i-MAB and solid solution phases. More importantly, beyond A = Al we also predict an additional 24 stable i-MAB phases and 36 stable solid solutions yet to be experimentally confirmed. We investigate the impact from choice of element (M′, M′′, and A) and the effect of size and electronegativity of M′, M′′ and A for the formation of chemically ordered i-MAB or solid solution MAB phases. We also propose guidelines for which elemental combinations to use in search for novel i-MAB and solid solution MAB phases. This study reveals that alloying is viable path towards novel elemental combinations to allow for potentially tuneable and advantageous properties.
Method
All first-principles calculations are performed by means of density functional theory (DFT) using the Vienna ab initio simulation package (VASP) version 5.4.1.38–40 The projector augmented wave method is used with the generalized gradient approximation (GGA) as parameterized by Perdew–Burke–Ernzerhof (PBE).41–43 In systems with Cr, Mn, Fe, and Co, we used the spin-polarized GGA version and energy is taken for the lowest ferromagnetic and antiferromagnetic configurations sampled within the unit cell. Further details are given in Fig. S1 and S2.† For structural relaxation, the Brillouin zone was integrated by Monkhorst–Pack special k-point sampling, with a density of 0.05 Å−1 with a plane-wave energy cut-off of 400 eV.44 The convergence of formation enthalpy with respect to the k-point density and plane-wave energy cut-off is shown in Fig. S3.† The convergence criterion for self-consistency is reached when satisfying an energy convergence of 10–6 eV per atom and force convergence of 10–2 eV Å−1. Density of states (DOS) and crystal orbital Hamilton population (COHP) were retrieved using the LOBSTER code45–48 where the calculated band-structure energy is reconstructed into orbital interactions. Positive COHP values indicate an antibonding interaction, and negative COHP values indicate a bonding interaction. Details about calculating elastic moduli are provided in ESI.†The composition in focus is M2AB2 which in its ternary form have been reported in an orthorhombic Cmmm and hexagonal Pm2 structure, where M is a transition metal and A an A-group element, see Table S1† for reported ternary M2AB2 phases. In this work we have considered M from group 3 to 9; Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, and Ni, along with A elements; Al, Ga, In, Si, Ge, and Sn. Solid solution or chemical disorder of M′ and M′′ are modelled on the M sublattice for the two known M2AB2 crystal structures, i.e., with Cmmm and Pm2 space group symmetries, using the special quasi-random structure (SQS) method.49 Convergency tests show that supercells with 120 atoms or more give a qualitatively accurate representation and a quantitative convergency in terms of calculated formation enthalpies, equilibrium volumes, and lattice parameters.
We assess the thermodynamic stability of a phase by comparing its energy relative to competing phases in the ternary or quaternary system of interest. This will give information whether the phase is energetically favoured or prefer decomposition into other competing phases. The most competing set of competing phases, denoted equilibrium simplex, is identified using a linear optimization procedure50,51 which have been proven successful to confirm already experimentally materials as well as predicting the existence of new ones.51–56 The stability of a phase is quantified in terms of the formation enthalpy ΔHcp by comparing its energy to the energy of the equilibrium simplex according to
| ΔHcp = E[(M′1−xM′′x)2AB2] − E[equilibrium simplex]. | (1) |
A phase is concluded stable when ΔHcp < 0. Here E[(M′1−xM′′x)2AB2] represent the chemical order of lowest energy being chemically ordered M′4/3M′′2/3AB2 or solid solution (M′1−xM′′2)2AB2. However, when T ≠ 0 K, the contribution from configurational entropy for a disordered distribution of M′ and M′′ on the M sublattice in (M′1−xM′′2)2AB2 will decrease the Gibbs free energy, ΔGsolid solutioncp, as approximated by
| ΔGsolid solutioncp[T] = ΔHsolid solutioncp − TΔS. | (2) |
The entropic contribution ΔS, assuming an ideal solution of M′ and M′′ on the M-sites, is given by
| ΔS = −2kB[yln(y) + (1 − y)ln(1 − y)], | (3) |
Visualization of atomic structures was done with the VESTA code.58
Results and discussion
Considered models for quaternary MAB phases
In a recent work, we investigated the thermodynamic stability upon metal alloying in M2AlB2 phases, and predicted 15 chemically ordered phases as well as 16 solid solutions to be stable. The study included experimental verification of chemically in-plane ordered Mo4/3Y2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases.34 Here we expand the possible compositions by also including A = Ga, In, Si, Ge, and Sn. We have considered an M′ and M′′ ratio of 2:1 based on the stoichiometry of the quaternary i-MAB phases.34 Moreover, this ratio also allows for a good description of possible solid solutions upon alloying in a general quaternary (M′1−xM′′2)2AB2 phase as exemplified by the reported synthesis of (Mn1−xCrx)2AlB2 with a Cmmm space group symmetry.25 We have considered three different distributions of M′ and M′′, illustrated in Fig. 1, motivated by to date reported quaternary (M′1−xM′′2)2AB2 structures and the detailed search for possible chemical ordering conducted in ref. 34; (i) solid solution in the orthorhombic M2AB2 structure with Cmmm space group symmetry, (ii) solid solution in the hexagonal M2AB2 structure with Pm2 space group symmetry, and (iii) in-plane chemical order in i-MAB with four different space group symmetries (P2m, P2c, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, C2). The Rm structure is shown in Fig. 1c and it is also the structure assigned for the synthesized Mo4/3Y2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases in ref. 34. Note that in the i-MAB phase, M′′ is extended toward the A-layer, in turn displaying an ideal Kagomé lattice while the boron layers are hexagonal. For further details, see ref. 34.
m, C2). The Rm structure is shown in Fig. 1c and it is also the structure assigned for the synthesized Mo4/3Y2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases in ref. 34. Note that in the i-MAB phase, M′′ is extended toward the A-layer, in turn displaying an ideal Kagomé lattice while the boron layers are hexagonal. For further details, see ref. 34.
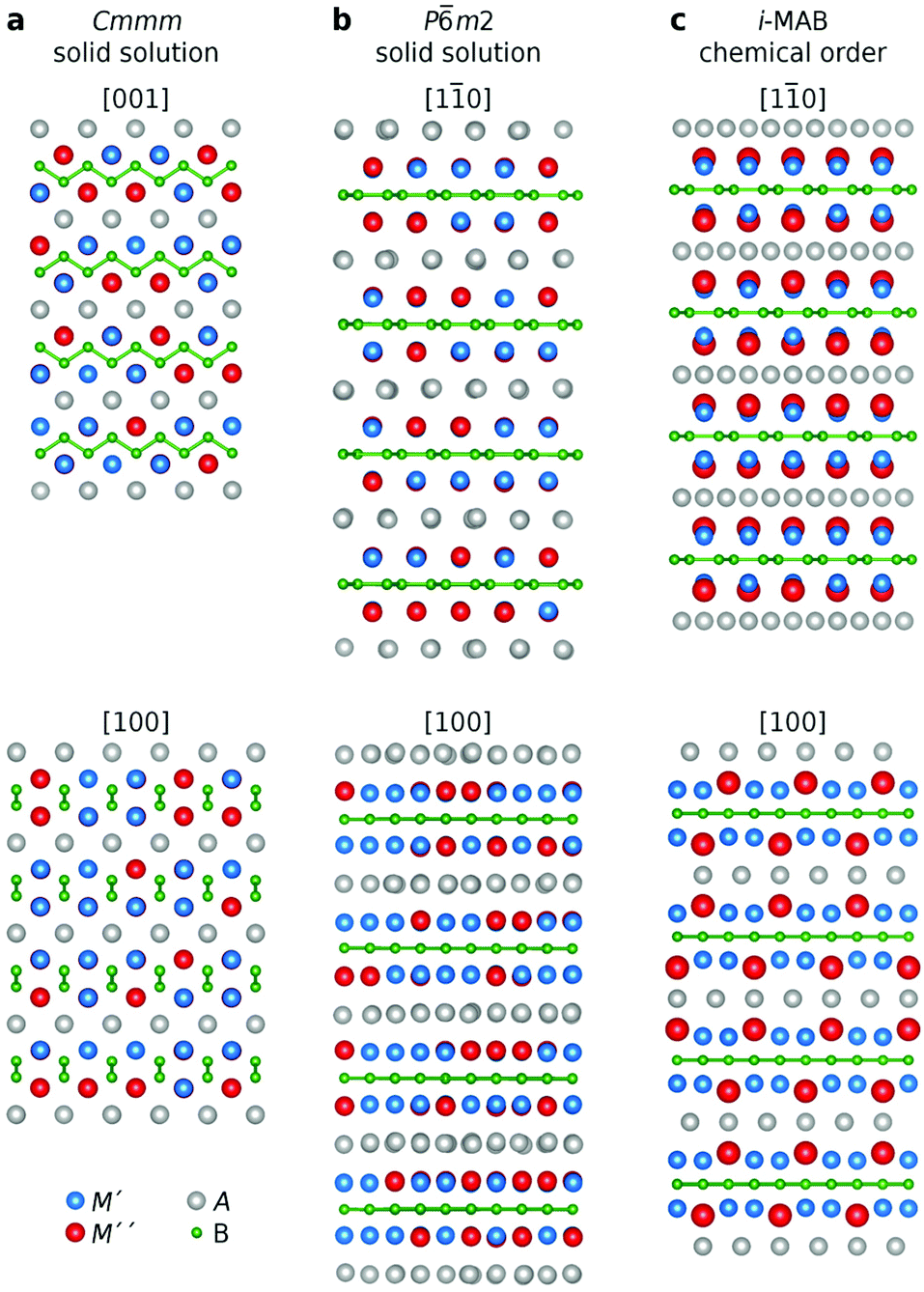 | ||
| Fig. 1 Schematic illustration of various elemental distributions in quaternary 212 MAB phases upon metal alloying of M′ and M′′. (a) solid solution with Cmmm space group symmetry, (b) solid solution with Pm2 space group symmetry, and (c) chemically ordered i-MAB with Rm space group symmetry. M′, M′′, A, and B elements are represented by blue, red, grey, and green atoms, respectively. | ||
Stability of quaternary MAB phases – chemical order vs. solid solution
Fig. 2a–f show the trends in thermodynamic stability for 1092 unique chemical compositions. Note that the results for A = Al have previously been presented in ref. 34. For each A element (A = Al, Ga, In, Si, Ge, Sn), a heatmap is shown where M′ and M′′ are assembled according to the periodic group of the M elements. The background color represents the calculated thermodynamic stability for chemical order of lowest energy, with a blue region representing stable phases (ΔHi-MAB or ΔGsolid solution < 0). ΔHi-MAB is evaluated at 0 K while ΔGsolid solution is evaluated at 2000 K by considering the contribution from configurational entropy to the Gibb's free energy, using eqn (3), for solid solution (M′0.67M′′0.33)2AB2. This choice is motivated by a typical synthesis temperature of 1000 to 1800 °C (1273 to 2173 K) for M2AB2 and (Mn1−xM′′0.2)2AlB2 (M′′ = Cr, Fe).5,17,25,26,59–61 We use a symbol representation to denote the structure of lowest energy at a temperature of 2000 K; solid solution Cmmm (open square), solid solution Pm2 (open hexagon), and i-MAB phases with P2m (filled right triangle), P2c (filled left triangle), Rm (filled up triangle) and C2 (filled down triangle) symmetries. In addition, experimentally known quaternary phases are marked by green squares for solid solution Cmmm MAB phases25,26,59 and orange squares for ordered i-MAB phases.34 A complete list of synthesized phases can be found in Tables S2 and S3.† Results evaluated at 0 K, i.e., without contribution from configurational entropy to the solid solution phases, is shown in Fig. S4.† Here it should be noted that experimentally known solid solution (M′1−xM′′x)2AlB2Cmmm phases are predicted to be not stable at 0 K (Fig. S4†), and stable when evaluated at 2000 K (Fig. 2). This emphasizes the importance of including configurational entropy for disordered solid solution phases when evaluating the stability.
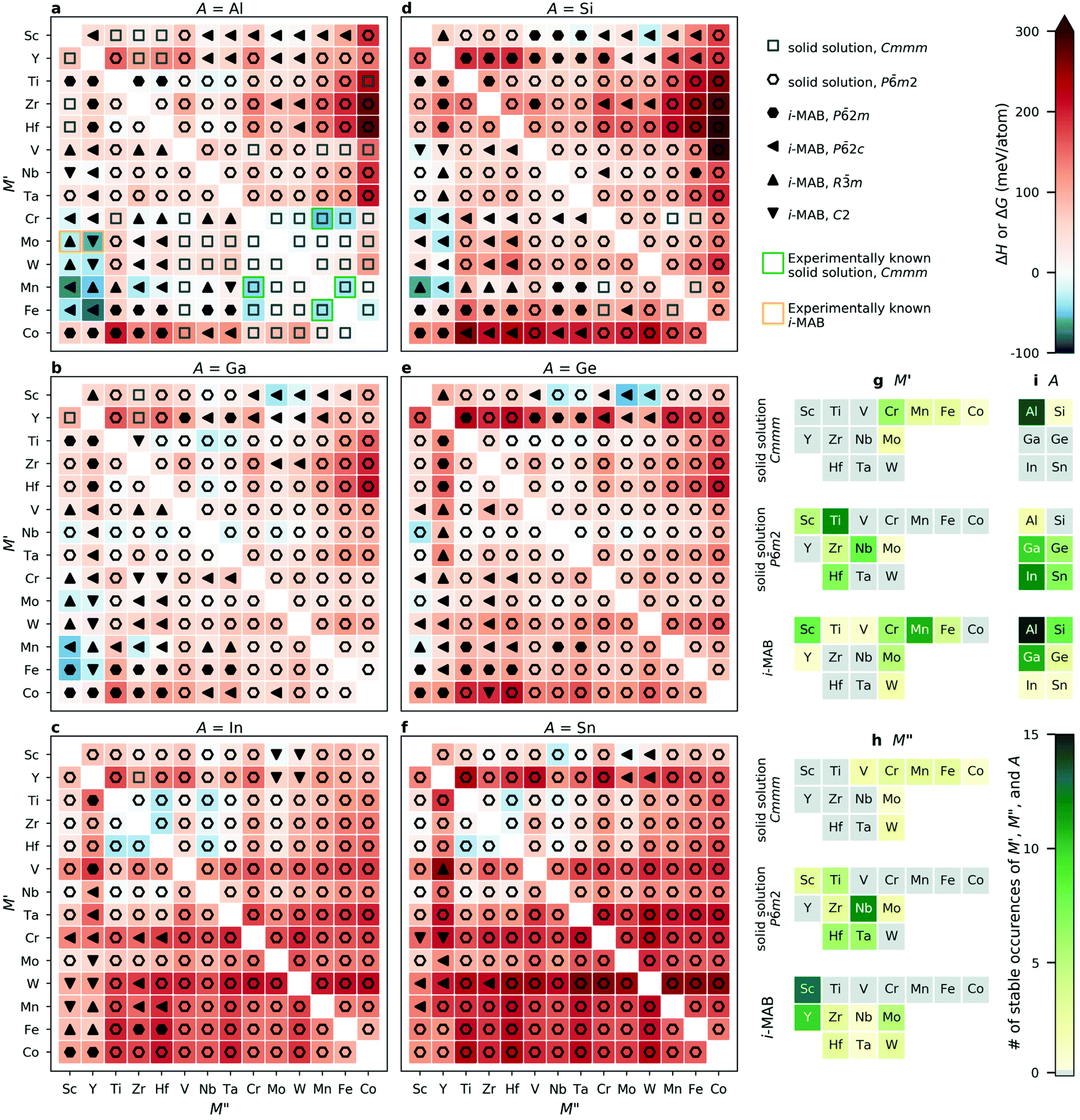 | ||
| Fig. 2 (a–f) Calculated formation enthalpy ΔH (order) or and Gibbs free energy of formation (solid solution) evaluated at 2000 K for A = Al, Ga, In, Si, Ge, and Sn. Number of atomic elements in stable i-MAB and solid solution MAB phases for (g) M′, (h) M′′, and (i) A. | ||
All four experimentally reported solid solution (M′0.67M′′0.33)2AlB225,26,59 are predicted to be stable, as well as the recently discovered Mo4/3Sc2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases.34Fig. 2g–i shows statistics for stable occurrences of M′, M′′, and A elements for all considered chemical configurations. In total, 39 i-MAB phases are predicted stable, i.e., with ΔHcp < 0, along with 52 stable solid solutions, i.e., with ΔGsolid solutioncp < 0 and ΔGsolid solutioncp < ΔHcp, out of which 15 have a Cmmm symmetry and 37 a Pm2 symmetry.
Results for A = Al, which have been published elsewhere,34 reveal 15 thermodynamically (Fig. 2a) and dynamically (Fig. S5–S19†) stable i-MAB phases with in-plane chemical ordering. Out of these, eight i-MAB phases have a calculated ΔHcp < −25 meV per atom, i.e. are herein judged to be significantly below zero, including M′4/3M′′2/3AlB2 (M′′ = Mo, Mn, Fe and M′′ = Sc, Y), W4/3Y2/3AlB2 and Mn4/3Zr2/3AlB2. The 16 solid solution Al-based phases predicted stable at 2000 K are dominated by the Cmmm symmetry, 14 compositions, combining metals mainly from group 6 to 9.
For the 11 i-MAB phases predicted thermodynamically stable (Fig. 2b) for A = Ga, nine is concluded to also be dynamically stable (Fig. S20–S30†). Out of these, three are identified with ΔHcp < −25 meV, including M′4/3Sc2/3GaB2 (M′ = Mn, Fe) and Fe/3Y2/3GaB2. These phases are accompanied by 10 stable solid solutions with Pm2 symmetry, exemplified by (Ti0.67Nb0.33)2GaB2 with ΔGsolid solutioncp = −24 meV per atom.
For A = In (Fig. 2c), only one i-MAB phase, Sc4/3Mo2/3InB2, is found to be stable, but just barely so, with ΔHcp < −3 meV per atom. Its dynamical stability is shown in Fig. S31.† More interesting are the 12 solid solutions with Pm2 symmetry predicted stable, out of which six have ΔGsolid solutioncp < −25 meV per atom, including (Ti0.67M′′0.33)2InB2, (Zr0.67Hf0.33)2InB2 (M′′ = Hf, Nb), and (Hf0.67M′′0.33)2InB2 (M′′ = Ti, Zr, Nb).
When A is from group 14, similar results are found. For A = Si, eight i-MAB phases are predicted to be thermodynamically stable (Fig. 2d), out of which four fulfil ΔHcp < −25 meV per atom, including M′4/3Sc2/3AlB2 (M′ = Cr, Mn) and M′4/3Y2/3AlB2 (M′ = Mo, Mn). Dynamical stability of these are demonstrated in Fig. S32–S39.† Only one solid solution is found stable, (Cr0.67Mn0.33)2SiB2, with Cmmm symmetry. For A = Ge and Sn, four i-MAB phases and 13 solid solutions with Pm2 symmetry are predicted stable (Fig. 2e and f). Out of these, two i-MAB phases are found with ΔHcp < −25 meV per atom, Sc4/3Mo2/3GeB2 and Sc4/3W2/3GeB2. Five solid solutions have ΔGsolid solutioncp < −25 meV per atom, including (Sc0.67Nb0.33)2AB2 (A = Ge, Sn), (Nb0.67Sc0.33)2GeB2, (Ti0.67Hf0.33)2SnB2, and (Hf0.67Ti0.33)2SnB2. All four thermodynamically stable i-MAB phases are dynamically stable (Fig. S40–S43†).
In summary, the 39 i-MAB phases predicted thermodynamically stable, i.e. with ΔHcp < 0, are mainly found for A = Al, Ga, and Si combined with M′ = Sc, Cr, Mn, Mo and M′′ = Sc and Y. Out of these, 36 i-MAB phases are also concluded as dynamically stable as seen by the calculated phonon dispersion (Fig. S5–S43†). The three dynamically unstable i-MAB phases have M′ = Sc in common (Fig. S20–S21 and S33†). Stable solid solutions with Pm2 symmetry are mainly found for A = Ga, In, Ge, Sn combined with M′ and M′′ from group 4 and 5. The 15 stable solid solution MAB phases with Cmmm symmetry are found for the traditional transition metals from group 6 to 9 and primarily for A = Al. These results illustrate that quaternary MAB phases, in the form of i-MAB or as solid solution, is a most prospective path towards expanding the attainable chemistries and structures which, in turn, allow for tuning potential. This highlights the value of computational materials discovery for accelerating exploratory synthesis in new chemical spaces. A complete list of stable i-MAB and solid solution phases are given in Table S4.†
Crystal symmetry and chemical order/disorder
An intuitive picture whether chemical order or solid solution is preferred for quaternary MAB phases is shown in Fig. 3 for the 1092 compositions considered, where the calculated stability, represented by the lowest value of ΔHi-MAB or ΔGsolid solution is shown as function of the energy difference between chemically ordered i-MAB and solid solution MAB (lowest energy of Cmmm or Pm2), ΔHi-MAB − ΔGsolid solution. The coloring represents the atomic size difference between M′ and M′′ (see Table S7† in for values used) while the symbol used represent the structure of lowest energy at 2000 K; hexagon for hexagonal i-MAB or solid solution, and square for orthorhombic solid solution structures. All reported synthesized i-MAB phases are marked by orange hexagons and solid solution MAB phases with Cmmm symmetry by green squares.
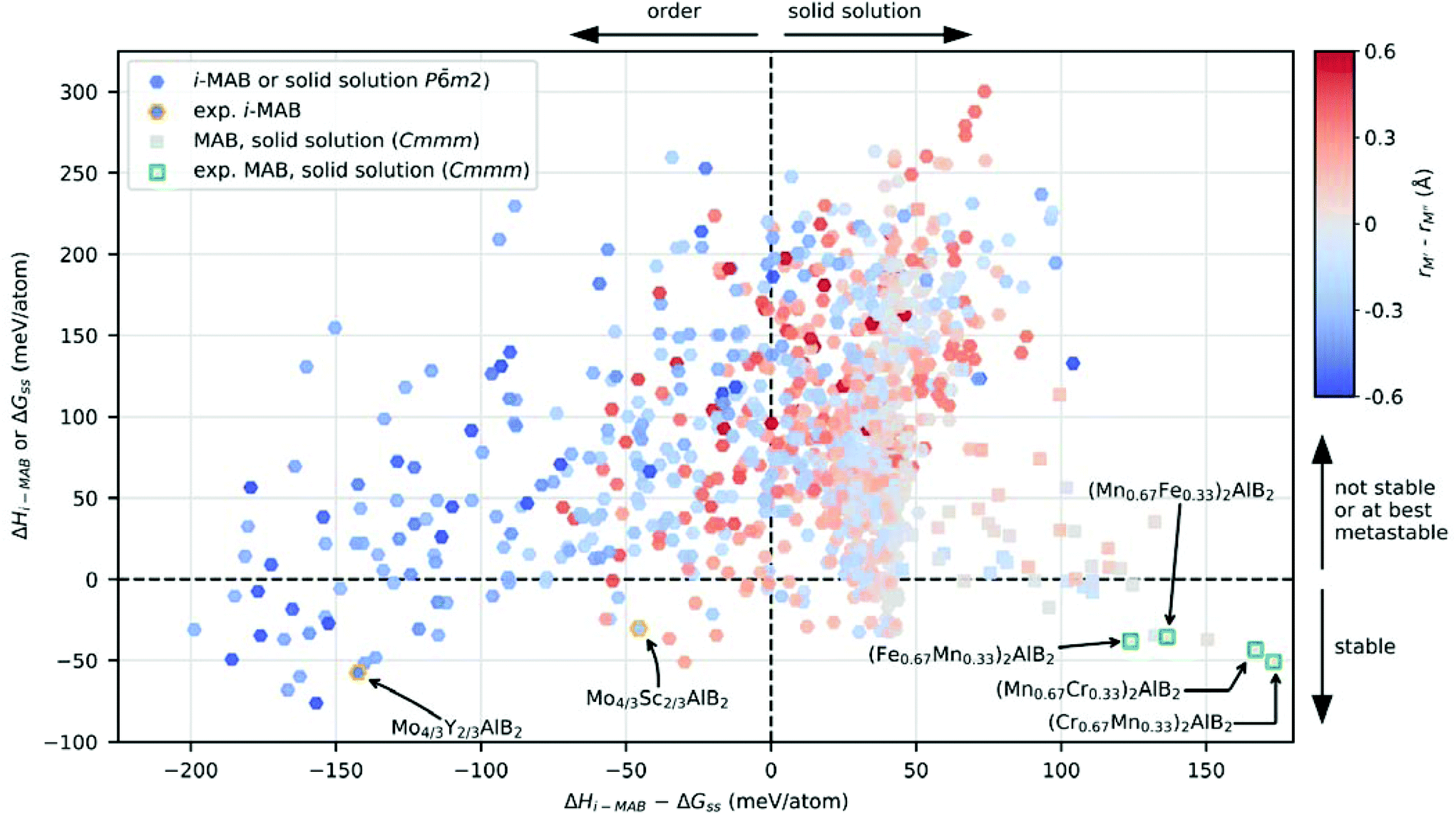 | ||
| Fig. 3 Calculated formation enthalpy (i-MAB) or Gibbs free energy of formation at 2000 K (solid solution MAB phase) of M′4/3M′′2/3AB2 as function of energy difference between i-MAB and solid solution MAB for all A. The symbol represents the symmetry of lowest energy and colouring represents the atomic size difference between M′ and M′′. Experimentally known i-MAB phases are indicated by orange hexagons and solid solution MAB phases with Cmmm symmetry by green squares. | ||
Thermodynamically stable i-MAB phases encompass 39 unique compositions which are found in the lower left quadrant of Fig. 3, among which a majority fulfill the atomic size criteria rM′′ > rM′ as indicated by the blue color. This includes the two synthesized i-MAB phases, both comprised of metal atoms with an atomic size difference, rM′′ > rM′, of at least 0.2 Å. Stable i-MAB phases are mainly represented by M′ = Sc, Cr, Mn, Mo and M′′ = Sc and Y, and A = Al, Ga, Si (see Fig. 2g–i). This results thus illustrates that there are many prospective i-MAB phases remaining to be experimentally discovered. A complete list of chemistries for stable i-MAB phases is found in Table S4.† Not stable, or at best metastable, i-MAB phases are found in the upper left quadrant of Fig. 3.
All of the reported synthesized MAB phases with solid solution of M′ and M′′ (Table S2†) are found stable and found in the lower right quadrant of Fig. 3. Here the atomic size difference of M′ and M′′ is small and typically in the range −0.2 Å < rM′ − rM′′ < 0.2 Å. There are more than 48 hypothetical solid solution MAB phases fulfilling this criterion, a complete list thereof is found in Tables S5 and S6,† which demonstrate that there are a vast number of phases remaining to be experimentally verified. The upper right quadrant represents not stable, or at best metastable, solid solution MAB phases.
Hexagonal symmetry – impact from atom size and electronegativity
To identify the attributes governing the preference for chemical order in the i-MAB structure or solid solution disorder in the Pm2 structure (both with hexagonal symmetry), we look at the size and electronegativity of M′ and M′′. Fig. 4a shows the energy difference ΔE of i-MAB compared to solid solution Pm2 evaluated at 2000 K as function of the size difference between M′ and M′′, ΔrM, where ΔE < 0 indicates favored i-MAB. Overall, ΔE is found to be correlated with the size difference, with a solid solution favored for similar size of M′ and M′′ while a larger difference favors i-MAB. This is most pronounced when M′′ is larger than M′, i.e., ΔrM < 0. The experimentally known i-MAB phases are found in the region ΔrM < −0.2 Å. For all six A elements, the i-MAB is in general energetically favored when M′ is smaller than M′′ as shown in Fig. 2b. Solid solution is typically favored when rM′ ≈ rM′′ or more specifically within a −0.2 Å < rM′ − rM′′ < 0.2 Å range.
 | ||
| Fig. 4 Energy difference ΔE between i-MAB and solid solution Pm2 as function of (a) size difference of M′′ and M′ and (c) electronegativity difference of M′′ and M′. Box- and swarmplots for ΔE categorized by difference in (b) size and (d) electronegativity of M′′ and M′ where negative and positive values are shown in blue and red, respectively. | ||
Next, in Fig. 4c we look at ΔE of i-MAB and solid solution as function of the electronegativity difference between M′ and M′′, ΔρM. Again, the region with ΔE < 0 indicates favored i-MAB and is mostly found in the region when ΔρM > 0. There is no apparent correlation between ΔE and ΔρM, as compared to ΔE and ΔrM in Fig. 4a, with data being more scattered. However, the majority of energetically favored i-MAB are identified for M′′ being more electronegative than M′, including the recently discovered Mo4/3Sc2/3AlB2 and Mo4/3Sc2/3AlB2i-MAB phases.34
Impact from choice of A, as shown in Fig. 4b and d, indicate a larger tendency for favouring i-MAB for A = Al, Ga, and Si, while both A = In and Sn favour formation of solid solutions. Note that both In and Sn have a larger atomic size than Al, Ga, and Si (Table S7†). This result resembles the impact from size of A as demonstrated for i-MAX phases.32
Solid solution MAB phases – impact from size and electronegativity
Next, we consider the solid solution MAB phases by comparing the orthorhombic Cmmm and hexagonal Pm2 symmetries. Again, we choose to look at the size and electronegativity differences of M′ and M′′ and possible impact from choice of A element. Fig. 5a shows the energy difference ΔE of solid solutions Cmmm and Pm2 as function of the size difference between M′ and M′′, ΔrM, where ΔE < 0 indicates favored Cmmm. Here we find that Cmmm in general and the synthesized phases in particular are all found within −0.2 Å < rM′ − rM′′ < 0.2 Å. Hexagonal Pm2 is almost always favored when rM′ − rM′′ < −0.2 Å or rM′ − rM′′ > 0.2 Å but is also found within −0.2 Å < rM′ − rM′′ < 0.2 Å. Impact for various A in Fig. 5b shows that A = Al is the element to go for if Cmmm symmetry is wanted, while Pm2 is favored for the other A elements. It should be noted that results in Fig. 5 do not give any information regarding the stability of the material. This can be found in Fig. 2 and explicitly in Tables S4E–S6.†
 | ||
| Fig. 5 Energy difference ΔE between solid solution MAB phases with Cmmm and Pm2 symmetry as function of (a) size difference of M′′ and M′ and (c) electronegativity difference of M′′ and M′. Box- and swarmplots for ΔE categorized by difference in (b) size and (d) electronegativity of M′′ and M′ where negative and positive values are shown in blue and red, respectively. Experimentally reported solid solution MAB phases with Cmmm symmetry are marked in panel (a) and (c). | ||
In Fig. 5c we look at ΔE as function of the electronegativity difference between M′ and M′′, ΔρM. Again, the region with ΔE < 0 indicates favored Cmmm while ΔE > 0 indicates favor for Pm2. Here no clear region of ΔρM is identified. Neither can any clear correlation be found for various A, as shown in Fig. 5d.
Summarizing key results governing the formation of chemical ordering in i-MAB or solid solution, we find that for all systems, i-MAB is statistically the preferred structure when the size of M′ is larger than M′′ combined with M′′ being more electronegative than M′. Comparing the difference in size (Fig. 3, 4a and b) and electronegativity (Fig. 4c and d) indicates that the former has a larger impact than the latter. The Hume-Rothery rules states that if there is a large enough difference in size and electronegativity between two atomic species, chemical order is preferred, whereas for minute differences solid solution is favored.62 Metal alloying in 212 MAB phases thus partly fulfills the Hume-Rothery rules as demonstrated by preference for i-MAB only when M′′ is larger than M′, as indicated by the blue colour in Fig. 3 and the left part of Fig. 4a. When M′ and M′′ is of similar size, solid solution is the energetically favoured order. It should be noted that the original Hume-Rothery rules are restricted to binary compounds. For higher order systems, specific rules need to be established for each system alone since they are dependent on the crystal structure, its sublattices and elements involved, as demonstrated herein and elsewhere.32,63,64
Properties of selected i-MAB phases
We choose to focus on the impact that different A-elements may have on the electronic and mechanical properties for selected i-MAB phases, namely Mo4/3Y2/3AB2 with A = Al, Ga, Si. These materials were chosen motivated by the M′ + M′′ combination being predicted stable for three different A-elements (Fig. 2) and that Mo4/3Y2/3AlB2 have been synthesized. Note that the space group symmetries of lowest energy – P2c, Rm, C2 – are close to degenerate in energy. Hence, for simplicity we here only include the Rm structure for a comparison of the properties. Data for P2c and C2 can be found in the ESI.†
The calculated electronic structure in terms of density of states (DOS) for selected i-MAB phases, with A = Al, Ga, Si in space group Rm, are plotted in the upper panels of Fig. 6. The change of A-element does not significantly affect the overall electronic structure, with the most notable change arising from a shift in the Fermi level for A = Si as compared to A = Al and Ga. This can be related to Si having one additional valence electron. For A = Al and Ga there are no significant qualitative difference which can be explained by the equivalent valency of Al and Ga. DOS for the other symmetries is shown in Fig. S44–S46 in ESI.† Due to the similarities in structure and layering, they are qualitatively equivalent with the results obtained for Rm.
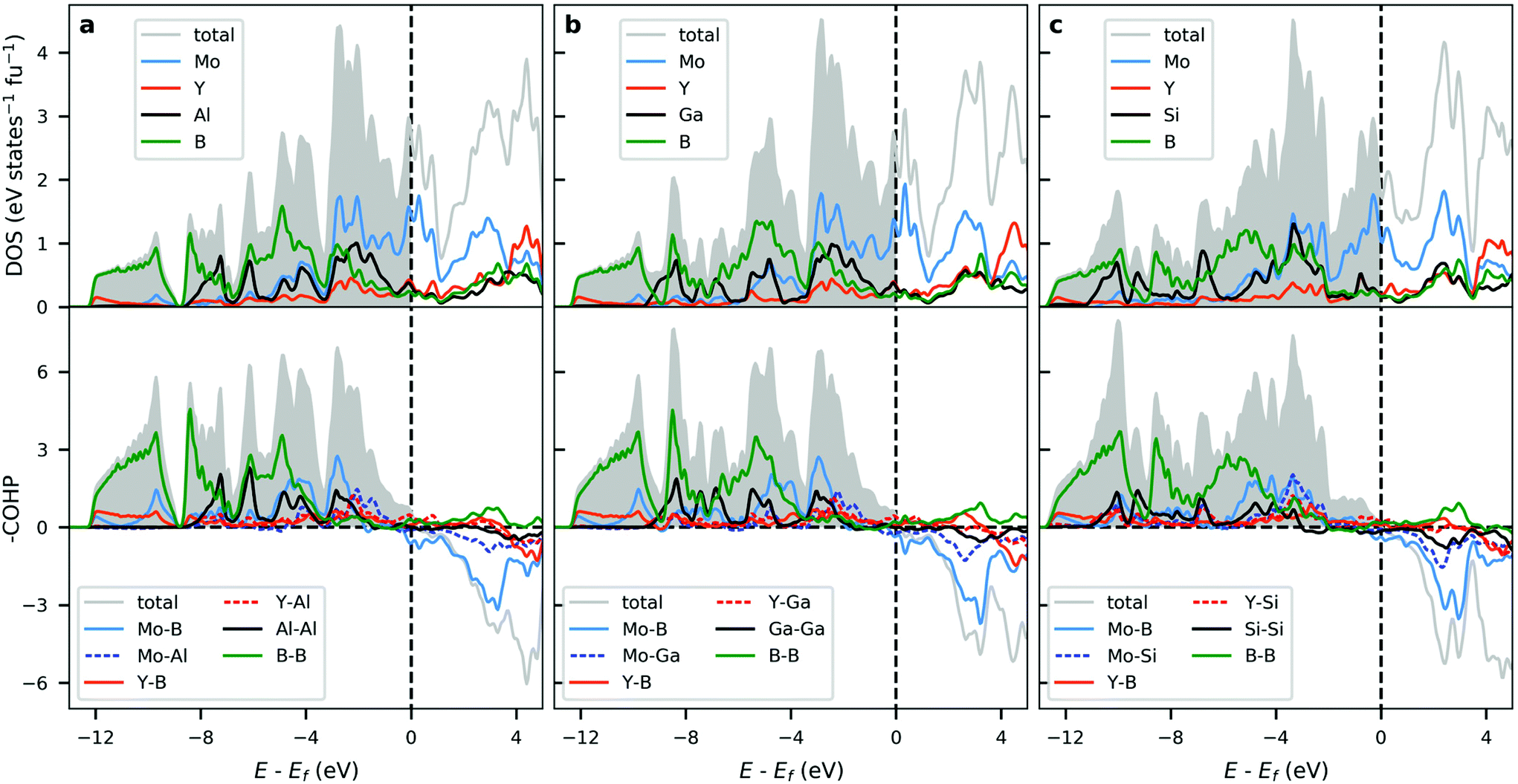 | ||
| Fig. 6 Calculated total and atomic density of states (DOS) and total and projected crystal overlap Hamilton population (COHP) analysis for (a) Mo4/3Y2/3AlB2, (b) Mo4/3Y2/3GaB2, and (c) Mo4/3Y2/3SiB2 of space group Rm. | ||
All i-MAB phases show an evident metallic character with a finite number of states at and around the Fermi level. This is also reflected in the electronic band-structures (Fig. S49–S57†) where a multitude of bands are crossing the Fermi level, along with a lack of significant anisotropy, which further confirms the metallicity of these materials.
The corresponding COHP analysis is plotted in the bottom panels of Fig. 6 for selected key interactions. The results show that all three i-MAB phases are found to be close to optimized from a bonding perspective. A slight but notable change in the distribution of bonding and antibonding states can be observed when going from Al and Ga to Si. Most states at and in the vicinity of the Fermi level can be attributed to non-bonding Mo-d. Additional bonding characteristics for the other symmetries is shown in Fig. S47–S48.†
The similarities and differences between the phases can also be quantified by integrating the occupied states of the COHP curve (ICOHP) and then comparing the contribution (in percentage) of the pairwise interactions with respect to the net (total) integrated bonding within each i-MAB phase. This is seen in Fig. 7a. The calculations reveal that the Mo–B and B–B interactions contributes the most as compared to Mo–A, Y–B, Y–A and A–A. Here it is also demonstrated that the bonding in Mo4/3Y2/3AlB2 and Mo4/3Y2/3GaB2 contribute similarly for all interactions. This in comparison with Mo4/3Y2/3SiB2 where a slight increase for Mo–Si and a decrease for Si–Si is observed. Note that the multiplicity for each type of interaction is included in the net contribution. Individual bond strengths and comparison between different interactions may differ, but these do not change the identified overall trends in bonding.
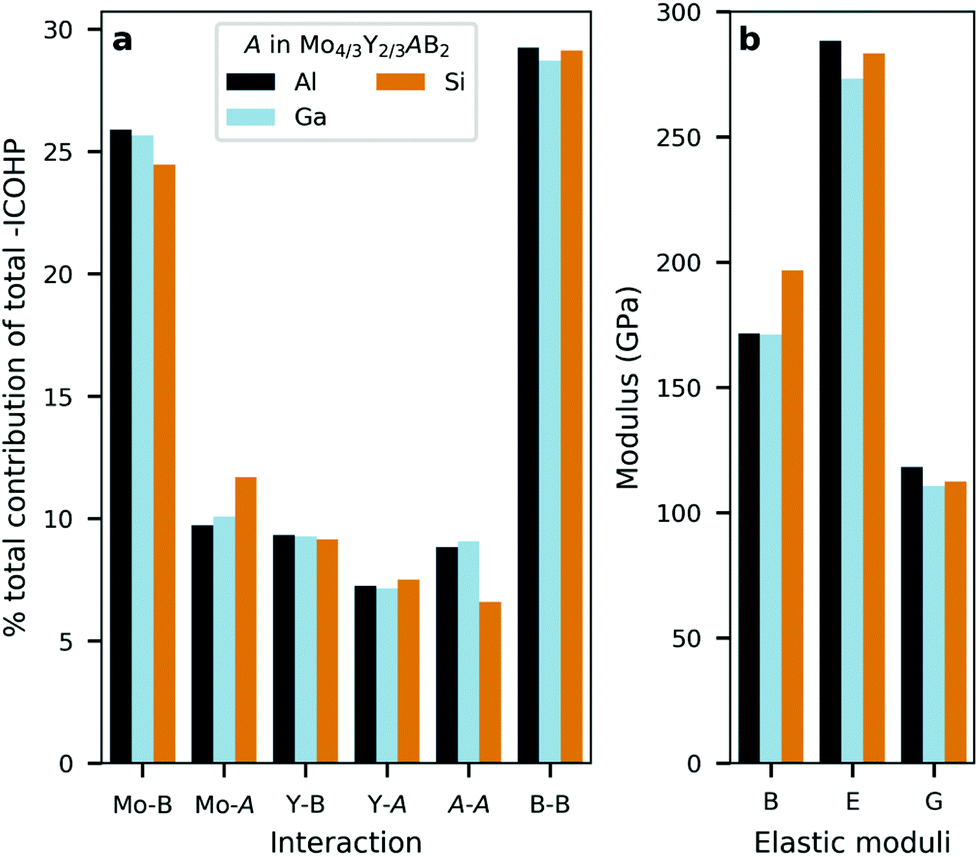 | ||
| Fig. 7 (a) The contributions, in percentage, of selected interactions with respect to the net bonding for selected and (b) calculated elastic moduli – bulk modulus B, Young modulus E and Shear modulus G. Data shown for Mo4/3Y2/3AB2 of space group Rm and A = Al, Ga, Si. | ||
The similar bonding characteristics among the three stable Mo4/3Y2/3AB2i-MAB phases – with A = Al, Ga, Si – is also reflected in Fig. 7b, where bulk modulus B, Young modulus E, and Shear modulus G is shown. When comparing Al, Ga and Si, we find only minor differences, although the bulk modulus is slightly larger for A = Si. Fig. S58 and Table S8† further shows that these results are independent of the assigned symmetry. A more detailed analysis of impact from different metals in M′4/3M′′2/3AlB2 on mechanical properties can be found in ref. 65.
Conclusion
In summary, through a systematic theoretical study of the phase stability of quaternary (M′1−xM′′x)2AB2 (212) MAB phases upon alloying between M′ and M′′ from group 3 to 9 (Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co) combined with various A-elements (Al, Ga, In, Si, Ge, Sn), we confirm all experimentally synthesized phases to date with both chemical order (i-MAB) and solid solution disorder. Importantly, beyond A = Al we also predict an additional 21 thermodynamically and dynamically stable i-MAB phases and 36 stable solid solutions (1 with Cmmm symmetry and 35 with Pm2 symmetry). These hypothetical phases remain to be experimentally verified, and synthesis is encouraged. Preference for order or solid solution upon metal alloying in 212 MAB phases are dictated mainly by the size difference between the alloying metal elements. Ordered i-MAB phases are energetically favoured when M′′ is larger than M′ by at least 0.2 Å. This is typically fulfilled for M′ = Cr, Mo, Mn, or Fe combined with M′′ = Sc or Y, and with A = Al, Ga, and Si. Phases with similar size of M′ and M′′ show preference for solid solution, with Cmmm being favoured for A = Al while Pm2 being favored for A = Ga, in, Ge, and Sn. Further investigation of selected i-MAB phases promising for synthesize reveals that different A-elements only have minor impact on the bonding characteristics and hence similar properties are expected. This study demonstrates that alloying is viable path towards novel elemental combinations to extend the frontier of MAB phase chemistries which, in turn, allows for potentially tuneable and advantageous properties.
Author contributions
The manuscript was written through contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Knut and Alice Wallenberg (KAW) Foundation for a Fellowship/Scholar Grant and Project funding (KAW 2020.0033). Support from the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No 2009 00971) is also acknowledged. The calculations were carried out using supercomputer resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC), the High Performance Computing Center North (HPC2N), and the PDC Center for High Performance Computing partially funded by the Swedish Research Council through grant agreement no. 2018-05973.References
- H. J. Becher, K. Krogmann and E. Peisker, Z. Anorg. Allg. Chem., 1966, 344, 140–147 CrossRef CAS.
- W. Jeitschko, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 163 CrossRef CAS.
- N. F. Chaban and I. U. B. KuzMa, Izv. Akad. Nauk SSSR, Neorg. Mater., 1973, 9, 1908–1911 Search PubMed.
- X. Tan, P. Chai, C. M. Thompson and M. Shatruk, J. Am. Chem. Soc., 2013, 135, 9553–9557 CrossRef CAS PubMed.
- M. Ade and H. Hillebrecht, Inorg. Chem., 2015, 54, 6122–6135 CrossRef CAS PubMed.
- L. Verger, S. Kota, H. Roussel, T. Ouisse and M. W. Barsoum, J. Appl. Phys., 2018, 124, 205108 CrossRef.
- Y. Zhou, H. Xiang, F.-Z. Dai and Z. Feng, Mater. Res. Lett., 2017, 5, 440–448 CrossRef CAS.
- S. Kota, W. Wang, J. Lu, V. Natu, C. Opagiste, G. Ying, L. Hultman, S. J. May and M. W. Barsoum, J. Alloys Compd., 2018, 767, 474–482 CrossRef CAS.
- W. Rieger, H. Nowotny and F. Benesovsky, Monatsh. Chem., 1965, 96, 844–851 CrossRef.
- W. Jeitschko, Monatsh. Chem., 1966, 97, 1472–1476 CrossRef CAS.
- Y. Zhang, S. Okada, T. Atoda, T. Yamabe and I. Yasumori, J. Ceram. Assoc. Jpn., 1987, 95, 374–380 CrossRef CAS.
- H. Zhang, F.-Z. Dai, H. Xiang, Z. Zhang and Y. Zhou, J. Mater. Sci. Technol., 2018, 35, 530–534 CrossRef.
- S. Kota, E. Zapata-Solvas, A. Ly, J. Lu, O. Elkassabany, A. Huon, W. E. Lee, L. Hultman, S. J. May and M. W. Barsoum, Sci. Rep., 2016, 6, 26475 CrossRef CAS PubMed.
- L. T. Alameda, C. F. Holder, J. L. Fenton and R. E. Schaak, Chem. Mater., 2017, 29, 8953–8957 CrossRef CAS.
- D. K. Mann, J. Xu, N. E. Mordvinova, V. Yannello, Y. Ziouani, N. González-Ballesteros, J. P. S. Sousa, O. I. Lebedev, Y. V. Kolen'ko and M. Shatruk, Chem. Sci., 2019, 10, 2796–2804 RSC.
- L. T. Alameda, P. Moradifar, Z. P. Metzger, N. Alem and R. E. Schaak, J. Am. Chem. Soc., 2018, 140, 8833–8840 CrossRef CAS PubMed.
- H. Zhang, F.-Z. Dai, H. Xiang, X. Wang, Z. Zhang and Y. Zhou, J. Mater. Sci. Technol., 2019, 35, 1593–1600 CrossRef.
- M. Khazaei, J. Wang, M. Estili, A. Ranjbar, S. Suehara, M. Arai, K. Esfarjani and S. Yunoki, Nanoscale, 2019, 11, 11305–11314 RSC.
- L. T. Alameda, R. W. Lord, J. A. Barr, P. Moradifar, Z. P. Metzger, B. C. Steimle, C. F. Holder, N. Alem, S. B. Sinnott and R. E. Schaak, J. Am. Chem. Soc., 2019, 141, 10852–10861 CrossRef CAS PubMed.
- K. Kim, C. Chen, D. Nishio-Hamane, M. Okubo and A. Yamada, Chem. Commun., 2019, 55, 9295–9298 RSC.
- J. Wang, T.-N. Ye, Y. Gong, J. Wu, N. Miao, T. Tada and H. Hosono, Nat. Commun., 2019, 10, 2284 CrossRef PubMed.
- N. Miao, J. Wang, Y. Gong, J. Wu, H. Niu, S. Wang, K. Li, A. R. Oganov, T. Tada and H. Hosono, Chem. Mater., 2020, 32, 6947–6957 CrossRef CAS.
- Y. Yu and T. Lundström, J. Alloys Compd., 1995, 226, 5–9 CrossRef CAS.
- S. Okada, K. Iizumi, K. Kudaka, K. Kudou, M. Miyamoto, Y. Yu and T. Lundström, J. Solid State Chem., 1997, 133, 36–43 CrossRef CAS.
- L. A. Hanner, H. O. Badr, M. Dahlqvist, S. Kota, J. Rosen and M. W. Barsoum, Mater. Res. Lett., 2021, 9, 112–118 CrossRef CAS.
- P. Chai, S. A. Stoian, X. Tan, P. A. Dube and M. Shatruk, J. Solid State Chem., 2015, 224, 52–61 CrossRef CAS.
- Q. Du, G. Chen, W. Yang, J. Wei, M. Hua, H. Du, C. Wang, S. Liu, J. Han, Y. Zhang and J. Yang, J. Phys. D: Appl. Phys., 2015, 48, 335001 CrossRef.
- S. Hirt, F. Yuan, Y. Mozharivskyj and H. Hillebrecht, Inorg. Chem., 2016, 55, 9677–9684 CrossRef CAS PubMed.
- F.-Z. Dai, H. Xiang, Y. Sun and Y. Zhou, J. Mater. Sci. Technol., 2019, 35, 1432–1438 CrossRef.
- P. Ma, S. Li, J. Hu, X. Lu, W. Yu and Y. Zhou, J. Alloys Compd., 2020, 814, 152290 CrossRef CAS.
- M. Naguib, G. W. Bentzel, J. Shah, J. Halim, E. N. Caspi, J. Lu, L. Hultman and M. W. Barsoum, Mater. Res. Lett., 2014, 2, 233–240 CrossRef.
- M. Dahlqvist, A. Petruhins, J. Lu, L. Hultman and J. Rosen, ACS Nano, 2018, 12, 7761–7770 CrossRef CAS PubMed.
- B. Anasori, M. Dahlqvist, J. Halim, E. J. Moon, J. Lu, B. C. Hosler, E. N. Caspi, S. J. May, L. Hultman, P. Eklund, J. Rosén and M. W. Barsoum, J. Appl. Phys., 2015, 118, 094304 CrossRef.
- M. Dahlqvist, Q. Tao, J. Zhou, J. Palisaitis, P. O. Å. Persson and J. Rosen, J. Am. Chem. Soc., 2020, 142, 18583–18591 CrossRef CAS PubMed.
- Q. Tao, M. Dahlqvist, J. Lu, S. Kota, R. Meshkian, J. Halim, J. Palisaitis, L. Hultman, M. W. Barsoum, P. O. Å. Persson and J. Rosen, Nat. Commun., 2017, 8, 14949 CrossRef PubMed.
- B. Ahmed, A. E. Ghazaly and J. Rosen, Adv. Funct. Mater., 2020, 30, 2000894 CrossRef CAS.
- A. Mockute, Q. Tao, M. Dahlqvist, J. Lu, S. Calder, E. N. Caspi, L. Hultman and J. Rosen, Phys. Rev. Mater., 2019, 3, 113607 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- A. Zunger, S. H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS PubMed.
- M. Dahlqvist, B. Alling, I. A. Abrikosov and J. Rosén, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024111 CrossRef.
- M. Dahlqvist, B. Alling and J. Rosén, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 220102 CrossRef.
- P. Eklund, M. Dahlqvist, O. Tengstrand, L. Hultman, J. Lu, N. Nedfors, U. Jansson and J. Rosén, Phys. Rev. Lett., 2012, 109, 035502 CrossRef PubMed.
- A. S. Ingason, A. Mockute, M. Dahlqvist, F. Magnus, S. Olafsson, U. B. Arnalds, B. Alling, I. A. Abrikosov, B. Hjörvarsson, P. O. Å. Persson and J. Rosen, Phys. Rev. Lett., 2013, 110, 195502 CrossRef CAS PubMed.
- A. S. Ingason, A. Petruhins, M. Dahlqvist, F. Magnus, A. Mockute, B. Alling, L. Hultman, I. A. Abrikosov, P. O. Å. Persson and J. Rosen, Mater. Res. Lett., 2014, 2, 89–93 CrossRef CAS.
- A. Mockute, M. Dahlqvist, J. Emmerlich, L. Hultman, J. M. Schneider, P. O. Å. Persson and J. Rosen, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 094113 CrossRef.
- A. Mockute, P. O. Å. Persson, F. Magnus, A. S. Ingason, S. Olafsson, L. Hultman and J. Rosen, Phys. Status Solidi RRL, 2014, 8, 420–423 CrossRef CAS.
- A. Thore, M. Dahlqvist, B. Alling and J. Rosén, Comput. Mater. Sci., 2014, 91, 251–257 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- D. Potashnikov, E. N. Caspi, A. Pesach, S. Kota, M. Sokol, L. A. Hanner, M. W. Barsoum, H. A. Evans, A. Eyal, A. Keren and O. Rivin, Phys. Rev. Mater., 2020, 4, 084404 CrossRef CAS.
- D. Potashnikov, E. N. Caspi, A. Pesach, A. Hoser, S. Kota, L. Verger, M. W. Barsoum, I. Felner, A. Keren and O. Rivin, J. Magn. Magn. Mater., 2019, 471, 468–474 CrossRef CAS.
- L. Ke, B. N. Harmon and M. J. Kramer, Phys. Rev. B, 2017, 95, 104427 CrossRef.
- W. Hume-Rothery, R. E. Smallman and C. W. Haworth, The structure of metals and alloys, The Institute of Metals, London, 1969 Search PubMed.
- M. Dahlqvist and J. Rosen, Nanoscale, 2020, 12, 785–794 RSC.
- M. Dahlqvist, J. Lu, R. Meshkian, Q. Tao, L. Hultman and J. Rosen, Sci. Adv., 2017, 3, e1700642 CrossRef PubMed.
- H. Lind, M. Dahlqvist and J. Rosen, J. Phys.: Condens. Matter, 2021, 33, 255402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr02552j |
| This journal is © The Royal Society of Chemistry 2021 |