
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradation of graphdiyne oxide in classically activated (M1) macrophages modulates cytokine production†
Guotao
Peng
 a,
Tianbo
Duan
b,
Mengyu
Guo
c,
Yurui
Xue
d,
Chunying
Chen
c,
Yuliang
Li
d,
Klaus
Leifer
b and
Bengt
Fadeel
*a
a,
Tianbo
Duan
b,
Mengyu
Guo
c,
Yurui
Xue
d,
Chunying
Chen
c,
Yuliang
Li
d,
Klaus
Leifer
b and
Bengt
Fadeel
*a
aNanosafety & Nanomedicine Laboratory (NNL), Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden. E-mail: bengt.fadeel@ki.se
bDepartment of Engineering Sciences, Uppsala University, Uppsala, Sweden
cCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, China
dCAS Key Laboratory of Organic Solids, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
First published on 19th July 2021
Abstract
Graphdiyne oxide (GDYO) is a carbon-based nanomaterial possessing sp2 and sp-hybridized carbon atoms with many promising applications. However, its biocompatibility and potential biodegradability remain poorly understood. Using human primary monocyte-derived macrophages as a model we show here that GDYO elicited little or no cytotoxicity toward classically activated (M1) and alternatively activated (M2) macrophages. Moreover, GDYO reprogrammed M2 macrophages towards M1 macrophages, as evidenced by the elevation of specific cell surface markers and cytokines and the induction of NOS2 expression. We could also show inducible nitric oxide synthase (iNOS)-dependent biodegradation of GDYO in M1 macrophages, and this was corroborated in an acellular system using the peroxynitrite donor, SIN-1. Furthermore, GDYO elicited the production of pro-inflammatory cytokines in a biodegradation-dependent manner. Our findings shed new light on the reciprocal interactions between GDYO and human macrophages. This is relevant for biomedical applications of GDYO such as the re-education of tumor-associated macrophages or TAMs.
1. Introduction
Graphdiyne oxide (GDYO) is a novel carbon allotrope composed of both sp2 and sp-hybridized carbon atoms.1 GDYO has been explored in many areas, not least in biomedicine, due to its many unique properties.2 For instance, in a recent study, excellent antibacterial properties of GDYO were demonstrated.3 However, there are only a limited number of reports on the biocompatibility of GDYO,4 and few if any studies have explored the interactions between GDYO and primary human macrophages. The present study addresses the biocompatibility and biodegradability of GDYO using primary human monocyte-derived macrophages (HMDMs) as a model.5Macrophages play critical roles in host defense, inflammation, and tissue homeostasis.6 Macrophages display considerable plasticity insofar as they can undergo phenotypic and functional changes in response to local stimuli; this dynamic process is referred to as polarization.7 Hence, the classically activated proinflammatory M1 macrophages mediate antimicrobial and antitumor responses, whereas the alternatively activated anti-inflammatory M2 macrophages are involved in tissue repair.8 Moreover, tumor-associated macrophages (TAMs) are frequently skewed into immunosuppressive M2 macrophages and this may further promote tumor progression.9 The activation status of macrophages residing in a solid tumor is influenced by the local microenvironment whereas primary macrophages cultured ex vivo can be influenced by the addition of various cytokines to achieve M1 or M2 activation.8
Growing evidence indicates that nanomaterials modulate macrophage polarization, whereas macrophage activation status, in turn, may determine nanomaterial internalization. For instance, Fuchs et al. found that polystyrene nanoparticles skewed the M2 macrophage polarization without affecting M1 markers.10 Furthermore, we demonstrated previously that mesoporous silica particles were internalized through distinct pathways and to a different extent in granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) activated macrophages.11 Kodali et al.12 reported that bone marrow-derived macrophages exposed to superparamagnetic iron oxide nanoparticles displayed an impaired ability to transition from an M1 to M2 activation state with diminished phagocytosis of bacteria. In another recent study, folic acid-modified silver nanoparticles were delivered to M1 macrophages causing M2 polarization.13 Overall, macrophage activation status is important to consider when studying nano(bio)material effects.
Previous studies have revealed the enzymatic biodegradation of carbon nanotubes (CNTs) by various peroxidases, including myeloperoxidase (MPO), eosinophil peroxidase (EPO), and lactoperoxidase (LPO).14–16 Furthermore, graphene oxide (GO) degradation by purified MPO or by activated human neutrophils releasing MPO has also been demonstrated.17,18 In addition to peroxidase-driven degradation, we and others have documented peroxynitrite-dependent biodegradation of CNTs19,20 as well as GO.21 The generation of peroxynitrite (ONOO−) relies on nitric oxide (NO) catalyzed by inducible nitric oxide synthase (iNOS) and superoxide (O2−) generated by the NADPH oxidase. Importantly, such oxidative pathways are predominantly found in classically activated M1 macrophages tasked with the clearance of microorganisms.6 However, cellular or acellular biodegradation of GDYO has not been explored to date. Here, we set out to investigate the biocompatibility and biodegradability of GDYO using primary human macrophages as a model. We demonstrated that GDYO polarized M2 macrophages towards M1 macrophages, and we documented peroxynitrite-driven biodegradation of GDYO in M1 macrophages. Furthermore, GDYO modulated the cytokine responses in M1 macrophages in a biodegradation-dependent manner. These findings provide important new insights regarding the reciprocal interactions between GDYO and macrophages which may guide future biomedical applications of GDYO.
2. Experimental
2.1. Synthesis and characterization
Graphdiyne (GDY) was synthesized as described previously.22 GDYO was synthesized by the oxidation of GDY powder using a mixture of H2O2 and H2SO4 as a complex oxidant. Briefly, 50 mg of GDY was gradually stirred into 30% H2O2 solution (1 mL) and 98% H2SO4 (2.5 mL) under an ice-water bath for 1 h. The oxidization was stopped by adding 50 mL double-distilled water, followed by dialysis (cutoff, 3000) for seven days to remove mixed acid. GDYO samples were obtained by sonication for 4 h in water. The physicochemical characterization of GDYO was performed using atomic force microscope (AFM), X-ray photoelectron spectroscopy (XPS), and Fourier-transform infrared spectroscopy (FTIR). AFM images were captured using the Bruker MultiMode V8-SPM (Bruker, Germany) with a ScanAsyst mode. XPS survey scans were measured by an ESCALAB 250 X-ray photoelectron spectrometer (Thermo Scientific). FTIR spectra were collected using a Nicolet™ iN™10 Infrared Microscope (ThermoFisher Scientific). One droplet of the GDYO solution was transferred onto the 15 × 15 × 0.5 mm CaF2 infrared window, and dried and the transmission spectrum of GDYO was obtained by subtracting the CAF2 background spectrum.2.2. Endotoxin assessment
The TNF-α expression test (TET) was used to determine the endotoxin content of GDYO.23 To this end, the cytotoxicity of GDYO towards human monocyte-derived macrophages (HMDMs) was evaluated and the test was then conducted at non-cytotoxic concentrations. First, peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats obtained from adult human blood donors (Karolinska University Hospital, Stockholm) by density gradient centrifugation using Lymphoprep™ (STEMCELL Technologies, Sweden). PBMCs were positively selected for CD14 expression using CD14 MicroBeads (Miltenyi Biotec, Sweden). To obtain HMDMs, CD14 monocytes were cultured in RPMI-1640 cell medium supplemented with 2 mM L-glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 10% heat-inactivated fetal bovine serum (FBS), supplemented with 50 ng mL−1 recombinant M-CSF (PeproTech, UK) for four days. HMDMs were exposed to GDYO at the indicated concentrations for 24 h and cell viability was determined using the CytoTox 96® assay (Promega) and the Alamar Blue™ assay (ThermoScientific). HMDMs were then incubated with GDYO at 25 and 50 μg mL−1 in the presence or absence of the lipopolysaccharide (LPS) inhibitor, polymyxin B (10 μM) (Sigma) for 24 h. LPS (0.01 μg mL−1) (Sigma) was used as a positive control. TNF-α production was quantified with the human TNF-α ELISA (Mabtech AB, Sweden) according to the manufacturer's instructions.2.3. M1 and M2 macrophages
Primary human CD14-positive monocytes were obtained as described above in section 2.2. Macrophages were differentiated from CD14 monocytes in medium containing either GM-CSF (10 ng mL−1, PeproTech) or M-CSF (50 ng mL−1, PeproTech) for 4 days to obtain precursors of M1 and M2 macrophages, respectively. M1 macrophages were then obtained by exposure to LPS (100 ng mL−1, Sigma-Aldrich) and interferon (IFN)-γ (20 ng mL−1, Sigma-Aldrich), whereas precursors of M2 macrophages were further polarized with interleukin (IL)-4 (20 ng mL−1, Sigma-Aldrich) for 24 h. At day 5, the cells were harvested using trypsin-EDTA solution (0.25%) and gently scraped, centrifuged at 1300 rpm at room temperature for 5 min, washed thrice with PBS, and then processed for flow cytometry. Macrophages stimulated with GM-CSF and M-CSF without the addition of further polarization stimuli were used for comparison (see below). The buffy coats were obtained from the blood bank at the Karolinska University Hospital, Stockholm, and all procedures were carried out in compliance with the biosafety committee at Karolinska Institutet as outlined in “Rules for the handling of blood and other human sample materials” (1–31/2019). The samples are completely anonymized, and data cannot be traced back to individual donors. For this reason, specific approval is not required (Ethical Committee for Human Studies in Stockholm).2.4. Flow cytometry analysis
Cell surface markers were analyzed by flow cytometry. In brief, flow cytometry staining buffer (BioLegend, San Diego, CA) was added to the cells and blocking was performed by adding 2 μL of FcR blocking reagent per 106 cells (Miltenyi Biotec, Sweden), followed by gently vortexing the samples, and allowing the tubes to stand at room temperature for 15 min. The cells were then incubated with the conjugated antibodies, FITC mouse anti-human CD86 (BD Pharmingen) and FITC mouse anti-human CD206 (BD Pharmingen), and the isotype-matched control antibody (mouse IgG1, Invitrogen), then gently vortexed, and incubated at room temperature for 30 min protected from light. The single cell suspensions were maintained by intermittent vortexing during the incubation. The cell pellets were resuspended in flow cytometry staining buffer for analysis using the BD LSRFortessa™ X-20 (BD Biosciences, San Jose, CA). Data were analysed using FCS Express™ v. 7.0 software (DeNovo Software, Pasadena, CA).2.5. Cytotoxicity assessment
The cell viability assessment of M1 and M2 macrophages following GDYO exposure was evaluated by the LDH release assay as previously described.24 Different concentrations of GDYO (6.25, 12.5, 25, 50, 75, and 100 μg mL−1) were added together with polarization stimuli and cells were incubated for 24 h. After the exposure, the cell supernatants were collected to measure the LDH release using the CytoTox 96® assay kit following the manufacturer's instructions (Promega, Madison, WI). The LDH release was normalized to the maximum LDH induced by the cell lysis buffer provided in the kit. We also applied the Alamar Blue™ assay to determine cellular metabolic capacity. To this end, the Alamar Blue™ reagent was prepared freshly in RPMI-1640 cell medium according to the manufacturer's instruction (ThermoScientific). The exposed cells were rinsed with PBS and Alamar Blue™ reagent was added to each well. Following incubation for 3 h at 37 °C, fluorescence was measured at the excitation and emission wavelengths of 531 nm and 595 nm using a Tecan Infinite F200 plate reader (Tecan, Stockholm, Sweden). The results were normalized to the untreated negative controls.2.6. Cellular uptake by TEM
Macrophage uptake of GDYO was evaluated by TEM.25 Briefly, cells were incubated with a non-toxic dose of GDYO (25 μg mL−1) during the polarization step for 2 h and 24 h. Cells were then washed, harvested using trypsin-EDTA (0.25%) and gently scraped, and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Following the primary fixation, samples were rinsed with 0.1 M phosphate buffer followed by post-fixation in 2% osmium tetroxide in 0.1 M phosphate buffer, pH 7.4 at 4 °C for 2 h. The samples were then stepwise ethanol dehydrated followed by stepwise acetone/LX-112 infiltration and embedded in LX-112. Semi- and ultra-thin sections were prepared on a Leica EM UC7 microtome. The ultra-thin sections were contrasted with uranyl acetate followed by Reynold's lead citrate and examined using a Hitachi HT7700 microscope operating at 100 kV. Digital images were acquired with a 2k × 2k Veleta CCD camera.2.7. Western blot analysis
M1 and M2 macrophages were harvested using trypsin-EDTA solution (0.25%) and cells were lysed on ice with RIPA lysis buffer (50 mM Tris HCl, 150 mM NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS, 1 mM EDTA). Protease- and phosphatase inhibitors (Mini EDTA-free Protease Inhibitor Cocktail, Sigma Aldrich; 1 mM PMSF, Thermo Fisher; PhosSTOP, Sigma Aldrich), and 1 mM DTT (Sigma Aldrich) were added to the RIPA buffer and whole cell extracts were collected by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 20 min. The total protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Scientific). Fifty μg of total protein was loaded in each well on an SDS-PAGE gel (Invitrogen). The membrane was probed with recombinant rabbit monoclonal anti-iNOS antibody (ab178945, Abcam) at 1:500 dilution overnight at 4 °C. GAPDH was used as loading control (Invitrogen). After washing, the membrane was probed with the IRDye 800CW goat anti rabbit secondary antibody (LI-COR Biosciences) for 1 h at room temperature and detection was performed on an Odyssey CLx scanner (LI-COR Biosciences, Lincoln, NE).
000 rpm at 4 °C for 20 min. The total protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Scientific). Fifty μg of total protein was loaded in each well on an SDS-PAGE gel (Invitrogen). The membrane was probed with recombinant rabbit monoclonal anti-iNOS antibody (ab178945, Abcam) at 1:500 dilution overnight at 4 °C. GAPDH was used as loading control (Invitrogen). After washing, the membrane was probed with the IRDye 800CW goat anti rabbit secondary antibody (LI-COR Biosciences) for 1 h at room temperature and detection was performed on an Odyssey CLx scanner (LI-COR Biosciences, Lincoln, NE).
2.8. Nitric oxide detection
NO production in macrophages was determined by the DAF-FM-DA assay (Sigma-Aldrich, Stockholm) as described previously.21N-Omega-nitro-L-arginine methyl ester hydrochloride (L-NAME, 15 mM; Sigma-Aldrich) was used to inhibit NO production. Briefly, cells were washed with PBS at 24 h, 48 h and 72 h post-addition of polarization stimuli and incubated with DAF-FM-DA (10 μM) for 60 min. Then, cells were washed with PBS to remove excess probe and incubated for 30 min to allow complete de-esterification. The fluorescence signal was captured at the excitation and emission wavelengths of 495 and 515 nm using a Tecan Infinite® F200 plate reader (Männedorf, Switzerland) operating with Magellan™ v7.2 software (Tecan). The fluorescent images were captured using the ZOE Fluorescent Cell Imager (BioRad, Sweden).2.9. RT-qPCR analysis
mRNA expression in M1 and M2 macrophages was determined by RT-qPCR. To this end, a non-toxic concentration of GDYO (25 μg mL−1) was added during the polarization step for 24 h. At day 5, the cells were harvested using trypsin-EDTA solution (0.25%) and gently scraped and centrifuged at 1200 rpm at 4 °C for 5 min. Cells were then washed with ice cold PBS and collected by centrifugation. The total RNA was extracted using the RNeasy Mini Kit (QIAGEN, Sweden). RNA concentration was quantified by NanoDrop™ spectrophotometer (ThermoScientific). Total RNA (500 ng) was reverse transcribed using the iScript Advanced cDNA Synthesis Kit (Bio-Rad, Sweden) following the manufacturer's protocol. Transcription levels of the target genes NOS2 (forward primer: AGCTCAACAACAAATTCAGG, reverse primer: ATCAATGTCATGAGCAAAGG) and ARG2 (forward primer: ATTGATGCATTTGACCCTAC, reverse primer: CCTGTATTGTGTATTTCCTCAG), as well as the housekeeping gene GAPDH (forward primer: ACAGTTGCCATGTAGACC, reverse primer: TTGAGCACAGGGTACTTTA) were determined by relative RT-qPCR using a QuantStudio 5 Real-Time PCR System. Reaction mixtures were formulated using SYBR Green PCR Master Mix (ThermoScientific). Thermal cycling conditions were set as 95 °C for 10 min, 40 cycles of 3-step amplification of 15 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. The transcription level of the respective target genes was normalized to human GAPDH and then calculated relative to control using the 2−ΔΔCt method.2.10. Cytokine analysis
To determine the effects of GDYO on the cytokine secretion profiles of M1 and M2 macrophages, M1 and M2 macrophages were incubated with a non-toxic concentration of GDYO (25 μg mL−1) during the polarization step for 24 h. At day 5, the cell supernatants were collected. TNF-α and IL-10 concentrations were quantified by human TNF-α ELISAPRO kit (Mabtech, Sweden) and human IL-10 ELISA kit (Invitrogen), respectively. To study inflammasome activation,26 M1 macrophages were preincubated for 2 h with the pan-caspase inhibitor, zVAD-fmk (20 μM, Sigma Aldrich) or the NLRP3 inhibitor, MCC950 (10 μM, Sigma-Aldrich); additionally, the aryl hydrocarbon receptor (AhR) antagonist, CH223191 (10 μM, Sigma-Aldrich) was used to explore the role of AhR signaling. Then, GDYO (25 μg mL−1) was added and cells were incubated for 24 h. The inhibitors were added again at 24 h and cell supernatants were collected at 48 h for IL-1β analysis, using the human IL-1β ELISA kit (Invitrogen).2.11. GDYO degradation
2.12. Statistical analysis
The experiments were conducted in cells from three individual donors, and statistical analysis was performed using Student's t-test (GraphPad, Prism 8). Results shown are mean values ± S.D. Statistically significant differences were considered when p < 0.05.3. Results
3.1. Physicochemical characterization of endotoxin-free GDYO
GDYO was prepared by oxidation of GDY as described,22 and samples were subjected to characterization by AFM, FTIR, XPS, and Raman spectroscopy (refer to ESI†). Hence, AFM images indicated that the thickness of GDYO is about 1 nm (Fig. S1A and B†), confirming the 2D nature of the material. The lateral size of as-synthesized GDYO was 58 ± 33 nm (Fig. S1C†). FTIR displayed the characteristic peak for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1596 cm−1 and a peak for C
O at 1596 cm−1 and a peak for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C at 2090 cm−1 (Fig. S2A†). The XPS survey scan of GDYO revealed the peaks of C 1s and O 1s (Fig. S2B†). Moreover, four separate binding energy peaks of C 1s were located at 284.6, 285.1, 286.2, and 288.5 eV, corresponding to C–C (sp2), C–C (sp), C–O, and CO, with the molar percentages of 20.45%, 50.34%, 15.86%, and 13.35%, respectively (Fig. S2C and D†). Raman analysis of GDYO revealed the characteristic D band (1360 cm−1), G band (1582 cm−1), and CC band (2100 cm−1) (Fig. S2E†). Endotoxin testing is necessary whenever nanomaterial interactions with immune-competent cells are evaluated.27 We therefore deployed the TNF-α expression test whereby TNF-α secretion is quantified in the presence or absence of the LPS inhibitor, polymyxin B.23 GDYO samples were non-cytotoxic for HMDMs at concentrations up to 100 μg mL−1 as determined by the LDH release and Alamar Blue assays (Fig. 1A and B), and were found to be endotoxin-free (Fig. 1C).
C at 2090 cm−1 (Fig. S2A†). The XPS survey scan of GDYO revealed the peaks of C 1s and O 1s (Fig. S2B†). Moreover, four separate binding energy peaks of C 1s were located at 284.6, 285.1, 286.2, and 288.5 eV, corresponding to C–C (sp2), C–C (sp), C–O, and CO, with the molar percentages of 20.45%, 50.34%, 15.86%, and 13.35%, respectively (Fig. S2C and D†). Raman analysis of GDYO revealed the characteristic D band (1360 cm−1), G band (1582 cm−1), and CC band (2100 cm−1) (Fig. S2E†). Endotoxin testing is necessary whenever nanomaterial interactions with immune-competent cells are evaluated.27 We therefore deployed the TNF-α expression test whereby TNF-α secretion is quantified in the presence or absence of the LPS inhibitor, polymyxin B.23 GDYO samples were non-cytotoxic for HMDMs at concentrations up to 100 μg mL−1 as determined by the LDH release and Alamar Blue assays (Fig. 1A and B), and were found to be endotoxin-free (Fig. 1C).
 | ||
| Fig. 1 Cell viability and cellular uptake of GDYO in macrophage subtypes. (A and B) The viability of human monocyte-derived macrophages (HMDMs) following GDYO exposure for 24 h was evaluated using the LDH release assay (A) and Alamar blue assay (metabolic activity) (B). (C) TNF-α secretion in the presence or absence of the LPS inhibitor, polymyxin B sulfate (Poly-B) (10 μM). HMDMs were exposed to GDYO at 25 and 50 μg mL−1 for 24 h and TNF-α secretion was quantified by ELISA. Student's t-test was used for the statistical analysis. ***p < 0.0001. (D and E) Characterization of M1 and M2 macrophages by surface marker expression of CD86 (D) and CD206 (E) determined by flow cytometry. Data are presented as mean fluorescence intensity (MFI) values ± S.D. from three individual donors. Student's t-test was used for the statistical analysis. *p < 0.05, **p < 0.01. (F) Cellular uptake of GDYO in M1 and M2 macrophages evaluated by TEM analysis. Cells were exposed to a non-cytotoxic dose of GDYO (25 μg mL−1) for 2 h and 24 h post polarization. Arrows point to GDYO agglomerates inside cells. | ||
3.2. Cytotoxicity assessment and macrophage uptake of GDYO
The phenotype of primary human M1 and M2 macrophages was characterized by flow cytometry of cell surface marker expression. M1 macrophages exhibited significantly higher expression of CD86 than M2 macrophages, the costimulatory receptor for T cell activation (Fig. 1D). M2 macrophages expressed a higher level of the mannose receptor CD206 when compared to M1 macrophages, but the difference was not significant (p = 0.1) (Fig. 1E). M-CSF- and GM-CSF-stimulated HMDMs were included for comparison.11 The cytotoxicity potential of GDYO towards M1 and M2 macrophages was then assessed by using the LDH release assay. GDYO was non-cytotoxic or displayed very little cytotoxicity up to 100 μg mL−1 in M1 macrophages (Fig. S3A†) as well as M2 macrophages (Fig. S3B†) when cells were evaluated at 24 h. Next, the cellular uptake of GDYO was investigated at 2 h and at 24 h by TEM. We observed a similar pattern of uptake of GDYO between the M1 and M2 macrophages (Fig. 1F). Moreover, there were no ultrastructural signs of cell death upon GDYO exposure (25 μg mL−1).3.3. iNOS-dependent NO production in human M1 macrophages
To further confirm the phenotypes of M1 and M2 macrophages, we studied the expression of enzymes involved in arginine metabolism and NO production. As shown in Fig. 2A, M1 macrophages expressed iNOS at the protein level, but not M2, as expected.7 Furthermore, M1 macrophages displayed significantly higher expression of NOS2, the gene encoding iNOS, but lower gene expression of ARG2, which encodes arginase, when compared to M2 macrophages (Fig. 2B and C). To validate whether these differences also resulted in differences in NO production, we applied the cell-permeable fluorescent probe, DAF-FM-DA. M1 macrophages generated NO with peak levels at 48 h. The decline of NO production at 72 h might be due to the feedback of NO on iNOS to prevent NO overproduction.28 Importantly, in the presence of L-NAME, the NO production was suppressed (Fig. 2D and F). In comparison, no induction of NO was observed in M2 macrophages (Fig. 2E and G). Collectively, these results confirmed the M1 and M2 status of the two cell models, providing a useful model to assess GDYO effects. | ||
| Fig. 2 iNOS-dependent NO production in M1 macrophages. (A) Western blot analysis of iNOS in M1 and M2 macrophages. GAPDH was used to control for equal loading. (B and C) Gene expression profiles of NOS2 (C) and ARG2 (D) determined by RT-qPCR. The relative mRNA expression levels of NOS2 and ARG2 were normalized to GAPDH. Data are presented as the average ± S.D. from three individual donors. Student's t-test was used to evaluate the statistical significance between M1 and M2 macrophages. *p < 0.05, **p < 0.01. (D and E) Quantification of NO production in M1 and M2 macrophages determined by DAF-FM-DA assay. L-NAME (15 mM) was used to inhibit iNOS. The results are presented as the average ± S.D. from three individual donors. Student's t-test was used for the statistical analysis. *p < 0.05, ***p < 0.001; #p < 0.05, ###p < 0.001. NS = no significant differences. (F and G) Representative fluorescence images of NO production in M1 (F) and M2 (G) macrophages at 48 h with or without L-NAME, represented as the fluorescence determined by DAF-FM-DA assay. Scale bars: 50 μm. | ||
3.4. GDYO polarization of M2 macrophages to M1 macrophages
Next, we asked whether a non-cytotoxic dose of GDYO would affect the polarization of M1 and M2 macrophages. To this end, cells were incubated with GDYO (25 μg mL−1) for 24 h post addition of the polarization stimuli. The activation/polarization status of macrophages was then assessed based on surface marker expression, cytokine secretion and gene expression, as determined by flow cytometry, ELISA and RT-qPCR, respectively. Our results demonstrated that incubation of GDYO polarized M2 macrophages towards M1 macrophages. Specifically, GDYO decreased the expression of CD86 and CD206 in M1 macrophages, but the effects were not significant (Fig. 3A), whereas the exposure to GDYO triggered significantly higher expression of CD86 in M2 macrophages (Fig. 3B). Moreover, GDYO exposure led to a remarkably elevated production of the pro-inflammatory cytokine TNF-α, but not IL-10, in M2 macrophages, while no significant differences in IL-10 or TNF-α production were observed in M1 macrophages (Fig. 3C and D). We also found that GDYO triggered a significant upregulation of NOS2 mRNA expression and a concomitant downregulation of ARG2 in M2 macrophages, while a moderate downregulation of ARG2 and no change in NOS2 expression were noted in M1 macrophages exposed to GDYO (Fig. 3E and F). | ||
| Fig. 3 GDYO polarization of human M2 macrophages to M1 macrophages. (A and B) Surface marker expression presented as mean fluorescence intensity (MFI) of M1 and M2 macrophages post exposure to GDYO (25 μg mL−1) for 24 h. (C and D) Cytokine secretion in M1 and M2 macrophages following the exposure to GDYO (25 μg mL−1) for 24 h, determined by ELISA. (E and F) Gene expression profiles of M1 and M2 macrophages upon exposure to GDYO (25 μg mL−1) for 24 h, determined by RT-qPCR. The relative mRNA expression levels of NOS2 and ARG2 were normalized to GAPDH and calculated relative to M1 and M2 samples, respectively, using the 2−ΔΔCt method. Results are presented as average values ± S.D. using cells from three individual donors. Student's t-test was used for the statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
3.5. Peroxynitrite-driven degradation of GDYO in M1 macrophages
We recently documented iNOS-dependent biodegradation of GO in a zebrafish model.21 Biodegradation of GDYO has not been shown previously. However, in light of the fact that M1, but not M2 macrophages, expressed iNOS and were capable of producing NO, we reasoned that M1 and M2 macrophages might differ in their capacity to digest GDYO. To test the hypothesis, M1 and M2 macrophages were incubated with a non-cytotoxic concentration of GDYO (25 μg mL−1) and biodegradation was assessed at different time-points by Raman mapping and AFM analysis. As shown in Fig. 4A, degradation of GDYO was evident at 48 h in M1 macrophages as shown by the marked decline of the characteristic Raman signal (D, G, and CC bands) and there was an almost complete loss of the Raman signature at 72 h. On the other hand, no changes were detected when M2 macrophages were incubated with GDYO up to 72 h (Fig. 4B). The statistical analysis of 50 representative spectra per sample is reported in Table S1.† The intensities of the D and G peaks detected in M1 macrophages decreased to 21.2 ± 2.4% (D peak) and 17.8 ± 2.1% (G peak) at 48 h compared to 24 h, and declined further to 6.4 ± 1.2% (D peak) and 5.8 ± 1.1% (G peak) at 72 h. In comparison, no significant difference was observed for the GDYO sheets present in M2 macrophages (Table S1†). The degradation of GDYO in M1 macrophages was confirmed by AFM analysis (Fig. 4C). AFM demonstrated that the lateral size of the GDYO sheets as well as the thickness decreased in a time-dependent manner (Fig. 4C). In contrast, in M2 macrophages, no differences were observed by AFM in terms of the thickness or the lateral dimensions of GDYO (for a survey of the entire scanning area, refer to Fig. 4C, and see Fig. S4† for further details). The statistical analysis of the lateral size and thickness of 50 GDYO sheets per sample as determined by AFM is reported in Table S2.† Thus, in M1 macrophages, the lateral size of GDYO decreased from 44.0 ± 14.9 nm (24 h) to 10.7 ± 5.1 nm (72 h), and the thickness decreased from 12.88 ± 5.93 nm (24 h) to 2.74 ± 1.66 nm (72 h). In comparison, no significant difference in lateral size or thickness of the GDYO sheets was observed in the case of M2 macrophages (Table S2†).
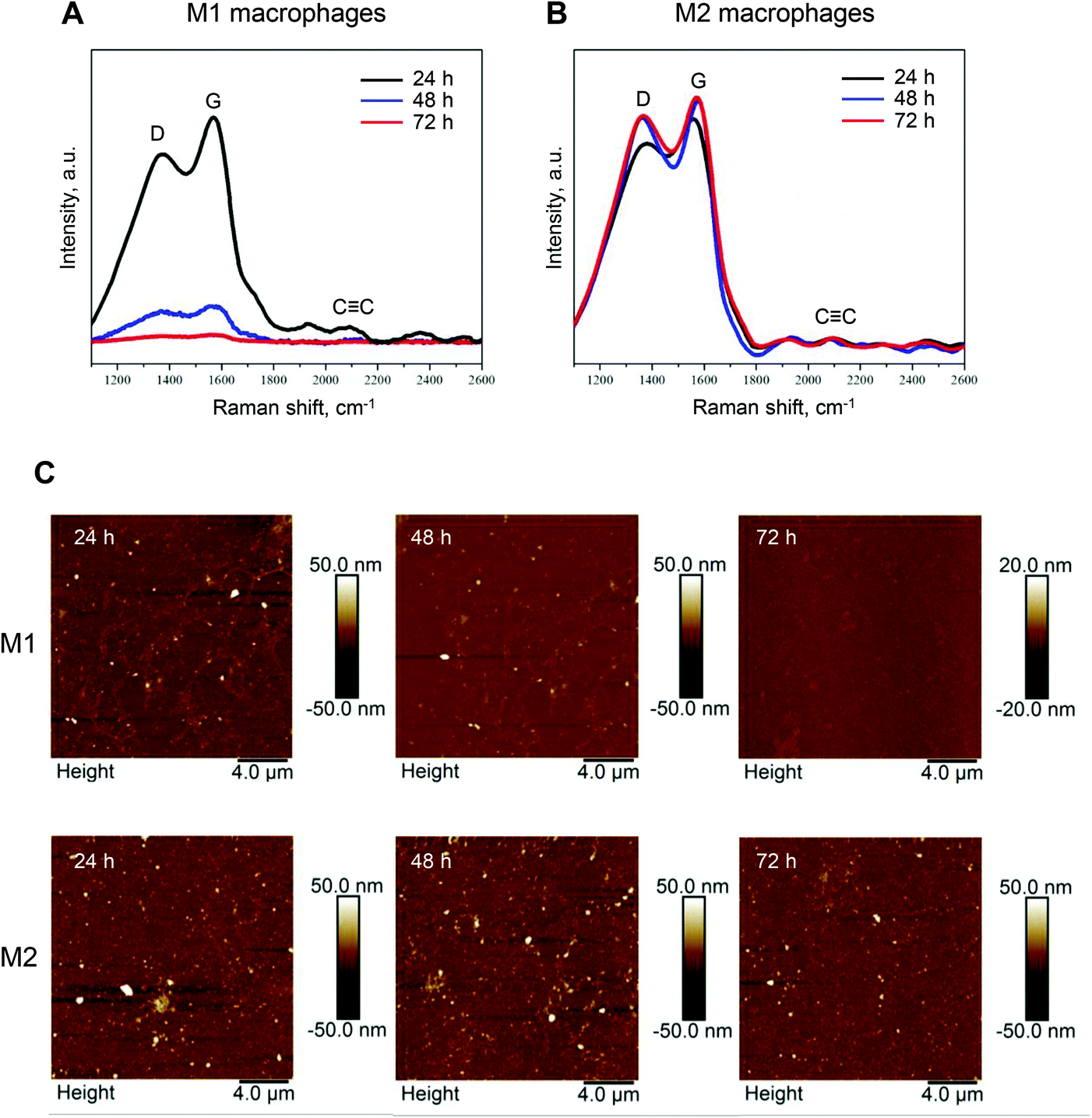 | ||
| Fig. 4 Biodegradation of GDYO in primary human M1 macrophages. (A and B) Raman mapping analysis of GDYO in M1 and M2 macrophages. Macrophages were incubated with a non-cytotoxic dose of GDYO (25 μg mL−1) during the polarization step for 24 h, 48 h and 72 h, as indicated. Each spectrum represents the average of 10.000 spectra across the sample. (C) Representative AFM images of M1 (upper row) and M2 macrophages exposed to GDYO for 24 h, 48 h, and 72 h. Refer to Fig. S4 and Tables S1, S2.† | ||
To identify the mechanism involved in the biodegradation of GDYO in M1 macrophages, we employed the pharmacological inhibitors, L-NAME and DPI, to block iNOS and NADPH oxidase, respectively. MPO, in turn, is predominantly expressed in neutrophils, but previous work has shown that GM-CSF regulates macrophage expression of MPO.29 Therefore, and in light of the fact that GO is known to undergo MPO-dependent degradation,17 MPO inhibitor I was also included to study its impact on the degradation of GDYO. Blocking iNOS and NADPH oxidase significantly suppressed the degradation of GDYO (Fig. 5A). In contrast, MPO inhibitor I showed no effect on biodegradation of GDYO. We further confirmed the peroxynitrite-driven degradation of GDYO in an acellular system using the peroxynitrite donor, SIN-1, according to a previously established protocol.21 After 5 days of incubation with SIN-1, the degradation of GDYO was proven by Raman analysis showing the loss of the characteristic D, G, and CC bands (Fig. 5B). These results clearly demonstrated that biodegradation of GDYO occurred through the NO/superoxide-peroxynitrite-driven pathway (Fig. 5C). AFM analysis confirmed that the lateral dimensions of the GDYO sheets were reduced from 46 ± 21.5 nm to 25.7 ± 16.6 nm in the presence of SIN-1, while no obvious differences were observed in terms of the thickness (Fig. 5D, E and Table S3†).
 | ||
| Fig. 5 Peroxynitrite-driven degradation of GDYO. (A) Raman analysis of GDYO (25 μg mL−1) in M1 macrophages after 48 h incubation in the presence or absence of the various inhibitors. L-NAME (15 mM), DPI (10 μM) and MPO inhibitor I (600 nM) were used to block iNOS, NADPH oxidase, and MPO, respectively. Each spectrum represents the average of 10.000 spectra. (B) Raman analysis of GDYO following incubation with the peroxynitrite donor, SIN-1 for 5 days (final concentration: 300 μM) showed degradation of GDYO as evidenced by loss of the characteristic D, G, and CC bands. (C) Schematic diagram showing the generation of NO by iNOS and superoxide radicals by NADPH oxidase, followed by the formation of the potent oxidant, peroxynitrite. The specific inhibitors, DPI and L-NAME are also shown. (D and E) Representative AFM images of GDYO with or without SIN-1 treatment for 5 days as above. Refer to Table S3.† | ||
3.6. GDYO modulation of cytokine responses in M1 macrophages
The consequences of biodegradation of nanomaterials are frequently overlooked.18 Therefore, we investigated the potential correlation between the biodegradation of GDYO and its modulation of cytokine responses in M1 macrophages. Remarkably, when the biodegradation of GDYO was blocked using L-NAME or DPI, IL-10 secretion was completely suppressed (Fig. 6A). The addition of MPO inhibitor I had no effect. Furthermore, a similar response was noted for TNF-α, albeit much less pronounced (Fig. 6B). | ||
| Fig. 6 GDYO modulation of cytokine responses in M1 macrophages. (A–C) M1 macrophage secretion of IL-10 (A), TNF-α (B), and IL-1β (C) upon GDYO exposure (25 μg mL−1, 48 h) in the presence or absence of the indicated inhibitors. L-NAME (15 mM), DPI (10 μM), and MPO inhibitor I (600 nM) were used to block iNOS, NADPH oxidase, and MPO, respectively. (D) IL-1β secretion in M1 macrophages upon GDYO exposure (25 μg mL−1, 48 h) in the presence or absence of zVAD-fmk (20 μM), MCC950 (10 μM), and CH223191 (10 μM) to block caspases, NLRP3, and AhR, respectively. Data shown are mean values ± S.D. using cells from three individual human donors. Student's t-test was used for the statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001. NS = no significant differences. (E) Schematic diagram summarizing the observed findings with respect to IL-1β production in M1 macrophages exposed to GDYO. The specific NLRP3 inhibitor, MCC950 and the pan-caspase inhibitor, zVAD-fmk are indicated. | ||
We then asked whether GDYO affected other pro-inflammatory cytokines such as IL-1β. Indeed, significant IL-1β secretion was triggered in M1 macrophages exposed to GDYO (Fig. 6C). This response was also degradation-dependent insofar as L-NAME and DPI abolished the GDYO-triggered IL-1β response (Fig. 6C). MPO inhibitor I partially decreased the IL-1β secretion, indicating a role of MPO in IL-1β production. To address whether IL-1β secretion was inflammasome-dependent,30 we incubated the cells with the pan-caspase inhibitor, zVAD-fmk, and the selective NLRP3 inhibitor, MCC950. As shown in Fig. 6D, IL-1β secretion was entirely prevented by these inhibitors, indicating that GDYO triggered NLRP3 inflammasome activation. Finally, to address whether the degradation products of GDYO could act via the aryl hydrocarbon receptor (AhR), a xenobiotic sensor,31 we also tested the effect of the AhR antagonist, CH223191. However, this did not affect IL-1β secretion in the current model (Fig. 6D). The schematic diagram in Fig. 6E summarizes our findings with respect to IL-1β secretion.
4. Discussion
GDYO, a novel carbon allotrope, contains both sp2 and sp-hybridized carbon atoms, which makes this a unique and appealing material for various applications,1,2 but this also necessitates more fundamental knowledge regarding its impact on biological systems. In the present study, we investigated the biocompatibility and biodegradability of GDYO using primary human macrophages as a model. We demonstrated that endotoxin-free GDYO is non-cytotoxic towards macrophages and that this material is able to repolarize M2 macrophages towards M1 macrophages; we also showed that peroxynitrite-driven degradation of GDYO occurs both in cellular and acellular systems. M1 macrophages are tasked with the clearance of microorganisms, and it may be speculated that a similar pro-oxidant response is activated in M1 macrophages that have internalized GDYO, leading to the degradation of the “offending” material. Furthermore, we demonstrated here that GDYO modulates cytokine responses in M1 macrophages in a biodegradation-dependent manner. In other words, biodegradation of GDYO was shown to drive the production of pro-inflammatory IL-1β, and when biodegradation was suppressed by blocking the NADPH oxidase or iNOS with pharmacological inhibitors, IL-1β production was abolished, along with IL-10 production. Whether the effects on cytokine production are mediated directly or indirectly by the degradation products of GDYO remains to be understood. Previous work has suggested that the AhR regulates NLRP3 inflammasome activation in peritoneal macrophages.32 This is relevant as previous studies have shown that the (partial) biodegradation of CNTs and GO may lead to the formation of oxidized polycyclic aromatic hydrocarbons (PAHs).33,34 PAHs, in turn, can trigger the AhR, an important sensor of xenobiotics as well as endogenous and dietary metabolites.31 However, the present findings implied that the impact of GDYO and/or its degradation products was AhR-independent, at least in our model. Notwithstanding, our study has provided evidence of reciprocal interactions between GDYO and primary macrophages.Macrophage degradation of carbon-based nanomaterials such as CNTs has been demonstrated by several laboratories.35 However, in most cases, macrophage-like THP-1 cells were used as a model.19,20 In contrast, we used primary human macrophages, and this has allowed us to demonstrate the fundamental differences between M1-activated and M2-activated macrophages in terms of their ability to “digest” GDYO. Moreover, few studies have been conducted to understand the relationship between biodegradation and its immunomodulatory effects. The present findings are therefore of significant value and further studies are warranted to assess whether the biodegradation (or the lack thereof) may be responsible for the immunological effects of other carbon-based nanomaterials. Biopersistence of a nano(bio)material is generally perceived as something problematic in the context of biomedical applications, but the degradation products also deserve close attention. We recently reported on the peroxynitrite-dependent degradation of GO using a similar acellular protocol,21 and it appears, based on the present study, that SIN-1 driven degradation of GDYO is less efficient as compared to GO. However, we found that the degradation of GDYO in M1 macrophages was highly effective, as shown by Raman analysis. The differences could perhaps be attributed to the different composition of GDYO (sp2 and sp-hybridized carbon atoms) and GO (sp2-hybridized carbon atoms),36 in that sp hybridization, in general, results in a stronger chemical bond than sp2 hybridization.37 Other factors may also come into play. Star and co-workers explored both pristine and carboxylated CNTs with respect to their degradation by horseradish peroxidase (HRP) and found that only the carboxylated CNTs were susceptible to degradation, possibly due to the fact that strong adsorption of HRP to carboxylated sites facilitated their degradation, while the hydrophobic nature of pristine CNTs affected the orientation of HRP, thus impeding the enzyme's ability to digest the CNTs.33 The same team reported that GO is more susceptible to HRP-driven degradation than reduced GO.38 However, as previously pointed out by Kagan et al.,19 peroxynitrite-driven degradation does not require close contact with enzyme(s), and it is likely that this mode of biodegradation is relatively insensitive to the initial oxidation state of the nano(bio)material. Nevertheless, the observation that GDYO is susceptible to peroxynitrite-driven biodegradation in M1 but not in M2 macrophages is useful in terms of guiding the application of this material in the biomedical setting. Indeed, it may also be pertinent to study whether macrophage polarization influences the degradation of other nanomaterials.
TAMs are relevant targets for anticancer therapy mediated, for instance, through the re-polarization away from tumor-promoting M2-like towards tumoricidal M1-like states.39 Several studies have shown that nanomaterials can be deployed to re-educate macrophages in the context of inflammatory or malignant diseases. Wang et al.40 prepared microenvironment-responsive polymeric nanoparticles with encapsulated IL-12 and showed that these nanocarriers could release IL-12 locally and subsequently re-educate TAMs in a mouse model of melanoma with little systemic toxicity. Han et al.41 showed that the administration of GO loaded with IL-4 plasmid DNA in a mouse model of myocardial infarction elicited macrophage polarization towards an M2-like state, mitigated fibrosis, and improved cardiac function. IL-4 is known to propagate M2 polarization. In the present study, GDYO alone (without any payload) enabled the repolarization of M2 towards M1 macrophages, as evidenced by the elevated markers of M1 activation including CD86 expression, TNF-α secretion and NOS2 gene expression. Importantly, we first confirmed that the GDYO samples were endotoxin-free. As discussed earlier, we also observed biodegradation-dependent modulation of IL-10 in M1 macrophages. IL-10 is an anti-inflammatory cytokine42 and an important player in controlling the immunosuppressive tumor microenvironment.43 The blockade of IL-10 was recently shown to enhance antitumor immunity in a mouse model of breast cancer.44 Further studies are warranted to explore whether GDYO could be harnessed for cancer treatment, potentially through the reprogramming of TAMs.
5. Conclusions
In summary, we have shown that GDYO is biocompatible towards primary human macrophages and polarizes M2 macrophages towards M1 macrophages. Moreover, classically activated M1 macrophages were shown to digest GDYO through a peroxynitrite-dependent pathway whereas no biodegradation was observed in M2 macrophages. This is the first demonstration of biodegradation of GDYO by immune-competent cells. We also provided first evidence of degradation-dependent modulation of cytokine responses in macrophages, with engagement of the NLRP3 inflammasome, a critical sensor of microbes and other danger signals.30 The biocompatibility and biodegradability of GDYO coupled with the ability to polarize or re-educate M2 macrophages into M1 macrophages are highly desirable properties in cancer therapy.45 Additionally, macrophage polarization also plays an important role in non-cancerous conditions (discussed above). Taken together, the present findings are relevant for biomedical applications of GDYO, a novel 2D material with considerable potential.2Author contributions
G.P. performed experiments, analyzed data, and drafted the paper; T.D. performed experiments, supervised by K.L.; M.G. characterized the test material, supervised by C.C.; Y.X. synthesized the test material, supervised by Y.L.; B.F. coordinated the study, analyzed data, and edited the paper; all co-authors approved the final version of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Commission through the Graphene Flagship (grant agreement no. 696656), and the Swedish Research Council (2016-05259).References
- H. Yu, Y. Xue and Y. Li, Adv. Mater., 2019, 31, 1803101 CrossRef CAS PubMed.
- J. Liu, C. Chen and Y. Zhao, Adv. Mater., 2019, 31, 1804386 CrossRef CAS PubMed.
- Y. Zhang, W. Liu, Y. Li, Y. W. Yang, A. Dong and Y. Li, iScience, 2019, 19, 662–675 CrossRef CAS PubMed.
- T. Zheng, Y. Gao, X. Deng, H. Liu, J. Liu, R. Liu, J. Shao, Y. Li and L. Jia, ACS Appl. Mater. Interfaces, 2018, 10, 32946–32954 CrossRef CAS PubMed.
- B. Fadeel, C. Bussy, S. Merino, E. Vázquez, E. Flahaut, F. Mouchet, L. Evariste, L. Gauthier, A. J. Koivisto, U. Vogel, C. Martín, L. G. Delogu, T. Buerki-Thurnherr, P. Wick, D. Beloin-Saint-Pierre, R. Hischier, M. Pelin, F. Candotto Carniel, M. Tretiach, F. Cesca, F. Benfenati, D. Scaini, L. Ballerini, K. Kostarelos, M. Prato and A. Bianco, ACS Nano, 2018, 12, 10582–10620 CrossRef CAS PubMed.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS PubMed.
- F. Ginhoux, J. L. Schultze, P. J. Murray, J. Ochando and S. K. Biswas, Nat. Immunol., 2016, 17, 34–40 CrossRef CAS PubMed.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS PubMed.
- D. I. Gabrilovich, S. Ostrand-Rosenberg and V. Bronte, Nat. Rev. Immunol., 2012, 12, 253–268 CrossRef CAS PubMed.
- A. K. Fuchs, T. Syrovets, K. A. Haas, C. Loos, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, Biomaterials, 2016, 85, 78–87 CrossRef CAS PubMed.
- A. Gallud, O. Bondarenko, N. Feliu, N. Kupferschmidt, R. Atluri, A. Garcia-Bennett and B. Fadeel, Biomaterials, 2017, 121, 28–40 CrossRef CAS PubMed.
- V. Kodali, M. H. Littke, S. C. Tilton, J. G. Teeguarden, L. Shi, C. W. Frevert, W. Wang, J. G. Pounds and B. D. Thrall, ACS Nano, 2013, 7, 6997–7010 CrossRef CAS PubMed.
- Y. Yang, L. Guo, Z. Wang, P. Liu, X. Liu, J. Ding and W. Zhou, Biomaterials, 2021, 264, 120390 CrossRef CAS PubMed.
- V. E. Kagan, N. V. Konduru, W. Feng, B. L. Allen, J. Conroy, Y. Volkov, I. I. Vlasova, N. A. Belikova, N. Yanamala, A. Kapralov, Y. Y. Tyurina, J. Shi, E. R. Kisin, A. R. Murray, J. Franks, D. Stolz, P. Gou, J. Klein-Seetharaman, B. Fadeel, A. Star and A. A. Shvedova, Nat. Nanotechnol., 2010, 5, 354–359 CrossRef CAS PubMed.
- F. T. Andón, A. A. Kapralov, N. Yanamala, W. Feng, A. Baygan, B. J. Chambers, K. Hultenby, F. Ye, M. S. Toprak, B. D. Brandner, A. Fornara, J. Klein-Seetharaman, G. P. Kotchey, A. Star, A. A. Shvedova, B. Fadeel and V. E. Kagan, Small, 2013, 9, 2721–2729 CrossRef PubMed.
- K. Bhattacharya, R. El-Sayed, F. T. Andón, S. P. Mukherjee, J. Gregory, H. Li, Y. Zhao, W. Seo, A. Fornara, B. Brandner, M. S. Toprak, K. Leifer, A. Star and B. Fadeel, Carbon, 2015, 91, 506–517 CrossRef CAS.
- R. Kurapati, J. Russier, M. A. Squillaci, E. Treossi, C. Ménard-Moyon, A. E. Del Rio-Castillo, E. Vazquez, P. Samorì, V. Palermo and A. Bianco, Small, 2015, 11, 3985–3994 CrossRef CAS PubMed.
- S. P. Mukherjee, A. R. Gliga, B. Lazzaretto, B. Brandner, M. Fielden, C. Vogt, L. Newman, A. F. Rodrigues, W. Shao, P. M. Fournier, M. S. Toprak, A. Star, K. Kostarelos, K. Bhattacharya and B. Fadeel, Nanoscale, 2018, 10, 1180–1188 RSC.
- V. E. Kagan, A. A. Kapralov, C. M. St. Croix, S. C. Watkins, E. R. Kisin, G. P. Kotchey, K. Balasubramanian, I. I. Vlasova, J. Yu, K. Kim, W. Seo, R. K. Mallampalli, A. Star and A. A. Shvedova, ACS Nano, 2014, 8, 5610–5621 CrossRef CAS PubMed.
- D. Elgrabli, W. Dachraoui, C. Ménard-Moyon, X. J. Liu, D. Bégin, S. Bégin-Colin, A. Bianco, F. Gazeau and D. Alloyeau, ACS Nano, 2015, 9, 10113–10124 CrossRef CAS PubMed.
- G. Peng, M. F. Montenegro, C. N. M. Ntola, S. Vranic, K. Kostarelos, C. Vogt, M. S. Toprak, T. Duan, K. Leifer, L. Bräutigam, J. O. Lundberg and B. Fadeel, Nanoscale, 2020, 12, 16730–16737 RSC.
- J. Xie, C. Wang, N. Wang, S. Zhu, L. Mei, X. Zhang, Y. Yong, L. Li, C. Chen, C. Huang, Z. Gu, Y. Li and Y. Zhao, Biomaterials, 2020, 244, 119940 CrossRef CAS PubMed.
- S. P. Mukherjee, N. Lozano, M. Kucki, A. E. Del Rio-Castillo, L. Newman, E. Vazquez, K. Kostarelos, P. Wick and B. Fadeel, PLoS One, 2016, 11, e0166816 CrossRef PubMed.
- N. Feliu, M. V. Walter, M. I. Montañez, A. Kunzmann, A. Hult, A. Nyström, M. Malkoch and B. Fadeel, Biomaterials, 2012, 33, 1970–1981 CrossRef CAS PubMed.
- K. Bhattacharya, C. Sacchetti, P. M. Costa, J. Sommertune, B. D. Brandner, A. Magrini, N. Rosato, N. Bottini, M. Bottini and B. Fadeel, Adv. Healthcare Mater., 2018, 7, 1700916 CrossRef PubMed.
- S. P. Mukherjee, K. Kostarelos and B. Fadeel, Adv. Healthcare Mater., 2018, 7, 1700815 CrossRef PubMed.
- M. Himly, M. Geppert, S. Hofer, N. Hofstätter, J. Horejs-Höck and A. Duschl, Small, 2020, 16, 1907483 CrossRef CAS PubMed.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed.
- S. Sugiyama, Y. Okada, G. K. Sukhova, R. Virmani, J. W. Heinecke and P. Libby, Am. J. Pathol., 2001, 158, 879–891 CrossRef CAS.
- P. Broz and V. M. Dixit, Nat. Rev. Immunol., 2016, 16, 407–420 CrossRef CAS PubMed.
- V. Rothhammer and F. J. Quintana, Nat. Rev. Immunol., 2019, 19, 184–197 CrossRef CAS PubMed.
- W. Huai, R. Zhao, H. Song, J. Zhao, L. Zhang, L. Zhang, C. Gao, L. Han and W. Zhao, Nat. Commun., 2014, 5, 4738 CrossRef CAS PubMed.
- B. L. Allen, G. P. Kotchey, Y. Chen, N. V. K. Yanamala, J. Klein-Seetharaman, V. E. Kagan and A. Star, J. Am. Chem. Soc., 2009, 131, 17194–17205 CrossRef CAS PubMed.
- H. Bai, W. Jiang, G. P. Kotchey, W. A. Saidi, B. J. Bythell, J. M. Jarvis, A. G. Marshall, R. A. S. Robinson and A. Star, J. Phys. Chem. C, 2014, 118, 10519–10529 CrossRef CAS PubMed.
- K. Bhattacharya, S. P. Mukherjee, A. Gallud, S. C. Burkert, S. Bistarelli, S. Bellucci, M. Bottini, A. Star and B. Fadeel, Nanomedicine, 2016, 12, 333–351 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- J. E. Bloor and S. Gartside, Nature, 1959, 184, 1313 CrossRef CAS.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- L. Cassetta and J. W. Pollard, Nat. Rev. Drug Discovery, 2018, 17, 887–904 CrossRef CAS PubMed.
- Y. Wang, Y. X. Lin, S. L. Qiao, H. W. An, Y. Ma, Z. Y. Qiao, R. P. Rajapaksha and H. Wang, Biomaterials, 2017, 112, 153–163 CrossRef CAS PubMed.
- J. Han, Y. S. Kim, M. Y. Lim, H. Y. Kim, S. Kong, M. Kang, Y. W. Choo, J. H. Jun, S. Ryu, H. Y. Jeong, J. Park, G. J. Jeong, J. C. Lee, G. H. Eom, Y. Ahn and B. S. Kim, ACS Nano, 2018, 12, 1959–1977 CrossRef CAS PubMed.
- M. Saraiva and A. O'Garra, Nat. Rev. Immunol., 2010, 10, 170–181 CrossRef CAS PubMed.
- V. S. Madamsetty, K. Pal, S. Keshavan, T. R. Caulfield, S. K. Dutta, E. Wang, B. Fadeel and D. Mukhopadhyay, Nanoscale, 2019, 11, 22006–22018 RSC.
- L. Shen, J. Li, Q. Liu, W. Song, X. Zhang, K. Tiruthani, H. Hu, M. Das, T. J. Goodwin, R. Liu and L. Huang, ACS Nano, 2018, 12, 9830–9841 CrossRef CAS PubMed.
- M. Ovais, M. Guo and C. Chen, Adv. Mater., 2019, 31, 1808303 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr02473f |
| This journal is © The Royal Society of Chemistry 2021 |