
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the drug loading mechanism of photoactive inorganic nanocarriers through molecular dynamics simulations†
Stefano
Motta
 a,
Paulo
Siani
b,
Andrea
Levy
b and
Cristiana
Di Valentin
*b
a,
Paulo
Siani
b,
Andrea
Levy
b and
Cristiana
Di Valentin
*b
aDipartimento di Scienze dell'Ambiente e del Territorio, Università di Milano Bicocca, Piazza della Scienza 1, 20126 Milano, Italy
bDipartimento di Scienza dei Materiali, Università di Milano Bicocca, via R. Cozzi 55, 2015 Milano, Italy. E-mail: cristiana.divalentin@unimib.it
First published on 20th July 2021
Abstract
Inorganic nanoparticles are gaining increasing attention as drug carriers because they respond to external physical stimuli, allowing therapy to be combined with diagnosis. Their drawback is low drug loading capacity, which can be improved by proper and efficacious functionalization. In this computational study, we take TiO2 spherical nanoparticles as prototype photoresponsive inorganic nanoparticles and we fully decorate them with two different types of bifunctional ligands: TETTs and DOPACs, which present different surface anchoring groups (silanol or catechol) but the same drug tethering COOH group, although in different concentrations (3 vs. 1), thus causing different steric hindrances. Then, we put these two types of nanocarriers in bulk water and in the presence of several DOX molecules and let the systems evolve through molecular dynamics (MD) simulations, clearly observing drug loading on the nanocarriers. This comparative MD study allows the investigation of the loading mechanism, performance of a conformational analysis and establishment of the guiding interactions through an energy decomposition analysis. We learn that DOX mostly interacts with the functionalized NPs through electrostatics, as a consequence of the protonated amino group, although several H-bonds are also established both with the ligands and with the oxide surface. Different ligands induce a different electrostatic potential around the NP; therefore, those which lead to the formation of more negative hotspots (here TETTs) are found to favour DOX binding. The leading role of electrostatics can provide a rational explanation for a pH-dependent drug release mechanism that is often invoked for DOX when reaching diseased cells because under anomalous acidic conditions both the NP surface and the carboxylate groups of the ligands are expected to get protonated, which of course would weaken, if not totally quench, the interaction of the nanocarrier with protonated DOX.
1. Introduction
Nanotechnology is currently playing a significant and quite successful role in advanced drug formulations, controlled drug release and targeted drug delivery, employing most frequently nanoparticles (NPs) of a small size and spherical shape,1,2 because they may remain in the blood circulatory system for a prolonged period, and thus penetrate the tumour blood vessel and access the tissue system, facilitating drug uptake by cells and ensuring that the drug action takes place at the targeted location.3 Their uptake by cells from the blood compartment is found to be much higher than that of larger particles of sizes 1 and 10 μm,4 which improves the efficiency and minimizes the side effects. Therefore, nanosystems, when capable of loading the critical quantity of drugs for their pharmaceutical effect on diseased cells, are excellent drug delivery vehicles.The very first generation of nanoparticle-based drug therapy involved lipid systems, such as liposomes and micelles, because they are able to encapsulate both hydrophilic compounds in the aqueous cavity and hydrophobic compounds with affinity to the phospholipid bilayer.5,6 These drug delivery systems are now approved by the U.S. Food and Drug Administration (FDA).5 Other successful organic drug carriers are based on chitosan, polymeric micelles, dendrimers, cellulose, alginate, etc.3
More recently, inorganic nanoparticles have been attracting increasing attention because they allow easier surface modification and controlled growth.3 Moreover, they are characterized by promising intrinsic properties, not present in organic nanoparticles. Most common inorganic drug delivery nanosystems include silver, gold, iron oxide, and silica nanoparticles or quantum dots (QDs).1,7 Metal nanoparticles of silver and gold have peculiar properties such as SPR (surface plasmon resonance) and present good biocompatibility, controllable morphology, size dispersion and easy surface functionalization.8–10 Iron oxide nanoparticles are the most studied magnetic nanosystems because of their biocompatibility, possibility to be guided to the desired positions by magnetic fields, and heat release under an alternate magnetic field (magnetic hyperthermia technique).11 QDs are known as semiconductor nanocrystals with diameters ranging from 2 to 10 nm presenting size-dependent optical properties, such as absorbance and photoluminescence.12,13
This ability of inorganic NPs to respond to external stimuli facilitates imaging and therapy and makes them an advanced class of multifunctional drug delivery systems (i.e. theranostic systems).7,14 They are designed for simultaneous diagnosis, tumor targeting, stimuli-controlled drug release and photodynamic therapy of cancer. For these reasons, they are currently among the most promising tools for nanomedicine. Despite this, due to the relative youth of this research field, little is known about the real mechanisms of action of these complex drug delivery systems, which opens opportunities for new studies.
The therapeutic efficiency of drug delivery systems is strongly affected by the drug loading mode. Non-covalent complexation (H-bonding, dispersion forces, and electrostatic interactions) is preferred because it does not only keep the drug in its pristine and therapeutic active state, but it is also a reversible type of conjugation.15,16 In this respect, low drug loadings constitute a critical issue for inorganic nanocarriers, because they are required to deliver the accurate amount of drugs to the diseased cells for the therapy to be effective. Hence, an atomistic understanding of the drug loading mechanism is a fundamental goal for the research community to design optimal and successful drug delivery vehicles based on inorganic nanomaterials. Among them, titanium dioxide (TiO2) nanoparticles stand out not only because they are cheap and easy-to-obtain, but especially because of their intrinsic ability to act as highly efficient photosensitizers and because of their easy functionalization with anchoring ligands for drug loading and improved biocompatibility.17 This is particularly true in the case of highly curved nanostructures <20 nm (spherical nanoparticles, nanorods, nanotubes, etc.).17 Therefore, TiO2 nanoparticles are excellent multifunctional systems that successfully combine pharmaceutical and photodynamic therapies. Examples of the use of TiO2 NPs in drug delivery include the work by Wang et al., who realized polyethylenimine (PEI)-modified TiO2 NPs with covalently bonded folate (FA) molecules for the delivery of the anticancer drug paclitaxel.18 PEI coating affects drug release, allowing it only upon UV irradiation, while FA ensures cancer-targeting since folate receptors are overexpressed in cancer cells. This is an efficient light-stimulated and site-specific drug carrier that allows control of the therapeutic effects upon manipulation of the irradiation time. Another example is the work by Wu et al., who synthesized highly biocompatible mesoporous TiO2 NPs functionalized with flavin mononucleotides, employed for the delivery of the anticancer drug doxorubicin (DOX).19 This drug-delivery system can be simultaneously used for intercellular imaging since FMN is a fluorescent probe.
DOX was also successfully loaded by Qin et al.20 on TiO2 spherical nanoparticles functionalized with TETT (N-(trimethoxysilylpropyl)ethylenediamine triacetic acid trisodium salt) ligands. This type of ligand can firmly anchor the metal oxide NP surface, induce its water dispersibility and allow for an easy and effective non-covalent drug loading. The DOX biological and pharmaceutical activity was found to be not affected. DOX is a conventional chemotherapy drug of the anthracycline family that intercalates in the DNA double helix and thereby prevents the DNA replication and cell division process.21
The aim of the present molecular dynamics study based on molecular mechanics (MM) force fields is to unravel the drug loading mechanism of DOX on functionalized TiO2 NPs, as a case study for the more general class of drug delivery systems based on inorganic nanoparticles. On the basis that TETT ligands exposed three deprotonated carboxylate groups (–COO−) each and DOX molecules exposed protonated amino groups (–NH3+), the interaction mechanism is expected to be mostly electrostatic. However, this has not been verified yet and other non-covalent interactions could be relevant in the interaction. Moreover, it is not yet known whether there is some contact between the drug and the NP surface. In order to evaluate the role played by the number of (–COO−) groups and by the steric encumbrance of the large TETT ligand, we performed a parallel study on a smaller ligand that can stably anchor the TiO2 surface and exposes only one (–COO−) group, which is the DOPAC (3,4-dihydroxyphenylacetic acid) ligand. Through this comparison, we want to unravel which are the crucial aspects of drug binding and how it is possible to control them by tailoring the optimal ligand. From this comparative study, we wish to prove that the drug loading estimated from experimental data is confirmed by the theoretical model. In other words, we want to establish whether the experimentally estimated number of loaded drug molecules can truly remain stably attached to the functionalized nanoparticles and to understand the mechanism of competition between solvated drug/drug or drug/nanocarrier interactions.
This work benefits from our previous studies, where we have developed chemically stable spherical TiO2 NP models with high-level quantum mechanical (QM) methods,22 have explored their water solvation with the QM/MM approach23 and have also assessed the validity and accuracy of the chosen MM force field parameters used in the present study for the description of bioinorganic nanohybrids against QM and QM/MM calculations.24–26 Moreover, for both TETT and DOPAC ligands, we have already obtained stable optimized structures of fully decorated TiO2 NPs by QM calculations,24,25 which have been an excellent starting point for the MD simulations.
Through this comparative molecular dynamics study, exploiting the simulated annealing procedure to achieve a more efficient sampling and exploration of the configurational space, we aim at obtaining a detailed atomistic picture of the drug loading mechanism by functionalized metal oxide nanoparticles, which will provide the basis and the criteria for the rational design of an optimal carrier system, where NP/ligand, ligand/ligand, NP/drug, ligand/drug and drug/drug interactions are properly balanced to ensure that the biochemical and structural properties of the drug are maintained during transport, the drug quantity is the therapeutic active dose and, finally, the drug binding provides sufficient stability without hampering the release kinetics.
The field of computational nanomedicine can still be considered to be in its infancy, stimulating the improvement of theoretical models towards more realistic conditions of dimensions and the surrounding environment for the research on nanomaterials with well-defined sizes, shapes and drug loading capacity, which are pivotal in the advancement of targeted drug delivery.
2. Computational details
2.1 Starting point geometry
The starting geometry of the TETT and DOPAC functionalized anatase TiO2 NP is the result of previous works,27,28 as it will be described in the following sections. TiO2 spherical nanoparticles were carved from large bulk anatase supercells, setting the radius of the sphere to the desired value of 2.2 nm. Only atoms within that sphere were considered. Some residuals with very low coordinated Ti atoms or monocoordinated O atoms were either removed or saturated with OH groups or H atoms, respectively. In other words, we used a small number of dissociated water molecules to achieve the chemical stability of the nanoparticle. The stoichiometry of the model is (TiO2)223·10H2O. After a simulated annealing process at 700 K, we have fully relaxed the equilibrated structure with both DFTB and DFT(B3LYP). Then, the DFTB-optimized NP was fully decorated either with TETT or with DOPAC molecules and then fully optimized in ref. 24 and 25 to obtain high coverage models, which have been used as the starting geometry in this work.2.2 Molecular dynamics simulations
Several studies using classical potentials to simulate TiO2 surfaces and nanoparticles are present in the literature.29–37 Here, the system was prepared using an in-house script that allows assigning suitable force-field parameters to the different species present in the system. For the TiO2 NP atoms, we used parameters developed by Brandt and co-workers38 according to the titanium and oxygen coordination sphere, improving the original ones by Matsui and Akaogi.39 TETT, DOPAC and DOX molecules were parametrized using the GAFF2 force-field with atomic partial charges obtained following the standard restrained electrostatic potential fitting protocol40 (RESP) on the single molecules using the basis set 6-31G* at the Hartree–Fock (HF) level of theory. The system was then immersed in a large cubic box (100.2 Å side for TETT and 89 Å side for DOPAC) of TIP3P41 water molecules (for simulations in water solvent) using the Gromacs solvate tool.42 All the subsequent energy minimization calculations and MD simulations were performed using the Gromacs MD engine.42 MD simulations in both vacuum and water environments were subjected to an equilibration protocol starting from 2000 steps of the steepest descent energy minimization, followed by a Berendsen NVT dynamics that heated the system from 0 to 300 K in 500 ps. During this simulation phase, all NP heavy atoms were restrained to their initial position with a force constant of 1000 kJ mol−1 nm−2. The particle mesh Ewald method43 was used to treat the long-range electrostatic interactions with the cutoff distance set at 12 Å. Short-range repulsive and attractive dispersion interactions were simultaneously described by a Lennard-Jones potential, with a cut off at 12 Å. A time step of 2.0 fs was used, together with the SHAKE algorithm, to constrain H-bonds. During the production phase (NPT in water), restraints were removed, the thermostat was switched to the V-rescale, and the pressure was set to 1 bar with the Parrinello–Rahman barostat (coupling constant of 2.0 ps). MD productions of 100 ns were performed both in a vacuum and in water.2.3 DOX loading
DOX was loaded in the water solvated system, using the following iterative protocol:1. A single DOX molecule was manually placed in the bulk solvent (distance from the closest NP/ligand atom >10 Å);
2. Overlapping water molecules were removed with VMD;44
3. A random TETT/DOPAC carboxylic group was deprotonated to preserve the neutrality of the system;
4. The number of water molecules and the parameters for the deprotonated TETT/DOPAC were updated in the topology file;
5. 2000 steps of the steepest descent minimization were performed to optimize the water shell around the added DOX molecule;
6. A 10 ns MD simulation was run to allow the DOX molecule to reach the NP surface and observe an initial binding.
This protocol was iterated for each DOX molecule, avoiding the presence of all the 10 DOX molecules simultaneously in the bulk solvent, which may have caused artifacts, such as the formation of large aggregates of DOX in the bulk solvent, which is ruled out by experimental spectroscopic and spectrometric studies45,46 unless the concentration is relatively high (>5 mM).47 Moreover, this somewhat reflects what is expected to happen also in experiments, given that, because of the low concentration of DOX, NP is approached by one DOX molecule at a time. After the DOX loading, the system was simulated for 500 ns.
2.4 Simulated annealing protocol
After the 500 ns production run, we used a three-step simulated annealing protocol to enhance the sampling of DOX binding conformations, as we noticed that most of the DOX remained stuck for a long time in their initial binding conformation. Thus, we decided to heat up the systems, since this allows the observation of transition across energy barriers and sampling new binding conformations. First, the system was heated up to 500 K in 20 ns. Then, the target temperature of 500 K was kept constant in the subsequent 50 ns. Finally, the system was cooled down back to 300 K in 20 ns (Fig. S1†). After that, a 160 ns simulation at 300 K was performed, considering only the last 150 ns as the production run for the analysis. This protocol was repeated twice, and the two production parts were used for the analysis.The two simulated annealing cycles were also performed on the system with higher deprotonation of the COOH groups. Starting from the last frame of the 500 ns production run, we deprotonated one carboxylic group per-ligand in both the TETT and DOPAC systems. The neutrality of the simulation box was preserved by adding Na+ counterions.
2.5 Other analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 script (figures in sections 3.2.1 and 3.2.2). The two variable subspace (distance and angle) was divided in a grid with a bin width and an angle of 0.1 Å and 0.9 degrees, respectively, and the population of each bin was then counted. The relative probability of each bin was then computed and the contour plot representing −log(Pi) was reported.
48 script (figures in sections 3.2.1 and 3.2.2). The two variable subspace (distance and angle) was divided in a grid with a bin width and an angle of 0.1 Å and 0.9 degrees, respectively, and the population of each bin was then counted. The relative probability of each bin was then computed and the contour plot representing −log(Pi) was reported.
3. Results and discussion
3.1 Functionalization of TiO2 NP with ligands
In this first section, we will present the dynamical behavior of fully decorated TiO2 nanoparticles with the two types of ligands considered in this work: TETT and DOPAC (Fig. 1). The starting structures for the MD simulations, before drug loading, are those obtained in two previous works by our group.25,49 At first, we analyze the ligand conformations around the nanoparticle in a vacuum and then in the presence of the water environment. This was a natural step to also evaluate changes as an effect of solvent, given that the DFTB-optimized NP decorated with ligands was previously investigated at the QM-level in a vacuum. | ||
| Fig. 1 2D structure of the TETT, DOPAC and DOX molecules. | ||
Using this starting model, in this work we performed the MD simulation and compared the dynamic behavior of the TETT-functionalized NP in a vacuum with that in water along a 100 ns production phase. A difference in the conformation of the ligands, presenting a higher interaction with the surface in the case of the system in a vacuum, is evident from the comparison of the last frames of the two parallel simulations (Fig. S4a†).
Under vacuum conditions, the system assumes a more compact conformation with TETT molecules in close contact with the NP surface. In contrast, in an aqueous environment, TETT chains become more dispersed in the solvent in a brush-like conformation. To be used as a descriptor of this effect, we measured the distance of the TETT nitrogen atom (the farthest from the anchoring Si group) from the NP surface (in particular from the closest surface Ti atom). This distance will be hereinafter referred to as “d(N⋯Ti)”. It is evident from Fig. 2a that in a vacuum the TETT molecules tend to maximize the interaction with the NP with the resulting short d(N⋯Ti) values. The comparison of distance distributions in the second half of the simulation in a vacuum revealed the presence of two small peaks at 4.0 Å and 4.8 Å and a high peak with a maximum probability around 6.2 Å (Fig. 2b). In this last conformation, the polar TETT tails directly interact with the NP surface. Only a few TETT molecules have reached distances greater than 8 Å under this condition.
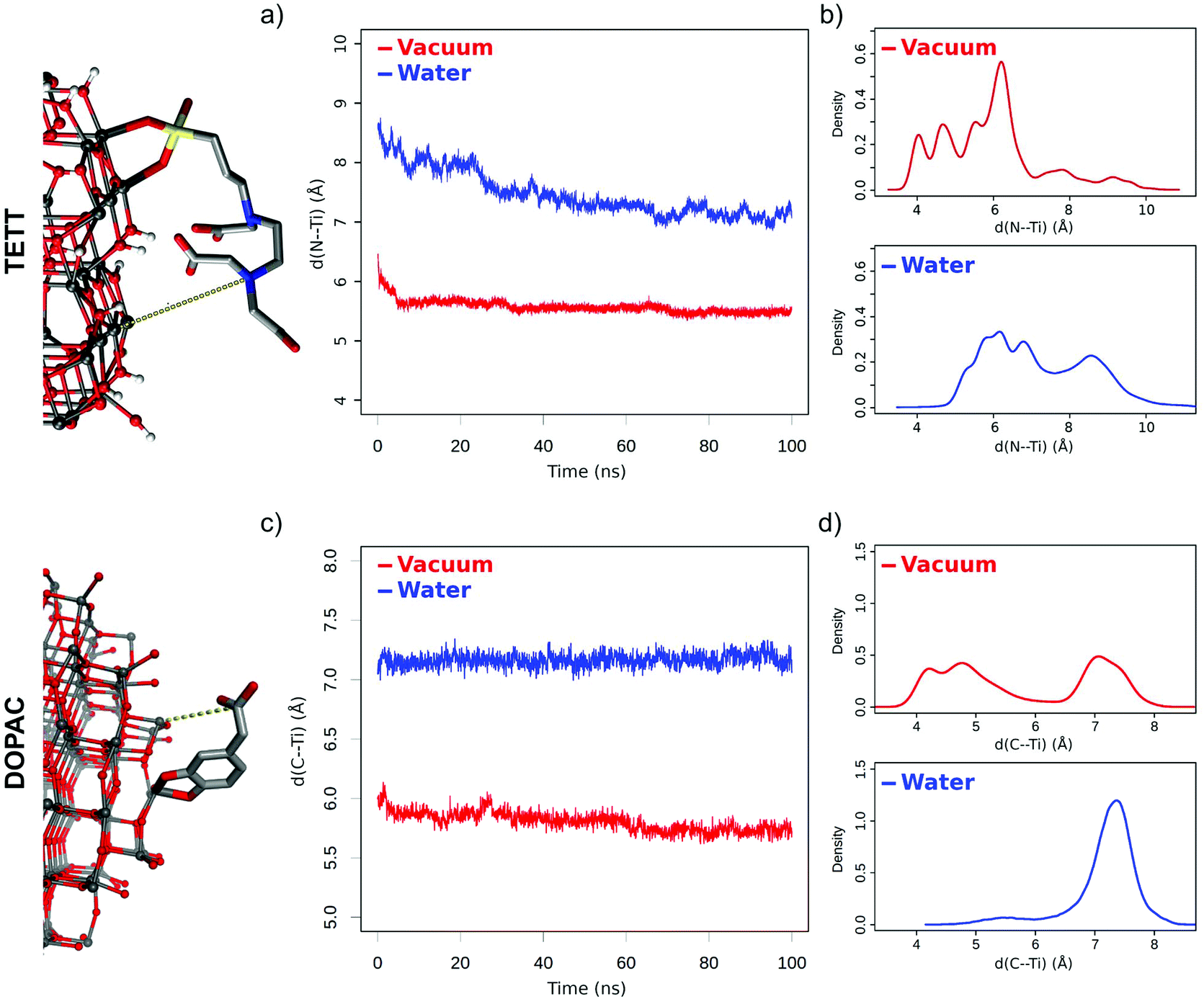 | ||
| Fig. 2 Distance between ligand tails and the NP surface. (a) Plot of d(N⋯Ti) distance values averaged over the 40 TETT molecules in time; (b) distribution of distances for each TETT molecule during the last 50 ns of simulation. (c) Plot of d(C⋯Ti) distance values averaged over the 46 DOPAC molecules in time; (d) distribution of distances for each DOPAC molecule during the last 50 ns of simulation. | ||
When the system is immersed in water, the occupied volume by TETT molecules increases because of their interaction with the solvent. Two possible TETT conformations can be found. A down conformation, with maximum distance probability at 6.0 Å, corresponding to the one sampled in a vacuum, and a more elongated up conformation (distance >7.7 Å), with the polar tail completely immersed in water, which has been sampled for about 35% of the TETT molecules and contributes to the increased overall average d(N⋯Ti) distance measured in water.
To gain atomistic insights into the interactions established between the TETT molecules and other components of the system (the NP and the water environment) we computed the average number of H-bonds formed (Table S2†). The most frequent interactions in a vacuum are between the carboxylic groups of TETT and the surface O atoms (OH|Ot and Ot|![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and appear in the down conformation. This type of interaction is drastically reduced in water (by about one-third with respect to vacuum) due to the high number of TETTs in the up conformation. When TETTs anchor in the bidentate or monodentate mode, the unbound –SiOH groups can establish H-bonds with the surface O atoms (SiO|Ot and Ot|SiO). However, this happens very rarely, both in a vacuum and in water. We notice that the lower total number of TETT interactions with the NP in water is compensated by a high number of H-bonds with the solvent molecules (Table S2,† right side). Water does not only interact with the carboxylic groups of TETTs (OH|water; water|O and water|OH) but also with the trisilanol groups (SiO|water and water|SiO).
O) and appear in the down conformation. This type of interaction is drastically reduced in water (by about one-third with respect to vacuum) due to the high number of TETTs in the up conformation. When TETTs anchor in the bidentate or monodentate mode, the unbound –SiOH groups can establish H-bonds with the surface O atoms (SiO|Ot and Ot|SiO). However, this happens very rarely, both in a vacuum and in water. We notice that the lower total number of TETT interactions with the NP in water is compensated by a high number of H-bonds with the solvent molecules (Table S2,† right side). Water does not only interact with the carboxylic groups of TETTs (OH|water; water|O and water|OH) but also with the trisilanol groups (SiO|water and water|SiO).
Finally, to understand the extent of interactions among and within TETT ligands, we estimated the number of intra- and inter-molecular H-bonds (Table S3†). We found a significant number of interactions in a vacuum, both between carboxylic groups of the same or different molecules (OH|O and OH|OH) and between carboxylic groups and trisilanol groups (OH|SiO and SiO|O). We register a reduction of about one-third in the number of these types of intra- and inter-ligand H-bonds in water.
DOPAC molecules can have up to two anchoring points (the catechol OH groups that dissociatively bind to surface Ti atoms). We found that 44 out of the total 46 molecules are bound to the NP in the bidentate mode, while only the remaining 2 in the monodentate mode. Also, in this case, the released protons bind to the undercoordinated O atoms on the surface. The atom types in the NP, according to the force-field parameters, are reported in Table S1.†
Similar to what was done in the previous section for the TETT-functionalized TiO2 NP, we investigated the structural dynamics of the DOPAC-functionalized one, both in a vacuum and in water, during a 100 ns production phase (Fig. S4b†). We observed different features in some respects, but we registered a similar behavior in some others, as detailed below.
DOPAC molecules have shorter chains compared to TETT ones. For this reason, even if they are present in a slightly higher number on the NP (46 DOPAC vs. 40 TETT molecules), the oxide surface is more exposed to the water solvent. Like TETTs, DOPAC molecules acquire a more compact conformation in a vacuum, whereas their chains become more dispersed when simulated in water. These conclusions are based on the analysis of the distance of the DOPAC carboxyl group (C atom) from the NP surface (from the closest surface Ti atoms), which we used as a descriptor for chain dispersion in the solvent (Fig. 2c) and named “d(C⋯Ti)”. Interestingly, we found that the lower average d(C⋯Ti) value obtained in a vacuum originates from two different conformations, i.e. one (down) with a low d(C⋯Ti) distance (around 4.5 Å) and the second one (up) with a higher d(C⋯Ti) distance (around 7.1 Å) (Fig. 2d). In constrast, the higher average distances computed in water originate from a single conformation with a high d(C⋯Ti) distance (around 7.2 Å) (Fig. 2d).
This analysis is different from that for the system decorated with TETTs, for which a single conformation with low distance values was observed in a vacuum, and two (up and down) conformations were observed in water (Fig. 2b). We attribute this effect to the different chain lengths: indeed, while TETTs have long and flexible tri-branched chains that allow the carboxylic groups to reach the NP surface, DOPACs have short chains stiffened by the aromatic ring that makes it difficult for the carboxylic group to reach the NP surface.
We monitored the average number of H-bonds formed between DOPAC and the NP surface but also between DOPAC and water (Table S4†). The most frequent interaction in a vacuum is that between the DOPAC carboxylic group and the surface hydroxyl groups, with the latter acting as an H-bond donor (Ot|O). The situation dramatically changes when we considered the system in water: the total number of interactions between DOPAC and NP is reduced by about 75%, and an even larger reduction is found among the carboxylic groups of different DOPACs. This is because the ligands prefer an up conformation when immersed in water. Indeed, a high number of DOPAC-water H-bonds is registered, mostly involving the COOH groups or the catechol O atoms as acceptors. A comparison of the H-bond analysis with the TETT-functionalized system (Table S2†) reveals some interesting differences. When comparing Tables S2 and S4,† one should keep in mind that the total number of carboxylic groups in the two systems is different (46 for DOPACs and 120 for TETTs). It follows that in a vacuum, the TETT carboxylic groups act as H-bond donors towards the NP O atoms (OH|Ot) more frequently than for DOPAC. On the other side, DOPAC carboxylic groups act as H-bond acceptors from the OH groups on the NP surface more frequently than for TETT. Moreover, water seems to have a different influence on the two systems: it interacts more with the carboxylic groups in DOPAC and causes a reduction of the number of DOPAC–NP interactions to a greater extent.
Finally, as it was done for TETT above, the intra- and intermolecular H-bonds of DOPAC molecules were computed (Table S5†). In a vacuum, we found a significant number of interactions between carboxylic groups (OH|O) and between carboxylic groups and catecholic oxygens (OH|Oc). Due to the presence of a single carboxylic group in the molecule, no intramolecular H-bonds could be found. Interestingly, in water, the H-bonds between pairs of DOPAC molecules are almost quenched, highlighting a stronger effect of the solvent here than for TETT in the previous section.
3.2 Drug loading on the ligand decorated TiO2 nanoparticles
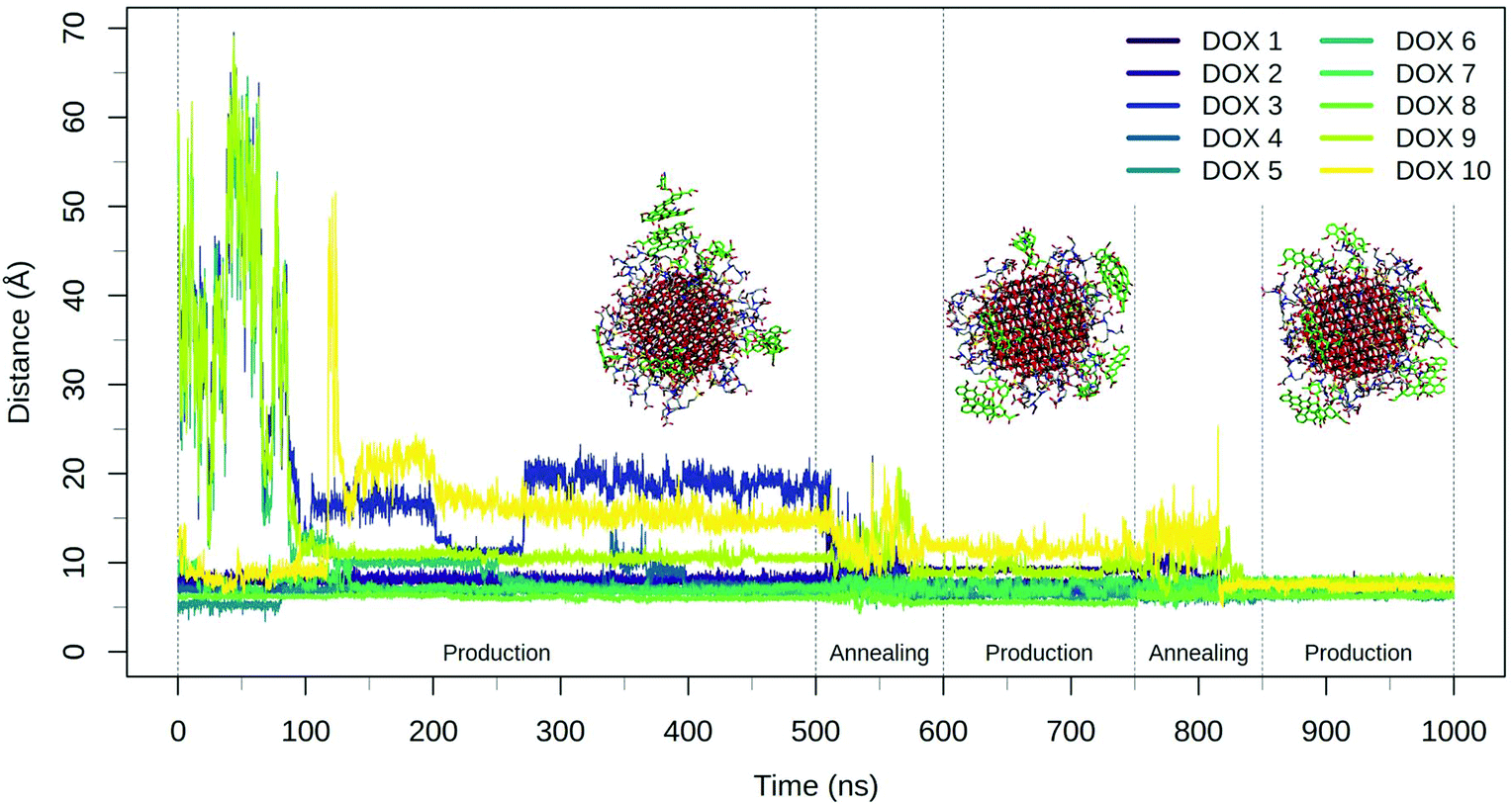 | ||
| Fig. 3 Plot of the DOX center of mass distance from the NP surface during the 500 ns production phase and the two consecutive annealing-production cycles. Representative frames of the system at the end of the three production phases are reported with TETT represented with grey sticks and DOX molecules drawn in green sticks. | ||
The annealing cycles have an evident effect bringing all DOX molecules closer to the NP. In particular, the DOX oligomer substructure was disrupted during the first annealing cycle, leading to one trimer and a dimer, with all the molecules strongly interacting with TETT molecules or with the NP surface. After the second annealing phase, only one DOX dimer is still observed and all the DOX molecules have reached a stable conformation with an average distance value of about 7 Å. Certainly, these results point out that the first 500 ns production phase was not sufficient to reach the most stable DOX conformation for all the molecules.
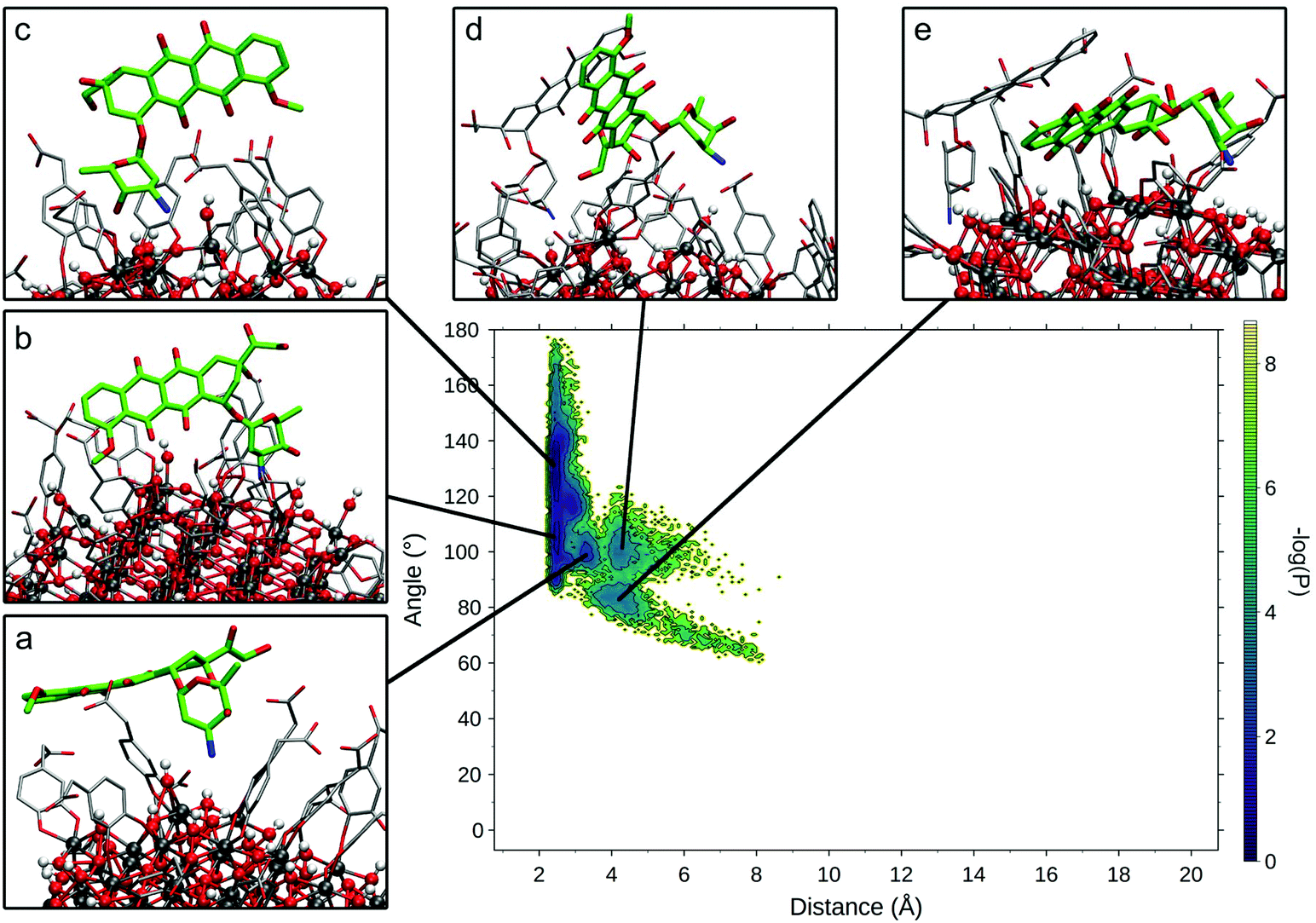
To compare the various binding modes of DOX on the functionalized NP, we designed two descriptors of the relative orientation of the molecules. The first descriptor is the distance between the DOX amino-group and the NP surface (Fig. S3a†), while the second descriptor is the angle involving the center of the NP, the N atom of DOX amino-group and one C atom on the opposite side of the DOX structure (Fig. S3b†). The resulting probability distribution map (Fig. 4) shows that the most sampled region presents DOX nitrogen very close to the NP surface (2.2 Å) with a variable angle ranging from 100 to 180 degrees.
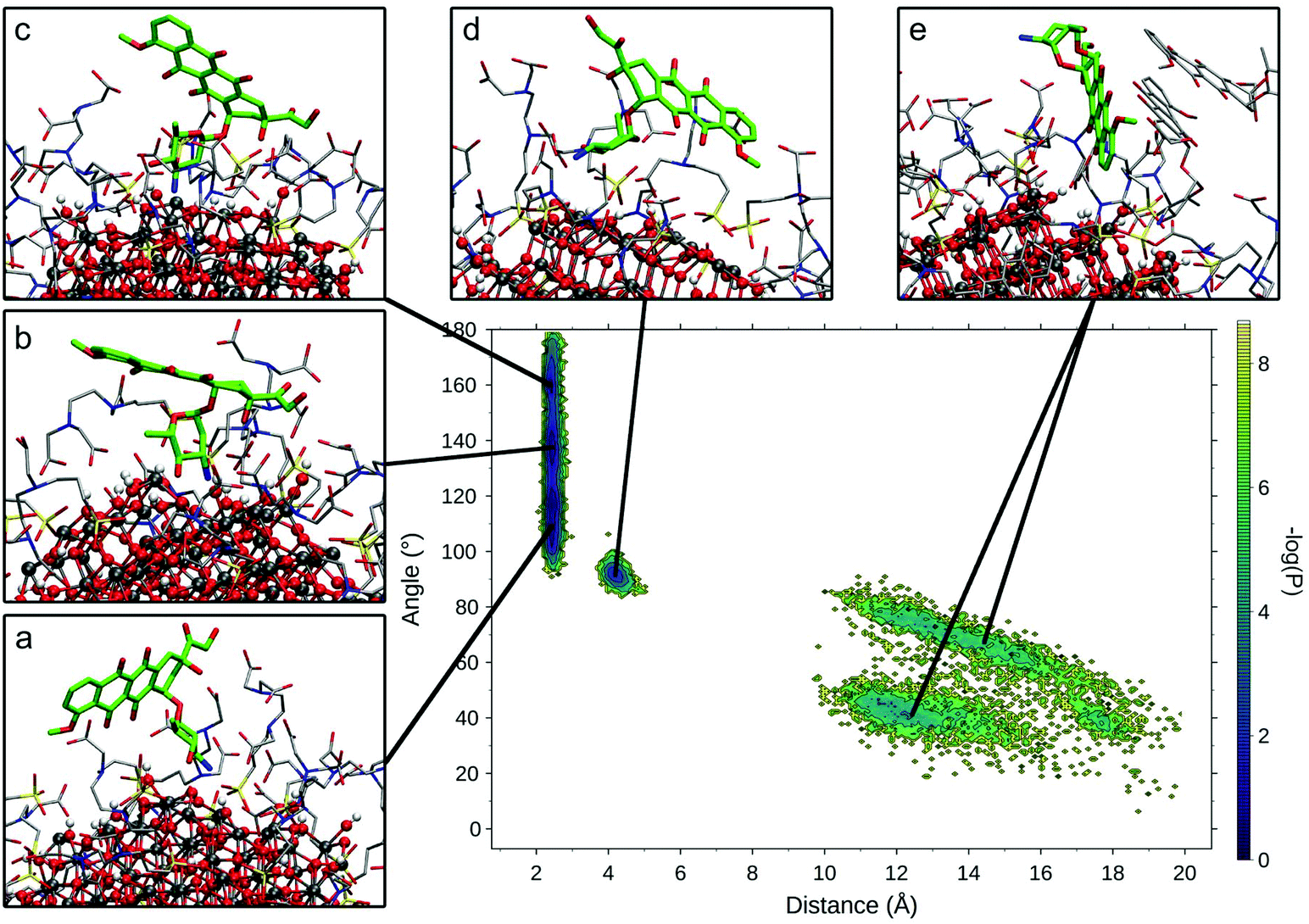 | ||
| Fig. 4 Relevant DOX binding states identified on the probability density map along two selected variables. Representative states for each conformational minimum identified were represented in insets (a)–(e), with DOX heavy atoms represented in bold green sticks, TETT and other DOX molecules heavy atoms in thin grey sticks and NP atoms as ball and sticks. | ||
Within this region, three different conformational states can be identified (Fig. 4a–c) presenting the same binding mode for the sugar moiety of DOX and a different orientation of the aglycon core, which interacts differently with the TETT sidechains and do not reach the NP surface. In these conformations, to maximize the interactions with NP, the sugar moiety is perpendicular to the surface, bringing both the amino and hydroxylic groups close to the surface. A similar binding mode is also observed for the conformation in Fig. 4d, with the DOX sugar group slightly further from the NP surface, interacting with an O atom of a nearby trisilanol group. In this conformation, the sugar moiety assumes an orientation parallel to the NP surface and the aglycon core stays far from NP atoms, due to the high density of TETT chains. Finally, conformations with the DOX sugar group far from the NP surface (distance >10 Å) have been identified (Fig. 4e). In this conformation, the sugar group points towards the solvent and may interact with nearby TETT chains, while the aglycon core is closer to the NP surface, but without establishing strong interactions with it. Gaps between different states originate from the fact that the distributions are derived from the production phase after the annealing, so high energy states (transitions between low-energy states) are not included in the map. Moreover, this is a combination of 10 different molecules, whose sampled space may not overlap.
We extracted frames belonging to each basin described in Fig. 4 and we characterized the electrostatic and VdW interactions between DOX and NP or TETT molecules (Table S6†). We noted that conformations a–c present very similar interaction profiles, mainly driven by the electrostatic interaction between the DOX sugar moiety and the NP surface. This is due to the highly negative NP oxygen atoms and the positively charged amino group on the DOX sugar group. The high variance observed is mainly due to the different geometry of the NP surface, which may change depending on the position on the NP. In conformations d and e, the interactions with NP are very low due to the higher distance of the amino group from the NP surface. Interactions with TETT have a value about 60% lower than the ones with NP, and VdW and Coulomb contributions are comparable. Conformation e significantly differs from the others for its low interaction with TETT. This lower contribution may be compensated by the interaction with other DOX molecules (two of the DOX in the trimer are part of the conformation in Fig. 4e).
Finally, we evaluated how TETT deprotonation may influence DOX binding, measuring the average Coulomb interaction energy of the TETT carboxylic groups in protonated and deprotonated TETT (Table 1). We found an increased interaction with deprotonated TETT molecules, also with their protonated carboxylic groups. This may be explained as a tendency of DOX to bind nearby deprotonated TETTs, increasing the interactions with their carboxylic groups. Moreover, we found a generally lower interaction with the carboxylic group located in the middle of the TETT carbon chain (see Fig. 1).
| TETT (kJ mol−1) | Deprotonated TETT (kJ mol−1) | |
|---|---|---|
| The three carboxylic groups were divided into tails (the two at the extreme of the molecule) and the middle. Deprotonation was always applied to the carboxylic tail 1 (in italics in the table). | ||
| Carboxylic tail 1 | −5.3 | −13.9 |
| Carboxylic tail 2 | −5.2 | −11.2 |
| Carboxylic middle | −3.3 | −7.8 |
During the 500 ns initial production phase, we have first noticed that all the DOX molecules immediately reach the NP surface but, in line with what observed for the TETT-functionalized NP, they remain stuck in the original conformation until the end of the simulation (Fig. 5), as a trimer, two dimers and three single DOX molecules. However, after the first annealing cycle, we observed a decrease in the DOX-NP distances and a disruption of the trimer, with the formation of three dimers. The second annealing cycle splits one of the dimers, but simultaneously a new one is formed, leaving a total of three dimers. During the annealing cycles, we do not observe a complete unbinding of DOXs, but most of them just change their binding sites moving around the functionalized NP surface.
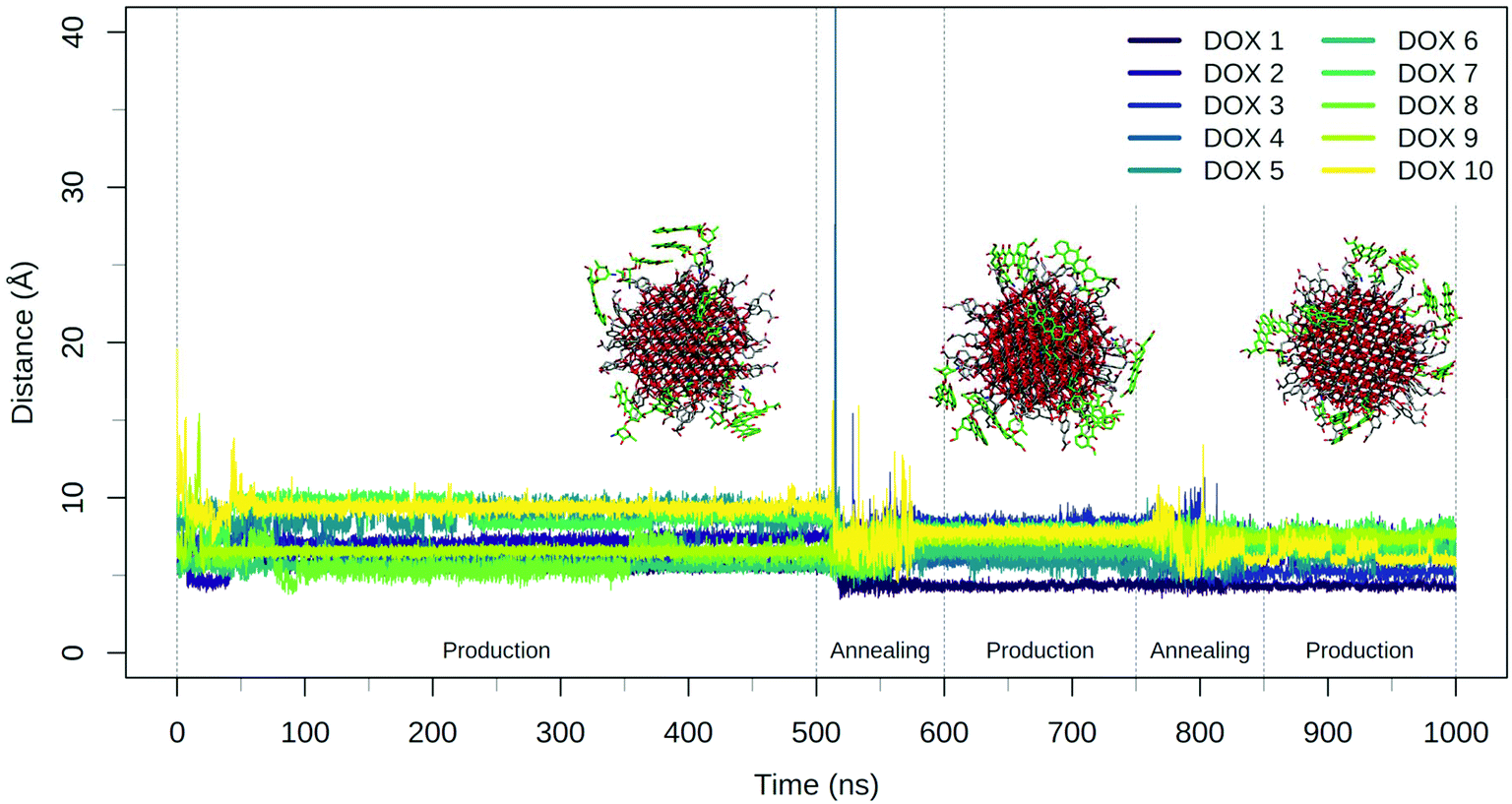 | ||
| Fig. 5 Plot of the DOX center of mass distance from the NP surface during the 500 ns production phase and the two consecutive annealing-production cycles. Representative frames of the system at the end of the three production phases are reported with DOPAC represented with grey sticks and DOX molecules drawn in green sticks. | ||
The DOX dimers appeared to be more persistent on the DOPAC-functionalized NP, compared to the TETT-functionalized one. We rationalize this effect with the different chain lengths of the two ligands. DOPACs are indeed too short to efficiently accommodate DOX, whose hydrophobic aglycon core remains mostly immersed in the solvent or packed with other DOX aglycon cores. Long TETT chains, on the other side, are able to adsorb the hydrophobic core of DOX molecules, promoting the formation of monomers.
We measured the values of the distance and angle descriptors of the DOX orientation on the surface (Fig. 6). We noticed that DOX preferentially binds with the sugar amino group, which directly interacts with the NP surface, similar to what was observed for the TETT-functionalized NPs (Fig. 6b and c). Only two minima were found in this region, one with DOX aglycon closer to the NP surface (Fig. 6b) and the second with DOX aglycon interacting with DOPACs (Fig. 6c). The other three minima are present in the map: one with the amino group of sugar towards the surface, but not in close contact with O atoms (the distance around 3.3 Å; Fig. 6a), and the other two with the sugar group farther from the surface and differently oriented (Fig. 6d and e). Interestingly, the last two conformations were only found when DOX is in a dimer with another DOX.
 | ||
| Fig. 6 Relevant DOX binding states identified on the probability density map along two selected variables. Representative states for each conformational minimum identified were represented in insets (a)–(e), with DOX heavy atoms represented in bold green sticks, DOPAC and other DOX molecules heavy atoms in thin grey sticks and NP atoms as ball and sticks. | ||
We extracted frames belonging to each basin described in Fig. 6 and we characterized the electrostatic and VdW interactions of DOX with the NP or with the DOPAC molecules (Table S7†). In conformation A, a small repulsive potential was found between DOX and the NP. This is probably due to the position of the sugar group, which is slightly farther from the NP surface, and for which the attractive and repulsive electrostatic potential sum up to a positive number. The lack of interaction with NP atoms is compensated by a strong interaction with DOPACs. Conformations b and c, the closest to the surface, present a strong electrostatic interaction between the sugar moiety and the NP but are also stabilized by interactions with DOPACs. Finally, conformations d and e, the farthest from the surface, present small positive electrostatic potential between the sugar moiety and the NP atoms. Interestingly, these conformations are the only two that present significant interactions between the aglycon core and the NP (especially conformation e). Moreover, both are stabilized by interactions with DOPAC and with other DOX molecules (values not reported in the table).
For both the TETT- and DOPAC-functionalized systems, we found that the sugar moiety interactions are the driving force for binding. This strongly interacts with the NP atoms forming a particular substructure in which both the amino group and the near hydroxyl group are involved in H-bonds with NP (Fig. S5†).
The main difference between the DOPAC- and the TETT-functionalized systems is their ability to stabilize DOX far from the NP surface. In the DOPAC-functionalized system, all the DOX molecules are bound very close to the NP surface (amino–surface distance below 10 Å), while in the TETT-functionalized system, conformations at a distance of over 10 Å can also be found, with basically no interaction with the NP. In both systems, at lower amino–surface distances (between 3 and 10 Å), we found a number of states where the DOX amino group is weakly interacting with the NP surface. However, in the region at low amino–surface distances (below 3 Å), both the TETT- and DOPAC-functionalized systems present conformations with strong interactions between the DOX sugar group and NP. Strong hydrogen bonds are formed during the simulations and the distances between the NP oxygens and the amino and hydroxyl groups on the DOX sugar moiety are very low, suggesting the possible formation of chemical bonds. Interestingly, in the case of TETT, a stable conformation at very high angle values (over 150°) was found, while such a conformation is absent in the case of DOPAC. Again, this discrepancy can be rationalized with the different ability of the two ligands in stabilizing the DOX aglycon core at a high distance from the NP. In the case of TETT, indeed, the conformation at high angle values presents the aglycon core far from the NP surface. This conformation was only possible because of the stabilization of the aglycon core by TETT ligands. The shorter DOPAC chains cannot stabilize the aglycon core at this large distance from the surface, and as a result, the DOX conformation at a high angle value is not observed. Overall, in all the bound conformations, a higher coulomb interaction between the sugar group and the NP for the TETT-functionalized system was also observed.
Finally, the DOPAC deprotonation enhancing effect on the DOX binding was also evaluated, measuring the average Coulomb interaction energy of the DOPAC carboxylic groups in protonated and deprotonated DOPAC. The average interaction of deprotonated DOPAC was about 7 kJ mol−1 stronger than that of the protonated one (−18.3 and −11.2 kJ mol−1, respectively). This result highlights the impact of pH on the stabilization of DOX over the functionalized NP and provides a rational basis for the pH-triggering release mechanism that is supposed for DOX delivery systems.
3.5 Effects of carboxylic group deprotonation
To explore the effect of a higher degree of deprotonation of the carboxylic groups, we changed the protonation state of the ligands and repeated two simulated annealing cycles. Determining that the protonation state of all the carboxylic groups on the NP is a non-trivial task: as the number of deprotonated COO− groups increases on the NP surface, the acidity of the remaining carboxylic groups decreases, making deprotonation progressively less favorable. For the present work, we limited our analysis to a system with one deprotonated carboxylic group per ligand unit, exploring the effect of this change on the drug loading process. A detailed study of the system under different pH conditions will be the object of a future specific work.We measured the values of the distance and angle descriptors of the DOX orientation on the surface for both the TETT and DOPAC systems (Fig. 7). Even in these simulations, DOX preferentially binds with the sugar amino group, directly interacting with the NP surface, similar to what was observed in the previous sections, at a lower degree of deprotonation. In the states at higher distance values, however, we spotted few main differences. The TETT-functionalized system presents several new minima at intermediate distances (4–6 Å) and one at slightly higher distances (6–8 Å). This latter conformation is stabilized by the interactions of the sugar amino group with deprotonated carboxylic groups (Fig. S6†).
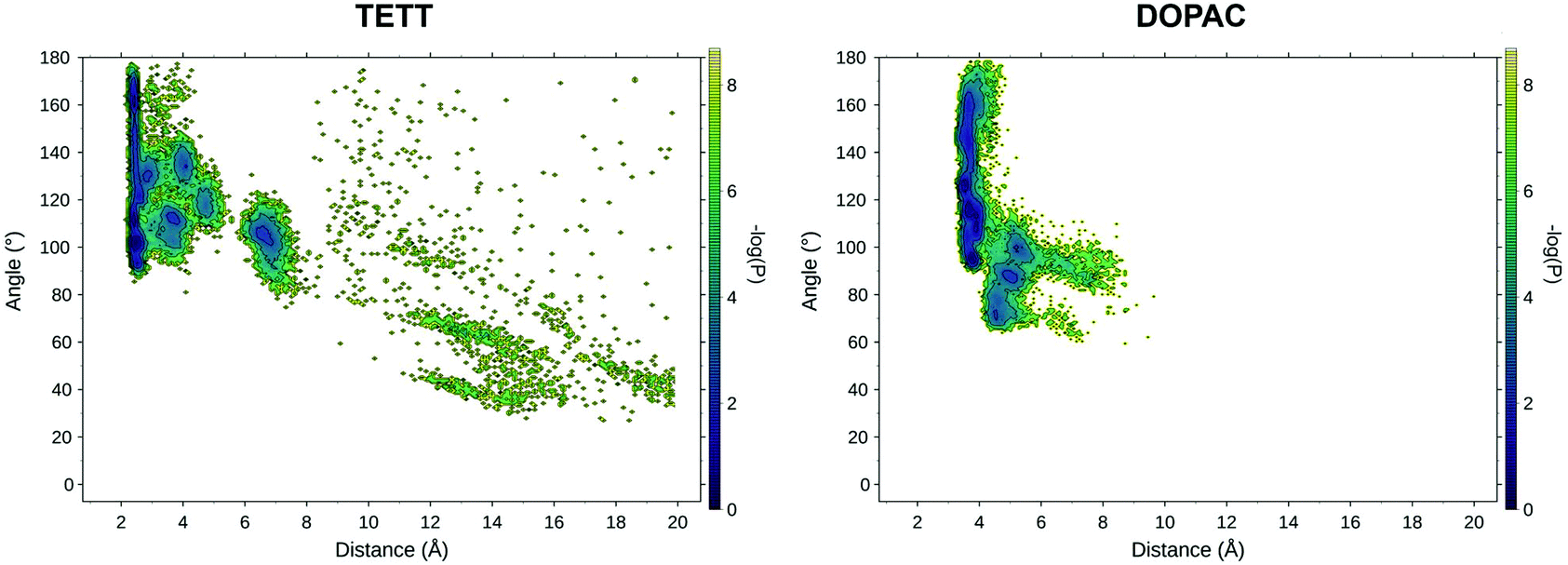 | ||
| Fig. 7 Relevant DOX binding states identified on the probability density map along two selected variables for the TETT (left) and DOPAC (right) systems. | ||
A similar but less pronounced trend was also found for the DOPAC-functionalized system, with an increased number of states found at distance values in the range between 4 and 8 Å, as an effect of the deprotonation of the COO− groups that can interact with the charged DOX amino group. Overall, in both systems, the effect of deprotonation was to stabilize conformational states with distances between the N atom of the protonated DOX amino group and the closest surface O atom of 6–10 Å, in which the ligand's carboxylic groups interact with the charged moiety of DOX.
3.6 NP surface composition effects on DOX binding
Here, we present a comparative analysis of the surface properties around the bound DOX for the TETT- and DOPAC-functionalized systems. The NP surface presents some undercoordinated Ti atoms, as already discussed and reported in Table S1.† As a consequence of the different binding modes of TETT and DOPAC, and the different number of ligands anchoring the NP (as reported in previous works25,49), the number of free undercoordinated Ti sites is higher in the TETT-functionalized system. Moreover, from the comparison of Tables S6 and S7,† we found that the DOX sugar moiety interaction with the NP is stronger when the NP is functionalized with TETTs. Given that the charges of NP atoms are the same on the two systems and that no significant differences were found in the binding geometry of DOX at low amino–surface distances (below 10 Å), we tend to ascribe the energetic discrepancy to a different surface composition. To verify this hypothesis, we first compared the number of oxygen atoms surrounding the DOX amino group at different shell radii for all the DOX molecules in the two production runs after the annealing cycles (Fig. S7†).In the TETT-functionalized system, most cases present a similar trend with an initial number of oxygens of about 3, which remain almost constant up to 4.5 Å. Interestingly, in a significant number of cases, we observe a rapid increase in the curves, and the number of oxygens within 4 Å from the DOX amino group exceeds the value of 5. In constrast, in the DOPAC-functionalized system, all the curves present a similar behavior, with no cases where the number of oxygens within 4 Å from the DOX amino group exceeds the value of 5. This analysis suggests that a different surface composition exists and that the TETT functionalized NP presents electrostatic hotspots with higher oxygen density that increase the interaction with DOX.
To further inspect the electrostatic surface composition of the two systems, we built the electrostatic potential surface using the APBS pymol plugin50 on the DFTB-optimized starting conformation (considered with 10 ligands deprotonated) and on the last frame of the simulation with ten DOX with both 10 ligand deprotonated, or with one carboxylic group per ligand deprotonated (Fig. 8). In both cases, with 10 ligands deprotonated (Fig. 8a and b), we observed that the TETT-functionalized system presents clear hotspots on the surface with high negative potential. These regions are indeed the preferential binding site for the positively charged sugar moiety of DOX molecules. In constrast, in the DOPAC-functionalized system, no regions with high negative charges were present in the starting conformation, and even after DOX binding, the electrostatic potential around the binding sites is less negative compared to the TETT case.
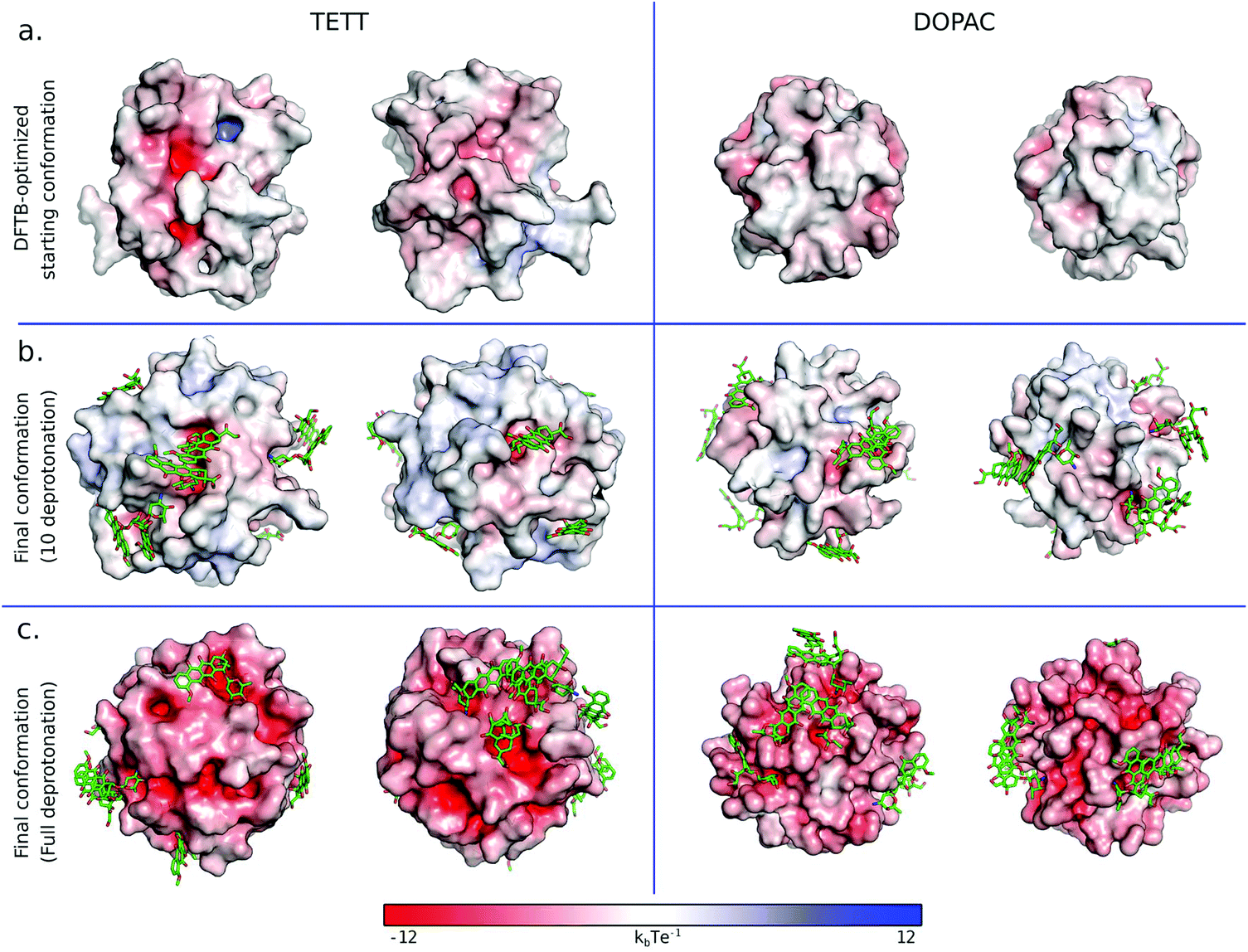 | ||
| Fig. 8 Electrostatic potential surface for the TETT (left) and DOPAC (right) functionalized systems in different configurations: DFTB-optimized starting conformation (a), the last frame of simulated annealing cycles with 10 ligands deprotonated (b) and the last frame of simulated annealing cycles with one carboxylic group per-ligand deprotonated (c). DOX molecules are reported in green sticks. | ||
This analysis highlights the important effect of the NP surface composition on the DOX binding. Given that this property is influenced by the ligand type and its binding mode, we suggest that it is not only the chemistry of the ligand tails that influences the DOX binding but also its NP anchoring group that, altering the NP surface composition, may create some favorable hotspots for DOX loading.
As expected, the increase in the number of deprotonated ligands (Fig. 8c) resulted in greater negative potentials on the system surface, which is at the basis of the pH-triggered mechanism of DOX release. These results are encouraging for future investigations of the drug release mechanism.
4. Summary and conclusions
The present MD study benefits from previous DFT-based investigations by our group, which produced optimized and chemically stable structures of TETT- and DOPAC-functionalized TiO2 nanocarriers of realistic sizes.28,29 The force field accuracy for the description of similar systems has been assessed in another previous work by some of us.26 This is an excellent starting point for our MM investigation on the drug loading capacity of these systems.Our first concern, here, was to investigate the effect of the surrounding water solvent on the conformation of the functionalized nanoparticles that were obtained by calculations in a vacuum. Since both TETT and DOPAC ligands present COOH ending groups, we first analyzed how the interaction of such groups with the NP surface varies upon moving from vacuum to aqueous conditions. As one would expect, the number of interactions is largely reduced but the effect is more pronounced for DOPAC (72%) than for TETT (52%), which retains several H-bond interactions with the NP surface atoms during all the simulation in water.
Then, we started our study on the drug loading capacity of these two different drug carriers. We added one by one, up to ten DOX molecules, to each system, performing short MD simulations at each step. In both cases, however, we observe that the DOX molecules tend to pile up, forming aggregates on the surface. Only after some simulated thermal annealing at 500 K we were able to observe redistribution of the DOX molecules all over the nanocarrier surface.
The MD simulations were then analyzed to identify the most common or recurring DOX tethering conformations on the NP through a probability density map based on two meaningful variables (the shortest distance of DOX from the NP surface and the angle of DOX orientation on the surface). We noticed that the most common is also the closest conformation and involves both electrostatic and H-bond interactions of the protonated amino from the DOX sugar group with surface O atoms. The distance between the N atom of the protonated amino group in DOX and the closest surface O is set at about 2–2.5 Å. This is true for both ligands. However, with DOPACs, there are some other configurations with distances between 3 and 8 Å, but nothing above 10 Å. In contrast, in the case of TETTs, some DOX molecules are stabilized at higher distances from the surface, above 10 Å, up to almost 20 Å. This is due to the larger size and the larger number of COOH catching groups of the TETT ligands. Indeed, through the analysis of the electrostatic potential surface, we have proved that the number and position of favorable hotspots, with high negative potential, on the surface of the functionalized NP, depend on the type of the ligand, in particular on the anchoring group and on the size of the ligand and those are the preferential binding sites for the positively charged sugar moiety of DOX molecules.
Increasing the number of deprotonated TETT resulted in the formation of additional states at higher distance values between the N atom of the protonated amino group in DOX and the closest surface O. These states are mainly stabilized by interactions between the ligand carboxylic tails and charged DOX amino group.
Through an energy decomposition analysis, we could also determine that DOX molecules tether the drug carrier mostly through electrostatic interactions by the protonated DOX amino group. This result provides a solid basis for the rationalization of a pH-triggered release mechanism of DOX, which is often proposed in the literature, because the increased protonation of both the NP surface and the carboxylate groups of the ligands is expected to quench their interaction with the protonated DOX amino group and, therefore, to favour the drug release in an acidic environment. The mechanism of drug release will be, however, the subject of a future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Lorenzo Ferraro, Martina Datteo and Laura Bonati. The project has received funding from the European Research Council (ERC) under the European Union's HORIZON2020 research and innovation programme (ERC Grant Agreement No [647020]).References
- C. P. Sharma, Drug delivery nanosystems for biomedical applications, Elsevier, 2018 Search PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- A. Puri, K. Loomis, B. Smith, J. H. Lee, A. Yavlovich, E. Heldman and R. Blumenthal, Crit. Rev. Ther. Drug Carrier Syst., 2009, 26, 523–580 CrossRef CAS PubMed.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef PubMed.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- J. Conde, J. T. Dias, V. Grazú, M. Moros, P. V. Baptista and J. M. de la Fuente, Front. Chem., 2014, 2, 48 Search PubMed.
- A. K. Gupta, R. R. Naregalkar, V. D. Vaidya and M. Gupta, Nanomedicine, 2007, 2, 23–39 CrossRef CAS PubMed.
- P. N. Prasad, Nanophotonics, Wiley, 2004 Search PubMed.
- Y. Volkov, Biochem. Biophys. Res. Commun., 2015, 468, 419–427 CrossRef CAS PubMed.
- G. Bao, S. Mitragotri and S. Tong, Annu. Rev. Biomed. Eng., 2013, 15, 253–282 CrossRef CAS PubMed.
- M. T. Morgan, Y. Nakanishi, D. J. Kroll, A. P. Griset, M. A. Carnahan, M. Wathier, N. H. Oberlies, G. Manikumar, M. C. Wani and M. W. Grinstaff, Cancer Res., 2006, 66, 11913–11921 CrossRef CAS PubMed.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643–10647 CrossRef CAS PubMed.
- T. Rajh, N. M. Dimitrijevic, M. Bissonnette, T. Koritarov and V. Konda, Chem. Rev., 2014, 114, 10177–10216 CrossRef CAS PubMed.
- T. Wang, H. Jiang, L. Wan, Q. Zhao, T. Jiang, B. Wang and S. Wang, Acta Biomater., 2015, 13, 354–363 CrossRef CAS PubMed.
- K. C. W. Wu, Y. Yamauchi, C. Y. Hong, Y. H. Yang, Y. H. Liang, T. Funatsu and M. Tsunoda, Chem. Commun., 2011, 47, 5232–5234 RSC.
- Y. Qin, L. Sun, X. Li, Q. Cao, H. Wang, X. Tang and L. Ye, J. Mater. Chem., 2011, 21, 18003–18010 RSC.
- C. M. Perou, T. Sørile, M. B. Eisen, M. Van De Rijn, S. S. Jeffrey, C. A. Ress, J. R. Pollack, D. T. Ross, H. Johnsen, L. A. Akslen, Ø. Fluge, A. Pergammenschlkov, C. Williams, S. X. Zhu, P. E. Lønning, A. L. Børresen-Dale, P. O. Brown and D. Botstein, Nature, 2000, 406, 747–752 CrossRef CAS PubMed.
- M. Setvin, J. Hulva, H. Wang, T. Simschitz, M. Schmid, G. S. Parkinson, C. Di Valentin, A. Selloni and U. Diebold, J. Phys. Chem. C, 2017, 121, 8914–8922 CrossRef CAS.
- G. Fazio, D. Selli, L. Ferraro, G. Seifert and C. Di Valentin, ACS Appl. Mater. Interfaces, 2018, 10, 29943–29953 CrossRef CAS PubMed.
- C. Ronchi, M. Datteo, M. Kaviani, D. Selli and C. Di Valentin, J. Phys. Chem. C, 2019, 123, 10130–10144 CrossRef CAS.
- M. Datteo, L. Ferraro, G. Seifert and C. Di Valentin, Nanoscale Adv., 2020, 2, 2774–2784 RSC.
- P. Siani, S. Motta, L. Ferraro, A. O. Dohn and C. Di Valentin, J. Chem. Theory Comput., 2020, 16, 6560–6574 CrossRef CAS PubMed.
- G. Fazio, L. Ferrighi and C. Di Valentin, J. Phys. Chem. C, 2015, 119, 20735–20746 CrossRef CAS.
- D. Selli, G. Fazio and C. Di Valentin, J. Chem. Phys., 2017, 147, 164701 CrossRef PubMed.
- A. V. Bandura and J. D. Kubicki, J. Phys. Chem. B, 2003, 107, 11072–11081 CrossRef CAS.
- P. K. Naicker, P. T. Cummings, H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2005, 109, 15243–15249 CrossRef CAS PubMed.
- V. N. Koparde and P. T. Cummings, J. Phys. Chem. B, 2005, 109, 24280–24287 CrossRef CAS PubMed.
- V. N. Koparde and P. T. Cummings, J. Phys. Chem. C, 2007, 111, 6920–6926 CrossRef CAS.
- M. Alimohammadi and K. A. Fichthorn, J. Phys. Chem. C, 2011, 115, 24206–24214 CrossRef CAS.
- S. Monti, C. Li, H. Ågren and V. Carravetta, J. Phys. Chem. C, 2015, 119, 6703–6712 CrossRef CAS.
- S. Monti, M. Pastore, C. Li, F. De Angelis and V. Carravetta, J. Phys. Chem. C, 2016, 120, 2787–2796 CrossRef CAS.
- A. M. Sultan, Z. C. Westcott, Z. E. Hughes, J. P. Palafox-Hernandez, T. Giesa, V. Puddu, M. J. Buehler, C. C. Perry and T. R. Walsh, ACS Appl. Mater. Interfaces, 2016, 8, 18620–18630 CrossRef CAS PubMed.
- T. Zheng, Y. Zhang, C. Wu, L. Zhou and P. T. Cummings, Appl. Surf. Sci., 2020, 512, 145713 CrossRef CAS.
- E. G. Brandt and A. P. Lyubartsev, J. Phys. Chem. C, 2015, 119, 18110–18125 CrossRef CAS.
- M. Matsui and M. Akaogi, Mol. Simul., 1991, 6, 239–244 CrossRef.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 2, 19–25 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Agrawal, S. K. Barthwal and R. Barthwal, Eur. J. Med. Chem., 2009, 44, 1437–1451 CrossRef CAS PubMed.
- P. Changenet-Barret, T. Gustavsson, D. Markovitsi, I. Manet and S. Monti, Phys. Chem. Chem. Phys., 2013, 15, 2937–2944 RSC.
- V. Barthelemy-Clavey, J. C. Maurizot, J. L. Dimicoli and P. Sicard, FEBS Lett., 1974, 46, 5–10 CrossRef CAS PubMed.
- R-Development-Core-Team, 2014.
- C. Ronchi, F. Soria, L. Ferraro, S. Botti and C. Di Valentin, arXiv.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with details of the NP atom-type composition, H-bond analysis and non-bonded interactions; figures representing the annealing ramp, the distance and angle variables, the groups of atoms used for the H-bond analysis and the DOX sugar moiety interaction with the NP surface. See DOI: 10.1039/d1nr01972d |
| This journal is © The Royal Society of Chemistry 2021 |