
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single residue can modulate nanocage assembly in salt dependent ferritin†
Mantu
Kumar‡
 ab,
Joanna
Markiewicz-Mizera§
a,
Julian David
Janna Olmos§
a,
Piotr
Wilk
a,
Przemysław
Grudnik
a,
Artur P.
Biela
ac,
Małgorzata
Jemioła-Rzemińska
ad,
Andrzej
Górecki
d,
Soumyananda
Chakraborti‡
*ae and
Jonathan G.
Heddle
*a
ab,
Joanna
Markiewicz-Mizera§
a,
Julian David
Janna Olmos§
a,
Piotr
Wilk
a,
Przemysław
Grudnik
a,
Artur P.
Biela
ac,
Małgorzata
Jemioła-Rzemińska
ad,
Andrzej
Górecki
d,
Soumyananda
Chakraborti‡
*ae and
Jonathan G.
Heddle
*a
aMalopolska Centre of Biotechnology, Jagiellonian University, Gronostajowa 7A, 30-392 Krakow, Poland. E-mail: soumyabiochem@gmail.com; Jonathan.heddle@uj.edu.pl
bPostgraduate School of Molecular Medicine, Żwirki i Wigury 61, 02-091 Warsaw, Poland
cDepartment of Cell Biology and Imaging, Institute of Zoology and Biomedical Research. Jagiellonian University, Gronostajowa 9, 30-387 Krakow, Poland
dFaculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Kraków, Poland
eNational Institute of Malaria Research, Delhi, India
First published on 1st July 2021
Abstract
Cage forming proteins have numerous potential applications in biomedicine and biotechnology, where the iron storage ferritin is a widely used example. However, controlling ferritin cage assembly/disassembly remains challenging, typically requiring extreme conditions incompatible with many desirable cargoes, particularly for more fragile biopharmaceuticals. Recently, a ferritin from the hyperthermophile bacterium Thermotoga maritima (TmFtn) has been shown to have reversible assembly under mild conditions, offering greater potential biocompatibility in terms of cargo access and encapsulation. Like Archeoglobus fulgidus ferritin (AfFtn), TmFtn forms 24mer cages mediated by metal ions (Mg2+). We have solved the crystal structure of the wild type TmFtn and several mutants displaying different assembly/disassembly properties. These data combined with other biophysical studies allow us to suggest candidate interfacial amino acids crucial in controlling assembly. This work deepens our understanding of how these ferritin complexes assemble and is a useful step towards production of triggerable ferritins in which these properties can be finely designed and controlled.
1. Introduction
Protein self-assembly in nature is utilised to generate a variety of sophisticated, often highly symmetric macromolecular architectures, such as protein cages.1,2 These self-assembled shapes are formed by a small number of subunits repeated many times to form hollow, and roughly spherical monodisperse structures. Typically, protein cages are highly stable and diverse in shape, size, and geometries3 and are amenable to genetic4,5 and chemical6,7 modification with production often achievable in high yield. The central cavities of protein cages can be used to carry cargoes while the exterior can be decorated with different groups such as targeting molecules or antigens.8,9 In recent years both natural and artificial protein cages have been developed for numerous potential applications including in biotechnology and as therapeutics.10–12 Among cages that have been developed in this way, ferritin has been perhaps the most extensively used including for cargo encapsulation, metal mineralisation, bio-sensors, and targeted drug delivery.13–15Ferritin is an iron storage protein found near ubiquitously across all forms of life. Generally, it is composed of 24 identical protein subunits assembled into an almost spherical octahedral symmetric protein cage with a hollow interior.13 The outer diameter of ferritin is 12 nm with an inner cavity of 8 nm. In nature ferritin's role is in iron homeostasis and to protect the cell from oxidative stress.16 Assembled ferritin contains several channels connecting the interior cavity to the external environment that include eight 3-fold axis channels, and six 4-fold axis channels, used for iron uptake and release.13,17
Ferritins are typically easy to purify with high yields and show high tolerance of modifications applied to all three surfaces of the assembly;18 outer surface19,20 interface,21–23 and interior cavity.24,25 The stability of ferritin is a useful property for protecting encapsulated cargoes, providing a physical barrier to potential degrading agents.26–28 Due to its unique qualities, ferritin has continued to be a major avenue of research for biotechnology and bio-nanoscience researchers and its potential as a drug delivery system has been a recent area of growing interest.29–32
A potential disadvantage of ferritins for applications including drug delivery is their high structural stability. Once the cages are formed, harsh conditions are often required to disassemble them. Known methods include extremes of pH (>12 or <3) and/or chaotropic agents such as urea and guanidine hydrochloride (GdnHCl).33–36 This may cause loss of viability of cargoes such as biological macromolecules which are precisely the type of cargo most likely to benefit from encapsulation in a stable cage.
Recently a new class of ferritin was discovered in hyper-thermophilic archaeon Archeoglobus fulgidus37–41 and in the bacterium Thermotoga martima.28,42 Both show salt mediated assembly of the protein cage from subunits i.e. in solution these ferritins almost exclusively exist as dimers, only assembling in the presence of certain monovalent or divalent metal cations. This was first shown in work using SEC demonstrating that ferritin 24mer cages formed at high salt concentration were dissociated after dialysing into low salt buffer.42 This was confirmed using Mg2+ as the divalent metal ion of choice and in further experiments including SEC, DLS and native PAGE. The ability of EDTA to cause disassembly of the cages was also demonstrated.12,28
To date these represent the only salt-dependent ferritins to have been significantly characterised. Ferritin from Archeoglobus fulgidus (AfFtn) is architecturally very different from any other known ferritins; it possesses tetrahedral symmetry with four large holes of approx. 4.5 nm diameter.37 In contrast, Thermotoga maritima ferritin (TmFtn) is structurally similar to classical ferritins, possessing octahedral symmetry and, as we show below, is highly stable.28 The controllability of ferritin assembly by salt concentration is an interesting feature with potential applications in facile cargo loading compared to classical ferritins. For TmFtn, particularly, this is interesting as the lack of large holes in the structure would offer more protection to sensitive cargoes compared to AfFtn. The possibility for TmFtn to undergo triggered disassembly has been shown using EDTA and presumably occurs by chelation of relevant metal ions.28
The structural details of TmFtn accounting for its unusual assembly/disassembly properties remain unknown. However, previous work on AfFtn has suggested that glutamate 65 plays a crucial role in its salt mediated-assembly with a point mutation (E65R) resulting in its complete abolition.41 Here we show that this same residue is also vital for salt-mediated assembly in TmFtn. Interestingly, we demonstrate that varying the residue at this position modulates the extent of salt dependence, with mutants E65K and E65R, forming cages in full i.e. with no observable dimer remaining in the reactions in absence of Mg2+ while E65Q, E65A and E65D have an intermediate requirement for Mg2+. Structural studies show the importance of the inter-dimeric interface interaction between side chains with possible involvement of divalent metal ions as the basis of the observed assembly characteristics.
2. Materials and methods
2.1. General
DNA modifying enzymes were purchased from New England Biolabs and Thermo Fisher Scientific. DNA primers were purchased from Eurofins and Sigma. E. coli BL21 (λ DE3) was purchased from Agilent Technologies. QIAquick gel extraction kit and QIAprep spin miniprep kits were purchased from Qiagen. Ancillary reagents and EDTA [2,2′,2′′,2′′′-(Ethane-1,2-diyldinitrilo) tetra acetic acid] were purchased from Sigma. All other chemical reagents used were either purchased from Sigma or Fisher Scientific, used as received unless otherwise specified. Water was purified using a Merck Millipore water purification system. Protein concentrations were determined by measuring absorbance at 280 nm using a NanoDrop spectrophotometer (Thermo Scientific).2.2. Cloning and purification of TmFtn mutants
The codon optimised synthetic gene encoding wild type TmFtn (UniProtKB – Q9X0L2 (Q9X0L2_THEMA)) cloned into a pET21a(+) plasmid, was purchased from Genscript (for sequence details see Table ST1†). To investigate the role of E65 in TmFtn assembly we designed several mutants carrying different charges at residue 65, namely E65Q, E65A (charge neutral), E65D (similar charge to wild type, but different side chain length) and E65K, E65R (opposite charge). Point mutation of the TmFtn gene was performed by mutant primers, using site directed mutagenesis, the details of primers are listed in Table ST2.† To check protein expression, the plasmids were transformed into E. coli strain BL21 (λ DE3) and cells were grown in LB medium at 37 °C with 100 mg L−1 ampicillin. Expression of constructs was induced by addition of 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) when the culture reached an optical density (600 nm) of 0.6. After induction, cells were grown for an additional 4 hours. Finally, cells were collected by centrifugation at 7900g (20 minutes) at 4 °C and the pellet kept at −80 °C until use. Subsequently pellet was resuspended in lysis buffer containing 1 μg ml−1 DNase I from bovine pancreas and one tablet of cOmplete™ Protease Inhibitor Cocktail (Roche), and incubated with stirring at room temperature for 30 minutes before employing a microfluidizer, where a constant pressure of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi was applied for complete cell wall and bi-layer disruption. Cell debris was removed via centrifugation at 48000g for 30 minutes. Next, the supernatant was collected from lysed cell samples and subjected to heating at 80 °C for 10 minutes while for E65Q and E65D mutants, heating time was reduced to 8 minutes, then centrifuged at 48000g for 30 minutes before supernatant was again collected and passed through 0.45 μm membrane filter to remove large impurities/aggregates. The supernatant fraction was purified by ion-exchange chromatography on an ÄKTA purifier (GE Healthcare Life Sciences) using HiTrap QFF columns with binding in 20 mM HEPES pH 7.4, 50 mM NaCl buffer and eluting with a 0.05–0.5 M NaCl gradient. The buffers were not supplemented with Mg2+. Fractions containing ferritin were pooled and concentrated using Amicon Ultra 10 or 50 kDa MWCO centrifugal filter units (Millipore) and the sample subjected to size-exclusion chromatography (SEC) on a HiLoad 26/600 Superdex 200 pg column in 20 mM HEPES pH 7.4 at room temperature. The purity of purified protein was assessed using SDS-PAGE (Fig. S1†), while a Superdex 200 increase 10/300 GL was used for SEC elution profile comparison. Protein concentration was measured using a NanoDrop UV–vis spectrometer (Thermo Scientific) using the theoretical extinction coefficient, 29910 M−1 cm−1 and molecular weight 19.4 kDa (calculated from the ExPASy online ProtParam tool).43 Typically, WT-TmFtn remains as a dimer in absence of metal ions and throughout our experiment 20 mM HEPES (pH 7.4) was used as standard buffer. In case of the E65D mutant an additional nonspecific mutation was found at position Ala 42 to Val (A42V).
000 psi was applied for complete cell wall and bi-layer disruption. Cell debris was removed via centrifugation at 48000g for 30 minutes. Next, the supernatant was collected from lysed cell samples and subjected to heating at 80 °C for 10 minutes while for E65Q and E65D mutants, heating time was reduced to 8 minutes, then centrifuged at 48000g for 30 minutes before supernatant was again collected and passed through 0.45 μm membrane filter to remove large impurities/aggregates. The supernatant fraction was purified by ion-exchange chromatography on an ÄKTA purifier (GE Healthcare Life Sciences) using HiTrap QFF columns with binding in 20 mM HEPES pH 7.4, 50 mM NaCl buffer and eluting with a 0.05–0.5 M NaCl gradient. The buffers were not supplemented with Mg2+. Fractions containing ferritin were pooled and concentrated using Amicon Ultra 10 or 50 kDa MWCO centrifugal filter units (Millipore) and the sample subjected to size-exclusion chromatography (SEC) on a HiLoad 26/600 Superdex 200 pg column in 20 mM HEPES pH 7.4 at room temperature. The purity of purified protein was assessed using SDS-PAGE (Fig. S1†), while a Superdex 200 increase 10/300 GL was used for SEC elution profile comparison. Protein concentration was measured using a NanoDrop UV–vis spectrometer (Thermo Scientific) using the theoretical extinction coefficient, 29910 M−1 cm−1 and molecular weight 19.4 kDa (calculated from the ExPASy online ProtParam tool).43 Typically, WT-TmFtn remains as a dimer in absence of metal ions and throughout our experiment 20 mM HEPES (pH 7.4) was used as standard buffer. In case of the E65D mutant an additional nonspecific mutation was found at position Ala 42 to Val (A42V).
2.3. pH and temperature stability
Different pH solutions were prepared from pH 2 to pH 13 using appropriate buffers as listed in Table ST3.† Protein samples 5 μl (6 mg ml−1) were incubated overnight at room temperature with 25 μl of pH solution (total reaction volume 30 μl, final concentration 1 mg ml−1). Protein samples were then run on native PAGE and the same samples were observed under transmission electron microscopy (TEM). Temperature stability tests were performed by incubating protein samples at 95 °C for 30 minutes in a dry heating block.2.4. Transmission electron microscopy
Samples were typically diluted to a final concentration 0.25–0.5 mg ml−1 and TEM images were obtained using a JEOL JEM-1230 microscope operated at an acceleration voltage of 80 kV. Briefly, 3.5 μl of sample was mounted on to glow-discharged copper-Formvar grids (FC400Cu100, EM Resolutions Ltd, UK) and incubated for 1 minute. Excess solution was removed. Subsequently 3.5 μl of uranyl acetate (2%) was added for staining, incubated for 1 minute and finally excess stain was blotted away.2.5. SDS-PAGE gels
Protein samples were mixed with 4× Laemmli sample buffer containing DTT and SDS. Samples were heated in a dry heating block for 10 minutes at 95 °C. After heating, samples were removed and centrifuged at maximum speed for 5 minutes using a bench-top centrifuge. Samples were run and resolved on a 12% Polyacrylamide gel using a constant voltage of 150–180 V for approximately one hour. Gels were stained using InstantBlue protein stain (Expedeon). PageRuler (Thermofisher) pre-stained protein marker was used to indicate the molecular weight of resulting protein bands. A single band at the expected molecular weight of TmFtn (∼20 kDa) was observed for all proteins. (Fig. S1†).2.6. Native-PAGE gels
TmFtn samples (4 μg protein) were run on precast 3–12% native Bis-Tris gels (Life Technologies – Invitrogen). Before running, samples were mixed with 4× native PAGE sample buffer containing 200 mM BisTris, pH 7.2, glycerol, and bromophenol blue, native PAGE sample buffer is devoid of DTT and SDS. The gel was run in a Mini Gel Tank (Life Technologies – Invitrogen) with 500 ml of 1× Novex Tris-Glycine Native Running Buffer at 150 volts constant for 1 hour 45 minutes to complete the electrophoretic migration. NativeMark unstained protein standard (Life Technologies – Invitrogen) was used to estimate the molecular weight of the migrated protein band. InstantBlue protein or Coomassie Brilliant Blue stain (Expedeon) were used for gel staining and visualisation.2.7. Dynamic light scattering (DLS)
DLS measurements were performed on a Malvern Zetasizer Nano ZSP with a temperature controller. Before experiments, samples were centrifuged at 25200g for 10 minutes. The experiments were carried out at room temperature in a sample volume of 50 μl. Buffers were filtered prior to use. All measurements were done in triplicate.
2.8. Circular dichroism (CD) spectropolarimetry
Secondary structural elements of TmFtn and its mutants were determined using a CD spectrophotometer (JASCO). The measurement was carried out at room temperature. The far-UV region was scanned between 190 and 250 nm with an average of three scans and a bandwidth of 1 nm. The final spectra were obtained by subtracting the buffer contribution (control) from the original protein spectra. The resulting spectra were almost identical to wild-type TmFtn, further indicating that proteins were correctly folded (see results section). CD spectra of TmFtn WT and mutants after being subject to different pHs were recorded at 1 mg ml−1 while CD temperature scans were performed at λ 222 nm from 25 °C to 95 °C in the quartz cuvette (pathlength 0.1 cm), measurements were recorded at each 0.2 °C increase.2.9. Iron mineralisation
Ammonium Iron(II) sulphate hexahydrate (Alfa Aesar, Formula weight 392.13 g mol−1) was used to mineralise WT and mutant TmFtn. In brief, 4 mg total protein was taken and 250 μl, 40 mM stock ammonium iron sulphate solution was slowly added at intervals with continuous stirring using a magnetic stirrer. Total volume was maintained at 5 ml using 20 mM HEPES buffer (pH 7.4). Total stirring time was 30–40 minutes. Then the final volume was reduced to 200 μl using a 100 kDa amicon filter and washed 3 times with 20 mM HEPES buffer pH 7.4.2.10. Crystallisation and structure solution
All investigated TmFtn protein variants were crystalised using the sitting drop vapor diffusion method. 1 μl drops of protein solution (10 mg ml−1) were mixed with an equal volume of the reservoir solution containing 1.8 M ammonium sulphate and 0.1 M MES, pH 5.8. No magnesium was added to the samples. Crystals were grown at 20 °C and appeared after 2 days. We also observed crystals of the same protein in alternative conditions such as 10% PEG (4000), 0.1 M MES, pH 5.8, however in this case crystal formation was slower with crystals appearing only after 7 days at 20 °C. Crystals for data collection were soaked in the reservoir solution supplemented with glycerol (20%) as a cryoprotectant and then flash-frozen in liquid nitrogen. Diffraction data were collected on BL 14.1 and BL 14.3 beamlines operated by the Helmholtz–Zentrum Berlin (HZB) at the BESSY II electron storage ring, Berlin-Adlershof, Germany.44 All data were indexed, integrated scaled and merged using XDS package with XDSAPP2.0 graphical user interface.45,46 Molecular Replacement was performed in Phaser using a structure of ferritin (KEGG database entry TM1128) from Thermotoga maritima (PDB ID:1VLG) as a starting model. Structures were refined using Phenix.47 Model building and completion were performed with Coot.48 Structure coordinates were deposited in Protein Data Bank with the accession codes: 6TXN for WT-TmFtn, 6TXK for TmFtn_E65K, 6TXM for TmFtn_E65R, 6TXI for TmFtn_E65A, 6TXL for TmFtn_E65Q, and 6TXJ for TmFtn_E65D.2.11. Computational analysis
For analysis, visualisation, and electrostatic map generation PyMOL (https://pymol.org) and UCSF chimera49 (http://www.rbvi.ucsf.edu/chimera/) were used. T-coffee server50 (https://tcoffee.crg.cat/) was used to generate structure-based multiple sequence alignment. ConSurf server51 (http://consurf.tau.ac.il/index_proteins.php) was used for the cross-species comparison of ferritins. PDBePISA server52 (https://www.ebi.ac.uk/pdbe/pisa/) was used to analyse the inter dimeric interface.2.12. Inflection temperature (Ti) measurement by nanoDSF
Tycho from Nanotemper (https://nanotempertech.com/tycho/) was used to measure the inflection temperature (Ti). 10 μl of protein sample (1 mg ml−1) was loaded into the capillaries, and subject to temperature scan from 25 °C to 95 °C. Inflection temperature (Ti) was measured for the different mutants of TmFtn using manufacturer's software.2.13. Differential scanning calorimetry measurements (DSC)
DSC measurements were carried out using a Nano DSC Series III device with Platinum Capillary Cell (TA Instruments), with an active volume of 0.3 ml. To avoid bubble formation during heating mode, the samples were degassed prior to being loaded by pulling a vacuum of 30–50 kPa on the solution for a period of 10–15 minutes. The sample cell was then filled with 0.3 ml of protein solution (1 mg ml−1 in 20 mM HEPES, pH 7.4) and an equal volume of buffer was used as a reference. The cells were sealed and thermally equilibrated for 10 minutes at the starting temperature of the run. The data were collected at a temperature range of 45–95 °C with a scan rate of 1 °C min−1 for heating. Measurements were carried out in duplicate. Data was analysed using software package provided by the manufacturer.2.14. Mass determination of proteins by mass spectrometry
Samples were analysed using a micrOTOF-Q II mass spectrometer (Bruker Daltonics, Germany) equipped with an electrospray ionisation source. The instrument was calibrated prior to measurements with ESI-L Low Concentration Tuning Mix (Agilent Technologies). Samples at a protein concentration of 1 mg ml−1 in 0.1% formic acid, 49.9% acetonitrile were directly infused into the mass spectrometer with a syringe pump at a flow rate of 6 μl minute−1. The mass spectrometer was operated in positive mode with a spray voltage of 4500 V with dry gas temperature of 180 °C. MS scans were acquired over a mass range of m/z 500–3000. The MS spectra were processed with maximum entropy deconvolution algorithm in Data Analysis 4.1 software (Bruker Daltonics, Germany) (Table ST4†).3. Results
3.1. In silico analysis of TmFtn and mutant design
Sequence alignment of TmFtn with AfFtn confirmed that the two proteins have overall 51% identity and 71% sequence similarity (calculated using online server EMBOSS Water53) despite the differences of domain (bacteria and archaea) and quaternary structure. Secondary structure alignment was performed using the T-coffee server50 (pipeline: proteins-structural alignments (Expresso)-ESPript 3.0 server,54 and AfFtn PDB ID:1S3Q used as input for secondary structure depiction). This analysis showed that residue E65 is conserved in helix B between the two ferritins (Fig. 1A). Whereas comparing large numbers of ferritins from different kingdoms using the ConSurf server,51 shows the same residue to have low conservation (Fig. S2A†). In the crystal structure of wild type TmFtn the subunit interface around E65 appears to be subject to repulsive interactions with E131 and D134 of a neighbouring subunit (Fig. 1B). TmFtn and AfFtn monomer structures were compared by using the DALI server55 followed by structural superimposition (Fig. S2B†). The results clearly show that structurally, E65 lies at an important position around the inter-dimeric interface, and in both protein cages they experience a similar microenvironment as shown by interface analysis by the PDBePISA server,52 (Fig. S2C†). | ||
| Fig. 1 Glutamate 65 is a key residue for TmFtn assembly. (A) Structure based sequence alignment of AfFtn and TmFtn using the T-Coffee server pipeline and EMBOSS water, showing secondary structure similarity and amino acid conservation. Glutamate 65 remains at the terminal of the helix B region which is also conserved between them. (B) Cartoon representation of WT-TmFtn cage and the magnified image (centre) clearly showing the position of glutamate 65 and its interacting partners, where the glutamate 65 residue remains at the dimeric interface (shown as green and blue colour helix), and is strongly repulsed by negative charge residues from neighbouring dimeric subunit residues (glutamate 131 and aspartate 134 shown in grey colour stick representation, while helix is highlighted in red). Rhs shows that the repulsive interaction is converted to an attractive interaction by a point mutation (arginine) at position 65. | ||
Working on the hypothesis that divalent metal ions could play a bridging role between these residues, thus accounting for the observed salt-dependent assembly, we in silico modelled the effect of E65, replacing it with R65 carrying the opposite charge (Fig. 1B). Results suggested that this mutation would result in favourable (salt bridge) interactions, potentially stabilising the interface. Similar predictions were carried out for a range of other mutations (K, A, Q, D) at position 65 which was then experimentally probed.
3.2. Amino acid changes at position 65 modulate salt-dependent assembly
In size-exclusion chromatography using an analytical SEC (Fig. 2A), we observed a single sharp peak for E65R and E65K eluting at ∼9 ml, corresponding to cages. These buffers were not supplemented with (Mg2+). WT TmFtn in absence of metal ions (Mg2+) elutes exclusively as dimers at around 12.5 mL (Fig. 2A). Other mutants apart from E65R and E65K eluted in two peaks, at 9 ml and 12.5 ml indicating a mixture of cages and dimers. The most abundant species in terms of dimer co-elution were WT and the mutant E65D (Fig. 2A and Fig. S1,† see peak at ∼230 ml on the preparative SEC) and E65A and E65Q to a lesser extent (Fig. 2A). For all the nanocages, the earliest eluting peak was collected and subjected to transmission electron microscopy (TEM). For all mutants, cage structures typical of ferritin were observed and cage formation was verified in transmission electron microscopy (TEM) which showed the particle diameter to be approximately 12–14 nm, with high homogeneity (Fig. 2B). In the case of WT, the cages were considerably weaker, and in order to promote full cage formation, 50 mM MgCl2 had to be added to avoid disassembly to dimers (As shown in Fig. 2C and D, WT tends to exist in solution mainly as dimers).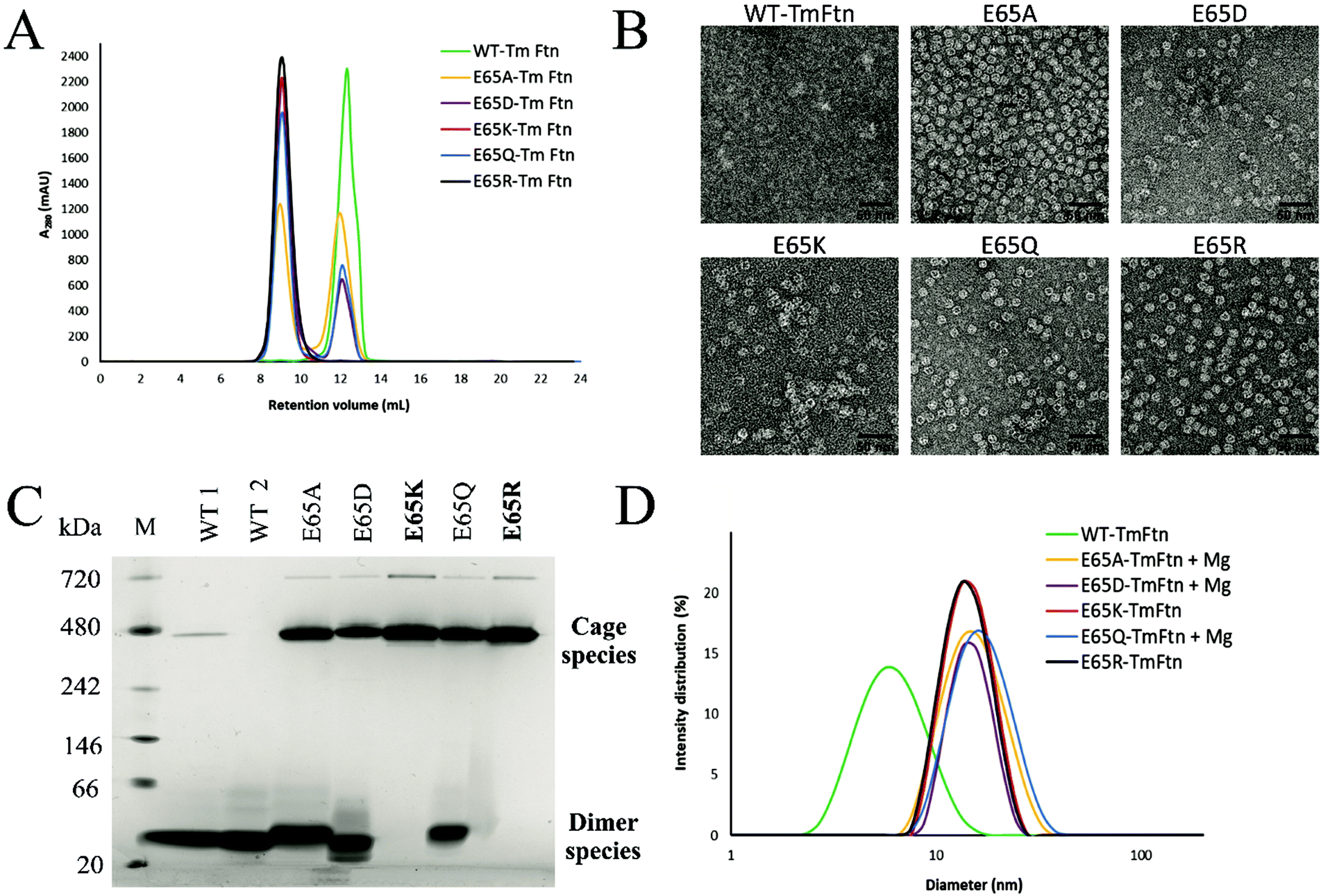 | ||
| Fig. 2 Characterisation and assembly of TmFtn E65 mutants. (A) Size exclusion chromatography (SEC) profile in absence of Mg2+ showing a single peak in the chromatogram for (E65K, E65R) mutants of TmFtn representing cage, whereas E65A, E65Q and E65D mutants show two peaks, one at ∼9 ml (cage) and another at ∼12.5 ml (dimer) representing the unassembled fraction. Wild-type protein shows a single peak at ∼12.5 ml (dimer) in absence of Mg2+. (B) TEM images The first SEC peak at ∼9 ml (cage) was collected and imaged in absence of Mg+2. Results show morphology of different TmFtn mutants and the fact that the particle size of all mutants is the same (∼12 nm). (C) Native PAGE shows typical ferritin like cage assembly bands for E65R, E65K in absence of metal ions whereas in other mutants and wild type TmFtn there are at least two clearly distinguishable protein bands corresponding to presence of assembled (cage) and unassembled (dimer) forms at 480 kDa and ∼40 kDa, respectively. Lane 1, protein marker; lane 2, wild type sample 1; lane 3, wild type sample 2 (all in dimer species); lanes 4–8, different TmFtn mutants. (D) TmFtn mutant cage assembly analysed by DLS in the absence and presence of Mg2+. Wild type TmFtn in absence of Mg2+ shows a diameter ∼8 nm indicating dimer while E65K and E65R in absence of Mg2+ shows a diameter ∼15 nm indicating cage. E65A, E65Q and E65D in the presence of minimal Mg2+ (10 mM) are homogeneous and show single peaks around ∼15 nm confirming complete cage formation. | ||
To identify the second eluting peak (Fig. S1†), we collected it in the case of E65D and carried out dynamic light scattering (DLS) analysis. This showed a lack of the expected peak (∼15 nm) corresponding to unassembled cages (Fig. S3A†). The cage structure could be recovered by the addition of 10 mM MgCl2 as demonstrated by both SEC and TEM (Fig. S3B†). To further verify the SEC data all TmFtn samples were subjected to native-PAGE analysis in the absence of MgCl2, which shows that E65K and E65R exclusively remained as cages, with an identifying prominent protein band around ∼480 kDa (Fig. 2C). In addition, we also observed another band close to ∼720 kDa, most probably corresponding to a dimer of ferritin cages, as is commonly found in other ferritins.56 The remaining mutant proteins showed at least three protein bands corresponding to presumed cage dimer (∼720 kDa); cage (∼480 kDa) and ferritin subunit dimer (∼40 kDa). To try to promote complete assembly, 10 mM Mg2+ was added to E65A, E65Q and E65D which were then subjected to DLS analysis. In the presence of Mg2+ they all showed a single peak around ∼15 nm diameter, indicating complete cage formation (Fig. 2D and Fig. S4†). Overall, the cage forming ability of different TmFtn mutants in absence of Mg2+ is as follows E65R ∼E65K > E65A ∼E65Q > E65D > WT.
3.3. Structural and thermal stability of mutant TmFtn cages
We previously showed that TmFtn can withstand a broad range of pHs (pH 5–10) and thermal stress, being apparently stable even at 95 °C for 30 minutes.28 Here we tested the stability of TmFtn mutants in the absence of Mg2+ as a function of pH and temperature by native PAGE and TEM (Fig. 3A–C). After purification, confirmation of correct size and folding of the proteins was carried out using SDS PAGE and CD (Fig. S1, S5 and ESI Table ST5†). In native PAGE the cage complex runs as a discrete band, any irreversible disassembly would be observable as the loss of this band and appearance of aggregates. Disassembly into smaller subunits would result in appearance of smaller molecular weight bands. All mutants showed the characteristic band corresponding to cages which appeared stable even after exposure to pH 2–10 and heating at 95 °C for 30 minutes. Additionally, mutants E65K and E65R seem resistant to the effects of EDTA even at 100 mM, confirming that the metal ion is unnecessary for cage formation in these two mutants. For other mutants and WT, some dependence on Mg2+ was observed with order of least to greatest dependence being E65Q ∼ E65A < E65D < WT (Fig. 3, S4). These results are based on conditions in which the band corresponding to cages was still observed on native PAGE. It is possible that the cages disassemble into constituent subunits in solution but then reassemble in the more favourable conditions of the native PAGE gel/buffer. Lack of such denaturation at high temperatures was confirmed by TEM experiments (Fig. 3C). The effect of pH was further addressed both by the TEM experiments which were carried out without change of buffer (Fig. 3B) and circular dichroism experiments over the range of pHs tested (Fig. S5A and B†). These results were in line with the CD results suggesting that in the mutant nanocages quaternary structure is completely lost only at pH 13 (Fig. S5A† and Fig. 3A). | ||
| Fig. 3 E65 mutants of TmFtn in absence of Mg2+ show high stability at different pHs and temperatures. (A) Native PAGE shows that E65 mutants of TmFtn at varying pH conditions (pH 2–13) run as expected for cages. Cage formation is not due to residual Mg2+ as shown by results after 100 mM EDTA treatment. (B) TEM images correspond to different TmFtn mutants incubated at two extreme pH conditions, pH 4 and 10. Even over such a broad pH range all the mutants (particularly E65R and E65K) retain their structural integrity. (C) Native PAGE shows retention of cage like architecture in E65 mutants after heating the sample at 95 °C for 30 minutes, the same samples were subjected to TEM analysis. TEM images also confirm retention of cage like structure after significant heat shock (all experiments performed in the absence of Mg2+). | ||
Native PAGE results revealed that mutant nanocages E65Q, E65D and E65A had a significantly smaller molecular weight band ∼40 kDa in addition to the band corresponding to cage over all pHs tested (Fig. 3A). This is consistent with the SEC results and is suggestive of an equilibrium between assembled cages and dimers. E65A and E65Q showed a much fainter smaller molecular weight band than E65D in both pH-varying and temperature-varying native PAGE gels. The stability of E65D was also measured using nano-DSC as this mutant appeared to form the least stable nanocage assembly. The results clearly showed at least two transitions with a major peak appearing at around 82.4 °C (Fig. S6 and S7†), significantly lower than the known denaturation temperature of horse spleen ferritin (∼93 °C57), the result was also in good agreement with CD measurements carried out at different temperatures (Fig. S6 and S7†). Having confirmed that the mutations at position 65 were able to affect stability for one mutant, we carried out a more comprehensive series of tests using nano-DSF which allowed a faster throughput. The results showed that TmFtn E65K and E65R are more stable compared to other three mutants (E65A, E65Q and E65D) (Table ST4†), in addition they can also assemble without salt/metal ions, like classical ferritins. E65A, E65Q and E65D showed low to intermediate/partial dependence of assembly on Mg2+.
3.4. Crystal structures of TmFtn mutants
To understand the detailed assembly process as well as to gain insights into effects on stability, we crystallised all TmFtn mutant variants. Crystals diffracted in the resolution range 1.8–2.5 Å. Crystal structures were solved using molecular replacement and deposited at the Worldwide Protein Data Bank (wwPDB), the details of data collection and refinement statistics with PBD IDs are shown in Table ST6.† The WT protein and all the mutants were crystallised in a rhombohedral space group of R32 with eight subunits in an asymmetric unit. TmFtn mutant monomers possess a typical ferritin-like four-helix bundle structural arrangement, consisting of 4 + 1 α-helices (four long helices (denoted as A to D)) and one short helix (E) (Fig. S8†). Detailed analysis reveals that they are typical ferritin structures, being oligomers composed of 24 subunits, with a cage-like architecture, possessing overall octahedral symmetry along with 3 and 4-fold axes of symmetry like canonical/maxi ferritin. Furthermore, eight 3-fold and six 4-fold symmetry channels were found to be common in all mutant crystal structures (Fig. 4A). Mutant and wild type proteins were compared and found to be structurally highly similar with a RMSDs between 0.26 Å for E65R and 0.31 Å in case of E65D (Table ST7†). Superimposition of mutant dimers further confirms that all the mutants are very similar to wild type TmFtn (Fig. 4B). As the channels are composed of identical residues (Table ST7†) the electrostatic potential values across the channel are the same in all mutants. We also measured the surface electrostatic potential inside and outside the lumen of wild type and mutant ferritins and they are comparable (−10 to +10), the only difference in electrostatic potential was observed at the inter-dimeric interface among different mutants (Fig. 4C). | ||
| Fig. 4 Crystal structure of TmFtn E65 mutants (A) Bottom: Cartoon representation of TmFtn cage where specific residues were represented as sticks, viewed along the 3-fold and 4-fold channels. Close-ups show the 3 and 4-fold channels. (B) Structural superimposition of WT and all mutant variant (dimer) structures of TmFtn. The low RMSD indicates no structural changes (Table ST7†). (C) Surface electrostatic representation of the WT-TmFtn dimer (left) and E65R mutant (right). The position of E65 is marked by a black circle. (D) A close -up image of the metal (Fe, orange sphere) binding core site shown in a TmFtn monomer helix (golden colour) with binding residues shown in stick representation. (E) Crystal structure of WT and mutant TmFtn and respective interacting partners at the dimeric interface, one monomer shown in green bears residue E65 (highlighted, shown in stick representation). The second monomer subunit is shown in blue with the position of residues interacting with residue 65 highlighted and represented as grey sticks. Interaction shown by dotted line between participating residues. All figures were generated using respective crystal structures. | ||
Ferritins including WT-TmFtn are known for their iron mineralisation ability. According to the crystal structure, individual TmFtn monomers consist of an iron (Fe) binding pocket known as site A.58 Site A is mainly constituted of two glutamic acid residues (E19 and E52) and one histidine residue (H55) (Fig. 4D and Fig. S8, S9†). As expected, the metal binding site was also found to be highly conserved among different TmFtn mutants (Fig. S9†). Further it was observed that the mineralisation site was located inside the core of the four-helix bundle and was distant (∼18 Å) from E65 residue. Though this does not negate the possibility of allosteric effect of mutations upon interaction with iron. This was ruled out by superimposition of mutant structures with the WT structure which showed no notable deviations (Fig. S8, S9 and ESI Table ST7†). This was further confirmed by iron mineralisation experiments in which purified mutant ferritin appeared as light brown in colour clearly indicating successful mineralisation. Iron absorbance was also detected at 380 nm and seen as coelution in SEC profile while the presence of an electron dense core was further confirmed by transmission electron microscopy (Fig. S10†). In all mutant crystal structures, an elongated electron density was found in the dimeric unit, most probably corresponding to the presence of fatty acid as previously been noted.59
4. Discussion
Metal ions are known to play significant roles in naturally occurring protein assemblies most notably as catalytic centres. Recently the concept of using metals not as catalysts but to direct protein self-assembly has been developed.60,61 A number of protein cages held together by such metal-protein interactions, wherein metals are located at well-defined positions, have been demonstrated.23,62,63In the case of TmFtn the situation appears more complex compared to designed protein cages: Residue E65 in the wild type protein appears to be strongly repulsed by two other neighbouring negatively charged residues, glutamate 131 and aspartate 134 (Fig. 1B and 4E). It seems likely that this repulsive interaction is sufficiently strong such that it cannot be overcome by opposing interactions across the dimer ensuring that, in absence of metal ions, WT-TmFtn does not assemble into the 24mer cage. An obvious explanation for the Mg2+-dependent assembly is that it acts to bridge these opposing negatively charged residues at the inter-dimer interface. This seems to be supported by the finding that replacing E65 with positively charged residues allows assembly in the absence of metal ions through salt bridge formation (Fig. 4E). Repulsive interactions among dimeric interfaces were also found to be important in E. coli non-haem-binding ferritin A (EcFtA), whose assembly was facilitated by increasing ionic strength.64 However, in the solved crystal structures of wild type TmFtn we could not detect evidence of Mg2+ near the inter-dimeric interface, close to E65. In many crystals it is difficult to distinguish Mg2+ ions from H2O. Therefore, we tried to substitute Mg2+ with other divalent metal ions but results were unable to provide any conclusive answer regarding a fixed Mg2+ ion position. Overall our results are consistent with those previously reported for AfFtn which also showed that the E65R mutation abolished salt dependent assembly resulting in a more stable complex41 although in this case the change was accompanied by a major rearrangement in which an open pore tetrahedral structure was converted into a closed pore octahedral structure. The AfFtn structures also showed no evidence of bound metal ions (Mg2+/Na+) in the vicinity of E65.41 In addition, the high structural similarity between different TmFtn mutants points to a lack of a significant allosteric effect stemming from changes at position 65. These data clearly suggest that a more subtle mechanism is responsible for TmFtn assembly. A summary of the effect of mutations at position 65 on the Mg2+ dependence of cage formation is given in Fig. 5.
 | ||
| Fig. 5 Schematic summary showing assembly requirements of different E65 mutants of TmFtn. TmFtn dimer (wild type) assembles to cage in presence of divalent metal ions (50 mM MgCl2) For TmFtn mutants E65K and E65R, no divalent cationic species is required for cage assembly, whereas in case of the other three mutants (E65A, E65Q and E65D) a lower concentration (10 mM Mg2+) of divalent cationic species is required to push the equilibrium of dimer and cage fully to cage. Figure prepared from the crystal structure (PDB ID: 1VLG). | ||
One possibility is that Mg2+ simply acts to initiate complex formation with the formed cage having sufficient additional stability such that loss of the metal ion does lead to cage disassembly. Alternatively, Mg2+ may have a more general, low affinity charge shielding effect allowing inter-dimer interaction to occur in the WT protein and that mutations to reverse the charge at position 65 provide an alternative interaction of sufficient strength to render this dispersed effect redundant. The fact that a neutral residue at the same position results in a protein able to form the 24mer cage at intermediate Mg2+ concentrations supports the idea of a more dispersed role for the metal ions in neutralising other repulsive interactions elsewhere in the complex. If so, we may expect other cations to potentially exert a similar effect. Recent work by Zhang et al.65 has shown structures of TmFtn cages formed in the presence of Mg2+ or Ca2+. Here the magnesium ion is coordinated by residues E51, E128 and E132 placing it close to the iron binding site (PDB ID 6LS3). It is also notable that ammonium sulphate is present in the crystallisation conditions suggest where it may be able to substitute for the action of Mg2+. Indeed, it can induce cage formation as seen in DLS (Fig. S11†) This would be unsurprising considering that it is present at a considerable concentration (1.8 M).
5. Conclusion
In this study we identified important key residues which are responsible for TmFtn assembly. We showed that we may tune the dependence of TmFtn cage assembly on Mg2+ concentration, achieving versions having no, intermediate or high dependence, with different versions having different stability characteristics. The ability to fine-tune TmFtn assembly via point mutations may prove a useful first step towards producing ferritins/protein cages able to capture/release useful cargoes under mild conditions.Author contributions
MK, JMM and JDJO carried out experiments, analysed data and wrote the manuscript, PW and PG collected X-ray diffraction data and solved the crystal structures, APB assisted in solving the crystal structures and overseeing experimental, MJR carried out DSC experiments and analysed DSC results, MK carried out CD experiments and AG analysed CD results, SC conceived and carried out experiments, analysed data, oversaw the work and wrote the manuscript, JGH oversaw the work and wrote the manuscript. All authors contributed to writing the manuscript.Data availability
All the data are described in Tables ST1–ST7 and Fig. S1–S11.† Crystallography data and deposited PDB IDs are listed in Table ST6 of the ESI.†Conflicts of interest
The authors declare the following competing financial interests: J.G.H. is the founder of and holds equity in nCage Therapeutics LLC, which aims to commercialise protein cages for therapeutic applications.Acknowledgements
J. G. H., J. D. J. O. and J. M. M. were funded by the Team Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM/2016-3/23). S. C. and M. K. were supported by the HOMING program (HOMING/2017-3/22) of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund awarded to S. C. S. C. also thankfully acknowledges the Ramalingaswami Fellowship (BT/RLF/Re-entry/09/2019) received from the Department of Biotechnology, India. Measurements of circular dichroism were carried out at the Department of Physical Biochemistry, Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University. We acknowledge Proteomics and Mass Spectrometry Core Facility of the Małopolska Centre of Biotechnology for protein mass determination and the MCB Structural Biology Core Facility (supported by the TEAM TECH CORE FACILITY/2017-4/6 grant from the Foundation for Polish Science) for valuable support. We also thank Olga Woźnicka of Institute of Zoology, UJ for TEM imaging. We thank HZB for the allocation of synchrotron radiation beamtime (supported from the European Union's Horizon 2020 research and innovation programme under grant agreement no 730872). The open-access publication of this article was funded by the BioS Priority Research Area under the program “Excellence Initiative – Research University” at the Jagiellonian University in Krakow.Notes and references
- N. F. Steinmetz, S. Lim and F. Sainsbury, Biomater. Sci., 2020, 8, 2771–2777 RSC.
- B. J. G. E. Pieters, M. B. Van Eldijk, R. J. M. Nolte and J. Mecinović, Chem. Soc. Rev., 2016, 45, 24–39 RSC.
- M. L. Flenniken, M. Uchida, L. O. Liepold, S. Kang, M. J. Young and T. Douglas, in Viruses and Nanotechnology, ed. M. Marianne and N. F. Steinmetz, Springer, Berlin, Heidelberg, 2009, vol. 327, pp. 71–93 Search PubMed.
- T. G. W. Edwardson and D. Hilvert, J. Am. Chem. Soc., 2019, 141, 9432–9443 CrossRef CAS PubMed.
- N. F. Steinmetz, EMBO Rep., 2019, 20, e48806 CrossRef PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- M. Rother, M. G. Nussbaumer, K. Renggli and N. Bruns, Chem. Soc. Rev., 2016, 45, 6213–6249 RSC.
- N. M. Molino and S.-W. Wang, Curr. Opin. Biotechnol., 2014, 28, 75–82 CrossRef CAS PubMed.
- D. Diaz, A. Care and A. Sunna, Genes, 2018, 9, 370 CrossRef PubMed.
- I. Stupka and J. G. Heddle, Curr. Opin. Struct. Biol., 2020, 64, 66–73 CrossRef CAS PubMed.
- J. G. Heddle, S. Chakraborti and K. Iwasaki, Curr. Opin. Struct. Biol., 2017, 43, 148–155 CrossRef CAS PubMed.
- S. Chakraborti, T.-Y. Lin, S. Glatt and J. G. Heddle, RSC Adv., 2020, 10, 13293–13301 RSC.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- B. Bhushan, S. U. Kumar, I. Matai, A. Sachdev, P. Dubey and P. Gopinath, J. Biomed. Nanotechnol., 2014, 10, 2950–2976 CrossRef CAS PubMed.
- S. Chakraborti and P. Chakrabarti, in Advances in Experimental Medicine and Biology, ed. S. Perrett, A. Buell and T. Knowles, Springer, Singapore, 2019, vol. 1174, pp. 313–329 Search PubMed.
- P. M. Harrison, Biochem. Educ., 1986, 14, 154–162 CrossRef CAS.
- Y. Zhang and B. P. Orner, Int. J. Mol. Sci., 2011, 12, 5406–5421 CrossRef CAS PubMed.
- Y. Jin, J. He, K. Fan and X. Yan, Nanoscale, 2019, 11, 12449–12459 RSC.
- S. Kang, P. A. Suci, C. C. Broomell, K. Iwahori, M. Kobayashi, I. Yamashita, M. Young and T. Douglas, Nano Lett., 2009, 9, 2360–2366 CrossRef CAS PubMed.
- K. Li, Z. P. Zhang, M. Luo, X. Yu, Y. Han, H. P. Wei, Z. Q. Cui and X. E. Zhang, Nanoscale, 2012, 4, 188–193 RSC.
- J. Swift, W. A. Wehbi, B. D. Kelly, X. F. Stowell, J. G. Saven and I. J. Dmochowski, J. Am. Chem. Soc., 2006, 128, 6611–6619 CrossRef CAS PubMed.
- T. A. Cornell, J. Fu, S. H. Newland and B. P. Orner, J. Am. Chem. Soc., 2013, 135, 16618–16624 CrossRef CAS PubMed.
- D. J. E. Huard, K. M. Kane and F. A. Tezcan, Nat. Chem. Biol., 2013, 9, 169–176 CrossRef CAS PubMed.
- R. M. Kramer, C. Li, D. C. Carter, M. O. Stone and R. R. Naik, J. Am. Chem. Soc., 2004, 126, 13282–13286 CrossRef CAS PubMed.
- M. T. Klem, J. Mosolf, M. Young and T. Douglas, Inorg. Chem., 2008, 47, 2237–2239 CrossRef CAS PubMed.
- S. Tetter and D. Hilvert, Angew. Chem., Int. Ed., 2017, 56, 14933–14936 CrossRef CAS PubMed.
- S. Deshpande, N. D. Masurkar, V. M. Girish, M. Desai, G. Chakraborty, J. M. Chan and C. L. Drum, Nat. Commun., 2017, 8, 1442 CrossRef PubMed.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stępień, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, C. Guo, H. Chen, X. Lin, G. Liu, B. Fei, X. Chen, B. Xu and J. Xie, ACS Nano, 2013, 7, 6988–6996 CrossRef CAS PubMed.
- P. Huang, P. Rong, A. Jin, X. Yan, M. G. Zhang, J. Lin, H. Hu, Z. Wang, X. Yue, W. Li, G. Niu, W. Zeng, W. Wang, K. Zhou and X. Chen, Adv. Mater., 2014, 26, 6401–6408 CrossRef CAS PubMed.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- L. Li, M. Muñoz-Culla, U. Carmona, M. P. Lopez, F. Yang, C. Trigueros, D. Otaegui, L. Zhang and M. Knez, Biomaterials, 2016, 98, 143–151 CrossRef CAS PubMed.
- M. Kim, Y. Rho, K. S. Jin, B. Ahn, S. Jung, H. Kim and M. Ree, Biomacromolecules, 2011, 12, 1629–1640 CrossRef CAS PubMed.
- X. Liu, W. Jin and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3653–3658 CrossRef CAS PubMed.
- R. Yang, Y. Liu, D. Meng, Z. Chen, C. L. Blanchard and Z. Zhou, J. Agric. Food Chem., 2017, 65, 1410–1419 CrossRef CAS PubMed.
- R. Yang, Y. Liu, C. Blanchard and Z. Zhou, Food Chem., 2018, 240, 935–939 CrossRef CAS PubMed.
- E. Johnson, D. Cascio, M. R. Sawaya, M. Gingery and I. Schröder, Structure, 2005, 13, 637–648 CrossRef CAS PubMed.
- J. Swift, C. A. Butts, J. Cheung-Lau, V. Yerubandi and I. J. Dmochowski, Langmuir, 2009, 25, 5219–5225 CrossRef CAS PubMed.
- B. Sana, E. Johnson, P. Le Magueres, A. Criswell, D. Cascio and S. Lim, J. Biol. Chem., 2013, 288, 32663–32672 CrossRef CAS PubMed.
- B. Sana, E. Johnson and S. Lim, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 2544–2551 CrossRef CAS PubMed.
- K. W. Pulsipher, J. A. Villegas, B. W. Roose, T. L. Hicks, J. Yoon, J. G. Saven and I. J. Dmochowski, Biochemistry, 2017, 56, 3596–3606 CrossRef CAS PubMed.
- P. Ceci, E. Forte, G. Di Cecca, M. Fornara and E. Chiancone, Extremophiles, 2011, 15, 431–439 CrossRef CAS PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, in The Proteomics Protocols Handbook, ed. J. M. Walker, Humana Press, Totowa, NJ, 2005, pp. 571–607 Search PubMed.
- U. Mueller, R. Förster, M. Hellmig, F. U. Huschmann, A. Kastner, P. Malecki, S. Pühringer, M. Röwer, K. Sparta, M. Steffien, M. Ühlein, P. Wilk and M. S. Weiss, Eur. Phys. J. Plus, 2015, 130, 141 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- K. M. Sparta, M. Krug, U. Heinemann, U. Mueller and M. S. Weiss, J. Appl. Crystallogr., 2016, 49, 1085–1092 CrossRef.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D, 2010, 66, 486–501 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- P. Di Tommaso, S. Moretti, I. Xenarios, M. Orobitg, A. Montanyola, J. M. Chang, J. F. Taly and C. Notredame, Nucleic Acids Res., 2011, 39, W13–W17 CrossRef CAS PubMed.
- H. Ashkenazy, S. Abadi, E. Martz, O. Chay, I. Mayrose, T. Pupko and N. Ben-Tal, Nucleic Acids Res., 2016, 44, W344–W350 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS PubMed.
- T. F. Smith and M. S. Waterman, J. Mol. Biol., 1981, 147, 195–197 CrossRef CAS PubMed.
- P. Gouet, E. Courcelle, D. I. Stuart and F. Métoz, Bioinformatics, 1999, 15, 305–308 CrossRef CAS PubMed.
- L. Holm and P. Rosenström, Nucleic Acids Res., 2010, 38, W545–W549 CrossRef CAS PubMed.
- B. Subhadarshanee, A. Mohanty, M. K. Jagdev, D. Vasudevan and R. K. Behera, Biochim. Biophys. Acta - Proteins Proteomics, 2017, 1865, 1267–1273 CrossRef CAS PubMed.
- S. Stefanini, S. Cavallo, C. Q. Wang, P. Tataseo, P. Vecchini, A. Giartosio and E. Chiancone, Arch. Biochem. Biophys., 1996, 325, 58–64 CrossRef CAS PubMed.
- A. Treffry, Z. Zhao, M. A. Quail, J. R. Guest and P. M. Harrison, Biochemistry, 1997, 36, 432–441 CrossRef CAS PubMed.
- S. Zanzoni, K. Pagano, M. D'Onofrio, M. Assfalg, S. Ciambellotti, C. Bernacchioni, P. Turano, S. Aime, L. Ragona and H. Molinari, Chem. – Eur. J., 2017, 23, 9879–9887 CrossRef CAS PubMed.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- A. S. Cristie-David and E. N. G. Marsh, Protein Sci., 2019, 28, 1620–1629 CrossRef CAS PubMed.
- A. D. Malay, N. Miyazaki, A. Biela, S. Chakraborti, K. Majsterkiewicz, I. Stupka, C. S. Kaplan, A. Kowalczyk, B. M. A. G. Piette, G. K. A. Hochberg, D. Wu, T. P. Wrobel, A. Fineberg, M. S. Kushwah, M. Kelemen, P. Vavpetič, P. Pelicon, P. Kukura, J. L. P. Benesch, K. Iwasaki and J. G. Heddle, Nature, 2019, 569, 438–442 CrossRef CAS PubMed.
- E. Golub, R. H. Subramanian, J. Esselborn, R. G. Alberstein, J. B. Bailey, J. A. Chiong, X. Yan, T. Booth, T. S. Baker and F. A. Tezcan, Nature, 2020, 578, 172–176 CrossRef CAS PubMed.
- D. Sato, S. Takebe, A. Kurobe, H. Ohtomo, K. Fujiwara and M. Ikeguchi, Biochemistry, 2016, 55, 482–488 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, B. Zheng, J. Zang, C. Lv, T. Zhang, H. Wang and G. Zhao, Res. Square, 2021 DOI:10.21203/rs.3.rs-170719/v1.
Footnotes |
| † Electronic supplementary information (ESI) available: Protein purification and characterization, bioinformatic analysis of TmFtn, DLS autocorrelation curve, thermal stability (DSC), iron mineralisation, circular dichroism analysis (Fig. S1–S11) and TmFtn gene, primer sequence, and buffers, physiochemical data, crystallographic data table, structure superimposition and metal binding core, (Tables ST1–ST7). See DOI: 10.1039/d1nr01632f |
| ‡ These authors contributed equally. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |