
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From spherical compartments to polymer films: exploiting vesicle fusion to generate solid supported thin polymer membranes†
Myrto
Kyropoulou
,
Saziye
Yorulmaz Avsar
,
Cora-Ann
Schoenenberger
 ,
Cornelia G.
Palivan
* and
Wolfgang. P.
Meier
*
,
Cornelia G.
Palivan
* and
Wolfgang. P.
Meier
*
Department of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058 Basel, Switzerland. E-mail: wolfgang.meier@unibas.ch; cornelia.palivan@unibas.ch
First published on 30th March 2021
Abstract
Solid supported polymer membranes as scaffold for the insertion of functional biomolecules provide the basis for mimicking natural membranes. They also provide the means for unraveling biomolecule–membrane interactions and engineering platforms for biosensing. Vesicle fusion is an established procedure to obtain solid supported lipid bilayers but the more robust polymer vesicles tend to resist fusion and planar membranes rarely form. Here, we build on vesicle fusion to develop a refined and efficient way to produce solid supported membranes based on poly(dimethylsiloxane)-poly(2-methyl-2-oxazoline) (PMOXA-b-PDMS-b-PMOXA) amphiphilic triblock copolymers. We first create thiol-bearing polymer vesicles (polymersomes) and anchor them on a gold substrate. An osmotic shock then provokes polymersome rupture and drives planar film formation. Prerequisite for a uniform amphiphilic planar membrane is the proper combination of immobilized polymersomes and osmotic shock conditions. Thus, we explored the impact of the hydrophobic PDMS block length of the polymersome on the formation and the characteristics of the resulting solid supported polymer assemblies by quarz crystal microbalance with dissipation monitoring (QCM-D), atomic force microscopy (AFM) and spectroscopic ellipsometry (SE). When the PDMS block is short enough, attached polymersomes restructure in response to osmotic shock, resulting in a uniform planar membrane. Our approach to rapidly form planar polymer membranes by vesicle fusion brings many advantages to the development of synthetic planar membranes for bio-sensing and biotechnological applications.
Introduction
Solid supported synthetic membranes play an important role in many scientific and technological processes. Due to their enhanced mechanical stability and lifetimes,1–5 these platforms, as a whole, are ideal candidates for complex applications such as for sensing specific biomolecules, studying physical membrane properties, and insertion of transmembrane proteins into synthetic mebranes.1–5 In practice, these platforms consist of an ultra-thin synthetic membrane (10–20 nm), self-assembled from amphiphilic molecules such as phospholipids or block copolymers, which is attached to a solid substrate, e.g. coated or bare silica, glass, and mica. Solid supported membranes are attractive for research and engineering mainly for two reasons. First, they can serve as matrix for mimicking natural membranes, offering a valuable tool to investigate the molecular mechanisms which underly the structure and function of natural membranes.6–11 Second, the fact that they are solid supported significantly enhances their stability and thus facilitates the engineering of biosensors, optoelectronics, coatings, tools for bio-analysis and microfluidic devices.4,12–18 Vesicle fusion is an established method for fabricating solid supported lipid membranes.7,8,19–21 Once the lipid vesicles adsorbing to the support reach a critical surface coverage, they spontaneously rupture and rearrange themselves into a lipid bilayer. While the latter process is extensively studied,7,8,19–21 there is limited information about bilayer formation with polymers, i.e., via polymer vesicle fusion or polymersome fusion. Initial attempts of charge mediated polymersome fusion on a solid support as well as some theoretical studies have been reported.22–24 Moreover, recent studies presented the polymerzation-induced (PISA) polymersome fusion and controlled fusion to induce tetrapod polymersome assembly.25,26 In the case of surface adsorbed polymer vesicles, rupture does not seem to happen spontaneously. Even though they share self-assembly principles with liposomes and also have a vesicular architecture, polymersomes have their thicker membranes (3–40 nm (ref. 20)) and thus differ in their physicochemical properties and behaviour20,27,28 which hampers the fabrication of planar polymer membranes via vesicle fusion. Alternatively, the Langmuir–Blodgett method is the most widely used approach for forming polymer membranes on solid supports.28–32 However, this method has considerable limitations: (i) it requires special equipment, (ii) it is time consuming, (iii) the creation of polymer bilayers is not necessarily straightforward, and (iv) residual organic solvent often remains in the membrane with adverse effects on biomolecules. Accordingly, creating a solid supported polymer membrane via induced-polymersome fusion, albeit challenging, is worth striving for.In the present study, we developed an improved strategy that overcomes inherent challenges of polymersome fusion; by considering the polymers’ physical properties and using an osmotic shock to trigger membrane formation, solid supported polymer membranes are created in a more reliable fashion. We consider our approach particularly advantageous for the following reasons: (i) it is refined and fast (ii) does not require specific equipment and it is compatible with a broad range of surface characterization methods and (iii) takes place under fully aqueous conditions which is essential for bio-applications. The motivation behind our study is the emerging potential of planar membranes for diverse applications, for example as biosensors, surface coatings, or biomimetic platforms that allow the study of membrane associated biomolecules.
For this purpose, we formed polymersomes from a small library of triblock copolymers composed of poly(2-methyl-2-oxazoline) (PMOXA, henceforth referred to as A block) and PDMS (B block) as the hydrophilic and hydrophobic blocks, respectively. We selected a small library of PMOXA-PDMS-PMOXA copolymers to test our approach because these amphiphilic copolymers have been already reported to generate planar membranes by other methods (e.g. Langmuir Blodgett method,32etc.). In addition, the membrane flexibility of such amphiphilic copolymers has been reported to be a key factor in enabling various bio-applications.20,27
Different triblock copolymers were mixed with a thiol-bearing ABA block copolymer and self-assembled in the presence of Ca2+, which is known to play an important role in Solid Supported Lipid Bilayer (SLB).8,33–35 To unravel the influence of the hydrophobic block length and thus, the thickness of the bounding membrane on the rupture of the polymersomes and subsequent planar membrane formation, we compared five triblock copolymers with a similar hydrophilic to hydrophobic block ratio but hydrophobic block lengths (units) ranging from 22 to 65 units. Specifically, we used: A3B22A3, A6B34A6, A7B42A7, A6B44A6 and A6B65A6, with thiol-bearing A18B47A18SH mixed in always at the same molar ratio to produce polymersomes that expose thiol groups on the outer membrane and contain high concentrations of Ca2+ ions in the aqueous cavity. We then attached the polymersomes on a gold coated substrate via gold–thiol36–39 chemistry and induced an osmotic shock to trigger the rupture of the polymersomes and the subsequent formation of a solid supported polymer membrane. We chose gold as substrate because besides enabling gold–thiol chemistry it is a material which is widely applied in material science, biosensing, electrochemical sensors and as platform for catalysis.40–47 To properly characterize our system, we first analysed self-assembly of the thiol-bearing polymersomes at a constant Ca2+ concentration (100 mM) and subsequently we examined the formation of solid supported membranes starting from surface-attached polymersomes. To monitor membrane formation, we used a combination of quartz crystal microbalance with dissipation (QCM-D) monitoring and atomic force microscopy (AFM). By probing the process of induced polymersome fusion, we present an efficient setup which after controlling: (i) the attachment of the polymersomes on the substrate and (ii) the osmotic shock conditions, leads to a planar polymer membrane formation. Particular emphasis was placed on studying and systematically characterizing the induced formation of solid supported polymer membranes as a versatile tool that provides a basis for various bioanalytical applications.
Materials and methods
PMOXA6-PDMS65-PMOXA6 was purchased by Polymer Source Inc.; gold coated silica wafers from Sigma Aldrich, gold coated QCM-D sensors from Q-sense; calcium chloride (CaCl2) from Merck. All chemicals were purchased in highest purity and used as received.Polymer synthesis and characterization
The block copolymers A3B22A3, A6B34A6, A6B44A6 and A7B42A7, the thiol-terminated PMOXA18-PDMS47-PMOXA18 were synthesized, fractionated with cosolvent fractionation and characterized as reported before.48 The thiol-bearing PMOXA18-PDMS47-PMOXA18SH was mixed at different molar ratios with the non-functionalized AxByAx block copolymers.Polymersome formation
Polymersome formation and purification were done according to the established film rehydration method.49,50 Briefly, 5 mg of the AxByAx amphiphilic triblock copolymers with or without 10% thiol-terminated A18-B47-A18 was dissolved in 1 mL of EtOH in a round-bottom glass flask and dried to a thin film on a rotary evaporator. The polymer film was rehydrated Ca2+ enriched HEPES buffer: with 1 mL of 10 mM HEPES, 100 mM CaCl2, pH 7.4 and stirred (∼300 rpm) overnight at room temperature. The turbid solution was extruded 21 times through a 0.2 μm Nucleopore Track-Etch membrane (Whatman) using a 1 mL syringe extruder (Avanti Polar Lipids, USA.Dynamic light scattering
Polymersomes obtained were characterized by dynamic light scattering measurements (DLS) and measured in a Zetaziser Nano (Malvern) at 25 °C equipped with a HeNe laser (λ = 633 nm). The samples were diluted ten times in 100 Ca2+ enriched HEPES buffer and left to equilibrate for 120 s. The data were fitted using the CONTIN method and averaged over up to 15 individual measurements. The same samples were then recovered and subjected to transmission electron microscopy (TEM).Transmission electron microscopy
Polymersome suspensions recovered from DLS measurements were diluted further 10 times (total dilution times 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 Ca+ enriched HEPES buffer and then deposited on glow-discharged carbon grids (Quantifoil, Germany) stained with 1.5% uranyl acetate solution and deposited on carbon-coated copper grid and consequently imaged with a transmission electron microscope (Philips Morgagni 268D) at an accelerating voltage of 80 kV.
:100 Ca+ enriched HEPES buffer and then deposited on glow-discharged carbon grids (Quantifoil, Germany) stained with 1.5% uranyl acetate solution and deposited on carbon-coated copper grid and consequently imaged with a transmission electron microscope (Philips Morgagni 268D) at an accelerating voltage of 80 kV.
Quartz crystal microbalance with dissipation monitoring
Real time membrane formation was studied by QCM-D on a Q-Sense E1 (Biolin Scientific, Sweden) instrument. All QCM-D measurements were performed in Ca2+ enriched HEPES buffer. The Au QCM-D sensor was stabilized under buffer flow at a flow rate of 100 μL min−1 until the frequency signal fluctuation was below ±1 Hz, when the baseline was recorded. Once the system was stable, polymersome solutions were injected at 5 μg mL−1 into the QCM-D chamber (flow rate 50 μL min−1). The formation of supported membranes was followed by a rinse with the Ca2+ enriched HEPES buffer for the removal of any non-absorbed polymersomes. Polymersome rupture was induced by a washing step with Ca2+-free HEPES buffer. In all measurements, the response of the quartz crystal resonance frequency and energy dissipation in shear oscillation mode was recorded as a function of time using odd overtones (3rd–13th). The viscosity and the shear elastic moduli were obtained by fitting the ΔF and ΔD based on four overtones 3rd–9th with Dfind Smart fit by the Q-Sense Dfind software for data analysis.Atomic force spectroscopy
The topography of the solid supported polymer membranes was characterized by atomic force microscopy (AFM) (JPK Nanowizard 3, Version 6.0.63). A Tap150 Al-G cantilever (Budget Sensors, resonance frequency 150 kHz, force constant 5 N m−1) was used for alternating current (AC) mode measurements in HEPES buffer. Images were analyzed with the JPK data processing software (Version spm_6.0.63).Spectroscopic ellipsometry
Spectroscopic ellipsometry (Accurion, Cauchy model) was used to determine the thickness of the gold coated surfaces before polymersome attachment and after potential polymersome rupture. The measurement took place in dry state and five separate measurements on each surface were averaged.Results and discussion
Thiol-bearing polymersome formation
Nano-assemblies with outer membranes exposing thiol functional groups were formed by thin-film rehydration39,50,51 with Ca2+ enriched HEPES buffer. We first tested the effect of adding thiol-bearing polymer on polymersome formation using one thin (A6B34A6) and one thick (A6B65A6) polymer to subsequently apply optimal formation conditions to all five ABA polymers. Therefore, we mixed A6B34A6 and A6B65A6 with increasing concentrations of the thiol-terminated A18B47A18 copolymer (0, 2.5, 5, 10, 15 and 20 mol%). Self-assembled structures were then extruded through 200 nm pore size filters to narrow the size distribution of the nano-assemblies which were then characterized with respect to increasing concentrations of thiol-terminated block copolymer by a combination of DLS and TEM analysis. When the thiol-bearing polymer was added at percentages from 0–10 mol%, the size and morphology of self-assembled structures barely differed (Fig. 1 and Fig. S1†). Using the intensity % distribution, the average diameter was 110 nm for A6B34A6 (Fig. 1a) and 120 nm for A6B65A6 (Fig. S1a†). | ||
| Fig. 1 (a) DLS graph and (b) TEM micrographs of A6B34A6 containing: 1. 0%, 2.5%, 3. 10%, 4. 15%, and 5. 20% SH-bearing polymer, scale bars = 200 nm. | ||
The polydispersity indices (PDIs) for A6B34A6 polymersomes range from 0.09–0.17 and for A6B65A6 from 0.11–0.21 (Table S1†). In addition, corresponding TEM micrographs are typical for hollow spherical structures, confirming the formation of polymersomes for all assemblies obtained with up to 10 mol% of thiol-bearing polymer.
On the other hand, increasing the molar percentage of thiol-bearing polymer above 10 mol%, specifically between 10–20 mol%, shifted and broadened the size peak to 200 nm for A6B34A6 (Fig. 1a) and 290 nm for A6B65A6. In both cases DLS graphs revealed additional small peaks that correspond to smaller objects around 30–40 nm in diameter (probably micelles) and larger objects with a diameter around 1 μm that we consider to be aggregates (Fig. S1a†). Consistently, TEM micrographs of the nano-assemblies formed with 10–20 mol% thiol-bearing block copolymers (Fig. 1b and Fig. S1b†) clearly show a prominent population of smaller, spherical micelles but also reveal the formation of large aggregates connected by worm-like micellar structures. The data obtained for the different concentrations of thiol-bearing A18B47A18 copolymer allowed us to optimize the conditions for producing thiol-bearing polymersomes. Intact polymersomes exposing sufficient amounts of thiol groups are a crucial prerequisite for an efficient surface attachment of the polymersomes via gold–thiol chemistry. Based on our data, we added 10 mol% thiol-bearing copolymer to different ABA block copolymers because this was the highest thiol concentration that did not interfere with polymersome formation. The main factor determining the response to the osmotic shock is the membrane thickness of the polymersome, specifically the length of the hydrophobic block, as the amount of thiol groups as well as all other experimental conditions for attachment and induced rupture were the same for the different types of polymersomes (Fig. S2 and S3†).
Polymersomes on gold coated surfaces: attachment and induced planar polymer membrane formation
We used QCM-D to investigate: (i) the attachment of polymersomes on gold coated sensors; (ii) the impact of osmotic shock induced by a flow of Ca2+-free HEPES buffer (the polymersomes were formed in Ca2+ enriched HEPES buffer) and if it is able to rupture the polymersomes and initiate the planar membrane formation; and finally, (iii) if the PDMS length (units) influences the formation of solid supported polymer membranes. QCM-D is a powerful technique for analyzing surfaces and commonly used for the study of solid supported membranes,7,24,27,28,52 especially lipid bilayer formation5,53,54 and the adsorption of compounds on a surface. QCM-D monitors frequency changes (ΔF) of special quartz sensors in response to changes of the adsorbed mass. In a liquid environment, the interaction of the QCM crystal surface with flexible molecular systems results in the formation of viscoelastic films that cause dissipation of oscillation (measured as dissipation change, ΔD). In addition, by monitoring both ΔF and ΔD, structural information (e.g., conformational changes in the film, crosslinking, and swelling) can be obtained. If polymersomes are induced to rupture in order to yield 2D membranes, their aqueous cargo, i.e. Ca2+ enriched HEPES buffer, is released and large structural rearrangements of the polymersome membranes are expected to occur, which should be reflected by a change in dissipation.52,55,56 We performed QCM-D measurements for thiol-bearing polymersomes based on five different ABA block copolymers (Fig. S4 and S5†). In Fig. 2, we present examples of the extremes where polymer membrane formation was triggered by osmotic shock and where the osmotic shock failed to trigger polymersome fusion and membrane formation. Fig. 2a shows the frequency and dissipation response of the 5th overtone during the addition and attachment of A6B34A6 thiol-bearing polymersomes (injection step), and illustrates the effect of rinsing the system with Ca2+-free HEPES (osmotic shock step) which results in an osmotic shock that leads to the rupture of polymersomes.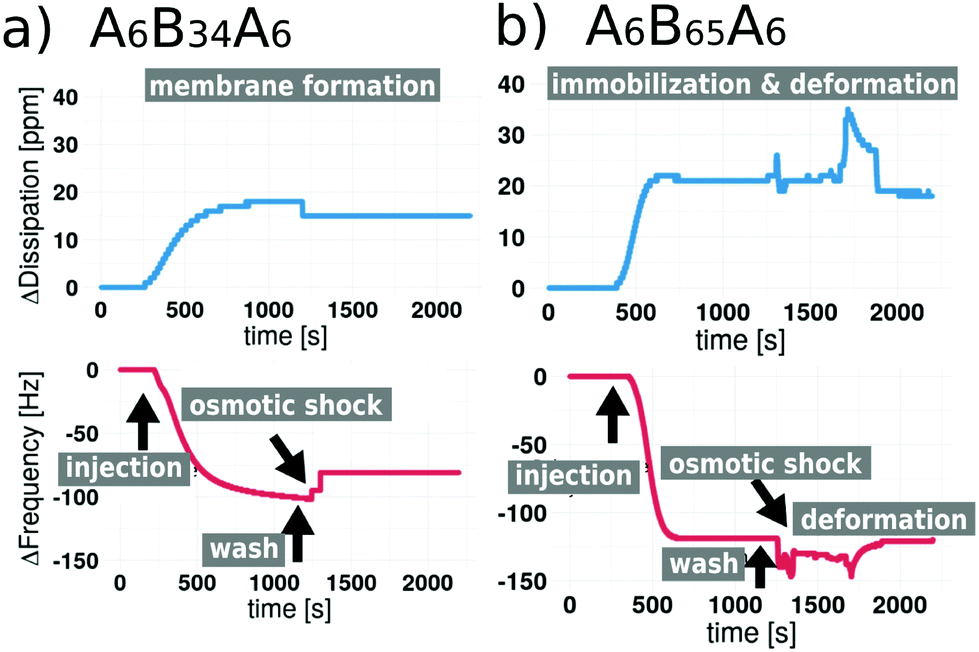 | ||
| Fig. 2 QCM-D frequency and dissipation shifts for (a) A6B34A6 polymersomes and (b) for A6B65A6 polymersomes containing thiol-modification and subjected to osmotic shock. | ||
The ΔF reaches a maximum of −120 Hz during polymersome attachment, which is reduced to ΔF −80 Hz in response to the osmotic shock. We recorded an increase of ΔD during attachment reaching a maximum of +18 ppm which slightly decreased to ΔD +13 ppm, reflecting changes in the assembly associated with membrane formation. As expected, these values differ from those obtained from QCM-D recordings of solid supported lipid bilayer formation, where ΔF typically is around −40 Hz.10,21,57 Conceivably, this difference can be related to the fact that polymersome membranes are thicker, so of higher mass.27,58 In the absence of an osmotic shock (Fig. S4a†), we observe attachment of the thiol-bearing polymersome on the sensor indicated by a decrease in ΔF. However, ΔF remains at −115 Hz, indicating that there is no membrane fusion if polymersomes are not ruptured. Similarly, ΔD reached +19 ppm and remained stable with a fluctuation of ±1 Hz. These data show that the presence of thiol-bearing polymer mediated the attachment of polymersomes to the surface of gold sensors. In addition, frequency and dissipation shifts related to solid supported membrane formation only occur in response to hypoosmotic conditions. QCM-D analysis of thiol-bearing A6B65A6 polymersomes (Fig. 2b and Fig. S4b†) revealed a different behavior in response to the osmotic shock. Following polymersome injection, we observed a shift in ΔF and ΔD (ΔF −121 Hz, ΔD 22 ppm) that was similar to the values observed for A6B34A6. Addition of Ca2+ free HEPES (osmotic shock) led to minor fluctuations of ΔF (max −150 Hz) and ΔD (max +36 ppm) but the values returned to the values indicative of polymersome attachment. While thiol-bearing A6B65A6 polymersomes successfully attach to the gold surface, hypoosmotic conditions neither induce rupture nor membrane formation. As observed for A6B34A6, attachment of A6B65A6 polymersomes depended on the thiol functional groups (Fig. 2 and Fig. S5†). By making the polymersomes very robust, the much longer PDMS block of A6B65A6 (65 PDMS units in comparison with 34 PDMS units) plays an essential role in hindering polymersome rupture and thereby preventing planar membrane formation. We further explored the relationship between hydrophobic PDMS block length (units) and membrane thickness derived from QCM-D experiments before and after osmotic shock (rehydrated conditions) (Fig. 3a). We distinguish a different behaviour depending on the membrane thickness: (i) polymersomes with a thin hydrophobic block (22–35 PDMS units) that form planar membranes after osmotic shock, (ii) polymersomes with an intermediate hydrophobic block (42–44 PDMS units) that appear to partly form membranes, and (iii) polymersomes assembled from ABAs with a long hydrophobic block (65 PDMS units), which stay intact after osmotic shock. Prior osmotic shock, layers of surface-attached polymersomes were around 100 nm thick with little variation, independent of the hydrohobic block length (units). This uniform thickness is in line with the diameter and PDI of the corresponding polymersomes in solution. After osmotic shock, however, we observed a big decrease in the thickness if polymersomes forming supported membranes had a thin hydrophobic block. Specifically, we obtained narrowly distributed thicknesses ranging from 18 nm to 25 nm which is a typical value for a planar conformation. For the second group of polymersomes with an intermediate length (units) to the hydrophobic block (semi-fusion or partial planar membrane formation), we observed a wide range of thicknesses from 17 nm to 101 nm. The broad range suggests that some polymersomes can rupture and form polymer membrane ‘patches’ whereas others are not affected by the osmotic shock and maintain their spherical shape. For the third group represented by A6B65A6 based polymersomes, we observed a narrow distribution of thicknesses between 83 and 90 nm, which is slightly lower that the thicknesses before osmotic shock. This might be caused by a small deformation of the polymersomes’ architecture in response to the osmotic shock. Using the QCM-D data (in particular ΔF and ΔD based on 3rd–9th overtones for the five types of polymersome tested) we were able to calculate the shear elastic modulus and viscosity of the assemblies obtained after osmotic shock as a function of the hydrophobic block length (units) (Fig. 3b). We performed the analysis of shear elastic modulus and viscosity to deepen the characterization of the viscoelastic properties of the solid supported polymersomes. The changes in response to osmotic shock observed by real-time QCM-D measurements confirm that the viscoelastic characteristics of an intact polymersome differ significantly from those of the planar polymer conformation.
 | ||
| Fig. 3 Changes of viscoelastic properties upon osmotic (a) thickness of the upper layer (nm) determined by QCM-D before and after osmotic shock, (b) shear elastic modulus after osmotic shock as a function of PDMS length (units), (c) thickness of the upper layer (nm) determined by SE before and after osmotic shock and (d) (left, “dry”) and (right, “wet”) difference of thickness. | ||
Moreover, the stiffer an assembly, the higher the shear elastic modulus,59,60 and according to the literature, planar assemblies are less flexible than the spherical ones.59,60 Assemblies after osmotic shock of polymers with thin hydrophobic blocks have roughly 5 times higher elastic moduli than those derived from the polymersomes with the thickest membrane. In addition, there is a wide distribution of elastic moduli for the group of intermediate polymers which after osmotic shock form inhomogenous planar membrane patches. The same trend is observed for the viscosity of the assemblies formed from the three types of polymersomes (Table 1 and Fig. S6†). The stiffer the solid supported polymer assembly after osmotic shock, the higher its viscosity. Polymersomes that are able to form planar membranes are significantly more viscous than the ones which remain intact. In addition, widely distributed, intermediate viscosity values are measured for the copolymers which only partially form a planar membrane. Both shear modulus and viscosity are in fact related to rigidity and as well as to the length (units) of the PDMS chains. To assess the effect of water on the formation of solid-supported membranes, we determined the assembly thicknesses in dry conditions by spectroscopic ellipsometry (SE; Fig. 3c). This is an established method to determine film thickness in the nm range based on the correlation between the refractive index of a material (η) and its thickness.61,62 We measured the thickness of gold-coated solid supports before and after polymersome attachment (Fig. S7†), and after subjecting the support with attached polymersomes to an osmotic shock (Fig. 3c). Polymersomes with a thin hydrophobic block showed a big decrease of thickness in the range of 12 nm. For the polymersomes based on intermediate PDMS lenth (units), we also found a big variation of thicknesses, even more pronounced than in hydrated conditions (Fig. 3a), which could reflect the fact that in dry state the overall scale of thicknesses is much smaller. Finally, the thickness of polymersomes with the longest hydrophobic block only slightly decreased after osmotic shock. Moreover, the membrane thicknesses measured by SE are in good agreement with those obtained by cryo-TEM and reported in the literature for corresponding copolymers.30,63,64 We then compared the membrane thicknesses obtained by SE (dry) to the ones obtained in real time by QCM-D (wet) (Fig. 3d) and plotted the difference of thickness values (Δthickness) for each polymersome type before and after osmotic shock as a function of the PDMS lenth (units). Taking into account the big influence of water, this comparison leads us to conclude that the longer the PDMS block, the smaller the Δthickness after osmotic shock. This data provides further evidence for the significance of the hydrophobic block length (units) in solid supported planar membrane formation: copolymers with large PDMS blocks self-assemble to polymersomes with thicker and thus more robust membrane boundaries. Another reason why the polymersome rupture becomes more unlikely with increasing hydrophobic block length (units) is that PDMS is a flexible polymeric compound and thus, the polymersomes withstand the osmotic pressure by slightly deforming but otherwise remain intact. In Fig. 4 we compare the average thickness obtained by cryo-TEM (values taken from the literature30,64) with the average thickness of solid-supported polymer films derived from QCM-D (wet thickness) and SE (dry thickness) measurements.
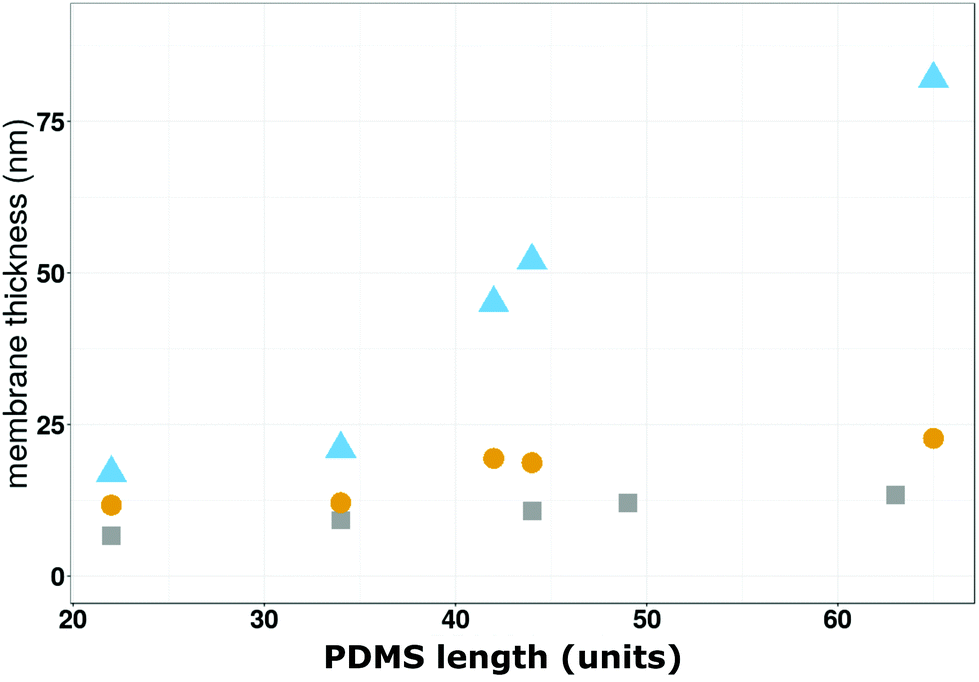 | ||
| Fig. 4 Comparison of the membrane thicknesses as a function of the PDMS length (units) obtained by cryo-TEM (grey squares),30,64 SE (yellow circles) and QCM-D (bleu triangles). | ||
| Polymer | ΔF (Hz) | ΔD (ppm) | Wet thickness (nm) | Dry thickness (nm) | Shear modulus (kPa) | Viscosity (μPa s) |
|---|---|---|---|---|---|---|
| A3B22A3 | −78 ± 5.1 | 13± 1.1 | 17 ± 4.5 | 11.7 ± 1.2 | 83 ± 7.9 | 3929 ± 741 |
| A6B34A6 | −82 ± 3.4 | 14 ± 1.3 | 21 ± 3.6 | 12.1 ± 0.99 | 80.9 ± 10.8 | 3092 ± 625 |
| A7B42A7 | −51 ± 17.1 | 8 ± 2.9 | 45 ± 22 | 19.4 ± 7.4 | 56.4 ± 27.8 | 2367 ± 1451 |
| A6B44A6 | −49 ± 16.2 | 6 ± 2.4 | 52 ± 18.2 | 18.7 ± 7.7 | 41.1 ± 21.5 | 2069 ± 1352 |
| A6B65A6 | −92± 4.2 | 18 ± 0.9 | 82 ± 5.1 | 22.7 ± 1.8 | 21.2 ± 6.6 | 944 ± 108 |
The first comparison reveals differences between the of thickness the polymersome bounding membrane and the planar, solid-supported polymer mebrane created by induced-polymersome fusion, both in a hydrated state. Comparison of the dry and wet thickness assessed by QCM-D and SE, respectively, are indicative of a swelling effect of water on the solid supported polymer membranes. Up to a block lenth (units) of 35 PDMS units, we obtain comparable thickness values by all three techniques because they correspond to planar conformations. For PDMS block lenth (units) between 35–70 units, membrane patches and polymersomes that do not rupture on the solid-substrate cause a broader range of thickness values and the divergence of the thickness values increases.
Topography of the solid supported assemblies determined by AFM
Immobilization of thiol-bearing polymersomes on gold surfaces via thiol–gold chemistry and the impact of osmotic shock was further investigated via AFM in liquid (Ca2+ enriched HEPES buffer). With AFM we obtained information about the surface topography of the solid supported polymer assemblies. After the immobilization reaction, AFM height images of the solid substrates reveal immobilized polymersomes of approximately ∼110 nm (Fig. 5A, Fig. S8 and S9†). | ||
| Fig. 5 Representative AFM micrographs (height channel) of (a) immobilized thiol-bearing A3B22A3, and (b) A6B34A6 planar membrane formation (left panel), A7B42A7 membrane ‘patches’ formation (middle panel) and A6B65A6 deformed polymersomes (right panel) after osmotic shock. | ||
Successful attachment of polymersomes on the gold coated substrate depended on thiol-bearing polymer as confirmed by polymersomes lacking thiol groups (Fig. S10†). After a rinsing step, we observed only residual polymers in the height range of 2.5–3.2 nm (Fig. S10†), which is way smaller than the height measured for both the immobilized polymersomes (90–110 nm) and for the mature planar membrane (19–20 nm). Also, the bare substrate, without deposition of polymersomes or any other treatment showed height profiles only in the picometer range (Fig. S11†) Consistent with the results from QCM-D and SE (Fig. 3), osmotic shock of A3B22A3 and A6B34A6 thiol-bearing polymersomes yielded a homogeneous surface, which looks flat and ranged between 19.9–21 nm in height (Fig. 5b, left and Fig. S8†). AFM height values are in good agreement with the values published for immobilized polymersomes and polymer films, respectively.14,24,30,38,65–68 For liposomes adsorbed on a surface and liposome-based planar membranes, the heights measured by AFM are lower, around 20–40 nm for the immobilized liposomes and ∼4–5 nm for the lipid membranes.55,57,69–72 This difference is expected due to the higher membrane thickness of the polymersomes compared to liposomes. For the polymers with intermediate PDMS length (units) (Fig. 5b, middle and Fig. S8†), AFM height images show an inhomogeneous surface topography (surface corrogation ∼31 nm) typical for the assembly of polymer membrane ‘patches’. A6B65A6 thiol-bearing polymersomes produced surfaces with a height around 175 nm which reflects intact albeit deformed polymersomes (Fig. 5b, right panel).
Conclusions
We investigated the formation of solid supported polymer membranes on gold coated surfaces governed by the following steps: (i) self-assembly of thiol-bearing polymersomes; (ii) polymersome immobilization on the surface via thiol–gold chemistry; and (iii) polymersome rupture induced by osmotic shock and subsequent polymer assembly rearrangement resulting in polymer membrane formation. We showed that the chain length (units) of the block-copolymers, in particular the hydrophobic PDMS block, from which the polymersomes are formed is crucial for their later rupture which in turn is a prerequisite for establishing planar membranes on solid support. More specifically, the longer the polymer blocks, the thicker is the bounding membrane of the polymersomes which renders the assemblies on the surface robust enough to remain unruptured after exposure to osmotic shock Although we cannot yet fully explain the mechanism underlying the process, our study based on various surface characterization techniques demonstrates that polymersomes under adequate conditions can form solid supported polymer membranes. We found out that only if we carefully combine the attachment of the polymersomes on a solid substrate with the osmotic shock we achieve induced polymersome fusion. This is of high importance since block copolymer membranes have extensive potential as platforms for bio-sensing, electrochemistry and screening applications. Moreover, considering that important scientific efforts have been focused on the functional reconstitution of biomolecules into polymersomes, we can now capitalize on that knowledge and create functional solid supported planar polymer membranes via induced-polymersome fusion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support of this project by the University of Basel, the Swiss National Science Foundation (SNSF) and the National Centre of Competence in Research Molecular Systems Engineering (NCCR-MSE). We thank Dr Samuel Lörcher (University of Basel) for the polymer synthesis and valuable scientific discussions and Gabrielle Persy (University of Basel) for TEM imaging.References
- R. Fetzer, K. Jacobs, A. Münch, B. Wagner and T. P. Witelski, Phys. Rev. Lett., 2005, 95, 127801 CrossRef CAS PubMed.
- E. Sackmann and M. Tanaka, Trends Biotechnol., 2000, 18, 58–64 CrossRef CAS PubMed.
- F. Brochard-Wyart, G. Debregeas, R. Fondecave and P. Martin, Macromolecules, 1997, 30, 1211–1213 CrossRef CAS.
- S. C. Thickett, C. Neto and A. T. Harris, Adv. Mater., 2011, 23, 3718–3722 CrossRef CAS PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- J. Jackman, W. Knoll and N.-J. Cho, Materials, 2012, 5, 2637–2657 CrossRef CAS.
- E. Reimhult, F. Höök and B. Kasemo, J. Chem. Phys., 2002, 117, 7401–7404 CrossRef CAS.
- E. Reimhult, F. Höök and B. Kasemo, Langmuir, 2003, 19, 1681–1691 CrossRef CAS.
- P. Losada-Pérez, O. Polat, A. N. Parikh, E. Seker and F. U. Renner, Biointerphases, 2018, 13, 011002 CrossRef PubMed.
- S. R. Tabaei, J. A. Jackman, S.-O. Kim, V. P. Zhdanov and N.-J. Cho, Langmuir, 2015, 31, 3125–3134 CrossRef CAS PubMed.
- G. R. Heath, P. L. Harrison, P. N. Strong, S. D. Evans and K. Miller, Soft Matter, 2018, 14, 6146–6154 RSC.
- A. M. Telford, S. C. Thickett and C. Neto, J. Colloid Interface Sci., 2017, 507, 453–469 CrossRef CAS PubMed.
- Q. Bao, S. Braun, C. Wang, X. Liu and M. Fahlman, Adv. Mater. Interfaces, 2019, 6, 1800897 CrossRef.
- J. You, Y. Liao, Y. Men, T. Shi, L. An and X. Li, Macromolecules, 2011, 44, 5318–5325 CrossRef CAS.
- L. Xu, H. Zhang and T. Shi, Polymer, 2016, 99, 185–192 CrossRef CAS.
- A. S. M. A. Haseeb, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016. ISBN 9780128035818 Search PubMed.
- Q. Bao, S. Fabiano, M. Andersson, S. Braun, Z. Sun, X. Crispin, M. Berggren, X. Liu and M. Fahlman, Adv. Funct. Mater., 2016, 26, 1077–1084 CrossRef CAS.
- G. Stoychev, S. Zakharchenko, S. Turcaud, J. W. C. Dunlop and L. Ionov, ACS Nano, 2012, 6, 3925–3934 CrossRef CAS PubMed.
- H. J. Kim, A. Kim, K. Miyata and K. Kataoka, Adv. Drug Delivery Rev., 2016, 104, 61–77 CrossRef CAS PubMed.
- S. Yorulmaz Avsar, M. Kyropoulou, S. Di Leone, C.-A. Schoenenberger, W. P. Meier and C. G. Palivan, Front. Chem., 2019, 6, 645 CrossRef PubMed.
- J. A. Jackman and N.-J. Cho, Langmuir, 2020, 36, 1387–1400 CrossRef CAS PubMed.
- Y.-L. Yang, H.-K. Tsao and Y.-J. Sheng, Soft Matter, 2016, 12, 6442–6450 RSC.
- S. Belegrinou, S. Menon, D. Dobrunz and W. Meier, Soft Matter, 2011, 7, 2202–2210 RSC.
- J. Dorn, S. Belegrinou, M. Kreiter, E.-K. Sinner and W. Meier, Macromol. Biosci., 2011, 11, 514–525 CrossRef CAS PubMed.
- S. Varlas, J. Am. Chem. Soc., 2019, 141(51), 20234–20248 CrossRef CAS PubMed.
- J. Xiao, J. Am. Chem. Soc., 2020, 142(14), 6569–6577 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- M. Mumtaz Virk, B. Hofmann and E. Reimhult, Langmuir, 2019, 35, 739–749 CrossRef CAS PubMed.
- C. Draghici, J. Kowal, A. Darjan, W. Meier and C. G. Palivan, Langmuir, 2014, 30, 11660–11669 CrossRef CAS PubMed.
- C. Draghici, V. Mikhalevich, G. Gunkel-Grabole, J. Kowal, W. Meier and C. G. Palivan, Langmuir, 2018, 34, 9015–9024 CrossRef CAS PubMed.
- J. Ł. Kowal, J. K. Kowal, D. Wu, H. Stahlberg, C. G. Palivan and W. P. Meier, Biomaterials, 2014, 35, 7286–7294 CrossRef CAS PubMed.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2020, 21, 701–715 CrossRef CAS PubMed.
- J. A. Jackman, J.-H. Choi, V. P. Zhdanov and N.-J. Cho, Langmuir, 2013, 29, 11375–11384 CrossRef CAS PubMed.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef CAS PubMed.
- M. Seitz, E. Ter-Ovanesyan, M. Hausch, C. K. Park, J. A. Zasadzinski, R. Zentel and J. N. Israelachvili, Langmuir, 2000, 16, 6067–6070 CrossRef CAS PubMed.
- G. Gunkel-Grabole, C. Palivan and W. Meier, Macromol. Mater. Eng., 2017, 302, 1600363 CrossRef.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- S. Rigo, G. Gunkel-Grabole, W. Meier and C. G. Palivan, Langmuir, 2019, 35, 4557–4565 CrossRef CAS PubMed.
- S. Rigo, M. Kyropoulou, C.-A. Schoenenberger and C. G. Palivan, in Racing for the Surface, ed. B. Li, T. F. Moriarty, T. Webster and M. Xing, Springer International Publishing, Cham, 2020, pp. 385–408 Search PubMed.
- K. Critchley, B. P. Khanal, M. Ł. Górzny, L. Vigderman, S. D. Evans, E. R. Zubarev and N. A. Kotov, Adv. Mater., 2010, 22, 2338–2342 CrossRef CAS PubMed.
- S. Ye, A. P. Brown, A. C. Stammers, N. H. Thomson, J. Wen, L. Roach, R. J. Bushby, P. L. Coletta, K. Critchley, S. D. Connell, A. F. Markham, R. Brydson and S. D. Evans, Adv. Sci., 2019, 6, 1900911 CrossRef CAS PubMed.
- J. Feng, W. Zhao, B. Su and J. Wu, Biosens. Bioelectron., 2011, 30, 21–27 CrossRef CAS PubMed.
- G. M. Santos, F. Zhao, J. Zeng and W.-C. Shih, Nanoscale, 2014, 6, 5718–5724 RSC.
- C. A. R. Chapman, H. Chen, M. Stamou, J. Biener, M. M. Biener, P. J. Lein and E. Seker, ACS Appl. Mater. Interfaces, 2015, 7, 7093–7100 CrossRef CAS PubMed.
- E. Seker, M. Reed and M. Begley, Materials, 2009, 2, 2188–2215 CrossRef CAS.
- A. M. Hodge, J. R. Hayes, J. A. Caro, J. Biener and A. V. Hamza, Adv. Eng. Mater., 2006, 8, 853–857 CrossRef CAS.
- D. Lee, X. Wei, X. Chen, M. Zhao, S. C. Jun, J. Hone, E. G. Herbert, W. C. Oliver and J. W. Kysar, Scr. Mater., 2007, 56, 437–440 CrossRef CAS.
- S. Lörcher and W. Meier, Eur. Polym. J., 2017, 88, 575–585 CrossRef.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, Macromolecules, 2017, 50, 1512–1520 CrossRef CAS.
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC.
- E. V. Konishcheva, D. Daubian, S. Rigo and W. P. Meier, Chem. Commun., 2019, 55, 1148–1151 RSC.
- J. A. Jackman, M. C. Kim, V. P. Zhdanov and N.-J. Cho, Phys. Chem. Chem. Phys., 2016, 18, 3065–3072 RSC.
- M. C. Kim, A. Gunnarsson, S. R. Tabaei, F. Höök and N.-J. Cho, Phys. Chem. Chem. Phys., 2016, 18, 3040–3047 RSC.
- M. Wallin, J.-H. Choi, S. O. Kim, N.-J. Cho and M. Andersson, Eur. Biophys. J., 2015, 44, 27–36 CrossRef CAS PubMed.
- R. P. Richter and A. R. Brisson, Biophys. J., 2005, 88, 3422–3433 CrossRef CAS PubMed.
- T. K. Lind and M. Cárdenas, Biointerphases, 2016, 11, 020801 CrossRef PubMed.
- K. Evans, Int. J. Mol. Sci., 2008, 9, 498–511 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- F. Höök, B. Kasemo, T. Nylander, C. Fant, K. Sott and H. Elwing, Anal. Chem., 2001, 73, 5796–5804 CrossRef.
- N.-J. Cho, K. K. Kanazawa, J. S. Glenn and C. W. Frank, Anal. Chem., 2007, 79, 7027–7035 CrossRef CAS PubMed.
- G. Gunkel-Grabole, A. Car, V. V. Naik, L. Marot, G. Ferk, C. Palivan and W. Meier, Macromol. Chem. Phys., 2016, 217, 966–973 CrossRef CAS.
- S. Rigo, D. Hürlimann, L. Marot, M. Malmsten, W. Meier and C. G. Palivan, ACS Appl. Bio Mater., 2020, 3, 1533–1543 CrossRef CAS.
- M. Lomora, I. A. Dinu, F. Itel, S. Rigo, M. Spulber and C. G. Palivan, Macromol. Rapid Commun., 2015, 36, 1929–1934 CrossRef CAS PubMed.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- I. Craciun, A. S. Denes, G. Gunkel-Grabole, A. Belluati and C. G. Palivan, Helv. Chim. Acta, 2018, 101, e1700290 CrossRef.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS PubMed.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- B. Iyisan, A. Janke, P. Reichenbach, L. M. Eng, D. Appelhans and B. Voit, ACS Appl. Mater. Interfaces, 2016, 8, 15788–15801 CrossRef CAS PubMed.
- E. I. Goksu, J. M. Vanegas, C. D. Blanchette, W.-C. Lin and M. L. Longo, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 254–266 CrossRef CAS PubMed.
- M. Li, M. Chen, E. Sheepwash, C. L. Brosseau, H. Li, B. Pettinger, H. Gruler and J. Lipkowski, Langmuir, 2008, 24, 10313–10323 CrossRef CAS PubMed.
- N. Buzhynskyy, M. Golczak, J. Lai-Kee-Him, O. Lambert, B. Tessier, C. Gounou, R. Bérat, A. Simon, T. Granier, J.-M. Chevalier, S. Mazères, J. Bandorowicz-Pikula, S. Pikula and A. R. Brisson, J. Struct. Biol., 2009, 168, 107–116 CrossRef CAS PubMed.
- P. Bao, M. L. Cartron, K. H. Sheikh, B. R. G. Johnson, C. N. Hunter and S. D. Evans, Chem. Commun., 2017, 53, 4250–4253 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr01122g |
| This journal is © The Royal Society of Chemistry 2021 |