
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the origin of serrated stacking motifs in planar two-dimensional covalent organic frameworks†
Christian
Winkler
 *a,
Tomas
Kamencek
ab and
Egbert
Zojer
*a
*a,
Tomas
Kamencek
ab and
Egbert
Zojer
*a
aInstitute of Solid State Physics, NAWI Graz, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria. E-mail: egbert.zojer@tugraz.at; christian.winkler@alumni.tugraz.at
bInstitute of Physical and Theoretical Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
First published on 7th May 2021
Abstract
Covalent organic frameworks (COFs) have attracted significant attention due to their chemical versatility combined with a significant number of potential applications. Of particular interest are two-dimensional COFs, where the organic building units are linked by covalent bonds within a plane. Most properties of these COFs are determined by the relative arrangement of neighboring layers. These are typically found to be laterally displaced, which, for example, reduces the electronic coupling between the layers. In the present contribution we use dispersion-corrected density-functional theory to elucidate the origin of that displacement, showing that the common notion that the displacement is a consequence of electrostatic repulsions of polar building blocks can be misleading. For the representative case of COF-1 we find that electrostatic and van der Waals interactions would, actually, favor a cofacial arrangement of the layers and that Pauli repulsion is the crucial factor causing the serrated AA-stacking. A more in-depth analysis of the electrostatic contribution reveals that the “classical” Coulomb repulsion between the boroxine building blocks of COF-1 suggested by chemical intuition does exist, but is overcompensated by attractive effects due to charge-penetration in the phenylene units. The situation becomes more involved, when additionally allowing the interlayer distance to relax for each displacement, as then the different distance-dependences of the various types of interactions come into play. The overall behavior calculated for COF-1 is recovered for several additional COFs with differently sized π-systems and topologies, implying that the presented results are of more general relevance.
1. Introduction
Covalent organic frameworks (COFs) are highly porous crystalline materials consisting of organic building blocks linked by covalent bonds.1–7 Because of their tunable structures, COFs have significant potential for various applications like gas storage,8–10 gas separation,11–14 catalysis,15–17 energy storage,18–20 or optoelectronics.21–27 Among the different topologies of COFs, two-dimensional (2D) systems have received particular attention. Here, the individual organic building units are linked via covalent bonds within a plane, forming highly regular 2D layers. These layers then stack on top of each other and the resulting stacks are held together primarily by comparably weak van der Waals interactions. The properties (electronic, optical, and catalytic) of the resulting three-dimensional (3D) stacks are then strongly determined by the packing motif of consecutive 2D layers,7,26,28–33 as it defines the shape of the pores and the overlap of the π-systems of neighboring layers. For example, depending on the symmetry and nodal structure of the involved orbitals/wavefunctions, the inter-layer shifts can determine, whether the resulting bulk systems are insulating, semiconducting, or even metallic.34,35 Furthermore, it has also been found that the band gaps of such layered COFs depend on the stacking motif of consecutive layers.32,36 Thus, it is of utter importance to unravel the actual packing motif of COFs and to understand, by which factors it is determined.The vast majority of the reported 2D COFs exhibit either eclipsed (cofacial) or serrated (shifted) AA-stacking,28,30,37–42 where the actual magnitude of the shift is hard to determine experimentally via X-ray diffraction due to the large peak broadening in the typically investigated powder samples.26,37,43 Furthermore, while there have been tremendous advances in the field of electron diffraction, also for this technique it remains a sizable challenge and takes quite some effort to determine the exact interlayer arrangement of COFs.44–49 In this context it is worth mentioning that in a recent study, by employing a combination of powder X-ray diffraction experiments, transmission electron microscopy, and electron diffraction, a shifted stacking motif has been successfully determined for a 2D COF.50 Different stacking motifs are found, for example, when the 2D sheets comprising the COF are not entirely planar.39,51 In the following we will, however, focus on planar COFs, as these systems allow a more straightforward analysis of the interplay between different geometric degrees of freedom and the energetic stability of the respective COF. Of particular appeal for such an analysis are COF-1 and COF-5 (the structures first reported in ref. 2). As far as modelling studies on these systems are concerned, Zhou et al.37 explicitly showed (employing density functional tight-binding methods) that the total energy of the COF becomes a minimum for shifted layer arrangements with displacements of around 1 Å. They hypothesized that the alignment of neighboring π-orbitals plays an essential role for these shifts and compared the stacking motif of the aromatic rings to the situation in graphite, albeit without determining the nature of the interactions that enforce the serrated structure.37 Lukose et al.38 also performed a computational study on the alignment of layered COFs, again considering COF-1 and COF-5. These authors also identified similar shifts of consecutive layers (∼1.4 Å) as the energetically favorable layer arrangements. In this work, as well as in ref. 36, the authors argued that repulsive Coulomb interactions between B and O linking units in neighboring layers would cause the eclipsed AA-stacking to be energetically unfavorable, but again without quantifying these interactions. Thus, to the best of our knowledge, a quantitative assessment of the different types of interactions as a function of the alignment of consecutive layers is still lacking. This lack prevents a fundamental understanding of the factors determining the packing motif in 2D COFs and also hinders the development of strategies for tuning the stacking arrangements of COFs (and their resulting electrical, optical, and catalytic properties).28,29,31 To generate such an understanding, in the present study we employ dispersion corrected density functional theory (DFT) calculations with a focus on decomposing the interlayer interactions in the prototypical model system COF-1 (see Fig. 1 and ref. 2 for the structure of this COF) into physically well-defined contributions. These comprise dispersion forces, electrostatic interactions, and exchange repulsions with orbital rehybridizations. To demonstrate the wider applicability of our findings, we extend our analysis to COF-5 and COFs containing porphyrin (Por-COF)52 and hexabenzocoronene cores (HBC-COF).40 The details of the structures of these COFs will be discussed below. Furthermore, by considering the example of COF-366![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 we briefly address how deviations from planarity influence the individual factors determining the packing motif.
35 we briefly address how deviations from planarity influence the individual factors determining the packing motif.
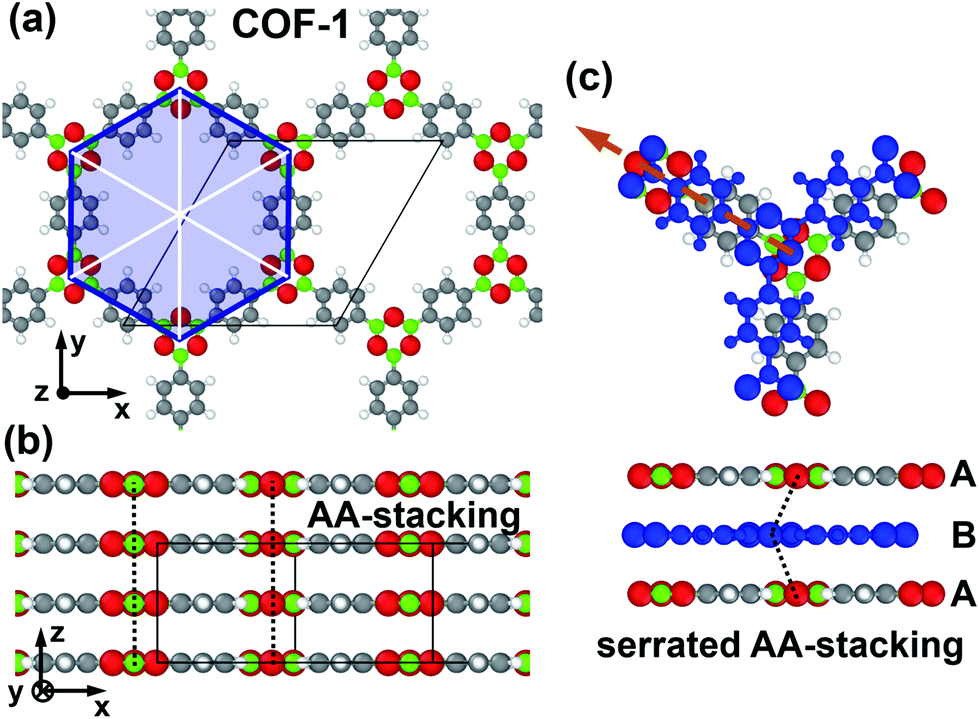 | ||
| Fig. 1 Structure of COF-1 and the primarily considered shift direction: panels (a) and (b) show top and side views of the structure of COF-1 for cofacial AA-stacking. The considered unit cell is shown by the thin, black, solid lines. The blue hexagon and the white lines highlight the hexagonal symmetry of the pores. In panel (c), a shifted (serrated) arrangement with a displacement of 1.75 Å parallel to one of the walls of the pore is shown. The shift direction is shown as a dashed, orange arrow and the shifted layer is marked in blue. The dashed black lines in panels (b) and (c) connect equivalent atoms in consecutive layers, indicating the stacking motif. Color code of the atoms: C … grey, H … white, B … green, O … red. | ||
2. Methods
For the investigations of the layered COFs considered in this study we employed dispersion corrected density functional theory, DFT, as implemented in the FHI-aims code.53,54 The PBE functional55,56 was used and van der Waals interactions were considered by using the Tkatchenko–Scheffler,57 TS, scheme. For comparison, also test calculations employing the computationally more costly many body dispersion correction,58–60 MBD, were performed. The electronic band structure of COF-1 was also calculated using the HSE0661,62 hybrid functional based on the PBE geometries. The corresponding data are shown in the ESI.† We used the conventional “tight” basis functions of FHI-aims with details for each atomic species described in the ESI.† For all bulk systems of the COFs a grid consisting of 3 × 3 × 6 k-points (4 × 4 × 8 for Por-COF) was employed for sampling reciprocal space, unless stated otherwise. Tests show that the total energy for this grid is well converged to within less than 1 meV for COF-1. To describe the occupation of the electronic states, a Gaussian type smearing function with a width of 0.01 eV was employed. For geometry relaxations, the positions of individual atoms were allowed to relax until the largest force component on any of the atoms was below 0.01 eV Å−1.The calculations of the potential energy surface for COF-1 as well as for shifts parallel to one of the pore walls were performed on structures employing the experimental lattice constants reported in literature (a = b = 15.420 Å and c = 3.328 Å).2 The unit cell of COF-1 was constructed such that it contains two layers in stacking direction (layers A and B, see Fig. 1). This allows displacing these layers along directions parallel to the xy-plane. In addition to considering systems with constant unit-cell height (and, thus, constant interlayer stacking distance), we also studied systems for which the unit-cell height was optimized for each displacement. Further details on the geometry relaxation and the determination of the optimal unit-cell heights and interlayer stacking distances can be found in the ESI.†
For the additional COFs considered in this work (COF-5, Por-COF, and HBC-COF), the in-plane lattice parameters had to be optimized, as for some of them no literature values are available. To find the optimal stacking arrangement in terms of in-plane shifts and (shift-dependent) stacking distance, we performed full geometry relaxations for the bulk systems. Details on these simulations are provided in the ESI† together with test calculations for COF-1 in which the in-plane lattice parameters were also optimized. The latter yields somewhat smaller lattice constants than in the experiments (in good agreement with previous computational studies63) but does not significantly impact the results.
To calculate the full potential energy surface for lateral displacements between layers in COF-1 we employed Gaussian process regression, GPR, as implemented in scikit-learn.64 The model vector consisted of the x and y positions of the shifted layer. As Kernel functions we used a combination of a constant kernel with the radial basis function kernel (RBF). To obtain the ideal hyper-parameters, the marginal log likelihood was maximized. The model was initially trained with 80 randomly chosen data points (i.e., displacements). Then 39 additional points were included at the x and y positions of the maximum model uncertainty. The final model uncertainty was estimated to be well below 10 meV for displacements smaller than 3.5 Å and to be below 50 meV for shifts around 6 Å. Details of the model and the obtained model error are reported in the ESI.†
In order to quantitatively assess the porosity of the various structures presented here, various characteristic measures like pore sizes and (non-)accessible surface areas have been calculated utilizing the Zeo++ code (version 0.2.2)65 based on a Voronoi tessellation66 and on statistical integration using probe spheres of different sizes,67,68 respectively. Details on the employed methodology can be found in the ESI.†
To determine the individual contributions to the interaction energy of the COFs, the system was split into two fragments, which consist of only one of the two layers in the unit cell each (layers A and B in Fig. 1). Technically, this was achieved by removing one of the layers from the unit cell, while not changing the unit-cell dimensions and the positions of the atoms in the other layer. Then the total energies of the COF containing both layers in the unit cell and of COFs comprising either only layers of type A or of type B were calculated. The resulting interaction energy between the fragments (i.e., layers) is given by
| ΔEint = EABtotal − (EAtotal + EBtotal). | (1) |
This interaction energy can be decomposed into the interaction energy resulting from the PBE calculations and the contribution due to the a posteriori correction for (long range) van der Waals interactions
| ΔEint = ΔEPBE + ΔEvdW. | (2) |
The individual contributions can be readily obtained from the FHI-aims output for the full system and the two sub-systems as
| ΔEPBE = EABPBE − (EAPBE + EBPBE) | (3a) |
| ΔEvdW = EABvdW − (EAvdW + EBvdW) | (3b) |
A more involved step is to decompose the PBE interaction energy, ΔEPBE, into the electrostatic contribution due to coulombic interactions between nuclei and electron clouds of the subsystems and into contributions due to exchange interactions and orbital rehybridizations. Various decomposition schemes that serve this purpose are available for finite-size systems, but for extended solids described by periodic boundary conditions such approaches are, unfortunately, rare. Therefore, a custom decomposition scheme was implemented as a post-processing tool for FHI-aims. This scheme is largely based on the periodic energy decomposition analysis (pEDA) scheme developed by Raupach and Tonner.69,70 Within this scheme, the authors basically extended the energy decomposition analysis (EDA) method developed by Ziegler/Rauk71,72 and Morokuma73 to periodic boundary conditions. The key idea in this method is to partition the interaction energy ΔEint,elec into well defined terms as shown in eqn (4).
| ΔEint,elec = ΔEelstat + (ΔEPauli + ΔEorb) | (4) |
As a first step, one can evaluate ΔEelstat by considering the charge densities of the individual, non-interacting fragments A and B and use them to construct a combined system {A,B}. This combined system contains the charge densities of the non-interacting fragments A and B at the positions these fragments exhibit in the combined system. Consequently, the sum of the non-distorted charge densities nA and nB is used to describe the combined system. The energy of system {A,B} can then be calculated by performing a single shot DFT calculation without a self-consistency cycle. This calculation yields the electrostatic energy E{A,B}elstat of the system as constructed from the fragments. The difference between this energy and the electrostatic energies of the individual fragments yields the quasiclassical electrostatic interaction between the layers, ΔEelstat, as:
| ΔEelstat = E{A,B}elstat − EAelstat − EBelstat. | (5) |
With the knowledge of ΔEelstat it is possible to assess, whether an electrostatic repulsion between the (unperturbed) charge densities of consecutive layers is actually responsible for the common appearance of shifted (serrated) structures of 2D COFs. Another consequence of the overlap between the charge densities of the interacting sub-systems is Pauli repulsion, which is strongly repulsive. Additionally, the wavefunction overlap triggers orbital rehybridization, which lowers the energy of the entire system. This effect is, however, comparably small in stacked π-systems between which no interlayer bonds are formed.74 In fact, Pauli repulsion and orbital rehybridization are intimately related, with a sizable part of the stabilizing effect of orbital rehybridization arising from a reduction of Pauli repulsion, especially in the absence of covalent interactions. Thus, in the following both energy contributions will be combined into a single term, ΔEorb,Pauli, which can be calculated from the overall interaction energy via:
| ΔEint = ΔEelstat + ΔEorb,Pauli + ΔEvdW. |
3. Results and discussion
For bulk structures of planar 2D COFs, the two parameters characterizing the relative arrangement of the layers are (i) the interlayer stacking distance and (ii) the direction and magnitude of the shift between consecutive layers parallel to the plane of these layers. Both factors play a significant role in determining the actual properties of a COF. An advantage of computer simulations is that they allow varying both parameters independently. In particular, one can first address the question, how shifts between consecutive layers impact the energetic stability and the properties of 2D COFs for a fixed interlayer distance. In a second step one can then address the question to what extent the situation is altered when the interlayer distance is allowed to adapt for each shift. In the following, for both scenarios the focus will be on analyzing the impact of the stacking geometry on the interaction energy split into contributions from van der Waals attraction, Coulomb interactions, and the impact of orbital rehybridization and Pauli repulsion. As far as the impact of the relative arrangement of neighboring layers on COF properties are concerned, we will consider the electronic structure of the COF as well as its porosity. Based on the electronic structure we will be able to draw qualitative conclusions with respect to charge transport within the COFs and regarding their optical properties. For the Por-COFs considered in section 3.5 we will even find that interlayer shifts can cause a metal-to-semiconductor transition in analogy to previous findings for metal–organic frameworks.753.1. Constant interlayer stacking distance
Before displacing consecutive layers in specific directions, it is important to identify the directions in which minima of the potential energy surface are to be expected. The corresponding potential energy surface, PES, for the stacking distance fixed to the experimental literature value (3.328 Å) is shown in Fig. 2. This PES has been obtained employing Gaussian process regression, GPR, as described in the Methods section and in the ESI.† It shows the expected six-fold symmetry. Interestingly, the cofacial arrangement of successive layers (Δx = Δy = 0.00 Å) is particularly unstable. The global minima of the PES are found for shift directions parallel to the pore walls (see Fig. 1) at displacements of around 1.75 Å. Therefore, in the following analysis we will focus on shifts along this direction. For the sake of comparison, we also calculated the total energy for a shift perpendicular to the pore wall (see ESI†) with the obtained data fully supporting the outcomes of the GPR fit. | ||
| Fig. 2 Panel (a) shows the total energy as a function of the displacements of consecutive COF-1 layers, i.e. the potential energy surface. The x- and y-axes are aligned in the same way as in Fig. 1. The layers are displaced along directions parallel to the xy-plane. The obtained values of the total energy are specified per unit cell containing two COF layers and are reported relative to the energy of the global minimum structure. The hexagonal symmetry of the system is indicated by the white lines (see also Fig. 1). These white lines also indicate directions parallel to the pore walls. Panel (b) shows a zoom into the region of smaller displacements for one of the symmetry inequivalent sections. Panel (c) shows the total energy as a function of the azimuthal angle ϕ (measured relative to the x-axis) at a constant radius set to 1.75 Å, i.e. the value at which the minimum of the energy is observed in panels (a) and (b). | ||
The evolution of the interaction energy, ΔEint, for the shift along one of the pore walls is shown in Fig. 3a, with the employed shifts reaching up to the AB-type stacking distance (∼8.9 Å). This energy is plotted relative to the value of the cofacially aligned layers, which amounts to −1290 meV per unit cell (see caption of Fig. 3). In passing we note that the evolution of ΔEint exactly follows that of the total energy, which is a consequence of its definition in eqn (1) and the fact that the energies of the individual fragments A and B for a fixed interlayer distance are independent of the shift. Notably, the value of ΔEint is highest (least negative) for the cofacial arrangement, which confirms the notion from the GPR data that this is a particularly unstable structure. Likewise, at a displacement of around 1.75 Å the interaction energy displays a pronounced minimum and rises again for larger displacements. This behavior is consistent with the observations reported in literature,37,38 although in our investigations the minimum occurs at slightly larger displacements. To understand the origin of that trend, the interaction energy is decomposed into contributions from van der Waals interactions, ΔEvdW, electrostatic interactions, ΔEelstat, and interactions due to Pauli repulsion and orbital rehybridization, ΔEPauli,orb, where all energies in Fig. 3a are plotted relative to their values for zero displacements (listed in the figure caption).
 | ||
| Fig. 3 Relative energies of COF-1 displaced parallel to a pore wall at a constant interlayer distance. (a) Comparison of interaction energy, vdW energy, electrostatic energy, and Pauli repulsion plus orbital rehybridization energy for the displacements. (b) Corresponding width of the valence band along a k-path parallel to the stacking direction for COF-1 layers as a function of the displacement. The bandwidth was determined as the difference between minimum and maximum of the respective electronic band. This evaluation of the bandwidth better mimics the more complex character of the valence band at larger displacements (>2.1 Å) than considering only the energies at high symmetry points. Energy values at 0.0 Å displacement: ΔEint = −1290 meV, ΔEint,elec = 2257 meV, ΔEvdW = −3547 meV, ΔEelectrostatic = −1344 meV, ΔEPauli,orb = 3601 meV, Etotal = −70442.671 eV. All energies are specified per unit cell containing two COF layers. The dash-dotted line indicates the shift at which an AB-type arrangement of the COF layers is reached. | ||
ΔEvdW is most attractive for the cofacial arrangement (−3547 meV per unit cell), and then increases (i.e., becomes less negative) upon displacing the layers. This behavior is not unexpected, as by displacing the layers parallel to a pore wall, parts of the layers are moved over open pore space. This increases the average interatomic distances and causes a drop in vdW attraction. This effect becomes particularly pronounced for displacements above ∼1.5 Å. Important to add is that we do not observe a dependence of this effect on the used van der Waals correction scheme, as is shown in the ESI† for MBD energy corrections instead of the otherwise employed TS scheme (see Methods section).
A similar trend compared to ΔEvdW is observed for the electrostatic energy shown by the blue open triangles in Fig. 3a, where it has to be stressed that also ΔEelstat remains negative (i.e., attractive) for all displacements with ΔEelstat amounting to −1344 meV for the cofacial case. As far as the evolution of ΔEelstat is concerned, it remains essentially constant for displacements up to around 2 Å and then becomes less negative (i.e., less attractive) for larger displacements. This means that the electrostatic attraction would be strongest in the cofacial case, and, thus, cannot be responsible for the serrated structure of COF-1, as hypothesized in ref. 36 and 38. This raises the question as to the origin of the overall attractive nature of electrostatic interactions in COF-1 despite the polarization of the heteroatoms in the boroxine linkage groups. In fact, the attraction can be attributed to so-called charge penetration effects, which have been described frequently for interacting organic molecules76–78 and which originate from the interpenetration of the charge clouds of non-bonded chemical entities. A more detailed discussion of charge penetration effects will be provided in section 3.4 below.
In contrast to the van der Waals and electrostatic interactions, the combined energy contribution due to Pauli repulsion and orbital rehybridization is always repulsive with a maximum of 3601 meV for the cofacial arrangement. This significantly destabilizes that structure. Another fundamental difference between ΔEorb,Pauli and the other energy contributions is that ΔEorb,Pauli changes significantly already for small displacements and then levels off for larger displacements. This is the primary reason, why the sum of ΔEvdW, ΔEelstat, and ΔEorb,Pauli, first drops with the displacement, then forms a minimum at 1.75 Å, and finally rises again for larger displacements. I.e., ΔEorb,Pauli is the factor that is actually responsible for the fact that energy minimum corresponds to a shifted rather than a cofacial arrangement of successive layers.
3.2. Band widths and Pauli repulsion
The crucial role of ΔEorb,Pauli raises the question, why Pauli repulsion is so large for a cofacial structure. To understand that, one has to keep in mind that when occupied orbitals of two molecules overlap, bonding and antibonding linear combinations are formed, where the bonding one is stabilized less than the antibonding one is destabilized. As the energies of the occupied bands (orbitals) enter into the expression of the total energy, wavefunction overlap involving fully occupied orbitals results in a repulsive contribution. This effect is particularly pronounced for large energetic splittings between the bonding and antibonding states and, correspondingly, for strong electronic couplings and large bandwidths. As a consequence of orbital symmetries, this is typically the case for cofacial arrangements of π-conjugated systems. A maximum bandwidth for a vanishing displacement is, indeed, observed also here, as is shown for the case of the valence band in Fig. 3b.When the layers are shifted relative to each other, the bandwidth decreases, essentially vanishes around a displacement of 2.5 Å and then slightly increases again, a trend that has been intensively discussed for organic semiconductors.63,74,79–83 The shift at which the bandwidth reaches its minimum value depends on the symmetry and nodal structure of the lattice periodic functions in the Bloch states constituting the different bands. Independent of that, bandwidths and, thus, Pauli repulsion are expected to be maximized for zero-displacement for all occupied bands. Additionally, both quantities should drop for small displacements, while the shift at which the minimum is reached is band-dependent. Therefore, one cannot expect a one-to-one correlation between the valence bandwidth and Pauli repulsion. This is discussed in more detail for quinacridone and pentacene in ref. 74. Consistently, as can be seen in Fig. 3, also in COF-1 the bandwidth and total energy adopt maximum values in the cofacial case, and both values drop at very small displacement, while there is no one-to-one correlation at larger displacements.
In the case of porous materials, there is an additional aspect which goes beyond what is observed in organic semiconductors. Its origin is sketched in Fig. 4, where one can see that different types of phenylene stacks, are affected differently by the displacement. Phenylene rings in the equivalent stacks 2 and 3 are shifted towards open pore space, such that the associated Pauli repulsion has to drop due to its dependence on wavefunction overlap. This is the primary reason, why ΔEPauli,orb drops over the entire range of displacements shown in Fig. 3a. Conversely, the phenylene units in stack 1 experience the more “traditional” situation that a shift reduces the overlap with one entity, but increase that with another (here the boroxines). This is also the situation typically observed in organic semiconductors and relates to the discussion in the previous paragraph.
 | ||
| Fig. 4 Illustration of the consequences of a shift between consecutive layers for a section of COF-1. The plot shows, how different types of phenylene stacks (denoted as stack 1, 2, and 3) are differently affected by the displacement. Here, the lattice vectors are omitted for clarity. Note that stack 2 and 3 are equivalent. Thus, both are affected equivalently by shifts along the indicated shift direction. | ||
To more directly illustrate the stack-dependent impact of the slip on the electronic structure of COF-1, it is useful to consider the associated band structures.32 From these band structures we then can derive charge transport relevant quantities like effective masses and bandwidths. First, we analyze the corresponding band structures shown in Fig. 5 in approximately the energy range spanned by the valence band. For the cofacial arrangement displayed in Fig. 5a one observes a three-fold degenerate valence band, which is essentially flat for k-vectors within the plane of the layers (with several additional bands between 0.0 and 0.35 eV). Such flat bands are actually expected for systems with three-arm cores and three-fold symmetry.84 The situation changes significantly in the ΓA direction, where the effective bandwidth of the valence band amounts to ∼1.8 eV, when considering the backfolding of the band due to the two layers in the unit cell. The densities of states projected onto the individual sub-units of the COF, namely the phenylene stacks 1, 2, and 3, and the boroxine stack (the PDOSs shown in Fig. 5c). They illustrate that for zero displacement the degenerate valence bands are dominated by equal contributions of states localized on the three phenylene linkers. The boroxine-related states are found at slightly more negative energies.
 | ||
| Fig. 5 Electronic band structure ((a) and (b)), projected density of states ((c) and (d)), and eigenstate density ((e)) of COF-1 for the cofacial arrangement ((a) and (c)) and for the minimum energy, serrated structure ((b), (d), and (e)) with successive layers shift by 1.75 Å along the direction of a pore walls. All quantities have been calculated for a structure with the interlayer distance fixed to 3.328 Å. The densities of states are projected onto the phenylene stacks 1, 2, and 3 (grey, purple, dark yellow, see Fig. 4) and the boroxine-based stack (red). The energy range in the PDOS plots is reduced compared to the band-structure plots and a gaussian broadening of 0.02 eV is employed. The dashed green line in panel (b) is the result of a fit of a simple 1D tight-binding model and serves to illustrate the structure of the valence band. In (e) an isodensity plot of the electron density associated with the valence band state at the Γ-point is shown. Color code of the atoms: C … grey, H … white, B … green, O … red. | ||
For the minimum energy conformation (with a displacement of 1.75 Å along the pore wall), the situation changes fundamentally (see Fig. 5b and d): the degeneracy of the valence bands is lifted and the topmost band is derived only from phenylene stack 1. This is apparent from the plot of the PDOSs in Fig. 5d and from the electron density corresponding to the highest occupied eigenstate in Fig. 5e. The width of that band is reduced to ∼1 eV. The band-width reduction is even more pronounced for most of the lower-lying bands associated with stacks 2 and 3 (consistent with the diminishing overlap of the respective phenylene units).
These changes in the band structure have a distinct impact on charge-transport related properties of the serrated system: the interlayer electronic coupling is reduced in line with the bandwidth by a factor of ∼1.8., which is detrimental for charge transport. The correspondence of the two quantities is a consequence of the coupling being directly proportional to the bandwidth for simple, cosine-shaped bands like the valence band of COF-1 (as can be inferred, for example, from a tight-binding description; see also dashed green line in Fig. 5b). Additionally, the effective mass for transport in π-stacking direction more than doubles from 0.61m0 in the cofacial case to 1.29m0 for the minimum energy structure (with m0 corresponding to the free electron mass). On more technical grounds, we note that the above trends prevail, when employing the hybrid HSE0661,62 functional instead of the PBE55,56 functional, as shown in the ESI.† Eventually, this means that the energetically favorable serrated packing motif corresponds to poorer charge transport properties. Furthermore, when analyzing the band gaps at the HSE06 level, we find that they are severely altered by changes in the stacking motif, varying from 2.2 eV at the cofacial arrangement to 3.7 eV at the serrated one.
3.3. Optimized interlayer stacking distance
While the above considerations provide relevant fundamental insights concerning the consequences of a displacement of neighboring 2D COF layers, they disregard the fact that an increased interlayer repulsion/attraction triggers an increase/decrease of the interlayer stacking distance. Therefore, as a next step, we studied, how the situation changes when the stacking distance between consecutive COF layers is optimized for each displacement. As this optimization is computationally rather demanding (see Methods section and ESI†), we here focus on shifts close to the energetic minimum determined above for a constant interlayer distance. In the ESI,† we additionally consider the AB arrangement and a recently observed ABC type stacking.51 These arrangements are significantly higher in energy than the favorable serrated stacking motif. In passing we note that now the evolution of the total energy no longer overlaps directly with that of the interaction energy, as due to the varying interlayer distance the energies of the sub-systems A and B slightly change as a function of the shift. Still, the evolutions of the total and the interaction energies run essentially in parallel, as shown in the ESI.†The data on the interaction energy in Fig. 6 reveal that the energy minimum for shifts parallel to the pore wall is still found around 1.75 Å, just like for the constant interlayer stacking distance. This can be rationalized by the observation that the calculated optimum interlayer distance (3.350 Å) considered in Fig. 6a is very close to the experimental value (3.328 Å) used for obtaining the trends in Fig. 3a (see also dash-dotted line in Fig. 6b). This is insofar rather surprising, as now the evolutions of all contributions to the interaction energy with displacement are fundamentally different from the situation discussed above. This goes far beyond a mere reduction of the magnitudes of the variations, which one would expect because of the additional “degree of flexibility” of the system.
 | ||
| Fig. 6 Relative energies, stacking distance, and valence bandwidth of COF-1 displaced parallel to one of the pore walls. Here, the stacking distance has been optimized for each displacement. (a) Comparison of interaction energy, vdW energy, electrostatic energy, and Pauli plus orbital rehybridization energy; (b) optimized stacking distance between consecutive COF-1 layers; (c) width of the valence band along a k-vector parallel to the stacking direction of COF-1 layers. The bandwidth was determined as the difference between minimum and maximum of the respective electronic band. Energy values at 0.0 Å displacement: ΔEint = −1957 meV, ΔEint,elec = 812 meV, ΔEvdW = −2769 meV, ΔEelstat = −385 meV, ΔEPauli,orb = 1197 meV, Etotal = −70443.275 eV. All energies are specified per unit cell containing two COF layers. | ||
Now, the van der Waals attraction no longer decreases continuously with displacement as in Fig. 3. Rather, the vdW energy becomes more negative up to a displacement of 1.75 Å and rises only afterwards. One can rationalize this behavior by a pronounced decrease of the interlayer stacking distance for small displacements relative to the cofacial case (Fig. 6b). This overcompensates the consequences of the decreased lateral overlap of the layers, as can be quantified by weighted distance histograms, which are contained in the ESI.† This overcompensation vanishes for displacements beyond 1.75 Å, where the change in stacking distance is less pronounced and where also larger portions of the COF layers come to lie above the (empty) pores of neighboring layers. In passing we note that, the decrease in interlayer distance with displacement roughly follows the evolution of ΔEPauli,orb in Fig. 3a. This is, in fact, sensible, considering that (i) Pauli repulsion is by far the largest contribution in Fig. 3a and that (ii) it depends on wavefunction overlap and should, thus, display a particularly pronounced distance dependence.
In line with this reasoning, the decrease in interlayer distance with layer displacement essentially inverts the situation for Pauli repulsion: ΔEPauli,orb remains strongly positive (i.e., repulsive) for all distances, but now, the decrease in wavefunction overlap due to the shape of the orbitals (vide supra) for shifted systems is overcompensated by an increase of the repulsion due to the larger wavefunction overlap at smaller inter-layer distances. Only for larger displacements ΔEPauli,orb drops again because of an increasing fraction of the COF coming to lie above the pores of neighboring layers. The consequences of the above-described compensation effects are visible also in the evolution of the valence bandwidth (see Fig. 6c), which now displays a pronounced decrease only for shifts beyond the equilibrium displacement of 1.75 Å. Therefore, the cofacial arrangement also loses part of its advantage over the energetic minimum structure as far as bandwidths (and the resulting interlayer electronic coupling) and effective masses are concerned. In fact, the bandwidth only decreases from 1.15 eV to 1.00 eV. For the effective mass, the effect is still a bit larger with an increase from 0.85m0 in the cofacial case to 1.31m0 for a layer displacement of 1.75 Å parallel to the pore wall.
Interestingly, also the evolution of the (attractive) electrostatic interaction is largely inverted compared to the situation for fixed interlayer distances and the magnitude of the changes is significantly increased. For small displacements, this evolution is again caused by the significantly decreasing interlayer distances. This amplifies the attractive charge-penetration effects for shifted layers. Only at larger displacements the diminishing overlap of significant parts of the COF layers becomes again the dominant factor and results in a reduction of the electrostatic interaction. As a consequence, when allowing the interlayer distance to relax for displaced structures, electrostatic interactions do favor the serrated configuration. This is, however, again not caused by a “conventional” electrostatic repulsion due to a high octupole moment of the B3O3 rings, as suggested by chemical intuition. Rather, the evolution is primarily a consequence of the changing inter-layer distance with small distances resulting in an increased electrostatic attraction. This calls for a more in-depth investigation of the role played by the B3O3 ring, which will be provided in the next section.
3.4. Attractive electrostatic energy and charge penetration effect
In all situations encountered so far, the electrostatic interactions between the layers were attractive (i.e., the electrostatic energy was negative). As mentioned before, this is commonly attributed to charge penetration, an effect that has been discussed extensively for interacting organic materials and organic semiconductor crystals.76–78 Conceptually, this effect describes that due to the interpenetration of charge/electron clouds the shielding of the positively charged nuclei is reduced and the attractive electron–nuclei interaction increases.As indicated above, for systems like COF-1 one would still expect that the significantly different electronegativities of the B and O atoms in the central B3O3 rings would result in sizable octupole moments resulting in a repulsion of cofacial B3O3 rings. To disentangle the roles of the phenylenes and the boroxine-based linkages, we split the individual COF-1 layers into two model systems consisting either of benzene (C6H6) or boroxine (B3O3H3) rings (i.e., into an only weakly polar and a highly polar unit; see insets in Fig. 7a). The dangling bonds of the model systems were saturated by H atoms and the rings were arranged at exactly the same positions they adopt in COF-1 with optimized interlayer distances. Then, ΔEelstat was calculated separately for each model system as a function of the shift of consecutive layers. The resulting energy evolution is shown in Fig. 7a for constant interlayer stacking distances and in Fig. 7c for optimized distances. For both cases, when considering only the benzene molecules, ΔEelstat is indeed primarily determined by charge penetration effects and is clearly attractive for all considered situations in analogy to the situation observed above for the full COF. Interestingly, when considering only the boroxine units, for small displacements the electrostatic interactions become repulsive, in line with the sizable octupole moments of the molecules. This is insofar relevant, as the electrostatic interaction between the layers in COF-1, in a first approximation, can be regarded as a superposition of the (largely independent) interactions of the phenylene subsystem and the boroxine subsystems, as shown in Fig. 7b and d (again for constant as well as for optimized interlayer stacking distances). This implies that there is, indeed, electrostatic repulsion between the boroxines at small displacements also in the actual COF-1. This repulsion is, however, overcompensated by the attractive interactions of the phenylenes, resulting in the final trends observed in Fig. 3 and 6. This overcompensation is clearly visible for both cases, comprising constant and optimized stacking distances. Interestingly, for constant interlayer stacking distance, this overcompensation is even strong enough that the cofacial arrangement of neighboring sheets, which is electrostatically unfavorable when considering only the boroxine units, becomes favorable for the full COF-1 layers.
 | ||
| Fig. 7 Panels (a) and (c) show the electrostatic energy contributions of the COF-1 sub-systems (boroxine unit and benzene) extracted from COF-1 with constant (a) and optimized (c) interlayer stacking distances. The nature of the sub-systems is indicated by the insets in panel (c). In panels (b) and (d) the sum of the electrostatic energies stemming from the two sub-systems is shown by the purple line and symbols and the electrostatic energy of COF-1 is given in blue. Panel (b) shows the results for constant and panel (d) for optimized interlayer stacking distances. Note that in contrast to Fig. 3 and 6, the electrostatic energies are not given relative to the situation for zero displacement. Rather, their absolute values are plotted, as, when separately considering the boroxines, the electrostatic energy changes sign as a function of the displacement. All energies are specified per unit cell containing two COF layers. | ||
3.5. Additional layered COFs
In the following, we will briefly address to what extent the above observations are specific to COF-1, or whether they are of more general nature. Therefore, we considered additional 2D COFs with π-systems of different size and nature and also with different pore topologies. These systems comprise COF-5, as a more extended analogue to COF-1, and the porphyrin- and hexabenzocoronene-based COFs Por-COF,52 and HBC-COF.40 Their structures are shown in Fig. 8. For Por-COF we consider two versions of that system, one consisting of Zn-metallated porphyrin (Zn) and one without a metal incorporated in the center of the molecule (NH). | ||
| Fig. 8 Optimized structures of the considered COFs: COF-1, COF-5, HBC-COF, NH-Por, Zn-Por. For each COF the respective unit cell containing two original layers is shown. The atoms of the top layer are colored according to their chemical nature (C … grey, H … white, B … green, O … red, N … blue, Zn … violet). The atoms in the displaced bottom layer are all plotted in dark green. The layer displacements of all COFs are indicated by purple arrows. Interestingly, the resulting packing motif of the aromatic systems of COF-1, COF-5, and HBC-COF, resemble that of graphite.37 | ||
When performing a full geometry optimization (for details see Methods section), all of these COFs adopt a serrated structure (see Fig. 8). This is consistent with the expectation from literature that apart from a few exceptions such shifted AA-stackings are the preferred stacking motif.37 The obtained lateral displacement vectors, Δvxy, between consecutive layers are reported in Table 1 and indicated by purple arrows in Fig. 8. Considering the absolute values of these vectors, we find that they are of similar magnitude for all COFs. Furthermore, from the interlayer stacking distances reported in Table 1 one can see that the energetically favorable serrated layer arrangements exhibit significantly smaller stacking distances compared to the cofacial arrangement.
| COF-1 | COF-5 | HBC-COF | NH-Por | Zn-Por | ||
|---|---|---|---|---|---|---|
| Δvxy/Å | (1.50, −0.86) | (1.49, −0.46) | (0.00, 1.60) | (1.18, 1.18) | (1.22, 1.22) | |
| z/Å | Cofacial | 3.62 | 3.59 | 3.62 | 3.54 | 3.56 |
| Optimal | 3.36 | 3.39 | 3.43 | 3.33 | 3.32 |
To understand, whether similar driving forces as in COF-1 are responsible for the serrated AA-stacking, Fig. 9 compares the changes in interaction, van der Waals, electrostatic and Pauli repulsion plus orbital rehybridization energies between cofacial structures with optimized interlayer distances and fully optimized structures for all considered COFs. The absolute values of the energies are contained in the ESI.† All COFs display the same behavior that has been discussed in section 3.3 for COF-1: the displaced structure is stabilized by an increased van der Waals and Coulomb interaction while the Pauli repulsion increases consistent with the observation in Fig. 6. Considering that also the relative magnitudes of the individual contributions are reasonably similar in all systems, one can confidently attribute these changes to a superposition of the effects due to the displacement and the concomitantly decreased interlayer stacking distance in the serrated structure (see Table 2) in analogy to the discussion in section 3.3. This is shown explicitly for COF-5 in the ESI,† where equivalent trends as in Fig. 6 are shown for the interaction energy and its individual contributions (van der Waals, Coulomb, and Pauli repulsion plus orbital rehybridization).
 | ||
| Fig. 9 Energy differences between the cofacial and the optimized structures of the COF-1, COF-5, HBC-COF, NH-Por, Zn-Por. Differences in the total interaction energy are compared to the contributions, van der Waals, electrostatic, and Pauli repulsion plus orbital rehybridization interactions. Minor differences in the values for COF-1 in Fig. 9 and 6 are due to the fact that for Fig. 6 we relied on the experimental lateral unit-cell parameters, while here, for the sake of comparability with the other COFs, all unit-cell parameters were optimized. | ||
| ΔEtotal/eV | ΔEint/eV | ΔEvdw/eV | ΔEelstat/eV | ΔEPauli,orb/eV | |
|---|---|---|---|---|---|
| Planarized | −3.89 | −4.78 | −1.74 | 2.63 | |
| Optimal | −5.73 | −7.09 | −2.26 | 3.61 | |
| Difference | −1.87 | −1.85 | −2.31 | −0.52 | 0.98 |
Another interesting observation can be made, when considering the hexabenzocoronene core of HBC-COF: there, the stacking motif of the C atoms is strongly reminiscent of the most common form of graphite (CCDC 918549).85 To find out, whether the stacking in this material is governed by similar driving forces as in the COFs considered here, we applied our decomposition scheme also to periodically stacked graphene flakes. Indeed, the obtained situation is fully analogous to all the considered COFs. I.e., Pauli repulsion acts as the driving force pushing the layers/flakes towards a serrated stacking motif (for details see ESI†). In passing we note that graphene flakes instead of graphite have been considered for this comparison to avoid a semi-metallic character of the individual fragments (i.e. graphene), for which the performance of the employed decomposition scheme is not well tested.
For COF-5 and HBC-COF the influence of the structural changes on the band gaps and the effective masses is very similar to that discussed for COF-1. For COF-5 (HBC-COF), the energetically favorable slip leads to an increase of the effective masses for holes from 1.14m0 (1.18m0) at the cofacial arrangement to 1.70m0 (2.82m0) at the serrated arrangement. Additionally, the band gap is increased due to the slip from 1.37 eV (0.58 eV) to 1.98 eV (1.13 eV) when using the PBE functional. As generalized gradient based functionals massively underestimate band gaps, we also calculated the gap for COF-5 using HSE06, for which a similar gap increase, albeit from 2.06 eV to 2.70 eV was observed (HBC-COF turned out to be too complex for HSE06 calculations; for band structures see ESI†). For the Por-COFs, the induced change in the electronic structure is even qualitative, as for the cofacial arrangement both systems have a vanishing band gap showing metallic character, which changes for the energetically favorable serrated arrangement, for which an indirect gap of ∼0.5 eV (at the HSE06 level) opens up. Consistent with our findings, such metal-to-semiconductor transitions as a function of interlayer shifts have recently also been discussed for metal–organic frameworks.75
As additional quantities, we also studied the impact of the serrated arrangement of consecutive layers on the porosity and available surface area of the systems shown in Fig. 8. Here the impact of shifts of consecutive COF layers, however, turns out to be only minor with changes of only ∼3% in the accessible surface areas per volume (as shown in detail in the ESI†).
3.6. Impact of the non-planarity of individual COF layers: the example of non-planar COF-366
All above findings were obtained for planar or almost planar (HBC-COF) systems. This raises the question, how the situation changes for 2D COFs, which are non-planar. Therefore, we considered the example of COF-366.35 The structures of this system were calculated analogously to HBC-COF considered in section 3.5, with the addition that for the cofacial arrangement the porphyrins of consecutive layers were forced to be coplanar, while the stacking distance was optimized as outlined in the ESI.† This constraint was removed when performing the geometry relaxation optimizing also the stacking distance, which then yields the optimal structure. The resulting structures are shown in Fig. 10 a and b. Decomposing the interaction energy of these two systems into the individual contributions (see Table 2) reveals a behavior consistent with the above findings. | ||
| Fig. 10 Geometries of COF-366 with (a) planarized porphyrin cores and when fully relaxing all atomic positions plus the stacking distance (b). The linkers in different spatial directions are colored in either purple or green for better visibility. In panel (b) the twists around the axes of the linkers are indicated. | ||
One aspect is different though: instead of a displacement of consecutive COF layers, the shift of consecutive porphyrin units to minimize the total energy is achieved by a rotation around the linking groups and a reduction of the stacking distance from 4.31 Å at cofacial to 3.86 Å at the optimal arrangement. So, while the shift of the entire layer was hindered by steric interactions between the linking groups of consecutive layers, the system exploited a different degree of flexibility in order to minimize the energy and to avoid a cofacial arrangement. This being said, COF-366 is, of course, only one possible example of how steric interactions can be used to modify the stacking of COFs and one cannot exclude that for other structural motifs steric effects could be used to enforce a cofacial arrangement of π-backbones with the resulting favorable properties.
4. Conclusions
Based on the prototypical example of COF-1, we identified the driving forces that result in shifted (serrated) AA-stacking arrangements to be energetically favorable in planar 2D COFs. A quantitative assessment of the individual interactions determining the evolution of the total energy (dispersion, electrostatic, Pauli repulsion plus orbital rehybridization) is provided employing dispersion-corrected density-functional theory. For both, fixed and variable interlayer distances, the actual equilibrium structure is a consequence of a subtle interplay between van der Waals and Coulomb attraction as well as Pauli repulsion. How these different interactions play out is, however, fundamentally different in the two cases: in the fixed-distance case, van der Waals and Coulomb attractions favor a cofacial arrangement as a displacement of consecutive layers increases the net inter-atomic distances and, thus, diminishes the attraction due to dispersion forces and charge penetration effects. Conversely, Pauli repulsion favors displaced structures, which can be correlated at least in part with a decrease of the width of the valence band (especially at small displacements). This also results in clearly deteriorated transport parameters for the minimum structure with a decrease of the electronic coupling and an increase of the effective mass of the holes in the valence band by a factor of roughly two.The role of the different contributions is essentially inverted when optimizing the interlayer distance at each displacement. The reason for that is that the effects resulting from a decreased geometrical overlap in the displaced structures are compensated by the impact of the concomitantly observed decreased interlayer distance. Considering the fundamentally different origins of Coulomb, Pauli, and van der Waals interactions, these compensation effects play out differently. Nevertheless, the optimum displacement is essentially the same for both situations with a shift of 1.75 Å parallel to one of the pore walls.
The above results, at first glance, suggest that “conventional” electrostatic repulsion between the highly polarized boroxine units, which has previously been held responsible for the serrated structure of COF-1,36,38 does not play a role. An in-depth analysis of the contribution of the boroxine fragments, however, reveals that they do cause electrostatic repulsion for a cofacial arrangement of the layers, in line with chemical intuition. Still, in the actual COF, this repulsion is overcompensated by the attractive interaction of the phenylenes due to charge penetration.
A similar behavior as in COF-1 is found for a series of additional COFs with differently sized π-systems and differently shaped pores. The serrated equilibrium arrangement of the considered COFs increases the effective masses of the holes and in some cases even results in a transition from a metallic (cofacial case) to a semiconducting (serrated case) character of the material. Conversely, parameters related to the porosity of the materials are hardly affected by the slip of consecutive layers.
Overall, our findings imply that the structure of layered COFs is to a significant extent determined by effects that are of quantum-mechanical nature (like Pauli repulsion) and that depend on the symmetry and nodal structure of extended electronic states. As a consequence, approaches relying, for example, on classical force fields should not be able to provide a qualitatively correct description of the relevant physics. Even more importantly, our findings suggest that for obtaining layered COFs with cofacial stacking arrangements (as would be ideal for charge-transport applications), one cannot rely on the self-assembly of the individual layers. Also, merely reducing the classical electrostatic repulsion between layers by eliminating polar functionalities will not resolve the issue. Instead, one has to think of introducing additional driving forces in order to realize the desired stacking by tipping the otherwise unfavorable balance between van der Waals, Coulomb, and Pauli interactions.
Author contributions
E. Zojer and C. Winkler conceptualized the present study of the stacking arrangements of covalent organic frameworks and the decomposition into the determining interactions. C. Winkler performed all quantum-mechanical calculations and did a primary analysis of the conceived data. T. Kamencek analyzed the impact of the slip of consecutive COF layers on parameters associated with the porosity of the material. C. Winkler wrote a first version of the manuscript and prepared all figures. This version of the manuscript was revised by E. Zojer and C. Winkler. The entire project was supervised by E. Zojer.Funding sources
TU Graz Lead Project “Porous Materials at Work” (LP-03); DOC Fellowship of the Austrian Academy of Sciences (25783).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been financially supported by the TU Graz Lead Project “Porous Materials at Work” (LP-03). T. Kamencek acknowledges additional funding as recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Solid State Physics. The computational results have been achieved using the Vienna Scientific Cluster (VSC3).References
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- R. W. Tilford, W. R. Gemmill, H. C. Zur Loye and J. J. Lavigne, Chem. Mater., 2006, 18, 5296–5301 CrossRef CAS.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- A. P. Côté, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef PubMed.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., 2020, 59, 5050–5091 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- R. W. Tilford, S. J. Mugavero, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS PubMed.
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748–754 CrossRef CAS PubMed.
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28, 1277–1285 CrossRef CAS.
- H. L. Qian, C. X. Yang and X. P. Yan, Nat. Commun., 2016, 7, 12104 CrossRef CAS PubMed.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- H. Li, Q. Pan, Y. Ma, X. Guan, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 14783–14788 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- C. R. Deblase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed.
- A. K. Mandal, J. Mahmood and J. B. Baek, ChemNanoMat, 2017, 3, 373–391 CrossRef CAS.
- V. K. Yadav, S. H. Mir, V. Mishra, T. G. Gopakumar and J. K. Singh, Phys. Chem. Chem. Phys., 2020, 22, 21360–21368 RSC.
- H. V. Babu, M. G. M. Bai and M. Rajeswara Rao, ACS Appl. Mater. Interfaces, 2019, 11, 11029–11060 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- X. Ding, X. Feng, A. Saeki, S. Seki, A. Nagai and D. Jiang, Chem. Commun., 2012, 48, 8952–8954 RSC.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- A. Kuc, M. A. Springer, K. Batra, R. Juarez-Mosqueda, C. Wöll and T. Heine, Adv. Funct. Mater., 2020, 30, 1908004 CrossRef CAS.
- W. Zhou, M. Wei, X. Zhang, F. Xu and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 16847–16854 CrossRef CAS PubMed.
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- B. Lukose, A. Kuc, J. Frenzel and T. Heine, Beilstein J. Nanotechnol., 2010, 1, 60–70 CrossRef CAS PubMed.
- W. Zhou, H. Wu and T. Yildirim, Chem. Phys. Lett., 2010, 499, 103–107 CrossRef CAS.
- B. Lukose, A. Kuc and T. Heine, Chem. – Eur. J., 2011, 17, 2388–2392 CrossRef CAS PubMed.
- R. van der Jagt, A. Vasileiadis, H. Veldhuizen, P. Shao, X. Feng, S. Ganapathy, N. C. Habisreutinger, M. A. van der Veen, C. Wang, M. Wagemaker, S. van der Zwaag and A. Nagai, Chem. Mater., 2021, 33, 818–833 CrossRef CAS PubMed.
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed.
- X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef.
- A. M. Pütz, M. W. Terban, S. Bette, F. Haase, R. E. Dinnebier and B. V. Lotsch, Chem. Sci., 2020, 11, 12647–12654 RSC.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427 Search PubMed.
- T. Sun, C. E. Hughes, L. Guo, L. Wei, K. D. M. Harris, Y. B. Zhang and Y. Ma, Angew. Chem., 2020, 59, 22638–22644 CrossRef CAS PubMed.
- T. Sun, W. Lei, Y. Ma and Y. B. Zhang, Chin. J. Chem., 2020, 38, 1153–1166 CrossRef CAS.
- T. Sun, L. Wei, Y. Chen, Y. Ma and Y. B. Zhang, J. Am. Chem. Soc., 2019, 141, 10962–10966 CrossRef CAS PubMed.
- H. Sen Xu, Y. Luo, X. Li, P. Z. See, Z. Chen, T. Ma, L. Liang, K. Leng, I. Abdelwahab, L. Wang, R. Li, X. Shi, Y. Zhou, X. F. Lu, X. Zhao, C. Liu, J. Sun and K. P. Loh, Nat. Commun., 2020, 11, 1434 CrossRef PubMed.
- C. Gao, J. Li, S. Yin, G. Lin, T. Ma, Y. Meng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 9770–9775 CrossRef CAS PubMed.
- L. Zhang, Y. Zhou, M. Jia, Y. He, W. Hu, Q. Liu, J. Li, X. Xu, C. Wang, A. Carlsson, S. Lazar, A. Meingast, Y. Ma, J. Xu, W. Wen, Z. Liu, J. Cheng and H. Deng, Matter, 2020, 2, 1049–1063 CrossRef.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- S. Thomas, H. Li, R. R. Dasari, A. M. Evans, I. Castano, T. G. Allen, O. G. Reid, G. Rumbles, W. R. Dichtel, N. C. Gianneschi, S. R. Marder, V. Coropceanu and J. L. Brédas, Mater. Horiz., 2019, 6, 1868–1876 RSC.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- A. Tkatchenko, R. A. Distasio, R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 236402 CrossRef PubMed.
- A. Tkatchenko, A. Ambrosetti and R. A. Distasio, J. Chem. Phys., 2013, 138, 074106 CrossRef PubMed.
- A. Ambrosetti, A. M. Reilly, R. A. Distasio and A. Tkatchenko, J. Chem. Phys., 2014, 140, 18A508 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- L. Zhu, Y. Yi, Y. Li, E. G. Kim, V. Coropceanu and J. L. Brédas, J. Am. Chem. Soc., 2012, 134, 2340–2347 CrossRef CAS PubMed.
- P. Fabian, M. Vincent, G. Olivier, B. Mathieu, P. Peter, W. Ron, V. Jake and D. Cournapeau, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- M. Pinheiro, R. L. Martin, C. H. Rycroft and M. Haranczyk, CrystEngComm, 2013, 15, 7531–7538 RSC.
- D. Ongari, P. G. Boyd, S. Barthel, M. Witman, M. Haranczyk and B. Smit, Langmuir, 2017, 33, 14529–14538 CrossRef CAS PubMed.
- Y. S. Bae, A. Ö. Yazayd'n and R. Q. Snurr, Langmuir, 2010, 26, 5475–5483 CrossRef CAS PubMed.
- M. Raupach and R. Tonner, J. Chem. Phys., 2015, 142, 194105 CrossRef PubMed.
- L. Pecher and R. Tonner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1401 Search PubMed.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 664 CrossRef.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- C. Winkler, A. Jeindl, F. Mayer, O. T. Hofmann, R. Tonner and E. Zojer, Chem. Mater., 2019, 31, 7054–7069 CrossRef CAS.
- M. E. Foster, K. Sohlberg, M. D. Allendorf and A. A. Talin, J. Phys. Chem. Lett., 2018, 9, 481–486 CrossRef CAS PubMed.
- S. M. Ryno, C. Risko and J. L. Brédas, Chem. Mater., 2016, 28, 3990–4000 CrossRef CAS.
- G. Gryn'ova and C. Corminboeuf, J. Phys. Chem. Lett., 2016, 7, 5198–5204 CrossRef PubMed.
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS PubMed.
- O. Kwon, V. Coropceanu, N. E. Gruhn, J. C. Durivage, J. G. Laquindanum, H. E. Katz, J. Cornil and J. L. Brédas, J. Chem. Phys., 2004, 120, 8186–8194 CrossRef CAS PubMed.
- V. Lemaur, D. A. Da Silva Filho, V. Coropceanu, M. Lehmann, Y. Geerts, J. Piris, M. G. Debije, A. M. Van De Craats, K. Senthilkumar, L. D. A. Siebbeles, J. M. Warman, J. L. Brédas and J. Cornil, J. Am. Chem. Soc., 2004, 126, 3271–3279 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- J. L. Bredas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef CAS.
- D. A. da Silva Filho, E.-G. Kim and J.-L. Brédas, Adv. Mater., 2005, 17, 1072–1076 CrossRef CAS.
- S. Thomas, H. Li, C. Zhong, M. Matsumoto, W. R. Dichtel and J. L. Bredas, Chem. Mater., 2019, 31, 3051–3065 CrossRef CAS.
- J. D. Bernal and W. L. Bragg, Proc. R. Soc. London, Ser. A, 1924, 106, 749–773 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr01047f |
| This journal is © The Royal Society of Chemistry 2021 |